

Abstract
Vanishing white matter disease (VWMD) is an inherited autosomal-recessive hypomyelinating disease caused by mutations in eukaryotic translation initiation factor 2B (eIF2B). eIF2B mutations predominantly affect the brain white matter, and the characteristic features of VWMD pathology include myelin loss and foamy oligodendrocytes. Activation of pancreatic endoplasmic reticulum kinase (PERK) has been observed in oligodendrocytes in VWMD. PERK activation in response to endoplasmic reticulum stress attenuates eIF2B activity by phosphorylating eIF2α, suggesting that impaired eIF2B activity in oligodendrocytes induced by VWMD mutations or PERK activation exploit similar mechanisms to promote selective white matter pathology in VWMD. Using transgenic mice that allow for temporally controlled activation of PERK specifically in oligodendrocytes, we discovered that strong PERK activation in oligodendrocytes during development suppressed eIF2B activity and reproduced the characteristic features of VWMD in mice, including hypomyelinating phenotype, foamy oligodendrocytes, and myelin loss. Notably, impaired eIF2B activity induced by PERK activation in oligodendrocytes of fully myelinated adult mice had minimal effects on morphology or function. Our observations point to a cell-autonomous role of impaired eIF2B activity in myelinating oligodendrocytes in the pathogenesis of VWMD.
Keywords: eIF2B mutations, hypomyelination, oligodendrocyte, pancreatic endoplasmic reticulum kinase, vanishing white matter disease
Introduction
Vanishing white matter disease (VWMD), also known as childhood ataxia with CNS hypomyelination (CACH), is an inherited autosomal-recessive hypomyelinating disease (Ainsworth, 2005; Scheper et al., 2006). The neurological signs of VWMD, including progressive cerebellar ataxia and spasticity, usually begin in late infancy or early childhood. The disease progresses rapidly and most patients die a few years after disease onset. VWMD is associated with cystic cavitations of the white matter and profound loss of myelin, whereas CNS gray matter and the other organs are generally spared (van der Knaap et al., 1997, 2006). Oligodendrocytes are predominantly affected in VWMD, exhibiting characteristic foamy morphology (Wong et al., 2000; Scheper et al., 2006). Oligodendrocyte numbers are reduced in the severely affected white matter, but are increased in the less affected areas.
VWMD is caused by mutations in any one of the ubiquitously expressed genes encoding the five subunits of eukaryotic translation initiation factor 2B (eIF2B; Leegwater et al., 2001; Scheper et al., 2006). eIF2B serves as an essential guanine nucleotide exchange factor required for the recycling of eIF2 with each round of translation initiation (Proud, 2005). VWMD mutations have been noted to reduce the guanine nucleotide exchange factor activity of eIF2B (Richardson et al., 2004; Scheper et al., 2006; Horzinski et al., 2009). Nevertheless, knock-in mice with VWMD mutations, although showing attenuated eIF2B activity in cells, do not exhibit myelin abnormalities under normal conditions (Geva et al., 2010). The role of attenuated eIF2B guanine nucleotide exchange activity in VWMD pathogenesis remains unknown and existence of a strict correlation between the two has even been challenged experimentally (Liu et al., 2011). The guanine nucleotide exchange activity of eIF2B is under strong negative regulation by the phosphorylation of the α subunit of its substrate, eIF2α (Pavitt and Ron, 2012). The latter, in turn, is effected by four known kinases, including pancreatic endoplasmic reticulum kinase (PERK), which couples endoplasmic reticulum (ER) stress to eIF2α phosphorylation (Harding et al., 1999). Interestingly, recent studies show that PERK signaling is enhanced in oligodendrocytes of VWMD patients (van der Voorn et al., 2005; van Kollenburg et al., 2006), suggesting that PERK activation synergizes with eIF2B mutations to inhibit protein synthesis in oligodendrocytes undermining cellular function and contributing to the pathogenesis of VWMD.
To explore this hypothesis experimentally, we have generated transgenic mice that allow for pharmacological temporally controlled activation of PERK with an otherwise inert compound (AP20187) specifically in oligodendrocytes, in the absence of any additional stress, and showed that AP20187 treatment enhances the activity of PERK signaling in oligodendrocytes of the transgenic mice in a dose-dependent manner (Lin et al., 2013). In this study, we exploit this mouse model to mimic the reduction of eIF2B activity to levels observed in oligodendrocytes of VWMD patients and measure the effects on CNS function and morphology, thus enabling a critical evaluation of the role of attenuated eIF2B activity in the pathogenesis of VWMD. Our findings indicate that selective impairment of eIF2B in oligodendrocytes during development can account for the features of VWMD pathology.
Materials and Methods
Mice.
Heterozygous PLP/Fv2E-PERK (PLP/Fv2E-PERK Tg/+) mice on the C57BL/6J background (Lin et al., 2013, 2014) were intercrossed to obtain homozygous PLP/Fv2E-PERK (PLP/Fv2E-PERK Tg/Tg) mice, and then PLP/Fv2E-PERK Tg/Tg mice were intercrossed to produce PLP/Fv2E-PERK Tg/Tg mice. To activate the Fv2E-PERK transgene in the oligodendrocytes of PLP/Fv2E-PERK Tg/Tg mice, the mice of either sex were given daily intraperitoneal injections of AP20187 (Ariad Pharmaceuticals); controls were injected with vehicle (4% ethanol, 10% PEG-400, 2.0% Tween 20 in water) only. All animal procedures were conducted in complete compliance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of South Alabama and the University of Minnesota.
Real-time PCR.
Deeply anesthetized mice were perfused with ice-cold PBS. RNA was isolated from the brain using Trizol reagent (Invitrogen) and treated with DNaseI (Invitrogen) to eliminate genomic DNA. Reverse transcription was performed using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories). TaqMan real-time PCR was performed with iQ Supermix (Bio-Rad Laboratories) on a Bio-Rad iQ Real-Time PCR detection system as previously described (Lin et al., 2013, 2014).
Western blot analysis.
Tissues harvested from mice were rinsed in ice-cold PBS and were homogenized using a motorized homogenizer as previously described (Lin et al., 2005, 2008, 2014). After incubating on ice for 15 min, the extracts were cleared by centrifugation at 14,000 rpm for 30 min twice. The protein content of each extract was determined by DC Protein Assay (Bio-Rad Laboratories). The extracts (40 μg) were separated by SDS-PAGE and transferred to nitrocellulose. The blots were incubated with a primary antibody against myelin basic protein (MBP; 1:1000, Covance, Catalog #SMI-99P-500; RRID: AB_10120130) or actin (1:5000, Sigma-Aldrich, Catalog #A2103; RRID:AB_476694), followed by an HRP-conjugated secondary antibody, and following incubation with the ECL Detection Reagents (GE Healthcare Biosciences), the chemiluminescent signal was detected.
Immunohistochemistry and TUNEL staining.
Anesthetized mice were perfused through the left cardiac ventricle with 4% paraformaldehyde in PBS. The tissues were removed, postfixed with paraformaldehyde, cryopreserved in 30% sucrose, embedded in optimal cutting temperature compound, and frozen on dry ice. Frozen sections were cut using a cryostat at a thickness of 10 μm. For immunohistochemistry, the sections were treated with −20°C acetone, blocked with PBS containing 10% goat serum and 0.1% Triton X-100, and incubated overnight with the primary antibody diluted in blocking solution. Fluorescein, Cy3, or enzyme-labeled secondary antibodies (Vector Laboratories) were used for detection. Immunohistochemical detection of CC1 (APC7, 1:50; EMD Biosciences, Catalog #OP80-100UG, RRID: AB_213434), phosphorylated eIF2α (p-eIF2α, 1:100; Cell Signaling Technology, Catalog #:9721L; RRID: AB_330952), MBP (1:1000; Covance), active-caspase-3 (1:100; Cell Signaling Technology, Catalog #9664L; RRID: AB_2070042); CAAT enhancer binding protein homologous protein (CHOP, 1:300; Ron and Habener, 1992), or aspartoacylase (ASPA; 1:1000, kindly provided by Dr M.A. Aryan Namboodiri at Uniformed Services University of the Health Sciences, Bethesda, MD; Madhavarao et al., 2004; Locatelli et al., 2012) was performed. Fluorescent-stained sections were mounted with Vectashield mounting medium with DAPI (Vector Laboratories) and visualized with a Nikon A1 confocal microscope or Olympus FV1000 confocal microscope.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining was performed using the ApopTag kit (EMD Millipore) according to the manufacturer's instructions. The sections were mounted with Vectashield mounting medium with DAPI (Vector Laboratories) and visualized with a Nikon A1 confocal microscope or Olympus FV1000 confocal microscope.
Toluidine blue staining and electron microscopy (EM) analysis.
Mice were anesthetized and perfused with PBS containing 4% paraformaldehyde and 2.5% glutaraldehyde. The sciatic nerve, the cervical spinal cord, the white matter of the cerebellum, and the corpus callosum were processed and embedded. Thin sections were cut, stained with toluidine blue, and analyzed as described previously (Lin et al., 2013, 2014). Moreover, ultrathin sections were cut, stained with uranyl acetate and lead citrate, and analyzed as described previously (Lin et al., 2013, 2014). We counted the total number of axons, calculated the total percentage of myelinated axons, and measured the diameter of axons and the thickness of myelin sheaths using NIH ImageJ software (http://rsb.info.nih.gov/ij/) as described previously (Lin and Lin, 2010; Lin et al., 2014). G-ratio was calculated as the diameter of axon divided by the diameter of axon and myelin.
Measurement of protein synthesis with SUnSET.
We used surface sensing of translation (SUnSET) to determine the rate of protein biosynthesis in oligodendrocytes of explanted cultures of optic nerve and in Schwann cells of explanted cultures of sciatic nerve (Schmidt et al., 2009; Goodman et al., 2011; D'Antonio et al., 2013). The optic nerves and sciatic nerves were dissected from 18-d-old and adult PLP/Fv2E-PERK Tg/Tg mice or 18-d-old PLP/Fv2E-PERK Tg/+ mice. They were placed in DMEM media supplemented with 10% fetal bovine serum for 30 min at 37°C, and then were treated with AP20187 (10 or 1 nm) or vehicle. After 30 min, 2 μm puromycin was added to the cultures, and the cultures were incubated for an additional 1 h. Eight optic nerves or sciatic nerves per each condition were rinsed in ice-cold PBS, pooled, and homogenized using a motorized homogenizer as previously described (Lin et al., 2005, 2008). Proteins were extracted, quantified, resolved by SDS-PAGE, and transferred to nitrocellulose. The blots were incubated with a primary antibody against puromycin (1:5000, EMD Millipore, Catalog #MABE343), followed by an HRP-conjugated secondary antibody, and following incubation with the ECL Detection Reagents (GE Healthcare Biosciences), the chemiluminescent signal was detected. The intensity of the recorded chemiluminescence signal (including the entire molecular weight range of puromyecin-labeled peptides) was quantified using NIH ImageJ software.
Statistics.
Data are expressed as mean ± SD. Multiple comparisons were statistically evaluated by a one-way ANOVA test using Sigmaplot 12 software (Systat Software). The relationship between myelin thickness and axon diameter was statistically evaluated by a linear regression using Sigmaplot 12 software. P values <0.05 were considered significant.
Results
Selective activation of PERK in oligodendrocytes of young myelinating mice leads to tremor and premature death
Fv2E-PERK is an artificial PERK derivative, generated by fusing the eIF2α kinase effector domain of PERK to a polypeptide containing tandem modified FK506 binding domains (Fv2E) and can be induced to oligomerize and activate by an otherwise inert ligand, AP20187 (Lu et al., 2004). We have generated PLP/Fv2E-PERK transgenic mice that express Fv2E-PERK under the control of the mouse proteolipid protein (PLP) transcriptional control region (Lin et al., 2013). These mice express the Fv2E-PERK transgene specifically in oligodendrocytes in the CNS. Treatment of PLP/Fv2E-PERK mice with Fv2E ligand AP20187 activates PERK signaling selectively in oligodendrocytes without causing ER stress. Moreover, we found that heterozygous PLP/Fv2E-PERK (PLP/Fv2E-PERK Tg/+) mice treated with a low dose of AP20187 (0.5–2 mg/kg) are phenotypically normal and do not display any abnormalities of oligodendrocytes or myelin in the CNS (Lin et al., 2013, 2014), indicating that moderate PERK activation specifically in oligodendrocytes does not affect the viability or function of the cells in young, developing mice or adult mice under normal conditions.
It is known that AP20187 can penetrate the blood–brain barrier (BBB); however, its BBB permeability is moderate (Burnett et al., 2004). To achieve strong PERK activation specifically in oligodendrocytes, PLP/Fv2E-PERK Tg/+ mice were intercrossed to obtain homozygous PLP/Fv2E-PERK (PLP/Fv2E-PERK Tg/Tg) mice, which were given intraperitoneal injections of a higher dose of AP20187 (5 mg/kg) daily. Robust myelination occurs in the mouse CNS between postnatal day (P)7 and P21 (Baumann and Pham-Dinh, 2001). Thus, PLP/Fv2E-PERK Tg/Tg mice were treated with either 5 mg/kg AP20187 or vehicle daily starting on P10. All the AP20187-treated PLP/Fv2E-PERK Tg/Tg mice exhibited a severe tremoring phenotype starting around P14, and died by P24 (Fig. 1A). In contrast, all PLP/Fv2E-PERK Tg/Tg mice treated with vehicle and age-matched wild-type mice treated with AP20187 (5 mg/kg) appeared healthy and survived. Moreover, we found that the effects of short-term AP20187 treatment on PLP/Fv2E-PERK Tg/Tg mice were reversible. PLP/Fv2E-PERK Tg/Tg mice that had been treated with 5 mg/kg AP20187 daily starting on P10 were released from the treatment on P15. All mice released from AP20187 treatment displayed a tremoring phenotype starting ∼P14, but gradually lost the tremoring phenotype starting ∼P24 and survived. Additionally, all PLP/Fv2E-PERK Tg/Tg mice treated with 5 mg/kg AP20187 daily starting on P7 died within 5 d. Nevertheless, PLP/Fv2E-PERK Tg/Tg mice treated with 5 mg/kg AP20187 daily starting on P28 did not display obvious tremoring phenotype, and survived for at least 2 weeks.
Figure 1.

Strong PERK activation specifically in oligodendrocytes during development reproduced VWMD clinical features in mice. A, Mouse survival curve; N = 8 animals. B, Real-time PCR analysis showed that AP20187 treatment significantly increased the expression of CHOP and GADD34 in the brain of 18-d-old PLP/Fv2E-PERK Tg/Tg mice, but did not alter the expression of immunoglobulin binding protein (BIP); N = 3 animals. C, D, CC1 and p-eIF2α double-labeling showed that p-eIF2α was undetectable in oligodendrocytes in the brain of 18-d-old PLP/Fv2E-PERK Tg/Tg mice treated with vehicle, but became detectable in oligodendrocytes (arrow) of PLP/Fv2E-PERK Tg/Tg mice treated with AP20187; N = 3 animals. E, F, CC1 and CHOP double-immunostaining showed that CHOP immunoreactivity was undetectable in oligodendrocytes in the brain of 18-d-old PLP/Fv2E-PERK Tg/Tg mice treated with vehicle, but became detectable in oligodendrocytes (arrow) of AP20187-treated mice; N = 3 animals. Error bars represent SD. *p < 0.05. Scale bars: C, E, 10 μm.
As expected, we found that PERK signaling was activated specifically in oligodendrocytes of PLP/Fv2E-PERK Tg/Tg mice treated with 5 mg/kg AP20187 daily starting on P10. Real-time PCR analysis showed that AP20187 treatment significantly increased the expression of PERK-responsive genes CHOP and growth arrest and DNA damage 34 (GADD34; Fig. 1B). CC1 and p-eIF2α double-immunostaining showed that AP20187 treatment increased the level of p-eIF2α in the majority of oligodendrocytes (77 ± 10% of CC1-positive cells were CC1 and p-eIF2α double-positive) in the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) of 18-d-old PLP/Fv2E-PERK Tg/Tg mice (Fig. 1C,D). Moreover, CC1 and CHOP double immunostaining showed that the immunoreactivity of CHOP was undetectable in oligodendrocytes in the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) of 18-d-old transgenic mice treated with vehicle, but became detectable in a number of oligodendrocytes (36 ± 12% of CC1-positive cells were CC1 and CHOP double-positive) of AP20187-treated mice (Fig. 1E,F).
Unfortunately, it is impossible to directly measure the guanine nucleotide exchange activity of eIF2B in oligodendrocytes in vivo. Thus to gauge the effect of Fv2E-PERK activation on eIF2B activity, we exploited a recently described procedure to measure rates of protein biosynthesis in explanted neural tissue by tracking the incorporation of puromycin into newly synthesized proteins (Schmidt et al., 2009; Goodman et al., 2011). Approximately 60% of glial cells in the optic nerve are oligodendrocytes (Dennis and Gerschenfeld, 1969; Vaughn, 1969). Therefore, we indirectly assessed the eIF2B activity in oligodendrocytes by measuring the rate of protein biosynthesis in the cells using explanted cultures of optic nerve (Omlin and Waldmeyer, 1989). Treatment with a high dose of AP20187 (10 nm, corresponding to 5 mg/kg AP20187 treatment in vivo) significantly suppressed protein biosynthesis in the optic nerve generated from 18-d-old PLP/Fv2E-PERK Tg/Tg mice (Fig. 2A,C). In contrast, treatment with a low dose of AP20187 (1 nm, corresponding to 0.5 mg/kg AP20187 treatment in vivo) slightly, but not significantly, decreased protein biosynthesis in the optic nerve generated from 18-d-old PLP/Fv2E-PERK Tg/+ mice (Fig. 2B,D).Thus, these data demonstrate that treatment with the high dose of AP20187 strongly activates the transgene product Fv2E-PERK and significantly inhibits eIF2B activity in oligodendrocytes of PLP/Fv2E-PERK Tg/Tg mice. Nevertheless, treatment with the low dose of AP20187 moderately activates Fv2E-PERK and slightly reduces eIF2B activity in oligodendrocytes of PLP/Fv2E-PERK Tg/+ mice. Collectively, these results suggest that impaired eIF2B activity by strong PERK activation specifically in oligodendrocytes during active phase of myelination leads to a severe tremoring phenotype and premature death of mice.
Figure 2.

Strong PERK activation suppressed protein synthesis in oligodendrocytes. A, C, SUnSET measurement of protein synthesis showed that treatment with 10 nm AP20187 significantly suppressed protein biosynthesis in the optic nerve generated from both 18-d-old and adult PLP/Fv2E-PERK Tg/Tg mice. B, D, SUnSET measurement of protein synthesis showed that treatment with 1 nm AP20187 slightly reduced protein biosynthesis in the optic nerve generated from 18-d-old PLP/Fv2E-PERK Tg/+ mice. The levels of puromycin-labeled peptides were reported relative to actin. The experiments were repeated at least three times. Error bars represent SD. *p < 0.05.
Impaired eIF2B activity specifically in oligodendrocytes during development reproduces VWMD pathology in mice
The tremoring phenotype was consistent with myelin abnormalities in the CNS of PLP/Fv2E-PERK Tg/Tg mice treated with 5 mg/kg AP20187 daily starting on P10. MBP immunostaining showed that AP20187 treatment noticeably reduced the degree of myelination in the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) of 18-d-old PLP/Fv2E-PERK Tg/Tg mice (Fig. 3A). Western blot analysis showed that the level of MBP was significantly decreased in the CNS of 18-d-old PLP/Fv2E-PERK Tg/Tg mice treated with AP20187 compared with vehicle-treated mice (Fig. 3B). Importantly, EM analysis showed that AP20187 treatment significantly reduced the percentage of myelinated axons in the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) of 20-d-old PLP/Fv2E-PERK Tg/Tg mice (Fig. 3C,D). AP20187 treatment also significantly decreased the thickness of the myelin sheaths and increased the G-ratio of myelinated axons in the CNS white matter of PLP/Fv2E-PERK Tg/Tg mice (Fig. 3C,E,F). Nevertheless, AP20187 treatment did not have noticeable effects on the diameter or morphology of axons in the CNS of PLP/Fv2E-PERK Tg/Tg mice. On the other hand, we found that treatment with 5 mg/kg AP20187 did not significantly affect the myelination process in the peripheral nervous system of PLP/Fv2E-PERK Tg/Tg mice (Fig. 3G). Although Schwann cells in the sciatic nerve of PLP/Fv2E-PERK Tg/Tg mice expressed the Fv2E-PERK transgene (data not shown), treatment with a high dose of AP20187 (10 nm) did not significantly alter protein biosynthesis in the cultured sciatic nerve generated from 18-d-old PLP/Fv2E-PERK Tg/Tg mice (data not shown). Thus, these results suggest that impaired eIF2B activity in myelinating oligodendrocytes of young, developing mice leads to hypomyelination in the CNS, resulting in the tremoring phenotype and premature death of mice.
Figure 3.
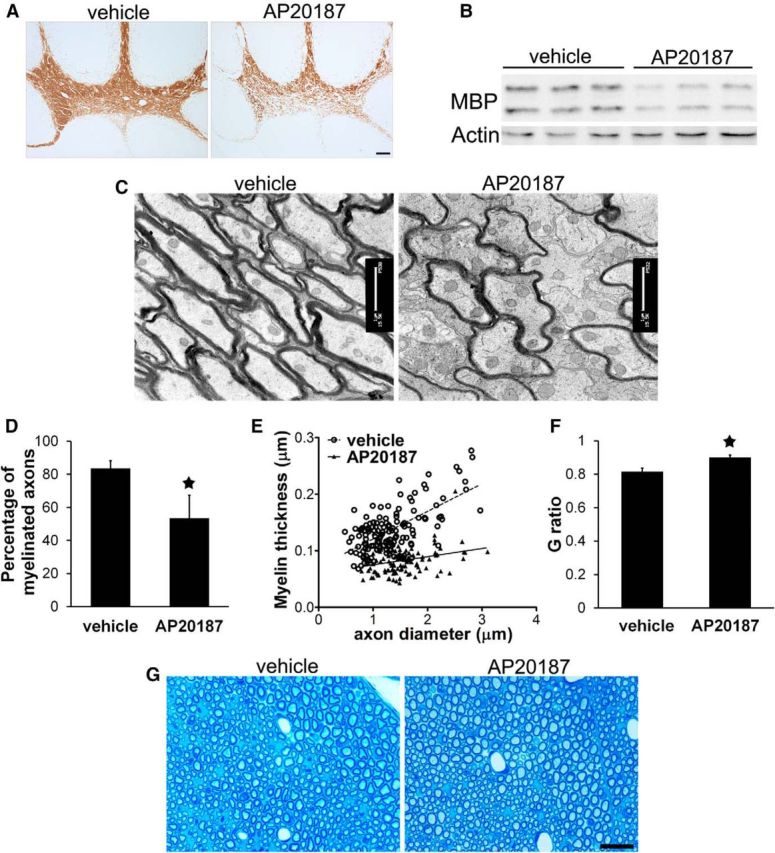
Impaired eIF2B activity specifically in oligodendrocytes during development led to hypomyelination in the CNS. A, MBP immunostaining revealed myelin loss in the cerebellum of 18-d-old PLP/Fv2E-PERK Tg/Tg mice treated with AP20187 compared with vehicle-treated mice. B, Western blot analysis showed that AP20187 treatment markedly reduced the level of MBP in the brain of 18-d-old PLP/Fv2E-PERK Tg/Tg mice. C, D, EM analysis showed that AP20187 treatment significantly decreased the number of myelinated axons in the brain of 20-d-old PLP/Fv2E-PERK Tg/Tg mice. E, Quantitative EM analysis showed that AP20187 treatment significantly reduced the thickness of the myelin sheaths in the brain of 20-d-old PLP/Fv2E-PERK Tg/Tg mice. F, Quantitative EM analysis showed that AP20187 treatment significantly increased the G-ratio of myelinated axons in the brain of 20-d-old PLP/Fv2E-PERK Tg/Tg mice. G, Toluidine blue staining showed that AP20187 treatment did not alter myelin integrity in the sciatic nerve of 20-d-old PLP/Fv2E-PERK Tg/Tg mice; N = 3 animals. Error bars represent SD. *p < 0.05. Scale bars: A, 100 μm; C, 1 μm; G, 20 μm.
Next, we determined whether impaired eIF2B activity specifically in myelinating oligodendrocytes during development induces the cell dysfunction that resembles oligodendrocyte pathology in VWMD. Immunostaining of both CC1 and ASPA, markers for oligodendrocytes, showed that the number of oligodendrocytes was moderately, but not significantly, reduced in the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) of 18-d-old PLP/Fv2E-PERK Tg/Tg mice treated with AP20187 compared with vehicle-treated mice (Fig. 4A,B). CC1 and TUNEL double- labeling showed that TUNEL-positive oligodendrocytes were undetectable in the CNS of 18-d-old PLP/Fv2E-PERK Tg/Tg mice treated with vehicle, and that there were few TUNEL-positive oligodendrocytes (<1% of CC1-positive cells were CC1 and TUNEL double-positive) in the CNS of PLP/Fv2E-PERK Tg/Tg mice treated with AP20187 (Fig. 4B). Similarly, CC1 and active-caspase-3 double labeling occasionally revealed apoptotic oligodendrocytes in the CNS of 18-d-old PLP/Fv2E-PERK Tg/Tg mice treated with AP20187 (data not shown). EM analysis revealed that the ultrastructural morphology of oligodendrocytes appeared normal in the CNS of 20-d-old PLP/Fv2E-PERK Tg/Tg mice treated with vehicle (Fig. 4C). Importantly, ∼35% of oligodendrocytes in the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) of AP20187-treated mice displayed the morphological features of the foamy oligodendrocytes observed in human VWMD patients (Wong et al., 2000). These cells had abundant cytoplasm and were filled with circular, membranous structures (Fig. 4D). The majority of foamy cells had an oval nucleus with condensed chromatin and extended processes that wrapped around axons to form myelin sheaths. Interestingly, there were few foamy cells that displayed the characteristics of apoptosis, including highly condensed chromatin mass, nuclear fragmentation, and shrunken cytoplasm (Fig. 4E,F). These data indicate that impaired eIF2B activity induces oligodendrocyte dysfunction and morphology change, resulting in severe myelin abnormalities and occasional cell apoptosis. Together, these results demonstrate that impaired eIF2B activity specifically in myelinating oligodendrocytes of young, developing mice is sufficient to reproduce the fundamental pathology of myelin and oligodendrocytes in VWMD.
Figure 4.

Impaired eIF2B activity in oligodendrocytes during development led to foamy oligodendrocytes and occasional cell apoptosis. A, CC1 immunostaining revealed moderate but not statistically significant reduction of oligodendrocyte numbers in the CNS of 18-d-old PLP/Fv2E-PERK Tg/Tg mice treated with AP20187 compared with vehicle-treated mice; N = 6 animals. B, CC1 and TUNEL double-labeling showed that CC1 and TUNEL double-positive cell was undetectable in the CNS of 18-d-old PLP/Fv2E-PERK Tg/Tg mice treated with vehicle, and there were few CC1 and TUNEL double-positive cells (arrow) in the CNS of PLP/Fv2E-PERK Tg/Tg mice treated with AP20187; N = 3 animals. C, EM analysis showed normal oligodendrocytes in the brain of 20-d-old PLP/Fv2E-PERK Tg/Tg mice treated with vehicle; N = 3 animals. D, EM analysis showed that a number of oligodendrocytes displayed foamy morphology in the brain of 20-d-old PLP/Fv2E-PERK Tg/Tg mice treated with AP20187. These cells had abundant cytoplasm filled with circular, membranous structures and an oval nucleus with condensed chromatin; N = 3 animals. E, F, Few foamy oligodendrocytes in the brain of AP20187-treated PLP/Fv2E-PERK Tg/Tg mice displayed the characteristics of apoptosis, including highly condensed chromatin mass, nuclear fragmentation, and shrunken cytoplasm; N = 3 animals. Error bars represent SD. Scale bars: B, 20 μm; C–F, 1 μm.
Impaired eIF2B activity in oligodendrocytes of adult mice induces minimal myelin abnormalities in the CNS
We further determined the effects of impaired eIF2B activity on oligodendrocytes in adult mice. Eight-week-old PLP/Fv2E-PERK Tg/Tg mice were treated with AP20187 (5 mg/kg) or vehicle daily for up to 3 weeks. Interestingly, AP20187-treated adult mice appeared healthy and were indistinguishable from vehicle-treated mice and from untreated wild-type mice. As expected, we found that AP20187 treatment activated PERK signaling specifically in oligodendrocytes of adult PLP/Fv2E-PERK Tg/Tg mice. Real-time PCR analysis showed increased expression of PERK-responsive genes CHOP and GADD34 (data not shown). CC1 and p-eIF2α double immunostaining, as well as CC1 and CHOP double-immunostaining showed that AP20187 treatment significantly increased the levels of p-eIF2α and CHOP specifically in oligodendrocytes of adult PLP/Fv2E-PERK Tg/Tg mice (data not shown). Moreover, we found that AP20187 treatment significantly suppressed protein biosynthesis in the optic nerve generated from adult PLP/Fv2E-PERK Tg/Tg mice (Fig. 2A,C).
ASPA immunostaining showed that AP20187 treatment did not significantly change the number of oligodendrocytes in the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) of adult PLP/Fv2E-PERK Tg/Tg mice (Fig. 5A,B). Neither ASPA and TUNEL double staining nor CC1 and active-caspase-3 double-staining showed any apoptotic oligodendrocytes in the CNS of adult PLP/Fv2E-PERK Tg/Tg mice treated with either vehicle or AP20187 (data not shown). Moreover, we found no foamy oligodendrocytes in the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) of adult PLP/Fv2E-PERK Tg/Tg mice treated with AP20187 (Fig. 5C). Although MBP immunostaining and Western blot analysis showed that AP20187 treatment did not significantly alter the degree of myelination in the CNS of adult PLP/Fv2E-PERK Tg/Tg mice (Fig. 6A,B), the sizes of the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) were slightly reduced in AP20187-treated mice compared with vehicle-treated mice (Fig. 6A). EM analysis showed that AP20187 treatment moderately but significantly reduced the percentage of myelinated axons in the CNS white matter (including the spinal cord, cerebellum, and corpus callosum) of adult PLP/Fv2E-PERK Tg/Tg mice (Fig. 6C,D). Interestingly, the diameter of axons in the CNS white matter of AP20187-treated mice was significantly decreased as compared with vehicle-treated mice, although AP20187 treatment had no effect on the thickness of the myelin sheaths (Fig. 6C,E). These data likely reflect that the reduction of axon diameter results in the shrinkage of the CNS white matter. Together, these results suggest that mature oligodendrocytes in adult mice are less sensitive to impaired eIF2B activity than myelinating oligodendrocytes in young, developing mice.
Figure 5.

Impaired eIF2B activity in oligodendrocytes had minimal effect on the cells of adult mice. A, B, ASPA immunostaining showed that AP20187 treatment did not change the number of oligodendrocytes in the CNS of adult PLP/Fv2E-PERK Tg/Tg mice. C, EM analysis showed that AP20187 treatment did not significantly alter the morphology of oligodendrocytes in the CNS of adult PLP/Fv2E-PERK Tg/Tg mice; N = 3 animals. Error bars represent SD. Scale bars: B, 10 μm; C, 1 μm.
Figure 6.

Impaired eIF2B activity in oligodendrocytes had minimal effect on myelin in the CNS of adult mice. A, MBP immunostaining revealed that the degree of myelination in the cerebellum of adult PLP/Fv2E-PERK Tg/Tg mice treated with AP20187 was comparable to vehicle-treated mice. B, Western blot analysis showed that AP20187 treatment did not significantly alter the level of MBP in the brain of adult PLP/Fv2E-PERK Tg/Tg mice. C, D, EM analysis showed that AP20187 treatment significantly but moderately decreased the number of myelinated axons in the brain of adult PLP/Fv2E-PERK Tg/Tg mice. E, Quantitative EM analysis showed that AP20187 treatment did not alter the thickness of the myelin sheaths, but significantly reduced the diameter of axons in the CNS of PLP/Fv2E-PERK Tg/Tg mice; N = 4 animals. Error bars represent SD. *p < 0.05. Scale bars: A, 500 μm; C, 1 μm.
Discussion
Correlative studies in yeast and cultured mammalian cells point to diminished eIF2B guanine nucleotide exchange activity in VWMD-associated eIF2B mutations (Richardson et al., 2004; Scheper et al., 2006; van der Knaap et al., 2006; Horzinski et al., 2009). However, to date, attempts to recapitulate the features of the disease in a relevant model system have been unsuccessful (Bugiani et al., 2010; Geva et al., 2010). A powerful genetic tool for selectively attenuating eIF2B activity in oligodendrocytes of young, myelinating mice has produced here a striking facsimile of the human disease, lending strong support for the primacy of diminished eIF2B exchange activity in its pathogenesis. We found that strong activation of PERK specifically in myelinating oligodendrocytes of young, developing mice suppressed eIF2B activity and led to severe hypomyelination, foamy oligodendrocytes, and occasional oligodendrocyte apoptosis in the CNS, which resembles oligodendrocyte and myelin pathology in VWMD patients (Scheper et al., 2006; van der Knaap et al., 2006). Moreover, these mice displayed a severe tremoring phenotype and premature death that recapitulates the hypomyelinating phenotype of VWMD. As such, this finding implies a cell-autonomous role of impaired eIF2B activity in myelinating oligodendrocytes in the pathogenesis of VWMD.
Interestingly, strong PERK activation in myelinating oligodendrocytes during development did not significantly alter the cell numbers. Moreover, strong PERK activation specifically in oligodendrocytes of adult mice suppressed eIF2B activity, but did not alter the number or morphology of oligodendrocytes in the CNS. Adult PLP/Fv2E-PERK Tg/Tg mice treated with AP20187 appeared healthy and exhibited modest reduction of myelinated axons in the CNS. Myelinating oligodendrocytes in young, developing mice must synthesize enormous amounts of membrane protein molecules to assemble myelin sheaths. Mature oligodendrocytes in adult mice, however, only synthesize just enough membrane protein molecules to maintain myelin integrity (Morell and Quarles, 1999). There is evidence that myelinating oligodendrocytes are far more sensitive to the disruption of protein translation than mature oligodendrocytes (Lin and Popko, 2009). Together, these data suggest that impaired eIF2B activity selectively disturbs the myelinating function of oligodendrocytes during the active phase of myelination. On the other hand, a recent study showed that myelinating oligodendrocytes are constantly generated from oligodendrocyte precursors in the CNS of adult animals, which replace dying oligodendrocytes and are engaged in myelin remodeling (Young et al., 2013). Therefore, it is possible that impaired eIF2B activity induced by strong PERK activation does not affect the viability of mature oligodendrocytes or their ability to maintain myelin integrity, but suppresses the myelinating function of newly generated oligodendrocytes, resulting in modestly increased unmyelinated axons in the adult CNS.
Although AP20187 treatment did not alter the thickness of the myelin sheaths in the CNS of adult PLP/Fv2E-PERK Tg/Tg mice, there were significant reductions of axon diameter and slight shrinkage of the CNS white matter in these animals. Recent studies have shown that oligodendrocytes support axon integrity and neuron survival by producing neurotrophic and nutritional factors (Dai et al., 2003; Ubhi et al., 2010; Fünfschilling et al., 2012; Lee et al., 2012; Bankston et al., 2013). Thus, there is a possibility that the suppressed production of oligodendrocyte-derived neurotrophic and/or nutritional factors induced by impaired eIF2B activity contributes to the reduction of axon diameter and the shrinkage of the CNS white matter observed in adult PLP/Fv2E-PERK Tg/Tg mice treated with AP20187.
Activation of PERK in response to ER stress coordinates an adaptive program to promote cell survival under stressful conditions (Walter and Ron, 2011; Pavitt and Ron, 2012). Nevertheless, PERK signaling is not exclusively beneficial to cells. In addition to suppressing global protein translation, strong PERK activation can be detrimental to cells through induction of CHOP, a proapoptotic transcription factor (Tabas and Ron, 2011; Hetz, 2012). Interestingly, it has been shown that the level of CHOP is increased in oligodendrocytes of VWMD patients (van der Voorn et al., 2005; van Kollenburg et al., 2006). A previous study, however, showed that CHOP induction promotes survival of ER-stressed myelinating oligodendrocytes in a mouse model of Pelizaeus–Merzbacher disease (Southwood et al., 2002). We showed here that strong PERK activation enhanced the expression of CHOP in a number of oligodendrocytes in both young, developing mice and adult mice. Although apoptotic oligodendrocytes were occasionally observed in the CNS of young, developing PLP/Fv2E-PERK Tg/Tg mice treated with AP20187, the number of oligodendrocytes in these mice were not significantly decreased compared with vehicle-treated mice. Moreover, there was no evidence that AP20187 treatment induced oligodendrocyte apoptosis or reduced the cell numbers in the CNS of adult PLP/Fv2E-PERK Tg/Tg mice. Additionally, we found that none of CHOP-positive oligodendrocytes were active-caspase-3-positive or TUNEL-positive in the CNS of AP20187-treated PLP/Fv2E-PERK Tg/Tg mice (data not shown). Therefore, it is unlikely that CHOP is involved in the detrimental effects of strong PERK activation on oligodendrocytes. On the other hand, EM analysis showed foamy oligodendrocytes in young, developing PLP/Fv2E-PERK Tg/Tg mice treated with AP20187, but not in adult AP20187-treated mice. Importantly, we found that all apoptotic oligodendrocytes in young, developing AP20187-treated mice were accompanied with foamy morphology, which indicates that the foamy morphology change precedes cell apoptosis. Collectively, these data suggest that suppression of protein biosynthesis induced by impaired eIF2B activity leads to foamy morphology change of oligodendrocytes and subsequent cell apoptosis during development. Nevertheless, the mechanisms by which disruption of protein biosynthesis causes foamy oligodendrocytes remain unknown and deserve additional study.
In contrast to dire consequences of strong PERK activation in myelinating oligodendrocytes, moderate PERK activation in response to ER stress preserves the viability and function of myelin-producing cells in various myelin disorders (Lin and Popko, 2009). It has been shown that PERK haploinsufficiency exacerbates apoptosis of both mature and myelinating oligodendrocytes in immune-mediated demyelinating diseases (Lin et al., 2005, 2006, 2007). Our recent studies showed that treatment with a low dose of AP20187 (0.5 mg/kg) moderately activates PERK signaling in oligodendrocytes of PLP/Fv2E-PERK Tg/+ mice and that moderate PERK activation slightly reduces eIF2B activity and promotes survival of both mature and myelinating oligodendrocytes in immune-mediated demyelinating diseases (Lin et al., 2013, 2014). We also showed that GADD34 inactivation moderately enhances PERK activation and protects myelinating oligodendrocytes against the cytotoxicity of IFN-γ in young, developing mice (Lin et al., 2008). Moreover, a recent report showed that GADD34 inactivation moderately enhances PERK activation in Schwann cells and improves myelination in the peripheral nervous system of a mouse model of Charcot-Marie-Tooth 1B disease (D'Antonio et al., 2013). The lesson emerging from these findings is that impaired eIF2B activity has biphasic effects on oligodendrocytes, which is highly context-dependent in the degree of the impairment. In the context of diseases associated with ER stress, modest attenuation of eIF2B activity (promoted by PERK activation or GADD34 inactivation) is protective, whereas in the context of VWMD, diminished eIF2B activity is highly detrimental.
VWMD is chronic and progressive, and most individuals with VWMD display episodes of rapid deterioration after febrile infections or minor head trauma (Scheper et al., 2006; van der Knaap et al., 2006). Several studies have shown that the levels of phosphorylated PERK and p-eIF2α are increased in oligodendrocytes of VWMD patients (van der Voorn et al., 2005; van Kollenburg et al., 2006). Our observations, which point to the primacy of diminished eIF2B activity in generating the VWMD phenotype, also suggest a role for transient intercurrent stress, such as that promoted by febrile infections or head trauma in the acute exacerbation of the disease observed clinically, as moderate activation of eIF2a kinases like PERK or double-stranded RNA-dependent protein kinase (Pavitt and Ron, 2012), that are normally well tolerated by normal oligodendrocytes promote a unsustainable drop in eIF2B activity in the oligodendrocytes of VWMD patients.
Footnotes
Wensheng Lin was supported by grants from the National Multiple Sclerosis Society (TA3026-A-1 and RG4813-A-2) and the National Institutes of Health (NS073132), and D.R. is a Principal Research Fellow of the Wellcome Trust. We thank Dr H. Brent Clark (Department of Laboratory Medicine and Pathology, University of Minnesota) for assistance in electron microscopy analysis, and Dr. A. M. Aryan Namboodiri (Uniformed Services University of the Health Sciences, Bethesda, MD) for providing the antibody against aspartoacylase.
The authors declare no competing financial interests.
References
- Ainsworth C. Molecular medicine: lost in translation. Nature. 2005;435:556–558. doi: 10.1038/435556a. [DOI] [PubMed] [Google Scholar]
- Bankston AN, Mandler MD, Feng Y. Oligodendroglia and neurotrophic factors in neurodegeneration. Neurosci Bull. 2013;29:216–228. doi: 10.1007/s12264-013-1321-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Bugiani M, Boor I, Powers JM, Scheper GC, van der Knaap MS. Leukoencephalopathy with vanishing white matter: a review. J Neuropathol Exp Neurol. 2010;69:987–996. doi: 10.1097/NEN.0b013e3181f2eafa. [DOI] [PubMed] [Google Scholar]
- Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM, Cohen DA. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol. 2004;75:612–623. doi: 10.1189/jlb.0903442. [DOI] [PubMed] [Google Scholar]
- Dai X, Lercher LD, Clinton PM, Du Y, Livingston DL, Vieira C, Yang L, Shen MM, Dreyfus CF. The trophic role of oligodendrocytes in the basal forebrain. J Neurosci. 2003;23:5846–5853. doi: 10.1523/JNEUROSCI.23-13-05846.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antonio M, Musner N, Scapin C, Ungaro D, Del Carro U, Ron D, Feltri ML, Wrabetz L. Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J Exp Med. 2013;210:821–838. doi: 10.1084/jem.20122005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MJ, Gerschenfeld HM. Some physiological properties of identified mammalian neuroglial cells. J Physiol. 1969;203:211–222. doi: 10.1113/jphysiol.1969.sp008860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, Nave KA. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva M, Cabilly Y, Assaf Y, Mindroul N, Marom L, Raini G, Pinchasi D, Elroy-Stein O. A mouse model for eukaryotic translation initiation factor 2B-leucodystrophy reveals abnormal development of brain white matter. Brain. 2010;133:2448–2461. doi: 10.1093/brain/awq180. [DOI] [PubMed] [Google Scholar]
- Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, Hornberger TA. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 2011;25:1028–1039. doi: 10.1096/fj.10-168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Horzinski L, Huyghe A, Cardoso MC, Gonthier C, Ouchchane L, Schiffmann R, Blanc P, Boespflug-Tanguy O, Fogli A. Eukaryotic initiation factor 2B (eIF2B) GEF activity as a diagnostic tool for EIF2B-related disorders. PLoS One. 2009;4:e8318. doi: 10.1371/journal.pone.0008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leegwater PA, Vermeulen G, Könst AA, Naidu S, Mulders J, Visser A, Kersbergen P, Mobach D, Fonds D, van Berkel CG, Lemmers RJ, Frants RR, Oudejans CB, Schutgens RB, Pronk JC, van der Knaap MS. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet. 2001;29:383–388. doi: 10.1038/ng764. [DOI] [PubMed] [Google Scholar]
- Lin W, Lin Y. Interferon-γ inhibits central nervous system myelination through both STAT1-dependent and STAT1-independent pathways. J Neurosci Res. 2010;88:2569–2577. doi: 10.1002/jnr.22425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Popko B. Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci. 2009;12:379–385. doi: 10.1038/nn.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Harding HP, Ron D, Popko B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J Cell Biol. 2005;169:603–612. doi: 10.1083/jcb.200502086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kemper A, Dupree JL, Harding HP, Ron D, Popko B. Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain. 2006;129:1306–1318. doi: 10.1093/brain/awl044. [DOI] [PubMed] [Google Scholar]
- Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007;117:448–456. doi: 10.1172/JCI29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kunkler PE, Harding HP, Ron D, Kraig RP, Popko B. Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-gamma. Am J Pathol. 2008;173:1508–1517. doi: 10.2353/ajpath.2008.080449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Lin Y, Li J, Fenstermaker AG, Way SW, Clayton B, Jamison S, Harding HP, Ron D, Popko B. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J Neurosci. 2013;33:5980–5991. doi: 10.1523/JNEUROSCI.1636-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Huang G, Jamison S, Li J, Harding HP, Ron D, Lin W. PERK activation preserves the viability and function of remyelinating oligodendrocytes in immune-mediated demyelinating diseases. Am J Pathol. 2014;184:507–519. doi: 10.1016/j.ajpath.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, van der Lei HD, Wang X, Wortham NC, Tang H, van Berkel CG, Mufunde TA, Huang W, van der Knaap MS, Scheper GC, Proud CG. Severity of vanishing white matter disease does not correlate with deficits in eIF2B activity or the integrity of eIF2B complexes. Hum Mutat. 2011;32:1036–1045. doi: 10.1002/humu.21535. [DOI] [PubMed] [Google Scholar]
- Locatelli G, Wörtge S, Buch T, Ingold B, Frommer F, Sobottka B, Krüger M, Karram K, Bühlmann C, Bechmann I, Heppner FL, Waisman A, Becher B. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat Neurosci. 2012;15:543–550. doi: 10.1038/nn.3062. [DOI] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004;23:169–179. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Moffett JR, Moore RA, Viola RE, Namboodiri MA, Jacobowitz DM. Immunohistochemical localization of aspartoacylase in the rat central nervous system. J Comp Neurol. 2004;472:318–329. doi: 10.1002/cne.20080. [DOI] [PubMed] [Google Scholar]
- Morell P, Quarles RH. Myelin formation, structure and biochemistry. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD, editors. Basic neurochemistry: molecular, cellular, and medical aspects. Philadelphia: Lippincott-Raven; 1999. pp. 69–93. [Google Scholar]
- Omlin FX, Waldmeyer J. Differentiation of neuron-like cells in cultured rat optic nerves: a neuron or common neuron-glia progenitor? Dev Biol. 1989;133:247–253. doi: 10.1016/0012-1606(89)90315-1. [DOI] [PubMed] [Google Scholar]
- Pavitt GD, Ron D. New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harb Perspect Biol. 2012;4:a012278. doi: 10.1101/cshperspect.a012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud CG. eIF2 and the control of cell physiology. Semin Cell Dev Biol. 2005;16:3–12. doi: 10.1016/j.semcdb.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Richardson JP, Mohammad SS, Pavitt GD. Mutations causing childhood ataxia with central nervous system hypomyelination reduce eukaryotic initiation factor 2B complex formation and activity. Mol Cell Biol. 2004;24:2352–2363. doi: 10.1128/MCB.24.6.2352-2363.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- Scheper GC, Proud CG, van der Knaap MS. Defective translation initiation causes vanishing of cerebral white matter. Trends Mol Med. 2006;12:159–166. doi: 10.1016/j.molmed.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus–Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/S0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, Whitney K, Masliah E. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci. 2010;30:6236–6246. doi: 10.1523/JNEUROSCI.0567-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Barth PG, Gabreëls FJ, Franzoni E, Begeer JH, Stroink H, Rotteveel JJ, Valk J. A new leukoencephalopathy with vanishing white matter. Neurology. 1997;48:845–855. doi: 10.1212/WNL.48.4.845. [DOI] [PubMed] [Google Scholar]
- van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol. 2006;5:413–423. doi: 10.1016/S1474-4422(06)70440-9. [DOI] [PubMed] [Google Scholar]
- van der Voorn JP, van Kollenburg B, Bertrand G, Van Haren K, Scheper GC, Powers JM, van der Knaap MS. The unfolded protein response in vanishing white matter disease. J Neuropathol Exp Neurol. 2005;64:770–775. doi: 10.1097/01.jnen.0000178446.41595.3a. [DOI] [PubMed] [Google Scholar]
- van Kollenburg B, van Dijk J, Garbern J, Thomas AA, Scheper GC, Powers JM, van der Knaap MS. Glia-specific activation of all pathways of the unfolded protein response in vanishing white matter disease. J Neuropathol Exp Neurol. 2006;65:707–715. doi: 10.1097/01.jnen.0000228201.27539.50. [DOI] [PubMed] [Google Scholar]
- Vaughn JE. An electron microscopic analysis of gliogenesis in rat optic nerves. Z Zellforsch Mikrosk Anat. 1969;94:293–324. doi: 10.1007/BF00319179. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wong K, Armstrong RC, Gyure KA, Morrison AL, Rodriguez D, Matalon R, Johnson AB, Wollmann R, Gilbert E, Le TQ, Bradley CA, Crutchfield K, Schiffmann R. Foamy cells with oligodendroglial phenotype in childhood ataxia with diffuse central nervous system hypomyelination syndrome. Acta Neuropathol. 2000;100:635–646. doi: 10.1007/s004010000234. [DOI] [PubMed] [Google Scholar]
- Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, Richardson WD. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron. 2013;77:873–885. doi: 10.1016/j.neuron.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]