

Abstract
Hippocampal CA1 pyramidal neurons receive inputs from entorhinal cortex directly via the temporoammonic (TA) pathway and indirectly via the Schaffer collateral (SC) pathway from CA3. NMDARs at synapses of both pathways are critical for the induction of synaptic plasticity, information processing, and learning and memory. We now demonstrate that, in the rat hippocampus, activity-dependent mGlu1 receptor-mediated LTD (mGlu1-LTD) of NMDAR-mediated transmission (EPSCNMDA) at the SC-CA1 input prevents subsequent LTP of AMPAR-mediated transmission. In contrast, there was no activity-dependent mGlu1-LTD of EPSCNMDA at the TA-CA1 pathway, or effects on subsequent plasticity of AMPAR-mediated transmission. Therefore, the two major pathways delivering information to CA1 pyramidal neurons are subject to very different plasticity rules.
Keywords: LTD, metaplasticity, NMDARs
Introduction
Pyramidal neurons in area CA1 of the hippocampus receive information from the entorhinal cortex (EC) via two different routes (van Strien et al., 2009). The direct temporoammonic (TA) pathway originates from layer III of EC and terminates on distal dendrites of CA1 pyramidal neurones in the stratum lacunosum moleculare (SLM). The indirect pathway originates from layer II of EC and passes via the trisynaptic circuit culminating in the Schaffer collaterals (SC) terminating on proximal dendrites of CA1 in the stratum radiatum (SR). The two inputs convey distinct spatial information and have different roles in learning and memory (Brun et al., 2002; Remondes and Schuman, 2002).
NMDARs are critical for induction of synaptic plasticity in numerous brain regions, including at the TA-CA1 (Remondes and Schuman, 2002) and SC-CA1 synapses (Collingridge et al., 1983). NMDARs are heteromeric assemblies (Cull-Candy et al., 2001) composed of two essential GluN1 subunits and two or three GluN2 subunits. There are four GluN2 subunits (A–D), but the GluN2A and GluN2B subunits predominate in the forebrain. There is evidence that NMDARs with different subunit composition can preferentially induce LTP or LTD (Liu et al., 2004; Massey et al., 2004), and NMDARs at the TA-CA1 and SC-CA1 inputs are reported to have different GluN2A/2B compositions (Arrigoni and Greene, 2004).
NMDARs themselves undergo synaptic plasticity (Bashir et al., 1991; Kwon and Castillo, 2008; Rebola et al., 2008). Although the functional consequences of LTP of EPSCNMDA are beginning to be understood (Rebola et al., 2010; Hunt et al., 2013), the effects of activity-dependent LTD of EPSCNMDA are still unclear. In the current study, we describe activity-dependent LTD of EPSCNMDA that relies on mGlu1 receptors (mGlu1-LTD) and occurs selectively at SC-CA1 but not TA-CA1 synapses. mGlu1-LTD alters the threshold for subsequent LTP of AMPAR-mediated transmission in the SC-CA1 input. These results show that there is a marked difference in plasticity of EPSCNMDA between two inputs to CA1 that has important functional consequences for plasticity of AMPAR-dependent transmission.
Materials and Methods
P14 male Wistar rats were killed by cervical dislocation in accordance with United Kingdom Animal (Scientific Procedures) legislation. Brains were removed and placed in ice-cold aCSF consisting of the following (in mm): NaCl 124, KCl 3, NaHCO3 26, NaH2PO4 1.25, CaCl2 2, MgSO4 1, d-glucose 10 (bubbled with 95% O2/5% CO2). Parasaggital slices (400 μm) were cut and the hippocampus isolated. Each “n” value is from a slice from a different animal.
Extracellular recording.
Standard techniques were used to record field potentials in response to alternate stimulation (100 μs, 3–10 V) of the SC and TA pathways. Responses were recorded and analyzed using WinLTP (Anderson and Collingridge, 2007). LTP was induced using theta burst (TBS; 2 × 4 stimuli at100 Hz, 200 ms interburst interval) or tetanic (100 stimuli at 100 Hz) stimulation. Picrotoxin was applied for LTP experiments in TA-CA1. Changes in synaptic strength were expressed to the normalized baseline (mean ± SEM) and significance tested using Student's t tests, 30 min after LTP induction. The Group II mGluR agonist DCG-IV was applied at the end of experiments to ensure selective stimulation of TA and SC inputs (Kew et al., 2001).
Whole-cell recording.
Electrodes (4–7 mΩ) were filled with (in mm): CsMeSO4, 130, NaCl 8, Mg-ATP 4, Na-GTP 0.3, EGTA 0.5, HEPES 10, QX-314 5, pH adjusted to 7.2–7.3 using CsOH and osmolarity to 275–290 mOsm with sucrose. CA1 pyramidal cells were voltage-clamped at −70 mV (EPSCAMPA) or −40 mV (EPSCNMDA). Picrotoxin (50 μm) and NBQX (5 μm) were applied to isolate EPSCNMDA. The decays of EPSCNMDA were fit with a double exponential using Clampfit (v10.2 for Windows; Molecular Devices). The value of the weighted time constant (τw) was then calculated using the following: τw = τ1*A1/(A1+A2) + τ2*A2/(A1+A2) (Bartlett et al., 2007) where A1 and A2 represent the amplitudes of the two components and τ1 and τ2 the decay constants. Differences were assessed using Student's t test.
Immunoblot analysis.
P14-P15 Wistar rats were killed by decapitation, and brains were dissected out and placed on ice-cold Krebs-Henseleit buffer. Brains were cut by a vibratome and two slices (600 μm thickness) containing the dorsal hippocampus were obtained. Under a stereomicroscope, the SR and SLM were dissected out and immediately frozen in dry ice and conserved at −80°C until processed. Samples were homogenized at 4°C in a lysis buffer composed of Tris-HCl 10 mm, pH 7.4, NaCl 150 mm, EDTA 5 mm, IGEPAL 1%, protease (Santa Cruz Biotechnology) and phosphatase (Sigma) inhibitor mixture. A total of 5 μl of tissue extracts was used for protein determination. Proteins (30 μg) were resuspended in SDS-bromophenol blue reducing buffer with 40 mm DTT and used for protein analysis. Immunoblotting was performed with the following primary antibodies: Homer 1a (1:200) and Homer 1b/c (1:200) (both from Santa Cruz Biotechnology), and β-actin (1:50,000, Sigma). After incubation in primary antibody overnight at 4°C, immunoblots were incubated with HRP-conjugated secondary antibodies (Calbiochem) and developed by ECL (Hybond ECL, GE Healthcare Europe). Statistical analysis was assessed by two-way ANOVA and Fisher's least squares difference.
Results
LTD of EPSCNMDA at SC-CA1 but not TA-CA1
Brief theta frequency stimulation (TFS; 5 Hz, 20 s) produced LTD of EPSCNMDA in the SC-CA1 input (depression to 61 ± 6% of control; n = 9; p < 0.05; Fig. 1A). LTD was not associated with a change in τw of EPSCNMDA (149 ± 11 ms and 171 ± 15 ms before and after LTD induction; n = 6; p > 0.05). TFS produced no significant change in EPSCAMPA (86 ± 5% of control; n = 6; p > 0.05; Fig. 1B). LTD of EPSCNMDA was not observed when TFS was applied to the TA-CA1 input (90 ± 8% of control; n = 12; p > 0.05; Fig. 1C), nor was there any effect on τw EPSCNMDA (92 ± 10 ms and 86 ± 8 ms before and after LTD, respectively, n = 12; p > 0.05).
Figure 1.
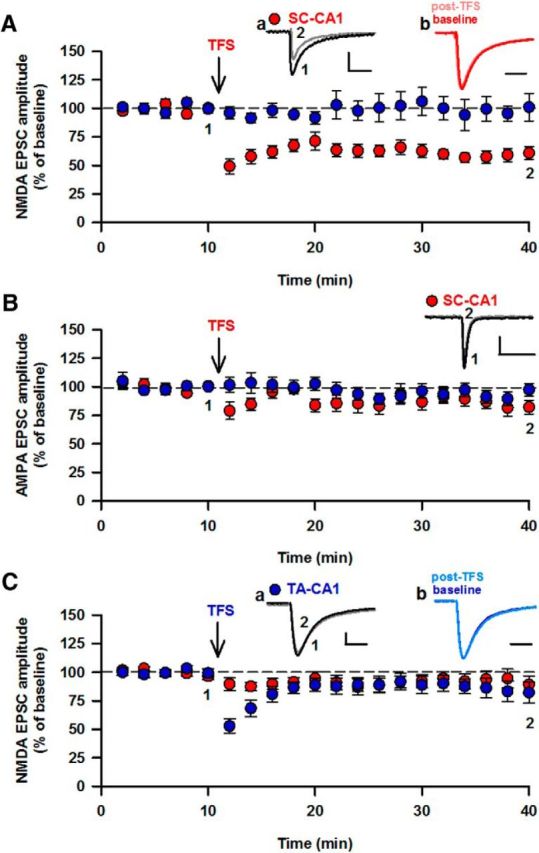
LTD of EPSCNMDA at the SC-CA1 but not the TA-CA1 input. A, Pooled data showing LTD of EPSCNMDA induced by 5 Hz, 20 s stimulation (TFS, indicated by ↓) delivered to the SC input. a, Superimposed responses before and after LTD. b, Peak scaled responses. B, TFS had no effect on AMPAR-mediated EPSCs in the SC-CA1 input. C, TFS did not induce LTD at the TA-CA1 input. a, b, Same as in A. Calibration: 20 pA, 100 ms.
mGlu1 receptor-dependent LTD of EPSCNMDA at SC-CA1
LTD of EPSCNMDA in the SC-CA1 was prevented by the pan-mGluR antagonist LY341495 (100 μm; 89 ± 9% of control; n = 7; p > 0.05; data not shown). The mGlu1 receptor antagonists LY367385 (30 μm; 97 ± 7% of baseline; n = 6; p > 0.05; Fig. 2A) and JNJ16259685 (100 nm; 87 ± 8% of baseline; n = 6; p > 0.05; data not shown) also prevented LTD of EPSCNMDA. However, the mGlu5 receptor antagonist MPEP did not prevent LTD induction (10 μm; depression to 65 ± 4; n = 6; p < 0.05; Fig. 2B). Thus, synaptic stimulation induces mGlu1-LTD of EPSCNMDA.
Figure 2.
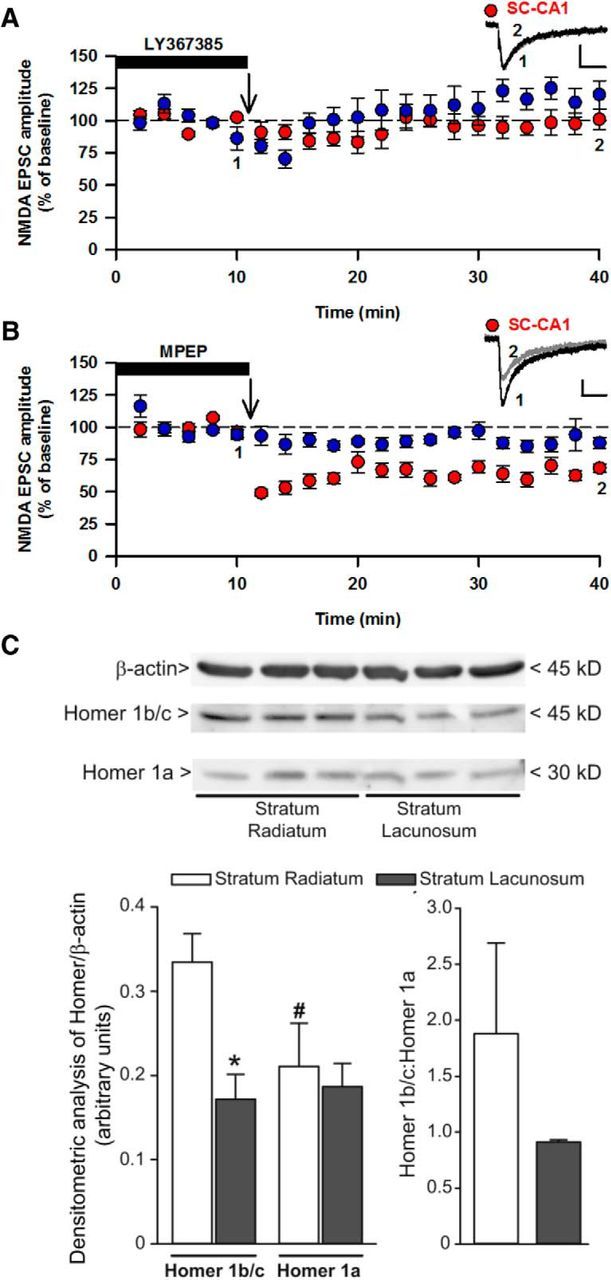
mGlu1-LTD of EPSCNMDA at the SC input. A, LTD of EPSCNMDA was blocked by the mGlu1 receptor antagonist LY367385 (30 μm). B, LTD was unaffected by the mGlu5 receptor antagonist MPEP (10 μm). Calibration: 20 pA, 100 ms. C, Homer 1b/c is reduced in SLM compared with SR. Densitometric values of Homer 1b/c and Homer 1a in the SR and SLM are mean ± SEM from three protein extracts pooled from 12 rats. The same filter was used for the detection of Homer 1b/c and Homer 1a with the same exposure time. *p < 0.05, Homer 1b/c: SLM vs SR. #p < 0.05, Homer 1a vs Homer 1b/c in SR.
No NMDAR subunit differences between SC- and TA-CA1 synapses
It has been reported that NMDARs at the TA-CA1 and SC-CA1 have different subunit composition (Arrigoni and Greene, 2004). However, there was no difference between τw of EPSCNMDA (SC: 124 ± 4; TA:117 ± 5 ms; n = 44; p > 0.05) in our experiments; and in addition, the GluN2B antagonist Ro25–6981 did not produce differential effects on EPSCNMDA (SC-CA1: depression of 59 ± 6%, n = 6; TA-CA1: 49 ± 7%, n = 6, p > 0.05). Thus, it is unlikely that differences in NMDAR subunits between the two inputs account for differences in LTD of EPSCNMDA.
Differences in Homer between TA-CA1 and SC-CA1 inputs
We investigated whether differences in mGlu-stimulated signaling could explain differences in LTD between TA- and SC-CA1. We found no difference in mGlu1- or mGlu5-mediated PI hydrolysis between the two inputs (data not shown). However, we found that levels of Homer 1b/c were significantly lower (p < 0.05) in SLM (0.17 ± 0.03) compared with SR (0.33 ± 0.03). In contrast, there was no difference (p > 0.05) in Homer 1a levels between SR (0.21 ± 0.05) and SLM (0.19 ± 0.03; Fig. 2C). Therefore, differences in homer 1b/c expression may explain, at least in part, the differences in mGlu1-LTD of EPSCNMDA between the two inputs.
Metaplasticity resulting from LTD of EPSCNMDA
Activity-dependent LTD of EPSCNMDA is predicted to significantly impact on subsequent plasticity of AMPAR-mediated transmission. To examine whether this is the case, fEPSPs were recorded in SR in response to stimulation of two independent SC inputs. TBS was first delivered to one input and resulted in LTP (132 ± 5% of control; n = 9; p < 0.05; Fig. 3A). Next the “priming protocol,” the same TFS that induces mGlu1-LTD of EPSCNMDA, was applied to the other input. This had no long-term effect on AMPAR-mediated fEPSPs (responses 92 ± 7% of control; n = 6; p > 0.05; Fig. 3A). However, following the priming protocol, TBS failed to induce LTP (101 ± 6% of control; n = 9; p > 0.05; Fig. 3A). Therefore, a stimulation protocol that results in mGlu1-LTD of EPSCNMDA prevents subsequent induction of LTP of AMPAR-mediated EPSPs.
Figure 3.
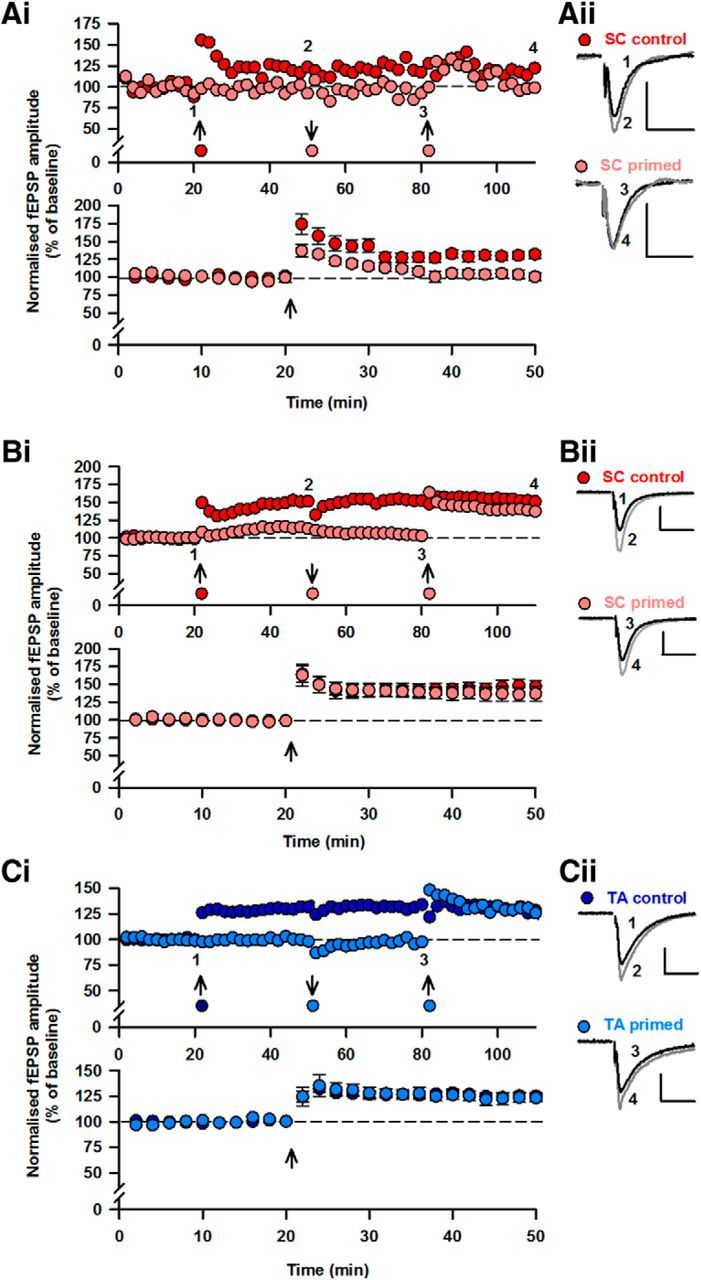
LTD of EPSCNMDA raises the threshold for LTP induction. Ai, Single two input experiment (top) and pooled data (bottom) showing that induction of SC-CA1 LTP by TBS is prevented by the priming stimulus (5 Hz, 20 s). LTP was induced in the control input (red) by TBS (↑). Priming to the second input (pink; ↓) prevented the subsequent induction of LTP by TBS (pink; ↑). For pooled data, experiments have been aligned to time of LTP induction in this and subsequent panels. Aii, Superimposed responses from the time points indicated. Bi, Single experiment (top) and pooled data (bottom) showing that induction of LTP by a strong stimulus (100 Hz, 1 s) is unaffected by the priming stimulus. LTP was induced in one input (red; ↑). Priming to the second input (pink; ↓) did not prevent subsequent induction of LTP (↑). Bii, Superimposed responses before and after LTP. Ci, Single experiment (top) and pooled data (bottom) showing that induction of LTP at the TA-CA1 input is unaffected by the priming stimulus. LTP was first induced in one input (dark blue; ↑). Priming to the second input (light blue;↓) did not prevent subsequent LTP induction. Cii, Superimposed responses before and after LTP. Calibration: 0.4 mV, 20 ms.
We next examined whether the effect of LTD of EPSCNMDA is to alter the threshold for LTP induction. Strong high-frequency stimulation (100 Hz, 1 s) was delivered to one input to induce LTP (146 ± 8%; n = 5; p < 0.05; Fig. 3B). Next, TFS was applied to the other input (Fig. 3B). High-frequency stimulation delivered to the primed input 30 min later resulted in LTP (137 ± 10%; n = 5; p > 0.05; Fig. 3B) that was similar to the control LTP (p > 0.05). Together, these results suggest that mGlu1-LTD shifts the threshold for induction of LTP, an effect that is overcome by a suprathreshold LTP induction paradigm.
The small depression (8 ± 7%, n = 6; p > 0.05) in fEPSPs produced by priming (Fig. 3B) may by itself reduce the probability of LTP induction. To ensure that this was not the case, we delivered a weak LTD induction (300 stimuli, 1 Hz) followed by TBS to one input while the other input received only TBS (data not shown). In the primed input (94 ± 2% of control, n = 5), the level of LTP (130 ± 5%, n = 5) was similar (p > 0.05) to control LTP (133 ± 4%, n = 5). Therefore, it is unlikely that any small change in AMPAR transmission by priming is responsible for reducing LTP.
Lack of metaplasticity at TA-CA1 input
Two independent inputs were stimulated to determine whether priming affected LTP at the TA-CA1 pathway. Priming did not prevent subsequent induction of LTP (control input 136 ± 5%; primed input 136 ± 7%; n = 5; p < 0.05; Fig. 3C). Therefore, priming, which has no effect on EPSCNMDA at the TA-CA1 input, has no effect on induction of LTP at this input.
Pharmacological depression of EPSCNMDA increases threshold for induction of LTP of AMPAR transmission
Because priming stimulation leads to LTD of EPSCNMDA, we examined whether a similar decrease in the contribution of NMDARs to synaptic transmission is sufficient to block the induction of TBS-induced LTP. Bath application of 0.3 μm d-AP5 resulted in depression of EPSCNMDA (65 ± 5% of control; n = 6; data not shown) that was not different (p > 0.05) from activity-dependent LTD of EPSCNMDA. In the presence of 0.3 μm d-AP5, LTP was not induced by TBS (101 ± 3%, n = 4; p > 0.05; Fig. 4Ai, bottom) in contrast to the effect of TBS in the absence of d-AP5 (125 ± 2%, n = 4; p < 0.05; Fig. 4Ai, top).
Figure 4.
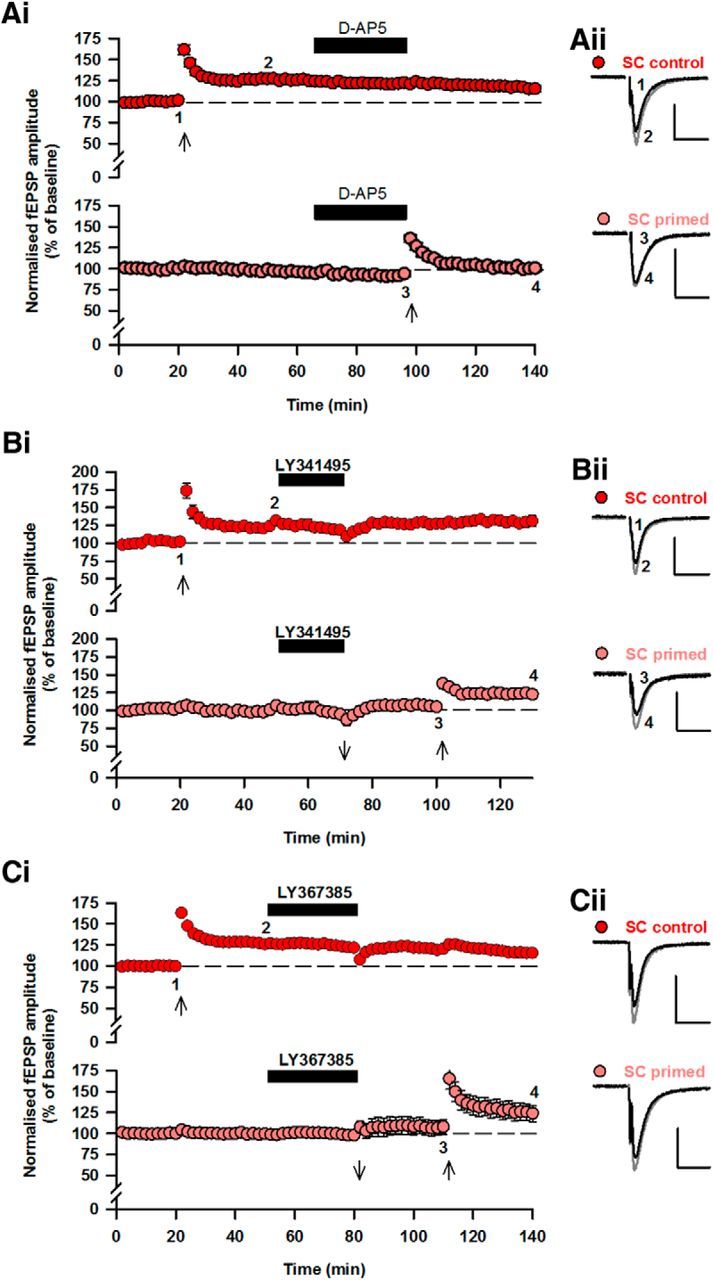
Priming-induced inhibition of LTP is mimicked by partial NMDAR antagonism and rescued by mGlu1 receptor blockade. Ai, Pooled data from two input experiments showing that 0.3 μm d-AP5 (which reduces EPSCNMDA to the same level as mGlu1-LTD) prevents LTP induction (bottom). Aii, Superimposed responses from the two inputs. Bi, Pooled data showing block of priming-induced inhibition of LTP by the mGluR antagonist LY341495 (100 μm) (top: control input; bottom: primed input). Bii, Superimposed responses before and after LTP. Ci, Pooled data showing rescue of priming-induced inhibition of LTP by the mGlu1 receptor antagonist LY367385 (30 μm) (top: control input; bottom: primed input). Cii, Superimposed responses before and after LTP. Calibration: 0.4 mV, 20 ms.
mGlu1 receptor-dependent metaplasticity at SC-CA1
Because LTD of EPSCNMDA was mGluR-dependent (Fig. 2), we examined whether the priming-induced block of LTP (Fig. 3) could be prevented by mGluR antagonists. TBS was delivered to induce LTP in the control input (132 ± 4%, n = 6; p < 0.05; Fig. 4Bi). Following this, the priming protocol was delivered to the second input in the mGluR antagonist LY341495 (100 μm) to block LTD of EPSCNMDA. Following washout of LY341495, TBS was delivered to the input primed previously in the presence of LY341495. Under these conditions, TBS resulted in LTP (123 ± 5%, n = 6; p < 0.05; Fig. 4Bi) that was not different from control LTP (p > 0.05; Fig. 4Bi).
Similarly, the effect of priming on LTP was blocked by the mGlu1 receptor antagonist LY367385 (30 μm). LTP induced by TBS following priming in LY367385 (126 ± 10%; n = 7; p < 0.05, Fig. 4Ci) was not different (p > 0.05) to LTP induced under control conditions (128 ± 4%; n = 7; p < 0.05, Fig. 4Ci). Therefore, antagonism of mGlu1 rescues the priming-dependent block of LTP.
Discussion
Activity-dependent changes in synaptic transmission are widely regarded as cellular correlates of learning and memory (Whitlock et al., 2006; Cooke and Bear, 2010). NMDARs play a critical role in the induction of synaptic plasticity of AMPAR-mediated transmission (Collingridge et al., 1983; Dudek and Bear, 1992) and in learning and memory (Morris et al., 1986). However, although it is established that LTP and LTD of NMDAR-mediated transmission occurs at a variety of different synapses (Bashir et al., 1991; Kwon and Castillo, 2008; Rebola et al., 2008, 2010; Hunt et al., 2013), this has not previously been examined at TA-CA1 synapses.
We now demonstrate in P14 rats that, although robust LTD was induced at the SC-CA1 input, we were unable to induce LTD of EPSCNMDA at TA-CA1. Therefore, SC-CA1 synapses appear to be far more susceptible to LTD of EPSCNMDA. Although our experiments measuring LTD of EPSCNMDA and those assessing metaplasticity were done under different recording conditions, our results strongly suggest that synaptically induced LTD of NMDAR-mediated transmission increases the threshold for subsequent LTP of AMPAR-mediated synaptic transmission. In this study, we have not examined other developmental ages and therefore cannot rule out that different results would occur at different ages.
LTD of EPSCNMDA and metaplasticity
Plasticity of NMDARs can confer an additional level of complexity (metaplasticity) to rules of synaptic modification, neuronal function, and behavior (Abraham, 2008; Hulme et al., 2013). However, there is a surprising paucity of data directly demonstrating that plasticity of NMDAR-mediated transmission can affect subsequent plasticity of AMPAR-mediated transmission (Hunt and Castillo, 2012). The supression of spontaneous glutamate release in cultured neurones was shown to upregulate NMDARs and facilitate AMPAR-LTP (Lee et al., 2010) and mossy fiber-CA3 LTP, which is not normally NMDAR-dependent, becomes dependent on NMDARs following LTP of EPSCNMDA (Rebola et al., 2010). In addition, heterosynaptic metaplasticity has recently been described at associational-CA3 synapses following NMDAR LTP at mossy fiber-CA3 synapses (Hunt et al., 2013). Regarding LTD of EPSCNMDA, previous work has shown that release of caged nitric oxide can result in a depression of NMDAR transmission that can impair subsequent LTP (Murphy and Bliss, 1999).
Metaplasticity prevented the induction of LTP by TBS but had no effect on LTP induced by a strong protocol (100 Hz, 1 s). It is likely that 100 Hz, 1 s stimulation produces large and sustained postsynaptic depolarization that allows sufficient NMDAR activation to induce LTP, despite the ∼40% LTD of EPSCNMDA. In contrast, TBS is likely to produce less depolarization; therefore, LTD of EPSCNMDA may result in sufficient reduction of NMDAR activation to prevent LTP induction. This is in keeping with previous studies that have modified NMDAR activation with different levels of AP5 (Cummings et al., 1996).
LTD of EPSCNMDA: role of mGlu1 receptors
Synaptically induced LTD was unaffected by the mGlu5R antagonist MPEP but was prevented by two distinct mGlu1R inhibitors. This is in contrast to a report demonstrating a role for mGlu5 receptors in LTD of EPSCNMDA in CA1 (Peng et al., 2010). An investigation (Volk et al., 2006) of mGluR-dependent LTD of EPSCAMPA suggested that mGlu1 receptor activation was required for expression of LTD of EPSCAMPA. In contrast, however, in our study mGlu1 receptor antagonism was necessary only during the induction of LTD, suggesting a direct role in the induction, rather than the expression, of LTD.
Expression of LTD of EPSCNMDA
LTD was not associated with a change in EPSCNMDA τw, suggesting no change in NMDAR subunit composition with LTD; this is in agreement with previous work on Group I mGluR-mediated LTD of NMDAR (Ireland and Abraham, 2009). In contrast, an activity-dependent switch from GluN2B-containing to GluN2A-containing NMDARs (Bellone and Nicoll, 2007; Yashiro and Philpot, 2008), which may rely on mGlu5 but not mGlu1 (Peng et al., 2010), has been demonstrated. Therefore, it is possible that at least two different forms of plasticity of EPSCNMDA exist: one dependent on a subunit switch in NMDARs driven by mGlu5 receptors and a second that does not rely on subunit changes and is driven by mGlu1 receptors.
Differences between plasticity of EPSCNMDA at TA-CA1 and SC-CA1
LTD of EPSCNMDA was induced in the SC-CA1 pathway but not in the TA-CA1 pathway. Interestingly, previous work has demonstrated that mGlu receptor-LTD of AMPAR-mediated transmission is significantly smaller at TA-CA1 synapses compared with SC-CA1 synapses (Xu et al., 2010). It has been suggested that EPSCNMDA at SC-CA1 has a greater GluN2B contribution compared with TA-CA1 (Arrigoni and Greene, 2004), which might contribute to the difference in LTD between the two pathways. However, we found no difference in sensitivity to Ro25-6981 and no difference in EPSCNMDA weighted decay time, suggesting it is unlikely that there are differences in NMDAR subunits between the two inputs.
Differences in mGlu1R signaling via Homer proteins may contribute to mechanistic differences in LTD of EPSCNMDA between the SC- and TA-CA1 inputs. Unlike Homer 1b/c, the shorter Homer1a cannot form Homer dimers, which are required for signaling. Therefore, Homer 1a can act as a dominant negative by disrupting mGlu1R signaling protein complexes mediated by long Homers (Kammermeier and Worley, 2007). Thus, the function of mGlu1Rs in LTD critically depends on the ratio between Homer1b/c and Homer1a. Hence, the differential expression of Homer1b/c between the SR and the SLM may, at least in part, explain the difference between LTD of EPSCNMDA between the two pathways. The impact of these differences on signaling mechanisms for mGlu1-LTD of EPSCNMDA is not known but is unlikely to be through PI pathways.
The lack of LTD of EPSCNMDA at the TA-CA1 input correlated with a lack of metaplasticity (i.e., priming stimulation did not affect subsequent LTP). However, given the different conditions required for induction of LTP in TA-CA1 versus SC-CA1, we cannot rule the possibility that the conditions of our experiments precluded metaplasticity from occurring in TA-CA1.
Although the consequences for hippocampal function of a difference in metaplasticity between the inputs is unclear, it is tempting to speculate that these differences may contribute to their distinct roles in memory. For example, the TA-CA1 is necessary for place cell maintenance (Brun et al., 2002) and memory consolidation (Remondes and Schuman, 2002), and it is possible that these functions rely less on metaplasticty but depend on TA firing of CA1 neurones that requires stable NMDAR transmission (Branco and Häusser, 2011). In contrast, the SC-CA1 input is involved in the development of new firing fields and memory acquisition (Nakazawa et al., 2003), properties that may be more reliant on plasticity of EPSCNMDA.
Footnotes
P.A.F. was supported by a Biotechnology and Biological Sciences Research Council PhD studentship. M.B. was supported by a Marie Curie International training network PhD (Symbad-ITN Marie Curie, Grant 238608, 7th Framework Programme of the European Union). This work was supported by the Wellcome Trust and Medical Research Council.
The authors declare no competing ancial interests.
References
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387–399. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Anderson WW, Collingridge GL. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J Neurosci Methods. 2007;162:346–356. doi: 10.1016/j.jneumeth.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Arrigoni E, Greene RW. Schaffer collateral and perforant path inputs activate different subtypes of NMDA receptors on the same CA1 pyramidal cell. Br J Pharmacol. 2004;142:317–322. doi: 10.1038/sj.bjp.0705744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI, Collingridge GL, Lodge D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology. 2007;52:60–70. doi: 10.1016/j.neuropharm.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Alford S, Davies SN, Randall AD, Collingridge GL. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature. 1991;349:156–158. doi: 10.1038/349156a0. [DOI] [PubMed] [Google Scholar]
- Bellone C, Nicoll RA. Rapid bidirectional switching of synaptic NMDA receptors. Neuron. 2007;55:779–785. doi: 10.1016/j.neuron.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Branco T, Häusser M. Synaptic integration gradients in single cortical pyramidal cell dendrites. Neuron. 2011;69:885–892. doi: 10.1016/j.neuron.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun VH, Otnass MK, Molden S, Steffenach HA, Witter MP, Moser MB, Moser EI. Place cells and place recognition maintained by direct entorhinal-hippocampal circuitry. Science. 2002;296:2243–2246. doi: 10.1126/science.1071089. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SF, Bear MF. Visual experience induces long-term potentiation in the primary visual cortex. J Neurosci. 2010;30:16304–16313. doi: 10.1523/JNEUROSCI.4333-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/S0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/S0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci U S A. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme SR, Jones OD, Abraham WC. Emerging roles of metaplasticity in behaviour and disease. Trends Neurosci. 2013;36:353–362. doi: 10.1016/j.tins.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Hunt DL, Castillo PE. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol. 2012;22:496–508. doi: 10.1016/j.conb.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DL, Puente N, Grandes P, Castillo PE. Bidirectional NMDA receptor plasticity controls CA3 output and heterosynaptic metaplasticity. Nat Neurosci. 2013;16:1049–1059. doi: 10.1038/nn.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland DR, Abraham WC. Mechanisms of group I mGluR-dependent long-term depression of NMDA receptor-mediated transmission at Schaffer collateral-CA1 synapses. J Neurophysiol. 2009;101:1375–1385. doi: 10.1152/jn.90643.2008. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ, Worley PF. Homer 1a uncouples metabotropic glutamate receptor 5 from postsynaptic effectors. Proc Natl Acad Sci U S A. 2007;104:6055–6060. doi: 10.1073/pnas.0608991104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew JN, Ducarre JM, Pflimlin MC, Mutel V, Kemp JA. Activity-dependent presynaptic autoinhibition by group II metabotropic glutamate receptors at the perforant path inputs to the dentate gyrus and CA1. Neuropharmacology. 2001;40:20–27. doi: 10.1016/S0028-3908(00)00118-0. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron. 2008;57:108–120. doi: 10.1016/j.neuron.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Yasuda R, Ehlers MD. Metaplasticity at single glutamatergic synapses. Neuron. 2010;66:859–870. doi: 10.1016/j.neuron.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Murphy KP, Bliss TV. Photolytically released nitric oxide produces a delayed but persistent suppression of LTP in area CA1 of the rat hippocampal slice. J Physiol. 1999;515:453–462. doi: 10.1111/j.1469-7793.1999.453ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Sun LD, Quirk MC, Rondi-Reig L, Wilson MA, Tonegawa S. Hippocampal CA3 NMDA receptors are crucial for memory acquisition of one-time experience. Neuron. 2003;38:305–315. doi: 10.1016/S0896-6273(03)00165-X. [DOI] [PubMed] [Google Scholar]
- Peng Y, Zhao J, Gu QH, Chen RQ, Xu Z, Yan JZ, Wang SH, Liu SY, Chen Z, Lu W. Distinct trafficking and expression mechanisms underlie LTP and LTD of NMDA receptor-mediated synaptic responses. Hippocampus. 2010;20:646–658. doi: 10.1002/hipo.20654. [DOI] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Rebola N, Srikumar BN, Mulle C. Activity-dependent synaptic plasticity of NMDA receptors. J Physiol. 2010;588:93–99. doi: 10.1113/jphysiol.2009.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remondes M, Schuman EM. Direct cortical input modulates plasticity and spiking in CA1 pyramidal neurons. Nature. 2002;416:736–740. doi: 10.1038/416736a. [DOI] [PubMed] [Google Scholar]
- van Strien NM, Cappaert NL, Witter MP. The anatomy of memory: an interactive overview of the parahippocampal-hippocampal network. Nat Rev Neurosci. 2009;10:272–282. doi: 10.1038/nrn2614. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Daly CA, Huber KM. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J Neurophysiol. 2006;95:2427–2438. doi: 10.1152/jn.00383.2005. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Xu JY, Chen R, Zhang J, Chen C. Endocannabinoids differentially modulate synaptic plasticity in rat hippocampal CA1 pyramidal neurons. PLoS One. 2010;5:e10306. doi: 10.1371/journal.pone.0010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]