

Abstract
Reovirus is an icosahedral, double-stranded (ds) RNA virus that uses viral polymerases packaged within the viral core to transcribe its ten distinct plus-strand RNAs. To localize these polymerases, the structure of the reovirion was refined to a resolution of 7.6 Å by cryo-electron microscopy (cryo-EM) and three-dimensional (3D) image reconstruction. X-ray crystal models of reovirus proteins, including polymerase λ3, were then fitted into the density map. Each copy of λ3 was found anchored to the inner surface of the icosahedral core shell, making major contacts with three molecules of shell protein λ1 and overlapping, but not centering on, a five-fold axis. The overlap explains why only one copy of λ3 is bound per vertex. λ3 is furthermore oriented with its transcript exit channel facing a small channel through the λ1 shell, suggesting how the nascent RNA is passed into the large external cavity of the pentameric capping enzyme complex formed by protein λ2.
All viruses with either a single-stranded, minus-sense RNA genome (such as influenza and measles viruses) or a dsRNA genome (such as reoviruses and rotaviruses) must package virally encoded RNA-dependent RNA polymerase (RdRp) molecules within virions to transcribe the genome into single-stranded, plus-sense RNA for translation and replication during infection. The dsRNA viruses are distinct, however, in having icosahedral particles within which the RdRp molecules are regularly bound and the genome remains enclosed throughout transcription. dsRNA virus particles thus provide useful models in which to study structural aspects of RNA-dependent transcription.
Reovirus, a member of the Reoviridae family of animal and plant viruses, has a genome comprising ten dsRNA segments, which are surrounded in virions by a multilayered, icosahedral protein capsid (diameter ~850 Å; mass ~110 MDa1). The outer capsid layer is arranged with quasi T = 13(laevo) symmetry and includes 200 heterohexamers of the μ1 and σ3 proteins (μ13σ33) (refs. 2,3) and 12 homotrimers of the σ1 protein4–7. These proteins are released during cell entry after fulfilling their roles in stability (σ3), attachment (σ1) and membrane penetration (μ1) (refs. 8–10). The 15-MDa, 23.5-kilobase pair genome is encased by an inner capsid layer having T = 1 symmetry and composed of 60 dimers of the λ1 protein11. This inner layer remains surrounding the genome after cell entry and may protect it from eliciting dsRNA-dependent host responses12,13. Also in the subviral ‘core’ that enters the cytoplasm are two proteins external to the λ1 shell (150 monomers of σ2 (nodules) and 12 pentamers of λ2 (turrets)) and two structurally minor proteins internal to the shell (12 copies of λ3 and ~24 copies of μ2) (refs. 14,15). These proteins constitute the enzymatic machinery for synthesizing and exporting the viral plus-strand RNAs.
Core protein λ3 (142 kDa) is the viral RdRp16–18. Core protein μ2 (83 kDa) has a genetic influence on the transcriptase and nucleoside triphosphatase (NTPase) activities of cores19,20 and functions as both an NTPase and an RNA 5′-triphosphatase in vitro (J. Kim, J.S.L. Parker and M.L.N., unpublished data). Shell protein λ1 (142 kDa) also has a genetic influence on the NTPase activities of cores21 and mediates NTPase, RNA 5′-triphosphatase and RNA and DNA helicase activities in vitro22,23. Despite these in vitro findings, the specific roles of μ2 and λ1 in viral mRNA synthesis remain unclear. The roles of turret protein λ2 (144 kDa) are more certain, in that it mediates the last three of four reactions in RNA capping24: RNA 5′-guanylyltransferase, 7-N-methyltransferase and 2′-O-methyltransferase11,25–29. The λ2 turrets also serve as the conduits through which plus-strand RNAs are exported from the core cotranscriptionally30–32. Both μ2 and λ1 are candidates to mediate the first reaction in capping, the RNA 5′-triphosphatase activity, based on their in vitro activities discussed above. The series of reactions in reovirus cap formation is the same as that used by eukaryotes and many other viruses33,34.
The RdRp λ3 has been localized beneath each of the five-fold axes of the λ1 shell11,15, placing it in an approximate position to export mRNA into the λ2 turret for completion of capping and export. The RdRps in other members of the Reoviridae family, such as rotavirus35–36, bluetongue virus (orbivirus)37–38 and cytoplasmic polyhedrosis virus (cypovirus)39, are also located beneath the five-fold axes of the inner shell. However, in none of these viruses have the positions and orientations of the RdRps been precisely determined. For all these viruses, a generally accepted, though largely unproven, model is that each of the 10–12 genome segments is dedicated to its own transcriptase complex within the particle15,18,31,35–39.
X-ray crystal structures have been reported for the reovirus core particle11 and the recombinant λ3 protein18. The 3.6-Å core structure was notable for showing how the λ1 shell is organized and how the capping enzyme domains in the pentameric λ2 turrets are arranged. It did not, however, reveal the λ3 and μ2 proteins, perhaps because those components are asymmetrically or irregularly positioned in cores. The 2.5-Å λ3 structure was notable for showing the organization of the globular protein with a central catalytic cavity and four channels for the entry and exit of substrates and products. The λ3 crystals were also remarkable because they remained intact after several rounds of RNA polymerization so that the roles of the different channels could be inferred from the structures of catalytic complexes. To understand more fully how cores mediate transcription and cotranscriptional capping and export of the viral plusstrand RNAs, we wished to know how the RdRp λ3 fits into the particle and interacts with other components. For this report, we identified the positions of λ3 by fitting the λ3 X-ray coordinates into a new, 7.6-Å map of the mammalian reovirion obtained by cryo-EM and 3D image reconstruction.
RESULTS
Refined cryo-EM reconstruction of the reovirion
A 3D reconstruction of the reovirus type 3 Dearing virion was computed from images of 7,939 particles by means of cryo-EM and image processing40. A conservative estimate of the final resolution, 7.6 Å, was determined by comparing two reconstructions computed from independent halves of the data set40 (Fig. 1). Inspection of the reconstruction (Fig. 2) revealed numerous rodlike features ~6 Å in diameter (suggestive of α-helices), and sheetlike features ~6 Å in thickness (suggestive of β-sheets) (Fig. 2b). The identification and reliability of these apparent secondary structure elements were assessed by comparing relevant portions of the cryo-EM map with X-ray crystal models of all five major capsid proteins, including each of the uniquely positioned copies of λ1, σ2, μ1 and σ3 (refs. 2,3) (Fig. 2c–e). Two distinct model-fitting programs, EMfit41 and SITUS42 (see Methods and Supplementary Methods online), were used for refining these comparisons. Of the 342 α-helical segments containing five or more residues in the X-ray models of these subunits, all were found to match rodlike features in the EM map contoured at 1.5 σ above noise, and all but one to match features at 2.0 σ (Table 1). Of 92 total β-sheets containing three or more β-strands in these subunits, all were found to match sheetlike features in the EM map at 2.0 σ (Table 1). This marked agreement between the EM- and X-ray-derived structures established the reliability of the cryo-EM data and also indicated that only limited shifts have occurred in any of these elements between the proteins in crystals and native virions.
Figure 1.

Assessment of the resolution limit in reovirion reconstruction. The set of 7,939 particle images was subdivided randomly into two subsets, and independent reconstructions were computed from these data. Fourier shell correlation (solid curve) and phase difference (dashed curve) criteria40 demonstrated that the resolution of the final reconstruction was at least 7.6 Å.
Figure 2.

Reovirion cryo-EM reconstructions. (a) Shaded-surface representation of complete virion, rendered at a resolution of 20 Å and viewed along a five-fold axis with a λ2 pentamer (blue) and a μ13σ33 heterohexamer (μ1, green; σ3, red) highlighted. The outlined region (dashed box) is shown at higher resolution in b. (b) Close-up view of the virion map rendered at a resolution of 7.6 Å illustrates fine details such as rodlike densities and also shows the interlocking arrangements of the five λ2 subunits in a pentamer (purple, green, magenta, greenish yellow and dark orange) and the three copies each of μ1 (red, yellow and light green) and σ3 (orange, dark blue and turquoise) that comprise a heterohexamer. (c) Equatorial section (2.2 Å thick) from the 20-Å rendering shows one quadrant of the view along a two-fold axis and identifies the approximate locations of viral components. (d) Same as c for the 7.6-Å rendering with locations of five-fold and three-fold axes indicated (the right and bottom edges of the panel coincide with two-fold axes). Numerous punctate features arise from α-helices viewed end on. (e) Stereo view of a small portion (N-terminal methyltransferase domain) of the λ2 X-ray model (ribbon) fitted into the corresponding density (wire cage) in the 7.6-Å EM map with α-helices in green and β-strands in yellow. Scale bars are labeled; bar in c also applies to d.
Table 1.
Secondary-structure elements in cryo-EM map of reovirion and quantitative fit of atomic models to the density map
| Protein | α-helicesa | β-sheetsa | LCCb |
|---|---|---|---|
| σ3(Q)c | 24 (24) | 9 (9) | 0.327 |
| σ3(R)c | 24 (24) | 9 (9) | 0.344 |
| σ3(S)c | 24 (24) | 9 (9) | 0.341 |
| σ3(T)c | 8 (8) | 3 (3) | 0.337 |
| μ1(Q)c | 45 (45) | 12 (12) | 0.248 |
| μ1(R)c | 45 (45) | 12 (12) | 0.244 |
| μ1(S)c | 45 (45) | 12 (12) | 0.241 |
| μ1(T)c | 15 (15) | 4 (4) | 0.242 |
| λ2 | 25 (25) | 10 (10) | 0.554d |
| σ2(5f)e | 11 (11) | 0 (0) | 0.409 |
| σ2(3f)e | 11 (11) | 0 (0) | 0.421 |
| σ2(2f)e | 11 (10) | 0 (0) | 0.283 |
| λ1-Af | 26 (26) | 6 (6) | 0.332 |
| λ1-Bf | 28 (28) | 6 (6) | 0.327 |
| Totalg | 342 (341) | 92 (92) | – |
| λ3h | 48 (33) | 2 (0) | 0.067 |
| λ3i | 48 (33) | 2 (0) | 0.146 |
| λ3j | 48 (5) | 2 (0) | 0.017 |
| λ3k | 48 (5) | 2 (0) | 0.021 |
Listed first is the number of α-helices with five or more residues or β-sheets with three or more strands in the atomic model of each protein, followed in parentheses by the number of each secondary-structure element that is clearly visible in the cryo-EM density map.
Laplacian correlation coefficient computed by SITUS for atomic model fitted into cryo-EM density map.
Based on nomenclature defined for bluetongue virus58, Q, R, S and T identify the four quasi-equivalent trimers in the icosahedral lattice. Total statistics are listed for all three, nonequivalent copies of the Q, R and S subunits of σ3 and μ1.
Value for λ2 pentamer.
Different types of σ2 subunits are close to the icosahedral five-fold (5f) axes, close to the icosahedral three-fold (3f) axes or at the icosahedral two-fold (2f) axes.
λ1-A subunits approach the icosahedral five-fold and two-fold axes; λ1-B subunits approach the icosahedral three-fold axes.
Totals for the major capsid proteins listed above in this table.
λ3 monomer.
λ3 pentamer constructed by five-fold replication of the single fitted molecule.
Randomly oriented λ3 monomer.
Randomly oriented λ3 pentamer.
The only rodlike densities in the cryo-EM map that did not precisely superimpose with α-helices in the X-ray crystal models were found in the same surface-exposed helix of the four uniquely positioned copies of σ3 (residues 225–231). Even in this case, however, the EM density was shifted by only ~1.1 Å from the X-ray model. In addition, some extended chains and loops in the crystal models, between the α-helices and β-sheets, could not be assigned to EM densities. These discrepancies suggest that some features may not be resolved at 7.6 Å or that those regions may be flexible in virions. A few other notable differences between the EM and X-ray structures of the outer capsid were observed. For example, the C-terminal portions of μ1 were only evident in the cryo-EM map (X.Z., M.L.N. and T.S.B., unpublished data). Despite small local differences, the overall agreement between the EM and X-ray structures provided a firm basis for further analyses.
Features of the λ1 shell
The two copies of λ1 in each of the 60 dimers composing the core shell occupy symmetrically distinct (that is, not quasi-equivalent) positions and have slightly different conformations (A and B)11. Each λ1-A subunit spans between one five-fold and one two-fold axis, whereas each λ1-B subunit approaches one three-fold axis. A pentamer of λ2 sits atop the λ1 shell around each icosahedral vertex. The σ2 monomers that also sit atop the shell occupy three unique positions: 60 are adjacent to the λ2 pentamers, 60 are near the icosahedral three-fold axes and 30 are in either of two possible orientations across the two-fold axes11.
As can be inferred from the preceding statements, all α-helices and β-sheets in both λ1 X-ray structures (A and B) were well fitted to densities in the cryo-EM map (Table 1). Despite this excellent match of major secondary structure elements, some loops and extended chains in either type of λ1 subunit did not have corresponding densities in the EM map. In addition, many of the λ1 residues that were missing from the X-ray structures11, presumably as a result of disordering or flexibility, were likewise not identifiable in the EM map. These included small portions of the N-terminal 210 residues of λ1-B. Two exceptions are noteworthy because they occur on the inner surface of the shell in regions that abut the RdRp λ3 (described below). In both cases, these regions are visible in the EM map as loops connecting α-helices. Residues 584–587 in λ1-A lie ~31 Å from the closest five-fold axis and connect two helices comprising residues 572–583 and 588–600. Residues 563–570 in λ1-B lie ~56 Å from the closest five-fold axis and connect two helices comprising residues 553–561 and 572–581. Densities in the EM map that are possibly attributable to portions of the N-terminal 240 residues of λ1-A, which are also missing from the core crystal structure11, are discussed below.
The excellent fits of both types of λ1 subunits demonstrate that the shell must be a generally stable and fixed structure in virions and non-transcribing cores. When virions and cores are compared, there is no evidence for expansion or contraction of the shell, and rotation of the λ1 subunits relative to the symmetry axes or each other is not detected.
Fitting the X-ray model of λ3 into the cryo-EM map
Density features at radii beneath the λ1 shell in the cryo-EM map (Figs. 2c,d and 3a–c) are mostly attributable to the genome, but must also include λ3 and μ2, as well as the N-terminal 240 residues of λ1-A. The 142-kDa RdRp λ3 is present in about one copy per five-fold axis14. Because they are inactive in virions43, we hypothesized that all of the λ3 subunits within those particles are probably in structurally homogeneous positions and conformations. This assumption, coupled with knowledge of the 2.5-Å X-ray structure of recombinant λ3 (ref. 18), stimulated our efforts to identify λ3 in the cryo-EM map.
Figure 3.

Fits of λ1 and λ3 X-ray models into reovirion cryo-EM map. (a) Slab through the cryo-EM map (10 Å thick), encompassing a five-fold axis and including core densities from low radii (~172 Å at bottom of panel) to the outer surface of the λ1 shell (radius ≈ 277 Å at top). (b) Same as a but with X-ray models of λ1 (black Cα backbone) and λ3 (dark orange, blue, orange, magenta and green Cα backbones) fit into the cryo-EM map. (c) Slab through the cryo-EM map (10 Å thick), viewed along a five-fold axis and toward the virion center, with the X-ray model of the λ3 pentamer fitted into the map. Thin dashed lines in b and c mark the boundaries of the slabbed regions shown, respectively, in c and a,b. Scale bar for a–c is labeled. (d) Plot of Laplacian correlation coefficients (LCC) computed by SITUS42, which quantify the fit of the λ3 monomer X-ray model into a defined search volume within the reovirion cryo-EM density map. For this calculation, the cryo-EM map was rotated to make a five-fold axis coincide with the z-axis of a Cartesian coordinate system, and a region encompassing more than one asymmetric unit was selected as the search volume, as described in Methods and Supplementary Methods online. The fit of the λ3 X-ray model into this volume was based on an exhaustive six-dimensional search with angular and translational sampling intervals of 5° and 2.2 Å, respectively. Here the LCCs are only plotted for the z-plane, yielding the best fit, and the value at each x and y position in the plane corresponds to the orientation of the model at that position that gives the highest LCC. The x- and y-axes of the plot identify the pixel coordinates within the search volume, with each pixel representing a step size of 2.2 Å. The value of the maximum peak (0.047) rose only to 0.052 upon Powell minimization refinement42. (e) Plot of the LCC for monomer and pentamer X-ray models of λ3 and λ2 fitted into the reovirion cryo-EM density search volume and for different rotation angles about the icosahedral five-fold axis. The 0° rotation angle for each model corresponds to the orientation and position of that model that gives the best fit as obtained in the experiments illustrated in d. (f) Stereo view of a small portion of the λ3 X-ray pentamer model (blue and orange ribbons distinguish adjacent monomers) fitted into the corresponding density (wire cage) in the 7.6-Å EM map. This region of the map includes minimal overlap between neighboring λ3 models.
We used the SITUS42 and EMfit41 programs for locating λ3 in the virion EM map (see Methods and Supplementary Methods online). Initial positioning of the X-ray model of monomeric λ3 (~180,000 Å3) into the reconstruction was done with SITUS. In experiments using different portions of the EM map as search volumes, we found that SITUS generated the same set of five nearly identical symmetry-related fits of λ3 to densities beneath the λ1 shell (Fig. 3d). To assess the uniqueness of the λ3 fits to these internal volumes, X-ray monomer models of λ1-A, λ1-B, λ2, μ1, σ2 and σ3 were also tested. The best fits of these six other X-ray models gave correlations much poorer than that of λ3 (Fig. 3e and data not shown). A pentamer model based on this fit for monomeric λ3 was constructed by replicating one molecule five times around the closest five-fold axis (Fig. 3b,c). SITUS fitting of this pentamer model showed that different rotational orientations were easily discriminated within a few degrees (Fig. 3e). This procedure was similarly sensitive to misalignments in the position or orientation of monomeric λ3 used to generate the pentamer models (data not shown). A pentamer model of λ2 was also tested against the internal search volume and gave a substantially poorer correlation than that of λ3 (Fig. 3e). Finally, the sensitivity of the SITUS fittings to noise in the EM map was assessed by means of several tests, all of which affirmed the reliability of the reported results. EMfit confirmed the SITUS result and identified essentially the same position for λ3 even for input models that were purposely misaligned (see Supplementary Methods online).
General features of the λ3 fit
The fit of the λ3 X-ray crystal model into the virion cryo-EM map is consistent with both structural and functional features expected for λ3. The intermolecular overlaps that occur in the λ3 pentamer model show that there is space for at most two, but more probably only one, λ3 molecule beneath each five-fold axis of the λ1 shell. For each λ3 monomer in the pentamer model, 7.4% of its structure overlaps with all four, 8.6% with three, 19.6% with two, 33.1% with one and 31.3% with none (Fig. 3f) of the other monomers. Thus, each monomer overlaps with all four others in the pentamer model, but the most extensive clashes occur with its two flanking neighbors. Small portions of each λ3 monomer cross the five-fold axis at several radii (Fig. 3b,c). Depending on the extent to which the X-ray model precisely mimics the λ3 structure in virions, this feature alone would preclude binding of all but one monomer at each vertex. This agrees with previous gel-based estimates of 12 copies of λ3 per particle14 and is consistent with modeling experiments indicating that the signal-to-noise ratio in the λ2 density exceeds that in the λ3 density by more than four-fold (see Supplementary Methods online).
The fitted λ3 model indicates extensive contacts between λ3 and the inner surface of the λ1 shell (Fig. 4). Each λ3 molecule, with ~47,000 Å2 of solvent-accessible surface in solution, interacts with one λ1-B and all five λ1-A subunits, creating a solvent-inaccessible surface of ~2,420 Å2 (Fig. 4b–d). Twelve residues of the λ1-B subunit are 5 Å or closer to λ3 in the fitted structures. For the five respective λ1-A subunits, 23, 13, 7, 2 and 2 residues approach within 5 Å of λ3 (Fig. 4c). Based on the fit, a substantial portion of the λ3-λ1 interactions results from electrostatic complementation, especially near the five-fold axis, where λ3 is basic and the λ1 shell is acidic (Fig. 4c,d). The broad interface between λ3 and λ1 suggests that λ3 is firmly anchored to the shell, consistent with the ‘fixed-polymerase–moving-template’ model of reovirus transcription15,18,44,45.
Figure 4.

Location of λ3 inside reovirus λ1 shell. (a) Space-filling representation of the inner surface of the λ1 shell viewed along a five-fold axis (white star). The five identical λ1-A subunits (those closest to the five-fold axis) are highlighted in yellow (A1), blue (A2), brown (A3), green (A4) and cyan (A5), and all five λ1-B subunits are shown in violet. Potential pores at the interfaces between neighboring λ1-A subunits are indicated by small red circles (shown also in c). The red line marks the position of a plane, ~6 Å from the five-fold axis, to one side of which over half of the pseudo atomic model of the reovirion was removed for the illustrations in e and f. (b) Same as a but with space-filling model of λ3 (red) added to show its position relative to the λ1 shell’s inner surface. The white arrow indicates the view direction in the cutaway illustrations in e and f. (c) Same as a but rendered to show the electrostatic surface potential of the λ1 shell inner wall. The λ3 footprint (black lines) outlines regions in λ1 within 5 Å of λ3 residues. (d) Electrostatic surface potential of λ3 overlaid with the footprint shown in c. The λ3 molecule (at right) and an enantiomeric version of it (at left) are viewed in a direction facing the mRNA exit channel and the surface that contacts the λ1 shell. The λ3 contact surface mainly consists of basic and neutral patches that seem complementary to the neutral and slightly acidic λ1 footprint seen in c. The λ3 enantiomer is included to help visualize the contact surface complementarity. (e) Shaded-surface, cutaway view of the pseudo atomic model of the reovirion, oriented with a five-fold axis vertical (red line) shows the arrangement of the globular λ3 molecules (red) on the inner surface of the λ1 shell. Most or all of the λ1-A1, -B2, -A2, -B3 and -A3 subunits, and small portions of the λ1-B1, -A5, -A4 and -B4 subunits, have been cut away. (f) Enlarged view of region outlined in e with λ2 in yellow, λ1 in blue and λ3 in red. Scale bars are labeled; bar in d also applies to a–c.
Despite the inherent high noise level in the central parts of the cryo-EM map, 33 of the 48 α-helices in the λ3 X-ray crystal model were clearly identified and fitted by both SITUS and EMfit (Table 1). Of the remaining 15 helices, 8 were displaced from adjacent unassigned densities and 7 were completely missing from the map. Of these 15 ‘unfit’ helices, 13 are found on the λ3 surface, with 7 (3 moved and 4 missing) on the surface that abuts λ1 (Fig. 5). These findings suggest that limited, local conformational changes occur on the λ3 surface during assembly into particles. Some of these changes seem to accompany interactions between λ3 and λ1, and others may reflect interactions between λ3 and μ2 or genomic RNA.
Figure 5.
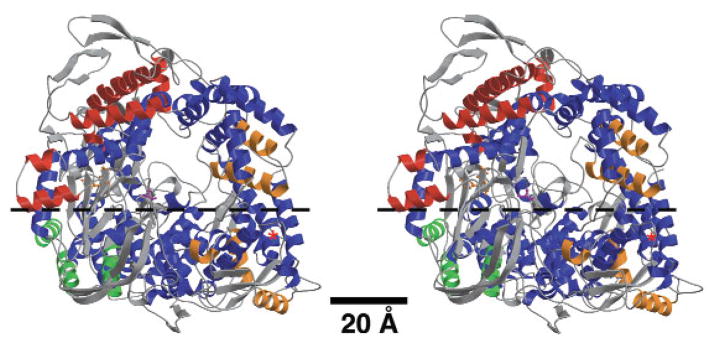
Stereo view of λ3 X-ray ribbon model with α-helices color-coded to signify position and correspondence with features in the cryo-EM map. The λ3 molecule is oriented with the mRNA exit channel facing the observer, a view opposite that for λ3 depicted in Figure 4b. Of the 48 total α-helices in λ3 (Table 1), the 33 that were well represented in the cryo-EM map are blue. The remaining 15 helices are distributed mainly at the periphery of the λ3 molecule, with three occurring in the N-terminal domain (green), seven in the polymerase domain (red) and five in the C-terminal domain (orange). A red asterisk marks the position of the nearest five-fold axis. Ball-and-stick figures highlight active site residues Asp585, Asp734 and Asp735 in orange and the priming NTP platform (Ser561) in magenta. A dotted line identifies the horizontal plane used to segment the λ3 molecule (the approximate one-half above the arrows was extracted) to reveal some of its internal features in Figure 4f.
DISCUSSION
Orientation of λ3 and route of transcript exit
The reovirus RdRp λ3 has been called a polymerase in a cage on the basis of its three-domain structure with four channels leading to the catalytic site18. The central polymerase domain has fingers, palm and thumb subdomains similar to those in other RNA polymerases46. N- and C-terminal domains create the ‘cage’ and enclose the catalytic site in a large central cavity18. Of the four channels, two correspond to entry paths: one for the single-stranded RNA template and one for NTPs. The other two channels provide exit paths for the RNA template (or the dsRNA product during replication) and the plus-strand (mRNA) transcript. An RNA cap-binding site on the surface of λ3, adjacent to the template entry channel, is thought to tether the capped 5′ end of the genomic plus strand throughout transcription, promoting the polymerase to reinitiate at the 3′ end of the minus strand after the end of each transcription cycle18.
The new fitting results show λ3 oriented such that its plus-strand RNA exit channel faces directly toward the λ1 shell, its NTP entry and template exit channels face approximately parallel to the shell and its cap-binding site and template entry channel face toward the particle center (Figs. 4e,f and 6). In addition, the bulk of λ3 lies to one side of the five-fold axis (Figs. 3b,c and 4b,c,e), with the mRNA exit channel directed toward the interface between two λ1-A subunits and one λ1-B subunit, ~37 Å from the five-fold axis (Fig. 4a–c). This region of λ1 has been identified as an opening to a small channel through the shell47,48 (Figs. 4a,c and 6). Although largely obscured by side chains in the core crystal structure, this channel is presumably widened during transcription to allow passage of the nascent transcripts into the large cavity of the λ2 turret, where they can be capped and released into the cytosol (Fig. 6). The outer end of this small channel opens directly beneath one λ2 subunit, and a groove on the underside of that subunit leads into the λ2 capping chamber11 (Fig. 6). Contacts between λ3 and the shell seem to create a seal around the mouth of the transcript exit channel in λ3 (Fig. 4c,d), limiting diffusion of transcripts away from the region of the interface between (λ1-A)2 and λ1-B. The simplest hypothesis is therefore that this small peripentonal channel, and not the similarly small channel directly at the five-fold axis11, is where the nascent RNA exits through the λ1 shell. There are at least four loops in λ1 that seem capable of moving by several angstroms each to create a shell-spanning channel wide enough for RNA transit near the interface between (λ1-A)2 and λ1-B. Two of these loops are in the λ1-A5 subunit (residues 609–618 and 662–687), and two are in the λ1-A1 subunit (residues 747–756 and 890–895). With the template exit channel facing approximately parallel to the λ1 shell and the cap-binding site and template entry channel facing toward the particle center, these channels are properly positioned for the dsRNA genome segments to be retained inside the particle interior throughout transcription, as is known to occur30–32,44,45.
Figure 6.

Space-filling, cutaway view of the reovirus core, showing a proposed exit pathway for newly synthesized plus-strand (+) RNA transcripts leading from λ3 through the λ1 shell to the λ2 cavity. The view direction and region depicted are approximately the same as shown in Figure 4f, but additional portions of λ2 were included to help identify the large exit cavity. The approximate locations of four RNA capping guanylyltransferases (GTase1–4), three N-terminal methyltransferases (N-MTase1–3) and three C-terminal methyltransferases (C-MTase1–3) in the λ2 pentamer subunits are shown. The minus and plus strands of the template dsRNA and the nascent plus-strand transcript are orange, black and yellow, respectively. The transcript is shown exiting the channel at the top of λ3, traversing a putative channel formed at the interfaces between two λ1-A subunits (A1 and A5) and between λ2 and the λ1-A5 subunit, and finally into the large λ2 channel. The cap structure (m7GpppG m2′) at the 5′ end of the dsRNA plus strand is shown tethered to the cap-binding region (green) on the λ3 molecule as previously proposed18. The λ1 subunits are colored according to the same scheme as in Figure 4a,b. The nascent transcript is shown as having already been modified with a 5′ cap (green) by the RNA 5′ triphosphatase (position unknown) and λ2 capping domains.
Possible locations of μ2 and the λ1-A N terminus
Melting and reannealing of the dsRNA genome segments during the course of reovirus transcription, or termination and reinitiation between transcription cycles, may require viral factors in addition to λ3 (refs. 18–22). As suggested for bluetongue virus49, the reovirus core may also require a diffusion-enhanced mechanism to pump NTPs into the particle interior for maintaining the observed rate of transcript elongation. Positions of the template entry, template exit and NTP entry channels identified by the λ3 fit in this study suggest that any additional factors would need to reside at the bottom or sides of λ3 beneath the λ1 shell. As suggested above, candidate factors are μ2 and the N-terminal 240 residues of λ1-A, neither of which have been precisely localized in the particle interior11. There are in fact unassigned densities in the 7.6-Å map that are internal to the shell, abut λ3 and do not seem attributable to dsRNA (Fig. 3b,c). Some of these are at the base of λ3, near the cap-binding site and template entry channel. Stronger and more extensive densities extend up the side of λ3, around the NTP entry channel. Portions of μ2 and the N-terminal residues of λ1-A might account for these unassigned densities. In the absence of good models (that is, without X-ray data for these components), however, we have no means to refine or to test the reliability of these assignments.
Comparisons with other Reoviridae viruses
RdRp molecules anchored to the inner surface of the 120-subunit T = 1 shell at or near each five-fold axis are probably a common feature of all Reoviridae members. Different members, however, may use channels at different positions in their inner shells for exporting the plus-strand RNAs produced by their RdRps. A reconstruction of rotavirus particles in the act of transcription has suggested that each of their transcripts exits through a peripentonal channel, not through the one directly at each five-fold axis50. In contrast, transcribing bluetongue virus particles are thought to use their pentonal channels, which are expanded during transcription, for RNA export49.
Some Reoviridae members, such as bluetongue virus, have all capping enzymes internal to the inner shell, unlike reoviruses, which have three capping enzymes external to that shell (Fig. 6). Thus, in the former cases, the viral transcripts must encounter all four capping enzymes before passing through the shell. This could be accomplished by several different arrangements, but two seem simplest. The capping enzymes could physically intervene between RdRp and shell, such that the latter do not make substantial contacts. In this case, the RdRp might orient as in reovirus particles, directing the transcript ‘upward’ through the capping enzymes and then the shell. Because bluetongue RdRp VP1 and capping enzyme VP4 can each bind independently to the shell within particles51, however, this stacked arrangement seems improbable. The capping enzymes could alternatively be positioned to one side of the RdRp so that the nascent transcript is passed out the side of the RdRp, into the adjacent capping enzymes, and then redirected ‘upward’ toward the shell. In this case, the RdRp would be rotated by ~90° relative to its orientation in reovirus particles.
METHODS
Cryo-EM and image processing
Reovirus type 3 Dearing virions were purified as described4, and small aliquots at a concentration of ~2 mg ml−1 were vitrified and maintained at −176 °C as described40. Micrographs were recorded at a range of defocus settings and under low-dose conditions (~20 e− Å−2) in FEI/Philips CM200 and CM300 FEG microscopes at nominal magnifications of 38,000× and 45,000×, respectively. A Zeiss PHODIS scanner was used to digitize each micrograph at 7-μm intervals, and these data were interpolated in RobEM (http://bilbo.bio.purdue.edu/~baker/programs/programs.html) to yield a pixel size corresponding to 2.3 Å in the specimen. The actual pixel size was later determined to be 2.21 Å based on scaling of the EM data to the X-ray crystal structure of the reovirus core particle (PDB entry 1EJ6)11.
A total of 54 micrographs with a defocus range of 1.3–3.2 μm under focus were selected for processing. RobEM was used to extract 8,996 individual particle images, each 511 × 511 pixels in size, from these micrographs. The origin and orientation parameters (x, y, θ, ϕ and ω) for each image were initially determined using polar Fourier transform (PFT) procedures52, with a reconstruction of the reovirion at a resolution of 18 Å used as the initial model. These parameters were refined by means of a modified version of the original PFT protocol: the Fourier transform of each image was adjusted to correct for effects of the microscope contrast transfer function (CTF) by means of FILTER2 (δ2 = 0.1) as described53, and the real and imaginary components of each image transform were correlated against unmodified components from corresponding data derived from a 7 × 7 search grid of model projections.
Initial refinement was restricted to the 4,859 particle images obtained from the 28 micrographs recorded at 200 kV and was conducted over 19 cycles with an orientation grid search of 0.2°. Each cycle consisted of PFT refinement of origin and orientation parameters, and subsequent computation of a 3D reconstruction and assessment of resolution40. This initial processing of the 200-kV data led to a model of the virion structure at a resolution of ~8.6 Å. The remaining 4,137 images, all recorded at 300 kV, were then added to the data set, and 15 more cycles were conducted over 0.1° search intervals. These operations were carried out on two dedicated Linux computers: a cluster with ten Intel Pentium III 500-MHz processors and a PC with dual AMD Athlon XP 1.4-GHz processors. Each cycle of refinement required 8 d.
The sensitivity and reliability of the orientation and origin refinement procedures were enhanced by limiting or modifying the structure factor data used to measure correlation coefficients between transforms of images and projections. First, data below a spatial frequency of 1/60 Å were omitted from the refinement calculations. Second, only data for which the absolute value of CTF exceeded 0.2 were used. Third, amplitude thresholds were imposed at each spatial frequency in both image and projection transforms such that data with an amplitude <0.6 of the average at a given frequency were omitted (for projection data, average amplitudes were computed from the entire set of 49 projections in the search grid for each image). Finally, transform data were multiplied by an inverse temperature factor to enhance the weight of the high spatial frequency components used in refinement, and its magnitude was gradually increased from 1/300 Å2 to 1/900 Å2 during the course of refinement.
At the end of each cycle of refinement, two independent reconstructions were computed using the EM3DR Fourier-Bessel program (http://bilbo.bio.purdue.edu/~baker/programs/programs.html) or a parallelized version of it (http://bilbo.bio.purdue.edu/~viruswww/Rossmann_home/river_programs/para-pft-em3dr.shtml), and the new resolution limit was assessed40,53. The subsequent cycle of refinement included all Fourier data to this resolution limit. Refinement progressed until no further improvement in resolution was realized with the current data or after more images were added. At the end of refinement, 7,939 of 8,996 particle images were included in the reconstruction reported here. During all cycles of refinement, the effects of the microscope CTF on each image were compensated in part by means of a pair of filtering operations (δ1 = 0.05, δ2 = 0.1) as described53. The distribution of particle orientations was sufficiently random as measured by the eigenvalue spectrum (all inverse eigenvalues <0.01) to allow computation of the final reconstruction to a resolution of 7.6 Å (ref. 40). The characteristic attenuation of high spatial frequency data for cryo-EM images was compensated in part by applying an inverse temperature factor of 1/400 Å2 to the final map53.
For resolution assessment, particle images were randomly sorted into two sets from which independent reconstructions were computed40. Densities attributed to genome (radius <243 Å) and solvent outside the capsid (radius >438 Å) were damped to background values with a Gaussian-type mask 26 Å wide54, and the 3D Fourier transform of each reconstruction was computed. Structure factors from each map were compared to compute Fourier shell correlation coefficients (FSCs) and phase differences as a function of spatial frequency (Fig. 1). A conservative estimate of the resolution limit was based on an FSC threshold of 0.5 and a phase difference threshold of 45°. At 7.6-Å resolution, the FSC of the reovirus reconstructions was 0.56 and the phase difference was 39.8° (Fig. 1).
Fitting atomic models into the cryo-EM density map
SITUS42 and EMfit41 were used to fit atomic models of monomeric reovirus subunits σ2, λ1 and λ2 (PDB entry 1EJ6)11, the μ13σ33 heterohexamer (PDB entry 1JMU)3 and monomeric λ3 (PDB entry 1N35)18 into the 7.6-Å EM map. To provide a basis for accurate fitting, the magnification of the reconstruction was scaled so that the core component of the virion matched the crystal model of the core11. The symmetry axes of the two structures were then superimposed to permit direct visualization of the fits of λ1, λ2 and σ2. The quality of averaged density in the outer capsid region of the reconstruction was assessed by manually docking, as a rigid body, the μ13σ33 crystal model into its four quasi-equivalent positions2,3. EMfit was then used to refine the fit at each position.
One-half of the entire reovirus map was used for the fitting done by EMfit. However, SITUS fitting was only carried out efficiently on smaller, local volumes encompassing the target density. Hence, for λ3 fitting, the SITUS search volume was initially done in a 200-Å cube (~8 × 106 Å3) and included a complete asymmetric unit that spanned a portion of the λ1 shell as well as the region inside the shell and overlapping the five-fold axis. The COLORES routine of SITUS42, which was used to fit the λ3 model into the cubic volume, generated a list of fits with the five best being related by five-fold symmetry coincident with the icosahedral five-fold axis contained in the search volume. More complete details of the fitting procedures and refinement and assessment of the fit obtained for λ3 are presented in Supplementary Methods online.
Graphic illustrations
Figure 1 was prepared with Xmgrace (http://plasma-gate.weizmann.ac.il/Grace/). Figure 2a,b was prepared with IRIS Explorer (http://www.nag.co.uk/Welcome_IEC.html), Figure 2c,d with RobEM (http://bilbo.bio.purdue.edu/~baker/programs/programs.html). Figures 2e, 3a–c, and f were prepared with BobScript55 and Raster3D56. Figure 3d,e was rendered with SPSS SigmaPlot (http://www.spss.com/spssbi/sigmaplot/spss.htm). Figure 4a,b was prepared with Raster3D, Figure 4c,d with Spock (http://quorum.tamu.edu/spock/) and Figure 4e,f with CNS57 and RobEM. Figures 5 and 6 were prepared with Raster3D. All figure labels were added with Adobe Photoshop 6.0 (http://www.adobe.com/products/photoshop/main.html).
Acknowledgments
We are especially grateful to S. Harrison and Y. Tao for providing the coordinates of the λ3 crystal structure before publication and also for discussions and comments on the manuscript. We also thank W. Zhang, C. Xiao, R. Ashmore, J. Johnson, A. McGough, R. Bernal, M. Sherman, M. Rossmann, P. Chacón and B. Bahlke for helpful discussions; V. Bowman, A. Simpson, P. Leiman, Y. Tao and J. Zhiu for assistance with figures; K. Reinisch and S. Liemann for providing crystal coordinates and discussion; and L. Szpankowski for digitizing micrographs. Work was supported in part by grants from the US National Institutes of Health to T.S.B. and M.L.N., a shared equipment grant from the US National Science Foundation to T.S.B., a Keck Foundation award to the Purdue Structural Biology group for purchase of the CM300 FEG microscope and a Purdue University reinvestment grant to the Structural Biology group. S.B.W. was additionally supported by the Purdue Biophysics Training Grant and a Purdue Research Foundation Fellowship.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
Note: Supplementary information is available on the Nature Structural Biology website.
References
- 1.Nibert ML, Schiff LA, Fields BN. Reoviruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. Raven; Philadelphia: 2001. pp. 1679–1728. [Google Scholar]
- 2.Dryden KA, et al. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol. 1993;122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liemann S, Chandran K, Baker TS, Nibert ML, Harrison SC. Structure of the reovirus membrane-penetration protein, μ1, in a complex with its protector protein, σ3. Cell. 2002;108:283–295. doi: 10.1016/s0092-8674(02)00612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furlong DB, Nibert ML, Fields BN. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol. 1988;62:246–256. doi: 10.1128/jvi.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strong JE, Leone G, Duncan R, Sharma RK, Lee PWK. Biochemical and biophysical characterization of the reovirus cell attachment protein σ1: evidence that it is a homotrimer. Virology. 1991;184:23–32. doi: 10.1016/0042-6822(91)90818-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larson SM, Antczak JB, Joklik WK. Reovirus exists in the form of 13 particle species that differ in their content of protein σ1. Virology. 1994;201:303–311. doi: 10.1006/viro.1994.1295. [DOI] [PubMed] [Google Scholar]
- 7.Chappell JD, Porta AE, Dermody TS, Stehle T. Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 2002;21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sturzenbecker LJ, Nibert M, Furlong D, Fields BN. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J Virol. 1987;61:2351–2361. doi: 10.1128/jvi.61.8.2351-2361.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandran K, Farsetta DL, Nibert ML. Strategy for nonenveloped virus entry: a hydrophobic conformer of reovirus penetration protein μ1 mediates membrane disruption. J Virol. 2002;76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandran K, Parker JSL, Ehrlich M, Kirchhausen Y, Nibert ML. The δ region of outer-capsid protein μ1 undergoes conformational change and release from reovirus particles during cell entry. J Virol. doi: 10.1128/JVI.77.24.13361-13375.2003. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reinisch KM, Nibert ML, Harrison SC. Structure of the reovirus core at 3.6 Å resolution. Nature. 2000;404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- 12.Samuel CE. Reoviruses and the interferon system. Curr Top Microbiol Immunol. 1998;233:125–145. doi: 10.1007/978-3-642-72095-6_7. [DOI] [PubMed] [Google Scholar]
- 13.Cullen BR. RNA interference: antiviral defense and genetic tool. Nat Immunol. 2002;3:597–599. doi: 10.1038/ni0702-597. [DOI] [PubMed] [Google Scholar]
- 14.Coombs KM. Stoichiometry of reovirus structural proteins in virus, ISVP, and core particles. Virology. 1998;243:218–228. doi: 10.1006/viro.1998.9061. [DOI] [PubMed] [Google Scholar]
- 15.Dryden KA, et al. Internal structures containing transcriptase-related proteins in top component particles of mammalian orthoreovirus. Virology. 1998;245:33–46. doi: 10.1006/viro.1998.9146. [DOI] [PubMed] [Google Scholar]
- 16.Drayna D, Fields BN. Activation and characterization of the reovirus transcriptase: genetic analysis. J Virol. 1982;41:110–118. doi: 10.1128/jvi.41.1.110-118.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Starnes MC, Joklik WK. Reovirus protein λ3 is a poly(C)-dependent poly(G) polymerase. Virology. 1993;193:356–366. doi: 10.1006/viro.1993.1132. [DOI] [PubMed] [Google Scholar]
- 18.Tao Y, Farsetta DL, Nibert ML, Harrison SC. RNA synthesis in a cage—structural studies of reovirus polymerase λ3. Cell. 2002;111:733–745. doi: 10.1016/s0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- 19.Yin P, Cheang M, Coombs KM. The M1 gene is associated with differences in the temperature optimum of the transcriptase activity in reovirus core particles. J Virol. 1996;70:1223–1227. doi: 10.1128/jvi.70.2.1223-1227.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noble S, Nibert ML. Core protein μ2 is a second determinant of nucleoside triphosphatase activities by reovirus cores. J Virol. 1997;71:7728–7735. doi: 10.1128/jvi.71.10.7728-7735.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noble S, Nibert ML. Characterization of an ATPase activity in reovirus cores and its genetic association with core-shell protein λ1. J Virol. 1997;71:2182–2191. doi: 10.1128/jvi.71.3.2182-2191.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bisaillon M, Bergeron J, Lemay G. Characterization of the nucleoside triphosphate phosphohydrolase and helicase activities of the reovirus λ1 protein. J Biol Chem. 1997;272:18298–18303. doi: 10.1074/jbc.272.29.18298. [DOI] [PubMed] [Google Scholar]
- 23.Bisaillon M, Lemay G. Characterization of the reovirus λ1 protein RNA 5′-triphosphatase activity. J Biol Chem. 1997;272:29954–29957. doi: 10.1074/jbc.272.47.29954. [DOI] [PubMed] [Google Scholar]
- 24.Furuichi Y, Muthukrishnan S, Tomasz J, Shatkin AJ. Caps in eukaryotic mRNAs: mechanism of formation of reovirus mRNA 5′-terminal m7GpppGm-C. Prog Nucleic Acid Res Mol Biol. 1976;19:3–20. doi: 10.1016/s0079-6603(08)60905-8. [DOI] [PubMed] [Google Scholar]
- 25.Cleveland DR, Zarbl H, Millward S. Reovirus guanylyltransferase is L2 gene product λ2. J Virol. 1986;60:307–311. doi: 10.1128/jvi.60.1.307-311.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao ZX, Joklik WK. Isolation and enzymatic characterization of protein λ2, the reovirus guanylyltransferase. Virology. 1991;185:377–386. doi: 10.1016/0042-6822(91)90785-a. [DOI] [PubMed] [Google Scholar]
- 27.Fausnaugh J, Shatkin AJ. Active site localization in a viral mRNA capping enzyme. J Biol Chem. 1990;265:7669–7672. [PubMed] [Google Scholar]
- 28.Luongo CL, Contreras CM, Farsetta DL, Nibert ML. Binding site for S-adenosyl-L-methionine in a central region of mammalian reovirus λ2 protein. Evidence for activities in mRNA cap methylation. J Biol Chem. 1998;273:23773–23780. doi: 10.1074/jbc.273.37.23773. [DOI] [PubMed] [Google Scholar]
- 29.Luongo CL, Reinisch KM, Harrison SC, Nibert ML. Identification of the guanylyltransferase region and active site in reovirus mRNA capping protein λ2. J Biol Chem. 2000;275:2804–2810. doi: 10.1074/jbc.275.4.2804. [DOI] [PubMed] [Google Scholar]
- 30.Gillies S, Bullivant S, Bellamy AR. Viral RNA polymerases: electron microscopy of reovirus reaction cores. Science. 1971;174:694–696. doi: 10.1126/science.174.4010.694. [DOI] [PubMed] [Google Scholar]
- 31.Bartlett NM, Gillies SC, Bullivant S, Bellamy AR. Electron microscopy study of reovirus reaction cores. J Virol. 1974;14:315–326. doi: 10.1128/jvi.14.2.315-326.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeager M, Weiner S, Coombs KM. Transcriptionally active reovirus core particles visualized by electron cryo-microscopy and image reconstruction. Biophys J. 1996;70:A116. [Google Scholar]
- 33.Furuichi Y, Shatkin AJ. Viral and cellular mRNA capping: past and prospects. Adv Virus Res. 2000;55:135–184. doi: 10.1016/S0065-3527(00)55003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shuman S. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog Nucleic Acid Res Mol Biol. 2001;66:1–40. doi: 10.1016/s0079-6603(00)66025-7. [DOI] [PubMed] [Google Scholar]
- 35.Prasad BVV, et al. Visualization of ordered genomic RNA and localization of transcriptional complexes in rotavirus. Nature. 1996;382:471–473. doi: 10.1038/382471a0. [DOI] [PubMed] [Google Scholar]
- 36.Pesavento JB, Lawton JA, Estes MK, Prasad BVV. The reversible condensation and expansion of the rotavirus genome. Proc Natl Acad Sci USA. 2001;98:1381–1386. doi: 10.1073/pnas.98.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gouet P, et al. The highly ordered double-stranded RNA genome of bluetongue virus revealed by crystallography. Cell. 1999;97:481–490. doi: 10.1016/s0092-8674(00)80758-8. [DOI] [PubMed] [Google Scholar]
- 38.Grimes JM, et al. The atomic structure of the bluetongue virus core. Nature. 1998;395:470–478. doi: 10.1038/26694. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, et al. Visualization of protein-RNA interactions in cytoplasmic polyhedrosis virus. J Virol. 1999;73:1624–1629. doi: 10.1128/jvi.73.2.1624-1629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker TS, Olson NH, Fuller SD. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol Mol Biol Rev. 1999;63:862–922. doi: 10.1128/mmbr.63.4.862-922.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rossmann MG, Bernal R, Pletnev SV. Combining electron microscopic with X-ray crystallographic structures. J Struct Biol. 2001;136:190–200. doi: 10.1006/jsbi.2002.4435. [DOI] [PubMed] [Google Scholar]
- 42.Chacón P, Wriggers W. Multi-resolution contour-based fitting of macromolecular structures. J Mol Biol. 2002;317:375–384. doi: 10.1006/jmbi.2002.5438. [DOI] [PubMed] [Google Scholar]
- 43.Farsetta DL, Chandran K, Nibert ML. Transcriptional activities of reovirus RNA polymerase in recoated cores. Initiation and elongation are regulated by separate mechanisms. J Biol Chem. 2000;275:39693–39701. doi: 10.1074/jbc.M004562200. [DOI] [PubMed] [Google Scholar]
- 44.Joklik WK. The reovirus particle. In: Joklik WK, editor. The Reoviridae. Plenum; New York: 1983. pp. 9–78. [Google Scholar]
- 45.Shatkin AJ, Kozak M. Biochemical aspects of reovirus transcription and translation. In: Joklik WK, editor. The Reoviridae. Plenum; New York: 1983. pp. 79–106. [Google Scholar]
- 46.Cheetham GM, Steitz TA. Insights into transcription: structure and function of single-subunit DNA-dependent RNA polymerases. Curr Opin Struct Biol. 2000;10:117–123. doi: 10.1016/s0959-440x(99)00058-5. [DOI] [PubMed] [Google Scholar]
- 47.Spencer SM, Sgro JY, Dryden KA, Baker TS, Nibert ML. IRIS explorer software for radial-depth cueing reovirus particles and other macromolecular structures determined by cryoelectron microscopy and image reconstruction. J Struct Biol. 1997;120:11–21. doi: 10.1006/jsbi.1997.3902. [DOI] [PubMed] [Google Scholar]
- 48.Luongo CL, et al. Loss of activities for mRNA synthesis accompanies loss of λ2 spikes from reovirus cores: an effect of λ2 on λ1 shell structure. Virology. 2002;296:24–38. doi: 10.1006/viro.2001.1258. [DOI] [PubMed] [Google Scholar]
- 49.Diprose JM, et al. Translocation portals for the substrates and products of a viral transcription complex: the bluetongue virus core. EMBO J. 2001;20:7229–7239. doi: 10.1093/emboj/20.24.7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lawton JA, Estes MK, Prasad BVV. Three-dimensional visualization of mRNA release from actively transcribing rotavirus particles. Nat Struct Biol. 1997;4:118–121. doi: 10.1038/nsb0297-118. [DOI] [PubMed] [Google Scholar]
- 51.Le Blois H, French T, Mertens PP, Burroughs JN, Roy P. The expressed VP4 protein of bluetongue virus binds GTP and is the candidate guanylyl transferase of the virus. Virology. 1992;189:757–761. doi: 10.1016/0042-6822(92)90600-t. [DOI] [PubMed] [Google Scholar]
- 52.Baker TS, Cheng RH. A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J Struct Biol. 1996;116:120–130. doi: 10.1006/jsbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 53.Bowman VD, et al. An antibody to the putative aphid recognition site on cucumber mosaic virus recognizes pentons but not hexons. J Virol. 2002;76:12250–12258. doi: 10.1128/JVI.76.23.12250-12258.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Heel M, et al. Single-particle electron cryo-microscopy: towards atomic resolution. Q Rev Biophys. 2000;33:371–424. doi: 10.1017/s0033583500003644. [DOI] [PubMed] [Google Scholar]
- 55.Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graph Model. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 56.Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 57.Brunger AT, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 58.Grimes JM, et al. An atomic model of the outer layer of the bluetongue virus core derived from X-ray crystallography and electron cryomicroscopy. Structure. 1997;5:885–893. doi: 10.1016/s0969-2126(97)00243-8. [DOI] [PubMed] [Google Scholar]