

Summary
Dengue virus, a member of the Flaviviridae family, has a surface composed of 180 copies each of the envelope (E) glycoprotein and the membrane (M) protein. The crystal structure of an N-terminal fragment of E has been determined and compared with a previously described structure. The primary difference between these structures is a 10° rotation about a hinge relating the fusion domain DII to domains DI and DIII. These two rigid body components were used for independent fitting of E into the cryo-electron microscopy maps of both immature and mature dengue viruses. The fitted E structures in these two particles showed a difference of 27° between the two components. Comparison of the E structure in its postfusion state with that in the immature and mature virions shows a rotation approximately around the same hinge. Flexibility of E is apparently a functional requirement for assembly and infection of flaviviruses.
Introduction
Many flaviviruses, such as dengue (DEN), yellow fever (YFV), West Nile (WNV), and tick-borne encephalitis virus (TBEV), are arthropod-borne, human pathogens. They can cause various diseases, including jaundice, encephalitis, hemorrhagic fevers, and shock syndrome (Lindenbach and Rice, 2001). Currently, there are no efficacious antiviral drugs against flavivirus infections, and vaccines are only available for YFV, TBEV, and Japanese encephalitis virus (JEV). Flaviviruses can be subdivided into those transmitted by ticks, by mosquitoes, and those that apparently lack an arthropod vector (Kuno et al., 1998).
In membrane-containing viruses, the external structural proteins function to bind to cellular receptors and to interact with host cell membranes for fusion and subsequent cell entry, as well as to direct viral assembly and budding. In addition, surface proteins normally possess antigenic determinants that elicit neutralizing antibodies. These functions are frequently distributed among several different viral proteins, but in the flaviviruses, the envelope glycoprotein (E) participates in all these functions. After entering cells by receptor-mediated endocytosis, fusion with the host membrane is initiated by low pH induced conformational change of the E protein (Heinz and Allison, 2000), followed by release of the genomic RNA into the cytoplasm. The genomic RNA is translated into a polyprotein in which capsid, precursor membrane (prM), and E structural proteins reside in the N-terminal region. The polyprotein is processed on the endoplasmic reticulum membrane into individual proteins by both viral and cellular proteases and assembled together with the genomic RNA into immature particles (Mackenzie and Westaway, 2001). These contain 60 asymmetric trimers of prM-E heterodimers (Zhang et al., 2003b). A substantial portion of the prM protein covers E, thereby protecting it against premature fusion while passing through the acidic environment of the trans-Golgi network (TGN) (Guirakhoo et al., 1992). The 100 N-terminal amino acids of the prM protein (the prepeptide, pr) are then released by furin cleavage (Stadler et al., 1997) in the final step of maturation. This cleavage induces a rearrangement of the E proteins and activates the virus to be fusogenic. In the mature virus, E proteins exist as homodimers that lie on the viral membrane, arranged as 30 “rafts” organized into a herringbone pattern (Figure 1). Each raft contains three parallel dimers, with the center of the raft coincident with an icosahedral 2-fold axis (Kuhn et al., 2002; Mukhopadhyay et al., 2003; Zhang et al., 2003a).
Figure 1. The Mature Dengue Virus Structure.
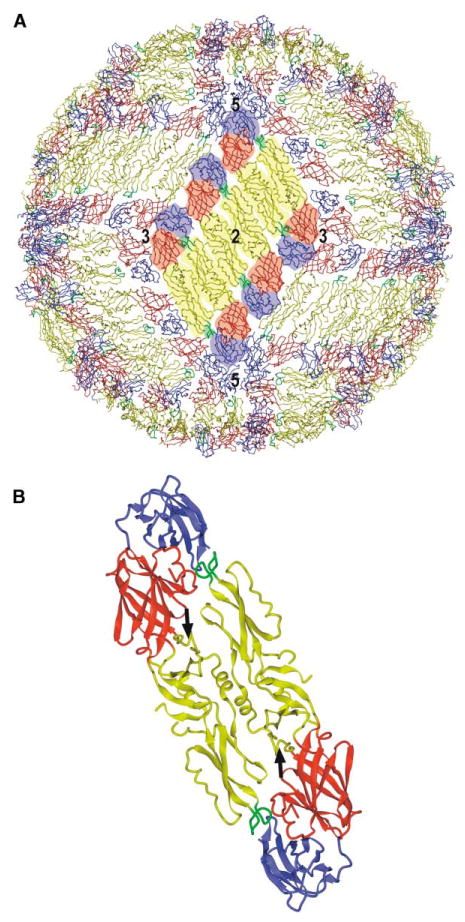
(A) Packing of the E proteins in the mature virus, showing the herringbone pattern. One of the 30 rafts, each containing three parallel dimers, is highlighted. Domains I, II, and III are colored red, yellow, and blue, respectively, with the fusion peptide in green. Symmetry axes are labeled.
(B) Ribbon diagram of the crystal dimer with the same color coding as in (A). Black arrows point to the kl β-hairpin in each monomer, where β-OG can bind.
A large tryptic fragment of the E protein (sE, also referred to here as the ectodomain of E) of TBEV (residues 1–395) was the first flavivirus component whose atomic structure was determined (Rey et al., 1995). The structure of sE consists of three domains: domain I (DI), the N-terminal, but structurally central, domain; domain II (DII), the fusion (or dimerization) domain containing the hydrophobic fusion peptide (residues 98–110) (Allison et al., 2001); and domain III (DIII), the putative receptor binding domain (Bhardwaj et al., 2001). The structure of a DEN serotype 2 sE dimer (DEN sE Harvard, DEN sE(H)) has also been determined both with and without n-octyl-β-D-glucoside (β-OG) detergent bound to sE (Modis et al., 2003). The difference between the two DEN sE(H) structures is a rearrangement of the “kl” hairpin (Figure 1), which is “open” on binding β-OG. This hairpin forms part of the “hinge” that connects domains DI and DII. Other structures of sE from flaviviruses, formed by treatment with acid and lipid to produce the postfusion state, have been reported very recently (Bressanelli et al., 2004; Modis et al., 2004). These structures show a trimer in which the fusion peptides of each monomer are at one end and domains DI and DIII are at the other. The “stem-anchor,” C-terminal 100 amino acids of DEN E are absent in sE, but they have been identified in virions using cryo-electron microscopy (cryo-EM) (Zhang et al., 2003a). The stem region (residues 396–451) consists of two helices, E-H1 and E-H2, connected by a highly conserved sequence (CS). The transmembrane anchor region (residues 452–495) consists of one pair of antiparallel coiled-coils, E-T1 and E-T2 (Zhang et al., 2003a).
We report here a different structure of DEN serotype 2 sE (DEN sE Purdue, DEN sE(P)), which is from the same isolate as DEN sE(H), and show that these structures differ by a hinge motion of ~10° between DII and DI + DIII. The two components of DEN sE(P) were docked into cryo-EM density maps of mature and immature DEN particles, which have been now determined to 9.5 and 12.5 A resolution, respectively. These results supercede our earlier analyses (Zhang et al., 2003a, 2003b) because the previous fitting was based on the sE structure of TBEV and because the resolution of the cryo-EM map of the immature virus has now been substantially improved. Our results demonstrate that the E molecule is highly flexible and undergoes considerable conformational changes during maturation. This inherent flexibility of E appears to be essential for the opposing processes of virus maturation and fusion.
Results and Discussion
The Crystal Structure of Dimeric DEN sE(P)
Recombinant sE protein from DEN PR159 strain (residues 1–395) (Cuzzubbo et al., 2001; Ivy et al., 1997) was used for crystallization experiments. The DEN sE(P) crystals were in space group P3221, with one molecule per asymmetric unit. The DEN sE(H) monomer was used as a molecular replacement search model to solve the DEN sE(P) structure, which was then refined to 2.6 A resolution (see Experimental Procedures and Table 1). Of the 395 amino acids in the polypeptide chain, 390 (residues 1–16, 19–224, and 228–395) could be visualized in the electron density. The DEN sE(P) monomers form head-to-tail dimers around the crystallographic 2-fold axes (Figure 1), similar to dimers of TBEV sE (Rey et al., 1995) and DEN sE(H) (Modis et al., 2003). Each monomer consists of three β strand dominated domains, as in TBEV sE with which it has 37% sequence identity (the nomenclature of the secondary structural elements is taken from Rey et al. [Rey et al., 1995] and is shown in Figure 2). Domains DI and DII are connected through four polypeptide strands, whereas DI and DIII are connected through only one strand. Two potential N-linked carbohydrate sites (at N67 and N153) are both glycosylated in the structure, as is also found in DEN virions produced in cell culture (Mukhopadhyay et al., 2003; Zhang et al., 2003a). N153, located in the E0F0 loop and involved in dimer contacts, is conserved among flavivirus E proteins, whereas N67 is unique to most strains of DEN viruses.
Table 1. Crystallographic Data and Refinement Statistics.
| Data Collection | Refinement | ||
|---|---|---|---|
| Cell dimensions (Å) | a = b = 71.9 | Number of reflections: working/free | 12,591/394 |
| c = 140.9 | R factor (%)c | ||
| Working set | 26.3 | ||
| Space group | P3221 | Test set | 29.4 |
| X-ray wavelength (Å) | 0.979 | Rmsd from ideality | |
| Resolution range (Å) | 50–2.6 | Bond length (Å) | 0.0252 |
| Number of observations | 62,468 | Bond angles (°) | 2.417 |
| Number of unique reflections | 13,020 | Most favored region (%)d | 77.0 |
| Completeness (%)a | 97.4 (77.7) | Additional allowed region (%)d | 23.0 |
| Rsym(%)a,b | 11.2 (48.6) | Disallowed region (%)d | 0.0 |
Numbers in parentheses represent values in the highest resolution shell.
, where I is observed integrated intensity and <I> is the averaged integrated intensity taken over n measurements for reflection h.
, where Fo is the observed structure factor amplitude and Fc is the calculated structure factor amplitudes based on the refined atomic positions, taken over the h reflections in the observed data set.
In the Ramachandran plot.
Figure 2. Sequence Alignment of TBEV and DEN sE Based on Structural Comparisons.

The numbering system is from DEN. The surface accessibility of the DEN sE(P) dimer, averaged over an 11 residue window, is plotted in violet. The temperature factor of DEN sE(P) is shown in dark blue. The plot of residue conservation between TBEV and DEN, measured in terms of point-accepted mutations (PAM) (Dayhoff, 1972) and averaged over an 11 residue window, is shown in light blue. The plot of Cα differences (brown) between TBEV and DEN sE is also shown. Black bars indicate regions with low sequence conservation. Residues of domains I, II, and III are in red, yellow, and blue, respectively. The four hinge peptides linking domains DI and DII are, therefore, at the red-yellow boundaries. Note the presence of conserved glycines in the hinge region. Secondary structural elements are shown below the sequences.
The interface between monomers within a dimer has an area of about 1800 A2 and is a mixture of hydrophobic (60%) and hydrophilic (40%) interactions (see Experimental Procedures). Most of these contacts are clustered in two regions. One is around the ij loop and the fusion peptide (the cd loop), which interacts with a hydrophobic “cavity” composed of the N-terminal loop of the A0 strand, the E0F0 loop, and the AB loop in DI and DIII from the opposing monomer. The other dimer contact region is around helix αB (residues 256–265) in DII, which interacts with the same residues across the dyad axis. Several surface-exposed loops are quite flexible, like the A0B0, F0G0, kl, and CD loops, which exhibit temperature factors over 100 A2 (Figure 2) relative to an average temperature factor of 56 A2.
Structural Comparison of DEN sE(P) with DEN sE(H)
The binding of β-OG in DEN sE(H) induces a local conformational change at the hinge region, with a root-mean-square deviation (rmsd) between equivalent Cα atom positions of 0.65 Å (Modis et al., 2003). In contrast, the DEN sE(P) structure is considerably different from both DEN sE(H) structures, with rmsds of about 1.4 A, even when 32 amino acids in the hinge region were excluded from the calculations. When the DI portion of DEN sE(P) is superimposed on the corresponding DI portion of DEN sE(H), atoms at the tip of DII are more than 10 A apart. However, separate alignments of the three domains give rmsd values of only 0.68, 0.69, and 0.47 Å for equivalent Cå atoms in DI, DII, and DIII, respectively (Figure 3). The large displacements between the DII domains are the result of a 10° hinge motion between DI and DII that correlates with different crystallization conditions. (The arbitrary zero of the hinge angle is defined here as that observed in the DEN sE(P) structure, with the positive direction being given by a rotation of DII that would superimpose DEN sE(P) onto DEN sE(H) [Figure 3].) DII pivots about a hinge that is roughly perpendicular to the dimer dyad (Figure 3). Thus, the differences in the crystal structures represent a hinge motion that would be required for domain DII to direct its fusion peptide toward the host cell surface in the prefusion process (Kuhn et al., 2002). The hinge motion is facilitated by the presence of conserved glycines in three of the four peptides that connect domains DI and DII (Figure 2).
Figure 3. Conformational Changes of the sE Structure.

(A and B) Stereo diagram showing superposition of TBEV (magenta), DEN sE(P) (yellow), and DEN sE(H) without β-OG (red) for (A) DI + DIII and (B) DII, viewed along the direction of the dimer 2-fold axis.
(C) Stereoview of superimposed E ectodomains showing their different hinge angles along the direction of the hinge axis, perpendicular to the view in (A) and (B). The alignment is based on DI, and the diagrammatic position of DIII corresponds to the prefusion structure.
Although the single polypeptide that connects the DI and DIII domains might be expected to provide a range of motion between them, the orientations of DIII relative to DI in the DEN sE structures are quite similar. The large number of hydrophilic interactions between these domains (Bressanelli et al., 2004) might restrict or eliminate interdomain flexibility at neutral pH. When the pH is lowered in the presence of liposomes, the interface between DI and DIII is disrupted, and DIII shifts more than 30 A compared to its position in the sE dimer to form, eventually, a postfusion trimer (Bressanelli et al., 2004; Modis et al., 2004). Furthermore, the rearrangement of DIII eliminates the hydrophobic “cavity,” which occurs in the sE dimer and serves to protect the fusion peptide.
When DEN sE(H) was incubated with β-OG (final concentration of 0.5% at 4°C), one detergent molecule bound to a hydrophobic pocket created by the kl β-hairpin in its “open” conformation (Modis et al., 2003). The kl β-hairpin in DEN sE(P) adopts a conformation that is intermediate in position between the β-OG-bound and -unbound sE(H) structures. Although β-OG was also added to the crystallization drop to obtain the DEN sE(P) crystals (final concentration of 0.3% at room temperature), no density was observed in the hydrophobic pocket. The absence of β-OG in DEN sE(P) might be a consequence of the lower concentration of β-OG, the higher temperature, or the presence of different precipitants used in the crystallization experiments.
Comparison of DEN sE with TBEV sE
Several regions of high sequence variability occur between TBEV and DEN sE (Figure 2). Most of these regions are exposed on the outside of the viral surface, consistent with receptor binding and neutralizing antibody eliciting functions. Four of these regions correspond to the polypeptides that connect DI and DII at the hinge region, and they have temperature factors greater than 75 A2. Other regions of variability occur in the putative receptor binding domain, DIII, and the F0 strand in domain DI.
The three domains of TBEV sE are similar to the corresponding domains in DEN sE, with most differences confined to a few surface loops and in the hinge polypeptides (Table 2). However, DEN and TBEV sE differ by about 10° in the angle between domains DI and DII, measured relative to an axis roughly parallel to the 2-fold axis of the dimer. This axis is perpendicular to the hinge axis between the DI + DIII and DII domains that is found when comparing DEN sE in different crystal forms.
Table 2. Structural Differences of DI and DII between TBEV and DEN sE(P).
| Sequence | B Factors (Å2)a | ||||
|---|---|---|---|---|---|
|
|
|
||||
| Loop | DEN (top) and TBEV (bottom) | DEN sE(P) | TBEV | Rmsdb (Å) | Comments |
| A0B0 | GVSGGSW GTQGTTR * | 106 | 85 | 2.0 | Very flexible region with high-temperature factors. |
| E0F0 | SGEEHAVGNDTGKHG TGD-YVAANETHSGR * | 64 | 28 | 3.6 | A conserved glycosylation site is marked with *. |
| F0G0 | KITPQSS SFTISSE | 100 | 24 | 2.0 | Close to the viral membrane. |
| de | CK--KN CEAKKK | 53 | 23 | 0.7 | |
| fg | MKD ------ K LDKTVEHLPT | 64 | 26 | 1.7 | Monomer-monomer contact region within dimers. |
| hi | PGADTQGSN HEG---AQN | 67 | 33 | 3.4 | |
| ij | PHAKKQ PHAVKM | 60 | 40 | 4.9 | In the DEN sE(P) dimer, this ij loop is in a monomer-monomer contact with a negatively charged region, whereas in TBEV sE dimer, the corresponding region is hydrophobic and not in contact with the ij loop. |
Mean overall temperature factor for DEN sE(P) and TBEV structures is 56 and 28 Å2, respectively.
Rmsd between equivalent Cα atoms.
The largest structural difference between DEN and TBEV sE in DIII occurs in surface loops located on the external face of the virus at the extreme ends of the dimers (Figures 2 and 4), possibly suggesting a receptor binding function. In addition, mutations in tissue culture-adapted mutants, in escape mutants to neutralizing antibodies, and in cell tropism-related mutants have also been mapped to this region (for review, see McMinn, 1997). Since DIII is the putative receptor binding domain, structural differences in DIII might be related to the difference in the arthropod vectors employed for virus transmission. The amino acid residues in some of the fully exposed loops (the AxA, CD, and FG loops) in DIII are likely to form a hydrogen bond network around the 5-fold axis of the mature DEN virion (Figure 4), implying that the proximity of several receptor binding sites may be required for an efficacious attachment and the subsequent receptor-mediated endocytosis.
Figure 4. The E Protein Structure in the Mature Virus.

(A) Stereoview of the E protein fitted to the cryo-EM density of the mature DEN around the 5-fold axis. Domains DI, DII, and DIII are colored as in Figure 1. The AxA, CD, and FG surface loops are shown as Cα traces.
(B) Stereo diagram showing sE fitted in the cryo-EM electron density map of the mature DEN virus. Molecules 1, 2, and 3 (see Table 3) are in red, green, and blue, respectively. Symmetry axes are labeled.
(C) Side view of the E dimer as determined by the independent fitting of DI + DIII and of DII into the density of the mature virus (red) superimposed onto the sE(P) dimer in the crystal (yellow). Note that the colors in (A) compared to (B) and (C) in both Figures 4 and 5 represent different structural components.
The E Protein in Mature Virions
The crystal structures of DEN sE showed that sE consists of two rigid bodies connected by a flexible hinge. Therefore, for the purpose of molecular fitting experiments, the crystal structure of DEN sE(P) was divided into two separate groups, DI + DIII and DII, which were then treated as independent rigid models. These were fitted into the 9.5 Å cryo-EM map of mature DEN (Zhang et al., 2003a) (see Experimental Procedures). This procedure gave a significantly better fit to the density than when the sE structure was fitted as a single rigid body (Table 3), not withstanding the increased number of parameters used in the fitting procedure. The improved fit reflects the natural tendency of the sE dimer to conform to the curved viral membrane surface instead of adopting the straighter conformation that occurs in the crystals (Figure 4). The structure that was found for each of the three sE monomers in the icosahedral asymmetric unit of the virus will necessarily be the same, because a simultaneous fitting procedure was employed (see Experimental Procedures). The angle between the two rigid bodies (DI + DIII and DII) was determined after the best fit to the EM density was obtained. Comparison of the resultant sE quasi-atomic monomer structure with the sE(P) crystal structure shows that the two rigid bodies are related by a hinge axis that roughly coincides with the axis relating these bodies in the different crystal structures of DEN sE. The hinge angle between the two fitted components was found to be 21° (Figure 3).
Table 3. Results Obtained by Fitting Crystallographic Structural Components into the Mature and Immature Dengue Virus Maps.
| Molecule 1d | Molecule 2d | Molecule 3d | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||||||
| Model | Sumfa | Clashb | –Denc | DIe | DIIe | DIIIe | DIe | DIIe | DIIIe | DIe | DIIe | DIIIe | |
| Mature | DEN sE(H) with β-OG | 38.2 | 0.7 | 2.4 | 36.5 | 39.0 | 36.0 | 38.8 | 39.4 | 37.4 | 38.9 | 39.2 | 36.5 |
| DEN sE(H) without β-OG | 38.6 | 0.4 | 1.9 | 36.8 | 39.7 | 36.0 | 39.3 | 40.3 | 37.3 | 39.1 | 39.7 | 36.3 | |
| DEN sE(P) | 37.3 | 1.1 | 3.0 | 36.6 | 37.2 | 35.8 | 38.7 | 37.6 | 36.8 | 38.8 | 37.5 | 36.1 | |
| DEN sE(P) DI + DIII and DIIf | 39.3 | 1.6 | 1.3 | 36.9 | 40.7 | 36.3 | 39.4 | 41.5 | 38.1 | 39.2 | 40.8 | 37.0 | |
| Immature | DEN sE(H) with β-OG | 46.1 | 3.1 | 2.5 | 43.8 | 52.0 | 41.4 | 41.9 | 51.3 | 39.5 | 42.5 | 52.7 | 40.8 |
| DEN sE(H) without β-OG | 46.6 | 2.3 | 1.8 | 45.0 | 52.6 | 40.9 | 43.2 | 50.9 | 40.4 | 44.0 | 51.9 | 41.8 | |
| DEN sE(P) | 47.0 | 2.1 | 1.5 | 45.3 | 52.9 | 41.0 | 43.4 | 52.3 | 39.8 | 44.4 | 53.3 | 40.6 | |
| DEN sE(P) DI + DIII and DIIf | 47.5 | 0.1 | 1.3 | 46.5 | 52.4 | 43.3 | 44.7 | 51.4 | 42.7 | 43.3 | 51.7 | 42.3 | |
Sumf is defined as the average value of density at all atomic positions normalized by setting the highest density in the map to 100. The higher Sumf values for the immature virus density are probably primarily the result of the extensive contact of the E and prM proteins, thus placing the atoms of E in the contact area into higher density. This is particularly true for DII.
Clash represents the percentage of atoms in the model that have steric clashes with symmetry-related subunits.
–Den denotes the percentage of atoms that are positioned in negative density.
Sumf values for domains DI, DII, and DIII, respectively.
Two rigid bodies, DI + DIII and DII, were fitted independently using both icosahedral and nonicosahedral symmetry as described in the Experimental Procedures section.
The symmetry operation that relates monomers within the dimer at the general position (referred to as the “general dimer”) is an almost exact 2-fold, with the approximate dyad axis oriented at an angle of 21° relative to the closest icosahedral 2-fold axis. The closest distance between the dyad axis of the general dimer and the icosahedral 2-fold axis is 9 A, with the point of closest approach being 70 A from the virus center. The general dimer has a very similar conformation to the dimer on the icosahetral 2-fold axis (the “icosahedral dimer”), with an rmsd of 0.4 A between equivalent Cα atoms, but differs from the sE(P) crystal dimer by an rmsd of 4.5 Å (Figure 4). The dimers in the mature virions are about 6 A longer than, but have the same width as, the dimers in the crystal structures. Such differences provide a closer packing association between rafts on the virus surface or might reflect inaccuracy of the microscope magnification calibration. However, the apparent asymmetry of the dimer's expansion might suggest a significant change in length.
Various interactions associate E monomers to form the herringbone pattern on the viral surface. DI, DII, and DIII of the E protein all contribute to the interdimer contacts (see Experimental Procedures for a description of how these interactions were identified), most of which are hydrophilic and conserved among flaviviruses. There are no interactions between the stem-anchor regions of neighboring dimers, which suggests that the interactions between E ectodomains are primarily responsible for the association of E dimers. The stem-anchor regions of E and the associated M proteins also interact with the corresponding E ectodomains within the same dimer. The inner surface of the E ectodomain (facing the membrane) has local regions of negative charge. The E-H1 and E-H2 helices in the stem region are both amphipathic, with one face slightly positively charged. Therefore, the stem-anchor regions may help to alleviate the electrostatic repulsion between the membrane-facing surface of the E ectodomain and the polar groups in the membrane's outer leaflet. However, during the fusion process, triggered by a lowered pH, the charge interactions between the membrane, the ectodomain of E, and the stem region of E will change and lead to a rearrangement of helices E-H1 and E-H2 (Bressanelli et al., 2004).
The E Protein in Immature Virions
The two rigid components (DI + DIII and DII) of the sE structure were fitted into the improved (12.5 A resolution) cryo-EM reconstruction of the immature virus using a procedure similar to the one used for the mature virus (see Experimental Procedures and Figure 5). The two rigid bodies were first fitted independently into each of the cryo-EM densities representing the three E molecules within one asymmetric unit. The relative orientation and position of the two rigid bodies to each other were very similar in all three monomers. The rmsd between equivalent Cα atoms for any pairwise comparison was about 0.8 A. The position and orientation of the hinge axis between DI + DIII and DII, that was found when comparing the E monomer in mature and immature DEN, was the same as when comparing DEN sE in different crystal structures. The hinge angle between DI + DIII and DII is –6° in immature particles, a difference of 27° from that in mature particles (Figure 3).
Figure 5. The E Protein Structure in the Immature Virus.
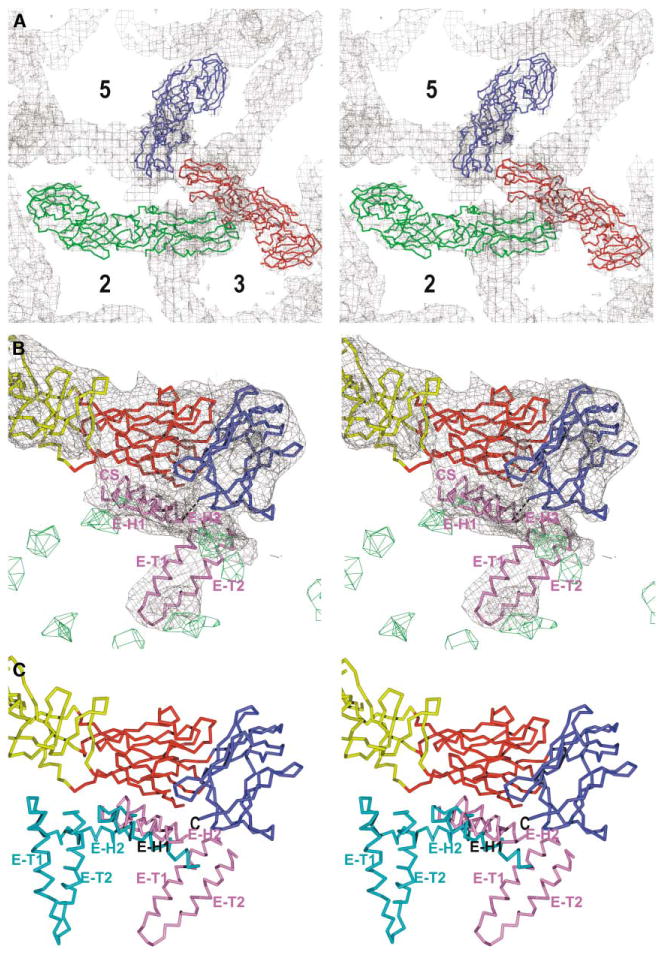
(A) Stereo diagram showing sE fitted into the cryo-EM electron density map of immature DEN. Molecules 1, 2, and 3 (see Table 3) are in red, green, and blue, respectively. Symmetry axes are labeled.
(B) Stereoview of the Cα trace of domains DI (red), DII (yellow), DIII (blue), and the stem-anchor region (violet) of E in the map of the immature virus. The electron density that corresponds to the membrane is colored green.
(C) The alignment of the E proteins in the mature and immature viruses. The superposition is based on DIII of the sE fragment. The ectodomain is shown as in (B). The stem-anchor regions in mature and immature viruses are colored cyan and violet, respectively. The C terminus of the E ectodomain is marked with “C.” Note the similar positions of helix H1.
The E-H1 and E-H2 helices were modeled into the cryo-EM density of the immature particle (Figure 5). The assignment of the stem-anchor regions in each icosahedral asymmetric unit was confirmed by their similar positional relationships to the corresponding E ectodomains. In both mature and immature virus particles, the position and orientation of E-H1 in the stem region relative to DI + DIII in the ectodomain are similar (Figure 5) and can be attributed to the close interactions between the stem and ectodomain. Furthermore, the density that can be ascribed to the hydrophobic, conserved sequence is closely associated with hydrophobic, conserved residues in stands B0 and H0 of DI in the E ectodomain. Although the interactions of E-H1 with DI and DIII may help to maintain their association at neutral or basic pH, these contacts are lost at low pH in forming the postfusion structure (Bressanelli et al., 2004; Modis et al., 2004). The positions and orientations of the amphipathic E-H2 helix and the transmembrane helices are completely different with respect to DI + DIII in mature and immature particles (Figure 6). In the mature virus, E-H2 is in close contact with DII, whereas in the immature virus DII is too far from the viral membrane to permit such a contact (Figure 5). The difference in orientation of the E-H2 helix in the mature and immature particles might be related to the absence of the pr peptide hiding a part of DII in the mature virus.
Figure 6. The Structure of the Stem and Anchor Regions.
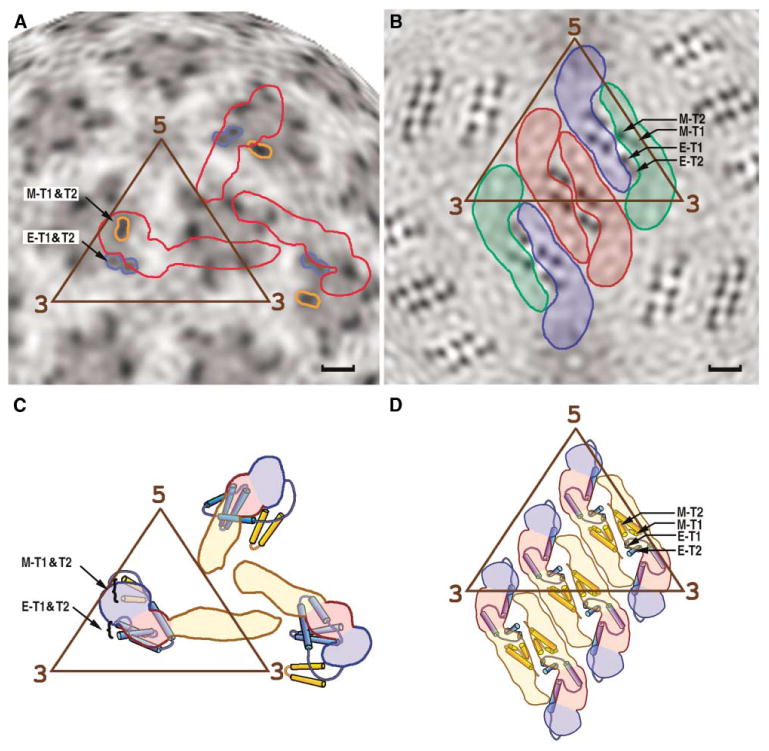
Position of the stem and anchor regions of the E and M proteins in the immature (A and C) and mature (B and D) particles. Radial projections of the three-dimensional cryo-EM densities at radius of 180 Å is shown for the (A) immature and (B) mature dengue virus particles, corresponding roughly to the middle of the lipid bilayer. The outlines of three E monomers, representing one asymmetric unit, are shown in red in (A). The three E monomers per asymmetric unit, arranged as a raft of dimers, are colored red, blue, and green in (B). Darker shading represents higher density regions resulting from the transmembrane protein helices E-T1 and E-T2 of the E glycoprotein and M-T1 and M-T2 for the membrane protein. Structural interpretations are shown in (C) and (D), respectively, where the color code for domains I, II, and III of the E protein is the same as in Figure 1. The scale bar is 20 Å long.
There are six pairs of antiparallel transmembrane regions per icosahedral asymmetric unit, one pair of E anchor helices and one pair of M anchor helices being associated with each E ectodomain (Figure 6). The inner and outer lipid leaflets are pinched closer together around each pair of transmembrane helices, in both the mature and immature particles. However, the surface of the membrane is roughly polygonal in the mature virus, whereas the membrane is essentially spherical in immature particles. This is probably related to the manner in which the E dimers hug the surface of the membrane in the mature virus compared with the less compact packing of the E monomers in immature particles.
Residues on the outer surface of DII, including the fusion peptide, are involved in the formation of the E-prM complex in immature virions. The fusion peptide is completely embedded in the prM protein and is protected from exposure to the acidic environment in the TGN during egress of virus particles (Zhang et al., 2003b). The prM protein runs from the top of the fusion peptide of the associated E molecule along the b and j strands to the αB helix of E. However, the prM density cannot be traced between this helix and the easily recognizable transmembrane helices of the M protein.
Role of the Hinge Motion of E in the Viral Life Cycle
Comparison of three different crystal structures of dengue sE and the structures of the E monomer in the immature and mature virions has shown that, at neutral or basic pH, the E molecule can bend about a hinge between DI + DIII and DII. It has also been shown that motion about this hinge occurs in the maturation process. Similarly, hinge motions are likely to be essential during the prefusion process when the fusion peptides are redirected toward the target cell membrane during or after the formation of E homotrimers (Bressanelli et al., 2004; Modis et al., 2004).
In immature virus particles, the E protein points away from the membrane with its fusion peptide at its extremity. In contrast, in the mature virions the entire E ectodomain lies on the viral membrane with the fusion peptide buried in the “cavity” formed by DI + DIII in the adjacent E molecule of an E dimer. During maturation, DI + DIII reorients with respect to the viral membrane and is accompanied by a 27° hinge motion between DII and DI + DIII. Furthermore, there is also a conformational change of the stem-anchor region, with E-H2 rearranged relative to DI + DIII.
In the fusion process, dimeric E molecules reassociate to form trimers after the exposure to acid pH (Allison et al., 1995). Kuhn et al. (Kuhn et al., 2002) have suggested a sequence of events required for trimerization. However, their proposal stops short of suggesting how the fusion peptides might become exposed for entry into the target cell membrane. The hinge movements of 37° between the E proteins in their postfusion state and in the mature virus might readily accomplish this task. The resultant radial extension and associated tangential contraction of each trimer would also require adjustment of the stem-anchor region. The sum of the lengths of the E-H1 and E-H2 helices is ~58 A in the mature virus, which is similar to the ~55 Å separation between the carboxyl end of the E ectodomain and the membrane surface in the postfusion complex (Bressanelli et al., 2004; Modis et al., 2004). Thus, the organization of the helices in the stem region during fusion is expected to remain mostly unchanged, but with the major rearrangement occurring between DI and DIII. In contrast, the maturation process rearranges the stem region from a roughly antiparallel organization of helices E-H1 and E-H2 to one that is roughly extended and parallel.
Experimental Procedures
Crystallization and Data Collection
Purified DEN sE (Cuzzubbo et al., 2001; Ivy et al., 1997) (residues 1–395) was dissolved in 20 mM Tris buffer (pH 8.0) at a concentration of 10–12 mg/ml. β-OG was mixed with protein stock solution to a final concentration of 0.3% prior to crystallization. The mother liquor used for hanging drop vapor diffusion crystallization consisted of 1.2 M sodium chloride, 0.1 M Tris buffer (pH 8.5), and 10%–15% (w/v) PEG 4000. Crystals, grown at room temperature, were soaked in a solution containing mother liquor plus 16% ethylene glycol, and then immediately flash-cooled in a nitrogen stream. Data were collected at beamline 14c at the Advanced Photon Source (Argonne National Laboratory, USA), and then processed using the HKL2000 program (Otwinowski and Minor, 1997). The crystals belonged to a primitive trigonal space group and had unit cell parameters of a = b = 71.9 Å, c = 140.9 Å (Table 1).
Structure Determination and Comparison
Attempts at using the TBEV sE structure (Rey et al., 1995) as a search model failed, probably due to the different hinge angle between the search model and DEN sE(P). Initial phases were then obtained by molecular replacement with the program AMoRe (Navaza, 1994, 2001) using the structure of the DEN sE(H) monomer derived from crystals which had a P3121 space group (Protein Data Bank [PDB] accession code 1OAM) (Modis et al., 2003) as a starting model. The space group of the DEN sE(P) crystals was determined to be P3221 by the translation search. Rigid body refinement, with each domain treated as a rigid unit, was performed with the CNS program (Brünger et al., 1998) using the maximum likelihood function as a target. Simulated annealing was used in the initial stages of atomic refinement, followed by cycles of manual rebuilding with O (Jones et al., 1991) together with positional refinement, and finally restrained, individual, B factor refinement (Table 1). Molecular contact calculations, structural alignments, and the determination of hinge angles were performed with programs in the CCP4 package (CCP4, 1994).
Fitting of sE into the Mature DEN Cryo-EM Density Map
The various crystal structures of DEN sE show variability in the hinge region between DI and DII, whereas DI and DIII remain associated as a rigid body. Hence, the fitting procedures were based on independent fitting of DII and of DI + DIII (see Results and Discussion). The fitting was based on DEN sE(P), which was divided into DI + DIII (residues 1–51, 133–190, and 276–395), and DII (all residues not in DI + DIII). These two rigid bodies were initially fitted independently into the 9.5 A mature DEN virion cryo-EM map (Zhang et al., 2003a) using the program EMfit (Rossmann et al., 2001) and constrained with the icosahedral symmetry of the virus. The relative position and orientation of the two rigid bodies within each of the three independent monomers within the icosahedral asymmetric unit were found to be closely similar, but differ significantly from those found in the crystal structures. The similarity among the three independent monomers also allowed the accurate determination of the relative position and orientation of these to each other.
Fitting of the two types of rigid bodies was then repeated, but using the now established nonicosahedral symmetry operators in addition to the icosahedral symmetry. The simultaneous fitting into the nonicosahedrally related densities improved the final accuracy of the fitting operation. The separation of Cα atoms at the N and C ends of the four polypeptides that form the hinge region between the two rigid bodies varies from 5.4 to 10.5 A with no steric clashes. Small adjustments of the polypeptide chains readily closed the gap between the “cut” ends. The quality of fit, measured in terms of Sumf, Clash, and –Den (Table 3), was roughly the same for each subunit at symmetry equivalent positions, indicating (i) that the symmetry operators had been well determined, (ii) that the separation of the monomer into two rigid bodies was justified, and (iii) that the map was an accurate representation of the virus structure. The original quasi-atomic structure determination of dengue virus (Kuhn et al., 2002; Zhang et al., 2003a) was based on eight parameters, with one rotational and one translational parameter for the icosahedral dimer and three rotational and three translational parameters for the general dimer. The present fitting involved a 12 parameter search, with 6 parameters used to define the orientation and position of each of the 2 rigid groups. Although a better fit might be expected owing to the use of more degrees of freedom in the present procedure, the results were significantly worse when either the dimers or the monomers found in the crystal structures were used for fitting (Table 3 and Figure 4).
Reconstruction and Analysis of Immature DEN Cryo-EM Map
The previous cryo-EM image reconstruction of DEN immature particles was limited to 16 Å resolution (Zhang et al., 2003b). The resolution of the reconstruction has been improved to 12.5 A (the resolution at which the Fourier shell correlation coefficient falls below 0.5), with the same images used as before, but sampled at a finer pixel size (2.12 A) and refined using the program POR (Ji et al., 2003). The rigid body DI + DIII (see above) was first fitted into the density map using the EMfit program, followed by fitting the rigid body DII while restraining connecting points to lie no further than 10 A apart. The two-step fitting procedure was otherwise performed as described above for the mature particle. The orientational and positional relationships between the two rigid bodies within the three monomers in each asymmetric unit of the immature virus were very similar. Thus, the fitting was constrained by imposing the nonicosahedral symmetry between the three E subunits on each of the two independent rigid bodies (DI + DIII and DII). A difference map was then generated by setting to zero all the density within a 4.5 A sphere of each sE atomic position. Therefore, all the remaining density outside the lipid bilayer should belong to the N-terminal part of prM, and the highest density in the membrane region should belong to the stem-anchor regions of E and prM. Atoms in sE that interact with other protein components in the virion were mapped by summing the density of the difference map within a 6.5 A radius sphere of each atom. A higher value for an atom indicated that the atom is close to a nonzeroed part of the map, which presumably should be a neighboring protein in contact with sE.
Acknowledgments
We thank Suchetana (Tuli) Mukhopadhyay for many helpful discussions, as well as Sharon Wilder and Cheryl Towell for the preparation of the manuscript. We are grateful to Narayanasamy Nandhagopal and Alan A. Simpson, as well as people working at APS BioCARS, for help in data collection. The work was supported by NIH Program Project grants to R.J.K., M.G.R., and T.S.B. (AI 55672, AI 45976).
Footnotes
Accession Numbers: Coordinates of the DEN sE(P) crystal structure and the fitted sE structures in both mature and immature virions have been deposited with the PDB (accession numbers are 1TG8, 1THD, and 1TGE, respectively).
References
- Allison SL, Schalich J, Stiasny K, Mandl CW, Kunz C, Heinz FX. Oligomeric rearrangement of tick-borne encephalitis virus envelope proteins induced by an acidic pH. J Virol. 1995;69:695–700. doi: 10.1128/jvi.69.2.695-700.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison SL, Schalich J, Stiasny K, Mandl CW, Heinz FX. Mutational evidence for an internal fusion peptide in flavivirus envelope protein E. J Virol. 2001;75:4268–4275. doi: 10.1128/JVI.75.9.4268-4275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj S, Holbrook M, Shope RE, Barrett ADT, Watowich SJ. Biophysical characterization and vector-specific antagonist activity of domain III of the tick-borne flavivirus envelope protein. J Virol. 2001;75:4002–4007. doi: 10.1128/JVI.75.8.4002-4007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J, Heinz FX, Rey FA. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004;23:728–738. doi: 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project, Number 4) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Cuzzubbo AJ, Endy TP, Nisalak A, Kalayanarooj S, Vaughn DW, Ogata SA, Clements DE, Devine PL. Use of recombinant envelope proteins for serological diagnosis of dengue virus infection in an immunochromatographic assay. Clin Diagn Lab Immunol. 2001;8:1150–1155. doi: 10.1128/CDLI.8.6.1150-1155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayhoff MO. Atlas of Protein Sequence and Structure 1972, Volume 5. Silver Spring, MD: National Biomedical Research Foundation; 1972. [Google Scholar]
- Guirakhoo F, Bolin RA, Roehrig JT. The Murray Valley encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology. 1992;191:921–931. doi: 10.1016/0042-6822(92)90267-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz FX, Allison SL. Structures and mechanisms in flavivirus fusion. Adv Virus Res. 2000;55:231–269. doi: 10.1016/S0065-3527(00)55005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy J, Nakano E, Clements DE. Subunit immunological composition against dengue infection. U.S Patent 6. 1997 Aug;165:477.
- Ji Y, Marinescu DC, Zhang W, Baker TS. Orientation Refinement of Virus Structures with Unknown Symmetry. Los Alamitos, CA: IEEE Computer Society; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, et al. Structure of the dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108:717–725. doi: 10.1016/s0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno G, Chang GJJ, Tsuchiya KR, Karabatso N, Cropp CB. Phylogeny of the genus Flavivirus. J Virol. 1998;72:73–83. doi: 10.1128/jvi.72.1.73-83.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 991–1041. [Google Scholar]
- Mackenzie JM, Westaway EG. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J Virol. 2001;75:10787–10799. doi: 10.1128/JVI.75.22.10787-10799.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMinn PC. The molecular basis of virulence of the encephalitogenic flaviviruses. J Gen Virol. 1997;78:2711–2722. doi: 10.1099/0022-1317-78-11-2711. [DOI] [PubMed] [Google Scholar]
- Modis Y, Ogata SA, Clements DE, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci USA. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis Y, Ogata SA, Clements DE, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Kim BS, Chipman PR, Rossmann MG, Kuhn RJ. Structure of West Nile virus. Science. 2003;302:248. doi: 10.1126/science.1089316. [DOI] [PubMed] [Google Scholar]
- Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- Navaza J. Implementation of molecular replacement in AMoRe. Acta Crystallogr D Biol Crystallogr. 2001;57:1367–1372. doi: 10.1107/s0907444901012422. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processingof X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature. 1995;375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- Rossmann MG, Bernal R, Pletnev SV. Combining electron microscopic with X-ray crystallographic structures. J Struct Biol. 2001;136:190–200. doi: 10.1006/jsbi.2002.4435. [DOI] [PubMed] [Google Scholar]
- Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. 1997;71:8475–8481. doi: 10.1128/jvi.71.11.8475-8481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003a;10:907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Corver J, Chipman PR, Zhang W, Pletnev SV, Sedlak D, Baker TS, Strauss JH, Kuhn RJ, Rossmann MG. Structures of immature flavivirus particles. EMBO J. 2003b;22:2604–2613. doi: 10.1093/emboj/cdg270. [DOI] [PMC free article] [PubMed] [Google Scholar]