

Abstract
The estrogen receptors (ERs) are ligand-inducible transcription factors that play key roles in the control of growth and differentiation in reproductive tissues. We showed that the novel Dbl family proto-oncoprotein Brx enhances ligand-dependent activity of ERα via a Cdc42-dependent pathway. Brx also significantly enhances ligand-dependent activity of ERβ. This enhancement is not affected by inhibition of p44/42 mitogen-activated protein kinase (MAPK) activation by PD98059. However, addition of the p38 MAPK inhibitor SB202190 abrogates the enhancement of ERβ activity by Brx, showing that p38 MAPK activity is required for the enhancement of ERβ function by Brx. In COS-7 cells, transfection of Brx leads to activation of endogenous p38 MAPK activity. Co-expression of the β2 isoform of human p38 MAPK and a constitutively active form of the p38 MAPK kinase MKK6 (MKK6-EE) synergistically augments ligand-dependent activity of ERβ. Our findings suggest that p38 MAPKs may be important regulators of ERβ activity.
The steroid hormone estrogen plays a critical role in the regulation of growth and development of reproductive tissues. The effects of estrogen in these tissues are due to changes in gene expression modulated by the estrogen receptors (ERs)1 that are members of the nuclear hormone receptor superfamily of transcription factors. After being activated by binding estrogen, the ER activates transcription of hormone-responsive genes. Prior to 1996 estrogen-dependent effects were thought to be mediated by a single estrogen receptor molecule, now designated ERα. Another estrogen receptor, ERβ, was cloned from human testis (1, 2). The ERs are modular transcription factors containing two transcription activation functions, AF-1 located in the N-terminal A/B domain, and AF-2 located within the C-terminal ligand-binding domain. Although AF-2 function is dependent upon ligand binding, AF-1 functions independently of ligand binding (reviewed in Ref. 3) but synergizes with AF-2 in the promotion of ligand-dependent transcription activation by the receptor (4).
The ER can also be activated by ligand-independent pathways involving signals originating from growth factor receptors. This pathway involves signaling from cell surface receptors and results in phosphorylation of the estrogen receptor (reviewed in Refs. 5, 6). Activation of ERα by epidermal growth factor was shown to involve phosphorylation of serine 118 in the AF-1 region of ERa through a Ras-Raf-MAPK signaling pathway (7, 8). Both ERα and ERβ are activated in transient transfection systems by co-expression of the oncogenic V12 mutant of Ras (RasV12). In both cases the activation depends on phosphorylation of target serine residues located within the receptors' AF-1 domains by p44/42 MAPK (ERK1/2) (9). Ras-dependent phosphorylation of the AF-1 domain of ERβ is thought to enhance ligand-independent transcriptional activation by enhancing recruitment of transcription co-activators such as SRC-1 (10).
We previously described the cloning and characterization of a cDNA encoding a protein Brx (breast cancer nuclear hormone receptor auxiliary factor) that interacts with ERα, is preferentially expressed in reproductive and immune tissues, and augments ligand-dependent transcriptional activation by ERα (11). Sequence analysis showed that Brx belongs to the Dbl family of proto-oncogenes (reviewed in Ref. 12) that function as guanine nucleotide exchange factors (GEFs) for Rho-GTPases, small Ras-related cytoplasmic proteins involved in signal transduction pathways governing growth and morphogenic transformation. Proteins acting as GEFs shift the equilibria of small GTPases toward an active GTP-bound state. We found that activation of ERα by Brx in transient transfection assays involves the Rho-related GTPase Cdc42. Our findings demonstrated for the first time that ERa function might be modulated by pathways dependent on small GTPases other than Ras.
Brx mRNA is highly expressed in the same human tissues that express ERβ mRNA. These include, specifically, ovary, uterus, testis, spleen, thymus, peripheral blood leukocytes, and specific areas of the brain, including the amygdala and hypothalamus,2 which are thought to be regulated by estrogen. The present study was performed to investigate the effect of Brx on ERβ. We show that Brx enhances ligand-dependent activity of ERβ. Furthermore, we demonstrate that Brx sends signals to ERβ through a pathway involving p38 MAPKs.
Experimental Procedures
Cell Culture and Transient Transfection Studies
Ishikawa human endometrial adenocarcinoma cells were cultured in phenol red-free Dulbecco's modified Eagle's medium/F-12 (Life Technologies, Inc.) supplemented with 5% charcoal-stripped fetal bovine serum (HyClone). For experiments examining the activation of the c-fos SRE, cells were serum-starved overnight prior to transfection. COS-7 cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. For experiments examining the activation of endogenous p38 MAPK activity, COS-7 cells were plated onto 100-mm dishes and transfected with 15 μg of expression vector for Brx, pBK-RSV, or MKK6-EE using FuGENE-6 (Roche Molecular Biochemicals) as described by the manufacturer. Twenty-four hours after transfection, cells were subjected to overnight serum starvation prior to lysis and immunoblotting. For transient transfection studies Ishikawa cells were plated onto 12-well plates and 100 ng of pRSV-ERβ, 1 μg of pRSV-Brx, 500 ng of GAL4-Elk-1, 500 ng of GAL4-ERβ or empty vector, and 1.5 μg of pBK-CMV, 1.0 μg of (ERE)2-tk-luciferase, 1.0 μg of SRE-tk-luciferase or 1.0 μg of G4E1b-luciferase were added to cells with FuGENE-6 as per the manufacturer's instructions. Cells were harvested 20 h after transfection, and luciferase assays were performed and normalized as described previously (11). In all transfection experiments, total amounts of DNA were made constant by the addition of empty vectors. 17-β-Estradiol (10 nm), 4-hydroxy tamoxifen (40 nm), PD98059 (10 μm, Cal-biochem), SB202190 (2 μm, Calbiochem) were added as described.
Plasmids
Expression vectors for Brx, (ERE)2-tk-luciferase, SRE-tk-luciferase, and MEKE, were described previously (11, 13–15). A full-length cDNA-encoding human ERβ (2) was amplified from a human prostate Marathon-Ready cDNA library (CLONTECH). The cDNA was subcloned into pBK-RSV (Stratagene) and sequenced to make RSV-ERβ. The ERβ cDNA beginning at the second codon was subcloned into the pM expression vector (CLONTECH) containing the GAL4 DNA binding domain to make GAL4-ERβ. A full-length cDNA encoding Elk-1 was amplified from a human peripheral blood leukocyte cDNA library (CLONTECH), subcloned into pM, and sequenced to make GAL4-Elk-1. Expression plasmids for MEKE and wild type MKK6 were kindly provided by Dr. Silvio Gutkind. MKK6-EE containing Ser to Glu and Thr to Glu mutations at codons 207 and 211, respectively, were derived from wild-type MKK6 by overlap amplification with primers containing site-specific mutations. The resulting cDNA was subcloned into pBK-RSV, and individual clones were sequenced to verify the presence of the mutations. Full-length cDNA encoding human p38β2 was amplified from a brain cDNA library (CLONTECH), subcloned into pBK-RSV, and sequenced.
Immunoblotting
Transfected COS-7 cells were harvested at 4 °C and lysed in SDS sample buffer by sonication. Following centrifugation, proteins were resolved by SDS-polyacrylamide gel electrophoresis and transferred to BA85 nitrocellulose (Schleicher and Schuell) by semi-dry electroblotting. Membranes were blocked in Tris-buffered saline containing 0.05% Tween-20 and 5% nonfat dry milk. Phospho-p38 MAPK primary antibody and HRP-conjugated anti-rabbit secondary antibody (both from New England BioLabs) were used and detected per the manufacturer's instructions. Epitope-tagged Brx and GAL4-ERβ were detected with HRP-conjugated anti-FLAG M2 monoclonal antibody (Sigma Chemical Co.) and HRP-conjugated GAL4 (DBD) monoclonal antibody (Santa Cruz Biotechnology, Inc.) per the manufacturers' instructions.
Protein Phosphorylation
Proteins were phosphorylated with active recombinant human p38β2 (Upstate Biotechnology Inc.) per the manufacturer's instructions. Twenty milligrams of protein substrates were incubated with 100 ng of active p38β2 in the presence of [γ-32P]ATP (1 mCi/ml) at 30 °C for 15 min with rotation in a final reaction volume of 40 ml. Reactions were stopped by addition of SDS sample buffer and heating to 95 °C for 5 min. Labeled proteins were resolved on 10% SDS-polyacrylamide gels and detected by autoradiography.
Results
Brx Enhances Ligand-dependent Transcriptional Activation by Estrogen Receptor β
Given our previous observation that Brx enhances ligand-dependent transcriptional activation by ERα in transiently transfected Ishikawa endometrial cells, we wanted to test whether Brx expression could also affect the ability of ERβ to activate a reporter. Transfection of ERβ resulted in severalfold ligand-dependent activation of the G4E1b-luciferase reporter (Fig. 1A). Co-transfection of an expression vector for full-length Brx enhanced ligand-dependent trans-activation by ERβ ∼ 10-fold.
Fig. 1.
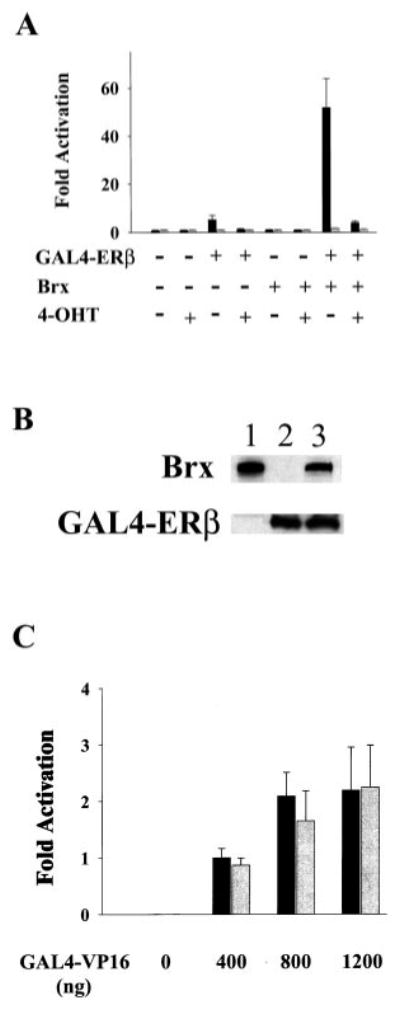
A, Brx co-expression augments ligand-dependent activation of a G4E1b-luciferase reporter plasmid by GAL4-ERβ. Augmentation of ERβ-mediated activation of G4E1b-luciferase by Brx co-expression is inhibited by the addition of 4-hydroxy tamoxifen. Ishikawa cells were transfected with 1 μg of G4E1b-luciferase and expression vectors for GAL4-ERβ3 (500 ng) and Brx (1 μg) or the respective amounts of empty vectors as control. Estradiol (10 nm, solid bars) or vehicle control (gray bars), and vehicle control or 4-hydroxytamoxifen (40 nm) was added to the cells as indicated. Cells were harvested after 20 h. Luciferase values represent -fold activation (mean) over control (RSV-0 with G4E1b-luciferase) in the absence of ligand, under each condition from three experiments performed in triplicate. Error bars represent standard deviations. B, Brx did not affect receptor expression levels. COS-7 cells were transfected with expression vectors for Brx (lane 1), GAL4-ERβ (lane 2), or both (lane 3). Brx and GAL4-ERβ were detected in lysates of transfected cells by Western blotting. Plasmid amounts were held constant by addition of empty expression vectors. C, Brx does not augment the activity of a chimeric GAL4-VP16 transcription factor. Ishikawa cells were transfected with 1 μg of G4E1b-luciferase reporter plasmid and expression vectors for GAL4-VP16 (0, 400, 800, or 1200 ng, as indicated) and Brx (1 μg, gray bars) or the same amounts of empty expression vectors (solid bars). Cells were harvested and assayed for luciferase activity after 20 h. Luciferase values represent means of-fold activation (with activation given by 400 ng of GAL4-VP16 arbitrarily assigned a value of 1.0) from two experiments performed in duplicate. Error bars represent standard deviations.
Although tamoxifen acts as an estrogen antagonist in the breast, in the uterus it acts as an estrogen agonist, and long term treatment of breast cancer with tamoxifen results in an increased risk of endometrial cancer (16, 17). We found that in Ishikawa cells 40 nm 4-hydroxytamoxifen blocked ligand-dependent activity of ERβ (Fig. 1A). Addition of 40 nm 4-hydroxytamoxifen completely eliminated ligand-dependent activation of the reporter by ERβ in the absence of Brx and greatly reduced ligand-dependent activity of the receptor in the presence of Brx. These results suggest that the full level of activity of ERβ in the presence of Brx was dependent on activation of the receptor by ligand binding. In our previous study we reported an antagonistic effect of tamoxifen on the ligand-dependent activity of ERα in Ishikawa cells (11).
We tested the possibility that Brx augments receptor activity through an effect on receptor expression levels. COS-7 cells were transfected with expression vectors for Brx and GAL4-ERβ. Western analyses showed that co-expression of Brx did not affect the level of GAL4-ERβ expression (Fig. 1B), suggesting that the observed augmentation of ligand-dependent activity of ERβ by Brx is not simply due to an increase in the amount of receptor protein expressed. We also tested the specificity of Brx action by assessing the ability of Brx to activate a chimeric GAL4-VP16 transcription factor. Under conditions in which Brx enhances the ligand-dependent activity of ERβ, Brx failed to enhance the activity of GAL4-VP16 (Fig. 1C). Brx expression also did not affect the activity of the G4E1b-luciferase reporter.
Brx Signaling to ERβ Does Not Involve p44/42 MAPK (ERK1/ERK2)
ERβ has been shown to be activated independently of ligand binding by a mechanism involving phosphorylation of Ser residues in AF-1 through the Ras-Raf-p44/42 MAPK (ERK1/ERK2) signaling pathway (9). It has also been shown that Cdc42 can activate ERK2 in cooperation with Raf (18). We therefore tested whether activation of ERβ by Brx involves p44/42 MAPK. Dual specificity mitogen-activated protein kinase kinases (MEK) activate p44/42 MAPK by phosphorylating specific Thr and Tyr residues. The inhibitor PD98059 is thought to block activation of p44/42 MAPK (19) by binding to inactive MEK1 and blocking activation by upstream kinases (20). In these experiments Brx only modestly affected activity of the receptor. Addition of 10 mM PD98059 did not affect the enhancement of ligand-dependent activity of ERβ by Brx (Fig. 2A). Addition of the inhibitor also did not affect ligand-dependent activity of ERβ in the absence of Brx. In control experiments the same concentration of PD98059 effectively inhibited activation of a SRE-tk-Luc reporter by mutationally activated MEK1 (21) (Fig. 2B).
Fig. 2.
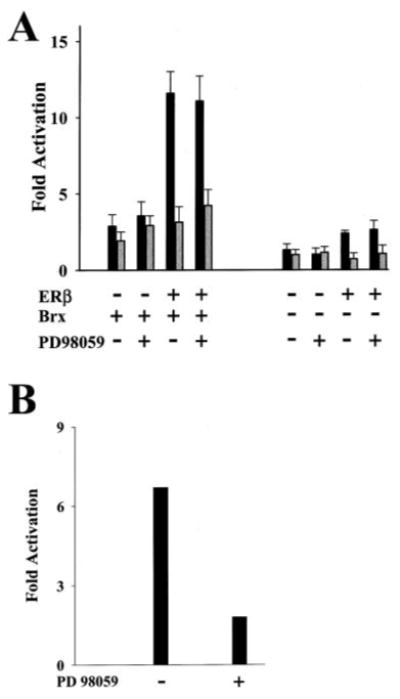
A, augmentation of ERβ activity by Brx is not affected by the inhibition of mitogen-activated protein kinase (MAPK) activity. The two panels show activation of (ERE)2-tk-Luc reporter plasmid by ERβ with co-expression of Brx (left panel) and without (right panel). Addition of PD98059, a mitogen-activated protein kinase kinase (MEK) inhibitor, did not attenuate the activation of ERβ with or without Brx co-expression. Ishikawa cells were transfected with 1 μg of (ERE)2-tk-Luc and expression vectors for ERβ (100 ng) and Brx (1 μg) or the respective amounts of empty vectors as control. Estradiol (10 nm, solid bars) or vehicle control (gray bars) and PD98059 (10 μm) (+) or an equal amount of vehicle (−) was added to the cells as indicated. Cells were harvested after 20 h. Luciferase values represent-fold activation (mean) over control (RSV-0 with (ERE)2-tk Luc in the absence of ligand) from three experiments performed in triplicate. Error bars represent standard deviations. B, PD98059 inhibits MAPK in Ishikawa cells. Activation of a serum response element reporter in Ishikawa cells is attenuated by the addition of PD98059. Ishikawa cells were serum-starved overnight and transfected with 1.0 μg of a c-fos serum response element SRE-tk-luciferase reporter. The two columns show activation of the SRE-tk-Luc reporter by co-transfection of an expression vector encoding MEKE, a constitutively active mutant form of MEK. PD98059 (10 μm) (+) or an equal amount of vehicle (−) was added to the cells. Cells were harvested after 20 h. Luciferase values represent -fold activation compared with reporter activity after transfection of empty expression vector (not shown).
Brx Activates ERβ by a p38 MAPK-dependent Pathway
We previously showed that Brx enhances ligand-dependent activity of ERα by a Cdc42-dependent pathway. Because activation of ERβ by Brx did not appear to involve p44/42 MAPK pathways, and because Cdc42 has been shown to activate p38 MAPKs (22), we tested whether the enhancement of ERβ activity by Brx could involve a p38 MAPK pathway. The pyridinyl imidazole compound SB202190 inhibits the α and β isoforms of p38 MAPKs by a mechanism involving direct binding to the kinases (23). In our transient transfection system using ERβ expressed as a GAL4 DBD fusion protein (GAL4-ERβ) and the G4E1b-luciferase reporter, transfection of GAL4-ERβ activated luciferase expression severalfold in the presence of ligand compared with control transfections lacking ligand. Co-transfection of Brx enhanced ligand-dependent activity of GAL4-ERβ ∼10-fold (Fig. 3A). Addition of 2 μm SB202190 inhibited activation of ERβ by Brx by ∼70%, suggesting that p38α or p38β activity is required for enhancement of ligand-dependent activity of ERβ by Brx. SB202190 did not inhibit the activity of a chimeric GAL4-VP16 transcription factor (Fig. 3B).
Fig. 3. Activation of ERβ by Brx is dependent on p38 MAPK activity.
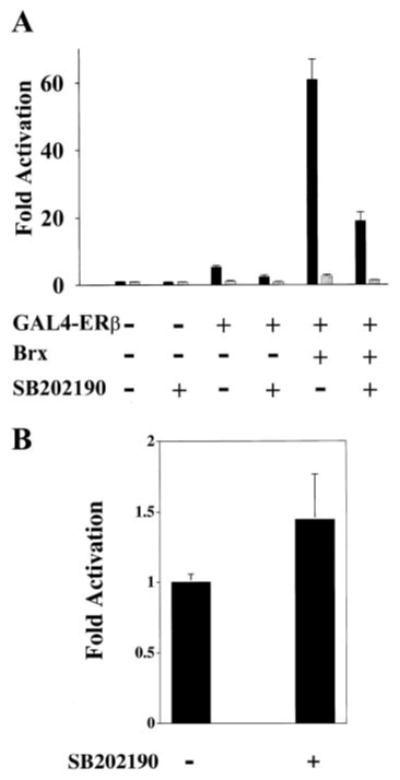
A, activation of ERβ by Brx is inhibited by the p38 MAPK inhibitor SB202190 (2 μm). Ishikawa cells were transfected with 1 μg of a G4E1b-luciferase reporter plasmid and expression vectors for GAL4-ERβ (100 ng) and Brx (1 μg) or the same amounts of empty expression vectors. Estradiol (10 nm, solid bars) or vehicle control (EtOH, gray bars) and SB202190 or control (SB202474) were added as indicated. Cells were harvested after 20 h. Luciferase values represent -fold activation (mean) over control (RSV0 with G4E1b-luciferase in the absence of ligand) from two experiments performed in duplicate. Error bars represent standard deviations. B, the p38 MAPK inhibitor SB202190 does not inhibit the activity of GAL4-VP16. Ishikawa cells were transfected with 1 μg of a G4E1b-luciferase reporter plasmid and 400 ng of an expression vector for GAL4-VP16. SB202190 (3 μm) was added as indicated. Luciferase values represent -fold activation (with activation given by 400 ng of GAL4-VP16 arbitrarily assigned a value of 1.0) from two experiments performed in duplicate. Error bars represent standard deviations.
Because our results suggested that p38 MAPK activity is required for augmentation of receptor activity by Brx, we tested whether p38 activation affects activity of ERβ in Ishikawa cells. The p38 MAPKs can be activated by several dual-specificity MAPK kinases, including MKK3, MKK6, MKK4, and MKK7 (24–27). MKK4 and MKK7 also efficiently activate JNK, and MKK3 activates only p38α, γ, and δ. Each of the p38 MAPKs (α, β, γ, and δ) has been shown to be phosphorylated and activated by MKK6 (28). A constitutively active mutant form of MKK6 (MKK6-EE,) contains Ser to Glu and Thr to Glu mutations at amino acid positions 207 and 211, respectively, and activates p38 MAPKs (29).
Transfection of Ishikawa cells with an expression vector for the β2 isoform of p38 MAPK or MKK6-EE alone did not activate GAL4-ERβ, but co-transfection of both synergistically enhanced ligand-dependent activity of GAL4-ERβ (Fig. 4A). The transcription factor Elk-1 is thought to be a physiological substrate of p38 MAPK (29). We therefore performed control experiments to test for activation of Elk-1 by MKK6-EE and p38β2 in Ishikawa cells. As we observed for ERβ, transfection of either p38β2 or MKK6-EE was not sufficient to activate a control GAL4-Elk-1 construct. Co-transfection of Ishikawa cells with both p38β2 and MKK6-EE synergistically activated GAL4-Elk-1 (Fig. 4B). To test directly whether Brx activates p38 MAPK, we transfected COS-7 cells and assessed the effect of Brx expression on p38 MAPK activity by immunoblotting. Transfection of Brx increased endogenous p38 MAPK activity approximately 3-fold (Fig. 4C). Transfection of MKK6-EE also activated endogenous p38 MAPKs severalfold in COS-7 cells (Fig. 4C). A relatively low transfection efficiency has prevented us from performing a similar analysis in Ishikawa cells. We tested whether p38 MAPK can utilize ERβ as a phosphorylation substrate. Active recombinant human p38β2 MAPK efficiently phosphorylated recombinant ERβ (Fig. 4D), suggesting that ERβ is a potential target substrate of the kinase.
Fig. 4.
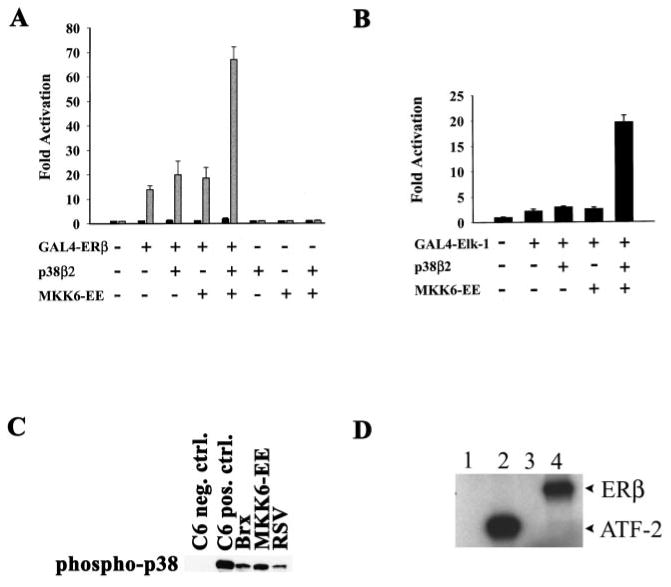
A and B, MKK6-EE and p38β2 synergistically activate ERβ (A) and Elk-1 (B) in Ishikawa. Cells were transfected with 1 μg of G4E1b-luciferase reporter and 500 ng of expression vectors for GAL4-Elk-1 or GAL4-ERβ, MKK6-EE, and p38β2 or the same amounts of empty expression vectors, as indicated. Cells were harvested for assay after 20 h. Values represent -fold activation (mean) over control (G4E1b-luciferase with empty vectors) from two experiments performed in duplicate. C, Brx activates endogenous p38 MAPK in COS-7 cells. Cells were transfected with expression vectors for Brx or MKK6-EE or with empty vector (RSV-0). Transfected cells were serum-starved overnight, and active p38 MAPK was detected in lysates of transfected cells by Western blotting. Positive and negative control lanes contained C-6 glioma cell extracts prepared with or without anisomycin treatment, respectively. D, ERβ is efficiently phosphorylated by p38β2. Active recombinant human p38β2 MAPK was incubated in vitro with [32P]ATP and substrate proteins for 15 min at 30 °C. Labeled proteins were resolved by electrophoresis and detected by autoradiography. Phosphorylation substrates were no protein (lane 1), recombinant GST-ATF-2 (lane 2), BSA (lane 3), and recombinant ERβ (lane 4).
Discussion
The estrogen receptors (ERs) are modular ligand-inducible transcription factors that play a key role in the regulation of growth and differentiation of reproductive tissues. An important issue in the study of ER signal transduction pathways is that of ligand-independent activation (5). Studies have documented effects upon ER-mediated gene activation by epidermal growth factor and other growth factors acting as ligand-independent receptor activators. Both ERα and ERβ can be activated independently of ligand binding by a Ras-Raf-p44/42 MAPK signaling pathway involving the phosphorylation of specific Ser residues in the receptors' AF-1 domains (9). In the case of ERβ, modification of the receptor was proposed to affect transcription by enhancing the recruitment of steroid receptor co-activator-1 (SRC-1) (10). The demonstration of signaling pathways linking Ras to regulation of ER activity was an important observation, because Ras has a critical role in cellular proliferation and oncogenesis.
Members of the Ras-related Rho family of small GTPases, including Rho, Rac, and Cdc42, regulate a variety of crucial cellular processes, including cytoskeletal organization, gene expression, cell cycle progression, cell adhesion, and intracellular membrane trafficking (30, 31). These proteins are regulated by GTP binding and are activated by guanine nucleotide exchange factors (Rho-GEFs) that catalyze the exchange of GDP for GTP. Rho-GEFs constitute a group of oncoproteins containing Dbl homology and pleckstrin homology domains that are required for the nucleotide exchange function (12). Different Rho-GEFs possess a variety of structural motifs presumably regulating function and specific interactions with other proteins. Although the Rho-GEFs are thought to be important targets of upstream activators of Rho GTPases, relatively little is known about the regulation of function of most of the GEFs. We recently described the cloning of brx, a novel Dbl family proto-oncogene, encoding Brx, a protein that enhances ligand-dependent activity of ERα by a Cdc42-dependent pathway (11). Our report was the first to implicate Rho GTPases in the regulation of ER function. Recently, Su et al. (32) demonstrated enhancement of ERa activity by the guanine nucleotide dissociation inhibitor GDIα, a finding that provided further support for a role for Rho GTPases in the regulation of ER activity. In this report we demonstrate that Brx enhances ligand-dependent activity of ERβ. The mechanism appears not to involve the p44/42 MAPK pathway, but instead involves activation of a p38 MAPK pathway (Fig. 5). This report represents the first report implicating p38 MAPK activation in the regulation of ERβ function.
Fig. 5.
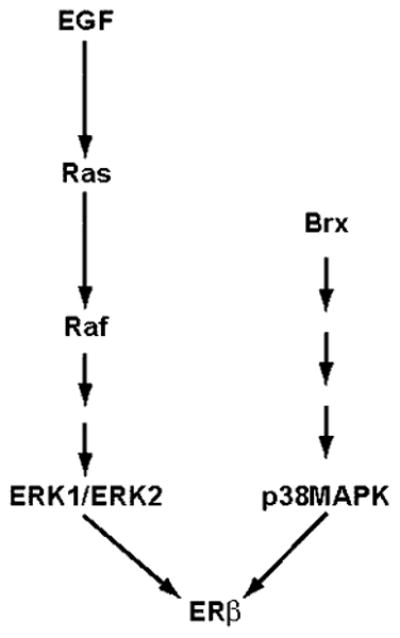
Brx augments ligand-dependent activity of ERβ via a p38 MAPK pathway.
Brx mRNA is highly expressed in human tissues that express ERβ mRNA, specifically including ovary, uterus, testis, spleen, thymus, peripheral blood leukocytes, and specific areas of the brain, including the amygdala and hypothalamus, which are thought to be regulated by estrogen (11).2 Immunohistochemical staining of human ovarian tissues showed high levels of Brx protein expression in granulosa cells of follicles (33). These data suggest that Brx is co-expressed in tissues expressing ERβ. We also found that Brx interacts with ERβ in binding assays in vitro.2 Given these findings, we wanted to know whether Brx affects ERβ function. In transient transfection assays using Ishikawa human endometrial adenocarcinoma cells, ligand-dependent activity of ERβ was markedly enhanced by co-transfection of a Brx expression vector. The full level of reporter activity required Brx, ERβ, the ERE, and estrogen. Addition of tamoxifen totally eliminated ligand-dependent activity of ERβ. In control experiments we demonstrated that activation of Brx is selective, because Brx did not affect the activity of the chimeric transcription factor GAL4-VP16. Additionally, analysis of protein expression levels in transfected COS-7 cells showed that Brx did not affect the level of GAL4-ERβ protein expression, a finding that suggests Brx does not augment ligand-dependent receptor activity simply by increasing receptor expression. Taken together, these results suggest that Brx affects the ligand-dependent activity of ERβ itself.
We examined the possibility that Brx enhances ERβ activity through a Ras-p44/42 MAPK pathway. Inhibition of MAPK activation by PD98059 did not affect the observed enhancement of ligand-dependent ERβ activity by Brx, suggesting that Brx affects ERβ function by a pathway that does not depend on activation of MAPK. We have also found that enhancement of ERa activity by Brx involves neither p44/42 MAPK nor phosphorylation of Ser-118.2 These findings showed that Brx must affect ERβ by signaling pathways distinct from the previously described Ras and p44/42 MAPK-dependent pathways (9).
We previously demonstrated that Brx augments ligand-dependent activity of ERα by a Cdc42-dependent pathway (11). Mutationally activated Cdc42 also strongly activates JNK and p38 MAPK (14, 22). One possibility, therefore, is that Brx activates a JNK or p38 MAPK that in turn augments ligand-dependent function of ERβ either directly or indirectly. We therefore tested for an effect of the p38 MAPK inhibitor SB202190 on ERβ activation by Brx and found that 2 μm SB202190 inhibited enhancement of ERβ activity by Brx by ∼ 70%, suggesting that p38 MAPK activity is required for activation of ERβ by Brx. We next tested directly whether a specific p38 MAPK could activate ERβ. Although transient transfection of either p38β2 or MKK6-EE was insufficient to activate the receptor, transfection of both synergistically activated the receptor. Similarly, for the GAL4-Elk-1 transfection control experiments, we found that the combination of both p38β2 and MKK6-EE cooperatively activated the luciferase reporter. We do not know why transfection of MKK6-EE was insufficient to activate ERβ. It is possible that MKK6-EE was unable to activate cellular p38 MAPK in Ishikawa cells, although it was able to do so in COS-7 cells in our control experiment. Unfortunately, our transfection efficiency for the Ishikawa cells has been too low to permit a similar analysis. Selectivity of p38 MAPK isoform activation by MKK6 has been described by Alonso (34). This group showed that, at relatively low concentrations both in vitro and in vivo, wild-type MKK6 activated p38α but failed to activate p38γ. At present, we do not know which p38 MAPK isoforms are expressed in Ishikawa cells and we have not yet determined whether the enhancement of ERβ activity by Brx exhibits selectivity for p38 MAPK isoforms. It is also possible that MKK6 is not involved in activation of ERβ by Brx in the Ishikawa cells. Our findings do suggest, however, that endogenous p38 MAPK activity in Ishikawa is required for activation of ERβ by Brx and that activation of p38β2 MAPK by MKK6-EE effectively enhances ligand-dependent activity of the receptor. We showed that Brx expression results in the activation of endogenous p38 MAPK in COS-7 cells.
p38 MAPKs have well established roles in the regulation and function of pro-inflammatory cytokines such as tumor necrosis factor-α and interleukin-1; they are also activated in cultured cells in response to environmental stresses such as exposure to UV radiation and hyperosmotic conditions (reviewed in Ref. 35). Recent experiments suggest that activation of p38 MAPK pathways may also play important roles in cellular differentiation processes. Differentiation in vitro of several cell lines has been shown to depend on p38 MAPK activity (reviewed in Ref. 36). It was recently reported that activation of p38 MAPKs is deficient in rhabdomyosarcoma cells (37). Activation of p38 MAPKs in these cells by ectopic expression of MKK6-EE resulted in growth arrest and terminal differentiation. Our findings suggest that p38 MAPKs may also play a role in the regulation of estrogen receptors, which are in turn key regulators of growth and differentiation in reproductive tissues.
During the preparation of this manuscript, Lee et al. (38) published a report showing activation of ERα by constitutively active MEKK1 in Ishikawa cells. These authors proposed that MEKK1 activates ERα through activation of JNK and p38 MAPK. They also demonstrated phosphorylation of ERα by p38 MAPK in vitro. These findings, considered together with our own results, suggest that p38 MAPKs may be important regulators of both forms of ER.
We do not know the mechanism for enhancement of ERβ activity by p38 MAPK and whether direct phosphorylation of the receptor by p38 MAPK is involved. However, we observed that p38 MAPK efficiently phosphorylated ERβ in vitro. By analogy with p44/42 MAPK-dependent activation of ERβ, direct phosphorylation of the receptor could affect the ability of liganded receptor to interact with or recruit a transcriptional co-activator such as SRC-1. Alternatively, p38 MAPK may indirectly affect the activity of ERβ by phosphorylating other factors required for ERβ function such as SRC-1 or p300.
Acknowledgments
We are grateful to Dr. Silvio Gutkind for the kind gifts of COS-7 cells and expression vectors for MKK6 and MEKE. We thank Drs. William Haffner and Prabir Chakraborty for their support during this project.
Footnotes
This work was supported by grants from the Uniformed Services University of the Health Sciences, the Elsa U. Pardee Foundation, and the Department of Defense Breast Cancer Research program.
The abbreviations used are: ER, estrogen receptor; MAPK, mitogenactivated protein kinase; ERK1/2, extracellular signal-regulated kinases 1 and 2; MEK, MAPK/ERK kinase; MKK, MAPK kinase; GEF, guanine nucleotide exchange factor; Brx, breast cancer nuclear hormone receptor auxiliary factor; RSV, Rous sarcoma virus; HRP, horseradish peroxidase; JNK, c-Jun N-terminal kinase; SRC-1, steroid receptor co-activator 1; DBD, DNA-binding domain; SRE, serum response element.
D. M. Rubino, unpublished.
References
- 1.Kuiper GGJM, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. J Biol Chem. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mosselman S, Polman J, Dijkema R. FEBS Lett. 1996;392:49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 3.Tsai M, O'Malley BW. Annu Rev Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 4.Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, Chambon P. Cell. 1989;59:477–487. doi: 10.1016/0092-8674(89)90031-7. [DOI] [PubMed] [Google Scholar]
- 5.O'Malley BW, Schrader WT, Mani S, Smith C, Weigel NL, Conneely OM, Clark JH. Recent Prog Horm Res. 1995;50:333–347. doi: 10.1016/b978-0-12-571150-0.50020-2. [DOI] [PubMed] [Google Scholar]
- 6.Kato S, Masuhiro Y, Watanabe M, Kobayashi Y, Takeyama K, Endoh H, Yanagisawa J. Genes to Cells. 2000;5:593–601. doi: 10.1046/j.1365-2443.2000.00354.x. [DOI] [PubMed] [Google Scholar]
- 7.Kato S, Endoh H, Mashuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 8.Bunone G, Briand PA, Miksicek RJ, Picard D. EMBO J. 1996;15:2174–2183. [PMC free article] [PubMed] [Google Scholar]
- 9.Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrie F, Giguere V. Mol Endocrinol. 1997;11:353–365. doi: 10.1210/mend.11.3.9902. [DOI] [PubMed] [Google Scholar]
- 10.Tremblay A, Tremblay G, Labrie F, Giguere V. Mol Cell. 1999;3:513–519. doi: 10.1016/s1097-2765(00)80479-7. [DOI] [PubMed] [Google Scholar]
- 11.Rubino D, Driggers P, Arbit D, Kemp L, Miller B, Coso O, Pagliai K, Gray K, Gutkind J, Segars J. Oncogene. 1998;16:2513–2526. doi: 10.1038/sj.onc.1201783. [DOI] [PubMed] [Google Scholar]
- 12.Cerione RA, Zheng Y. Curr Opin Cell Biol. 1996;8:216–222. doi: 10.1016/s0955-0674(96)80068-8. [DOI] [PubMed] [Google Scholar]
- 13.Nunez S, Medin J, Braissant O, Kemp L, Wahli W, Ozato K, Segars J. Mol Cell Endocrinol. 1997;127:27–40. doi: 10.1016/s0303-7207(96)03980-9. [DOI] [PubMed] [Google Scholar]
- 14.Coso O, Chiariello M, Yu J, Teramoto H, Crespo P, Xu N, Miki T, Gutkind J. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 15.Fromm C, Coso O, Montaner S, Xu N, Gutkind J. Proc Natl Acad Sci U S A. 1997;94:10098–10103. doi: 10.1073/pnas.94.19.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rutqvist LE, Johansson H, Signomklao T, Johansson U, Fornander T, Wilking N. J Natl Cancer Inst. 1995;87:645–651. doi: 10.1093/jnci/87.9.645. [DOI] [PubMed] [Google Scholar]
- 17.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 18.Frost JA, Steen H, Shapiro P, Lewis T, Ahn N, Shaw PE, Cobb M. EMBO J. 1997;16:6426–6438. doi: 10.1093/emboj/16.21.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pang L, Sawada T, Decker SJ, Saltiel A. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 20.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 21.Cowley S, Paterson H, Kemp P, Marshall CJ. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 22.Minden A, Lin A, Claret FX, Abo A, Karin M. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 23.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez I, Hughes RT, Mayer BJ, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 25.Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F, Johnson GL, Karin M. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 26.Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 27.Hu MCT, Wang YP, Mikhail A, Qiu WR, Tan TH. J Biol Chem. 1999;274:7095–7102. doi: 10.1074/jbc.274.11.7095. [DOI] [PubMed] [Google Scholar]
- 28.Keesler GA, Bray J, Hunt J, Johnson DA, Gleason T, Yao Z, Wang SW, Parker C, Yamane H, Cole C, Lichenstein HS. Protein Expr Purif. 1998;14:221–228. doi: 10.1006/prep.1998.0947. [DOI] [PubMed] [Google Scholar]
- 29.Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Aelst L, D'Souza-Schorey C. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 31.Kjoller L, Hall A. Exp Cell Res. 1999;253:166–179. doi: 10.1006/excr.1999.4674. [DOI] [PubMed] [Google Scholar]
- 32.Su LF, Knoblauch R, Garabedian MJ. J Biol Chem. 2001;276:3231–3237. doi: 10.1074/jbc.M005547200. [DOI] [PubMed] [Google Scholar]
- 33.Miller B, Rubino D, Driggers P, Haddad B, Cisar M, Gray K, Segars J. Am J Obstet Gynecol. 2000;182:286–295. doi: 10.1016/s0002-9378(00)70213-4. [DOI] [PubMed] [Google Scholar]
- 34.Alonso G, Ambrosino C, Jones M, Nebreda AR. J Biol Chem. 2000;275:40641–40648. doi: 10.1074/jbc.M007835200. [DOI] [PubMed] [Google Scholar]
- 35.Paul A, Wilson S, Belham CM, Robinson CJM, Scott PH, Gould GW, Plevin R. Cell Signal. 1997;9:403–410. doi: 10.1016/s0898-6568(97)00042-9. [DOI] [PubMed] [Google Scholar]
- 36.Nebreda A, Porras A. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 37.Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, Feramisco JR, Karin M, Wang JYJ. Genes Dev. 2000;14:574–584. [PMC free article] [PubMed] [Google Scholar]
- 38.Lee H, Jiang F, Wang Q, Nicosia SV, Yang J, Su B, Bai W. Mol Endocrinol. 2000;14:1882–1896. doi: 10.1210/mend.14.11.0554. [DOI] [PubMed] [Google Scholar]