

Abstract
Angiotensin II receptor type 1 blockers (ARBs), widely used antihypertensive drugs, have also been investigated for their anticancer effects. The effect of ARBs on prostate cancer in experimental models compared with meta-analysis data from clinical trials is conflicting. Whereas this discrepancy might be due to the use of supratherapeutic doses of ARBs in cellular and animal models as compared with the clinical doses used in human trials, further investigation of the effects of clinical doses of ARBs on prostate cancer in experimental models is warranted. In the current study, we sought to determine the effects of candesartan on prostate cancer cellular function in vitro and tumor growth in vivo, and characterize the underlying mechanisms. Our analysis indicated that clinically relevant doses of candesartan significantly inhibited growth of PC3 cell tumor xenografts in mice. Interestingly, the same concentrations of candesartan actually promoted prostate cancer cellular function in vitro, through a modest but significant inhibition in apoptosis. Inhibition of tumor growth by candesartan was associated with a decrease in vascular endothelial growth factor (VEGF) expression in tumors and inhibition of tumor angiogenesis, but normalization of tumor vasculature. Although candesartan did not impair PC3 cell viability, it inhibited endothelial-barrier disruption by tumor-derived factors. Furthermore, candesartan significantly inhibited expression of VEGF in PC3 and DU145 cell lines independent of angiotensin II type 2 receptor, but potentially via angiotensin II type 1 receptor inhibition. Our findings clearly demonstrate the therapeutic potential of candesartan for prostate cancer and establish a link between ARBs, VEGF expression, and prostate tumor angiogenesis.
Introduction
Prostate cancer is the most commonly diagnosed cancer among males according to the American Cancer Society (Siegel et al., 2012). About 68% of prostate cancer cases are diagnosed in the 55–74 year age group (Siegel et al., 2012), and this age group is also characterized by the high prevalence of comorbid conditions, most notably cardiovascular diseases (Roger et al., 2012).
Recently, a number of meta-analyses investigating a possible link between cancer incidence and cardiovascular disease drugs have been published (Sipahi et al., 2010; Mearns, 2011). One of the major targets of these analyses was the angiotensin II receptor type 1 blockers (ARBs), which are commonly prescribed for the management of cardiovascular diseases. The results of these analyses were controversial, with some suggesting a causal link between cancer (Sipahi et al., 2010) and ARBs, whereas others dispute such a link (Mearns, 2011). To further complicate the matter, there is a plethora of experimental evidence that suggests a possible beneficial role of ARBs in the management of multiple types of cancer, especially urogenital cancers (Miyajima et al., 2002; Kosaka et al., 2007; Takahashi et al., 2012). Experimental data demonstrated the ability of ARBs to inhibit progression and metastases in bladder, renal (Miyajima et al., 2002), and prostate cancer (Kosugi et al., 2006; Kosaka et al., 2007; Takahashi et al., 2012). This beneficial effect has been consistently reported both in monotherapy settings (Kosaka et al., 2007) and in combination with other antineoplastic agents. The antineoplastic effects of ARBs are believed to be due to their ability to inhibit cancer angiogenesis (Kosaka et al., 2007), which has been shown to be associated with severity and metastatic potential of prostate cancer (Kosaka et al., 2007).
Despite the solid experimental evidence supporting an antineoplastic effect of ARBs, the controversy between clinical and experimental data must be resolved. In the majority of experimental studies, the dose of ARBs used is supratherapeutic and always in combination with angiotensin II (AngII), which cannot be extrapolated to clinical practice (Uemura et al., 2003, 2005a; Takahashi et al., 2012). This point has been critically reviewed, and the importance of using clinically relevant doses of pharmacologic agents in preclinical studies has been noted (Reagan-Shaw et al., 2008). Another important consideration in investigating the effects of ARBs is the concomitant treatment with exogenous AngII (Uemura et al., 2003; Kosaka et al., 2007; Chen et al., 2013), which only blunted AngII-mediated effects. This paradigm ignores AngII-independent effects of candesartan as well as the role of locally produced AngII, which has been well characterized in a variety of tissues and cell types (Reid et al., 2011; Angeli et al., 2013; Lu et al., 2013). Recently, candesartan was shown to be proangiogenic in cerebral microvascular endothelial cells via activation of the angiotensin II type 2 (AT2) receptor (Alhusban et al., 2013). This effect occurred even in the absence of exogenous AngII. These two observations highlight the importance of investigating the potential direct effects of ARBs on tumor cell function and angiogenesis in the absence of exogenous AngII to gain better understanding of physiologic responses. In the current study, the focus was to systematically investigate the effect of clinically relevant concentrations of ARBs on the progression of prostate cancer both in vivo and in vitro. In addition, we investigated the effect of ARBs on tumor angiogenesis and vascular normalization, and the molecular mechanisms leading to ARB action on prostate tumor cells and tumor vasculature.
Materials and Methods
In Vivo Nude Mouse Tumor Xenograft Model.
All animal procedures listed in this article were performed as per the protocol approved by the Institutional Animal Care and Use Committee at the Charlie Norwood Veterans Affairs Medical Center (Augusta, GA; protocol #12-06-049). PC3 cells were grown to confluence in 250-ml flasks. Next, cells were suspended in sterile saline to a concentration of 5 × 106/ml. Cell suspension (100 µl/mouse) was injected subcutaneously in 8-week-old male nude mice (athymic nude mice; Harlan, Indianapolis, IN) (Kochuparambil et al., 2011). Mice were divided into two groups. The groups were subjected to intraperitoneal injections of candesartan (CV-11974 [see Alhusban et al., 2013] dissolved in 100 µl of 0.9% saline) at a dose of 6.5 mg/kg body weight every 24 hours for 18 days (treatment started 6 days after subcutaneous tumor injection when tumors of equal size were clearly visible), and the respective controls were injected intraperitoneally with 0.9% saline every 24 hours. CV-11974 is an activated form of candesartan that does not need to be activated by the liver, and hence can be readily used for both in vitro and in vivo experiments. Tumor diameters were measured with digital calipers on days 6, 12, 18, and 24, and the tumor volume in millimeters cubed was calculated by the modified ellipsoidal formula [tumor volume = ½(length × width2)] (Euhus et al., 1986). Mice were sacrificed on day 24 and tumors were dissected, weighed, and snap-frozen for further Western blot and immunohistochemistry analysis.
Reagents, Cell Lines, and Antibodies.
Human metastatic PC3 and DU145 prostate cancer cell lines were obtained from American Type Culture Collection (Manassas, VA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) high glucose (Hyclone, Logan, UT) with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin in 5% CO2 humidified atmosphere at 37°C. Primary antibodies against phosphorylated (p)–protein kinase B (Akt) (S473), total Akt, p-p38 mitogen-activated protein kinase (MAPK), and p–glycogen synthase kinase 3α/β were purchased from Cell Signaling Technology (Boston, MA). Anti–vascular endothelial growth factor (anti-VEGF) antibodies were purchased from Millipore (Billerica, MA). Primary antibodies against β-actin and laminin as well as AT2 receptor agonist (CGP-121141A; see Alhusban et al., 2013) were purchased from Sigma-Aldrich (St. Louis, MO). Primary antibodies against angiotensin II type 1 (AT1) and AT2 receptors were purchased from Abcam (Cambridge, MA). Anti-mouse and anti-rabbit horseradish peroxidase–conjugated secondary antibodies were obtained from Bio-Rad (Hercules, CA). Alexa-Fluor (488) secondary antibody was purchased from Invitrogen (Carlsbad, CA). Candesartan (CV-11974) was gifted to Dr. Fagan’s laboratory from AstraZeneca Pharmaceuticals (Wilmington, DE). Clinically relevant concentrations for the treatment of prostate tumor cells in vitro and athymic nude mice in vivo were calculated using the previously published protocol (Reagan-Shaw et al., 2008).
Candesartan Dose Calculations for In Vitro and In Vivo Experiments.
Two important points were considered when calculating the drug concentrations/doses for in vitro and in vivo studies: 1) dose calculations for intraperitoneal administration of candesartan in vivo, as compared with intravenous drug administration in mice, and 2) comparison of doses used in mice with that prescribed in humans. Therapeutic doses for mice as compared with humans were calculated using the following equation: human dose (milligrams per kilogram) = mouse dose × (mouse Km/human Km) (Reagan-Shaw et al., 2008). According to the manufacturer’s recommendation and the literature (Gleiter and Morike, 2002), average daily maintenance doses of candesartan for adults who weigh >50 kg are between 4 and 32 mg/day orally, and the bioavailability of candesartan is 15–40%. Since patients who are diagnosed with prostate cancer are elderly, and given the maximum prescribed dose of 32 mg/day orally, expected bioavailability (the fraction of dose that reaches the circulation) of candesartan is 40%, which is calculated as 32 × 40% = 12.8 mg/day for patients weighing 50 kg or more, and a comparable intravenous dose will be 12.8 mg/day for a 50-kg patient = 0.256 mg/kg. To reach this concentration by intraperitoneal administration, candesartan dose was doubled (0.512 mg/kg) considering the loss of drug through incomplete absorption into circulation and the drug removal through the hepatic system entering through mesenteric arteries. By using the mouse formula (aforementioned) based on human doses, dose of mouse (milligrams per kilogram) = dose of human/(Km of mice/Km of human): 0.512 mg/kg/(3/37) = 6.4 mg/kg (∼6.5 mg/kg).
For in vitro experiments, candesartan concentrations ranging from 0.5 to 25 µM are considered therapeutic, and anything above 25 µM was considered supratherapeutic. Comparisons between therapeutic and supratherapeutic concentrations of candesartan were included in in vitro studies.
Western Blot Analysis.
PC3 cells were cultured in DMEM in six-well plates to reach a monolayer and were serum starved. The wells were treated with serum-free DMEM containing candesartan at concentrations of 0.1–25 µM, or AT2 receptor agonist (CGP-121141A at concentrations of 100 nm to 1000 µM), and control cells were grown in DMEM alone. Whole-cell lysates were prepared using lysis buffer [50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 2 mM Na3VO4, and 1× complete protease inhibitors (Roche Applied Science, Indianapolis, IN)]. The protein concentration was measured by the DL protein assay (Bio-Rad). Western blot analyses were performed as described previously (Goc et al., 2013).
Reverse-Transcription Polymerase Chain Reaction Analysis of VEGF Expression.
Both PC3 and DU145 cell lines were cultured to reach a monolayer in DMEM in six-well plates and were serum starved. Wells were treated with serum-free DMEM containing candesartan, and control cells were grown in DMEM alone for 24 hours. Total RNA was extracted using TRIzol reagent (Invitrogen) following the manufacturer protocol. One microgram of RNA was used to make cDNA using an iScript cDNA synthesis kit (Bio-Rad) in a total volume of 20 µl for each reaction following manufacturer protocol. GoTaq green master mix 2× (Promega, Madison, WI) was used for polymerase chain reaction (PCR) amplifications in a total volume of 50 µl. Reverse-transcription PCR amplification was done using an Eppendorf thermal cycler (Eppendorf, Hauppauge, NY) using the following temperature and time periods: initial denaturation at 95°C for 3 minutes, followed by 25 PCR cycles using 1) denaturation at 94°C for 30 seconds, 2) primer annealing at 55°C for 30 seconds, and 3) extension at 72°C for 30 seconds and final extension for 72°C for 5 minutes. Two percent agarose gels were used to detect the RNA bands. The primer sequences used were as follow: VEGF forward, 5′-ctacctccaccatgccaagt-3′, and reverse, 5′-gcagtagctgcgctgataga-3′; glyceraldehyde-3-phosphate dehydrogenase forward, 5′-acccagaagactgtggatgg-3′, and reverse, 5′-agtagaggcagggatgatgtt-3′.
Cell Migration Assay.
Cell migration assay (wound healing assay) was performed as described previously (Goc et al., 2012b; Alhusban et al., 2013). Briefly, PC3 cells were grown on 12-well plates to reach confluence (24 hours), and then scratches were made in the cell monolayers using 1-ml pipette tips followed by treatment with different concentrations of candesartan (0.5, 5, 10, 25, and 200 µM) in serum-free DMEM. Control cells were incubated in DMEM alone. Images for scratches were taken at time 0 and 24 hours. The rate of migration was measured using the equation [(1 − T24/T0) × 100], where T24 is the area at the endpoint (24 hours) and T0 is the area at the start time (0 hours).
Transendothelial Migration Assay.
Transendothelial migration of prostate cancer (PC3) cells was measured using electric cell substrate impedance sensing (ECIS) equipment (Goc et al., 2012a) with human dermal microvascular endothelial cells (American Type Culture Collection) plated on 8W10E+ array chips (Applied Biophysics, Troy, NY). In brief, PC3 cells were plated in six-well plates. After 24 hours, the cells were incubated in serum-free DMEM containing candesartan (0.5, 25, and 200 µM) for 24 hours. To avoid the direct effect of candesartan in the conditioned medium on endothelial cell monolayer, medium with candesartan along with control dimethylsulfoxide containing medium was removed 1 hour after treatment and then supplemented with fresh serum-free medium and subsequent incubation for another 23 hours (a total of 24 hours post-treatment). Control cells were incubated in DMEM alone, then conditioned media were collected and live cells were collected from the plate using cell dissociation buffer [20 mM EDTA in phosphate-buffered saline (PBS; pH7.4)] to avoid integrin/receptor loss due to trypsin digestion. We then added either the cells or their conditioned media directly onto endothelial cell monolayers at a density of 5 × 104 cells/well in 50 µl of medium (or 50 µl of conditioned media). Real-time measurements on the transendothelial migration of PC3 cells were recorded by ECIS up to 10 hours.
Cell Proliferation Assay.
Proliferation of the PC3 cell line was determined using the nonradioactive bromodeoxyuridine (BrdU)-based cell proliferation assay (Roche Applied Science) according to the manufacturer’s protocol. In brief, PC3 cells were plated in 96-well flat-bottom plates at a density of 5 × 103 cells per well and allowed to grow for 24 hours. Cells were then treated with candesartan (0.5, 5, 10, 25, and 200 µM) for an additional 24 hours in serum-free conditions. Control cells were incubated in DMEM alone. Cells were then subjected to 5-bromo-2-deoxyuridine assay using the BrdU Labeling and Detection Kit III (Roche Diagnostics, Indianapolis, IN) as done previously (Goc et al., 2011, 2012a). BrdU incorporation into the DNA was determined by measuring the absorbance at both 450 and 690 nm on an enzyme-linked immunosorbent assay (ELISA) plate reader.
Cell Viability Assay.
Number of viable cells was assessed indirectly by means of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay using tetrazolium salt conversion into formazan crystals (Al-Azayzih et al., 2012). In brief, PC3 cells were plated in 96-well plates at 5 × 103 cells per well, and allowed to grow for 24 hours. Medium was then replaced with fresh serum-free DMEM containing candesartan (0, 0.5, 5, 10, 25, and 200 µM). After 24-hour treatment, cell viability was measured using the Cell Proliferation Kit I (MTT; Roche Applied Science) according to the manufacturer’s protocol and as standardized in the laboratory. The absorbance at 570 nm was measured using an ELISA reader and used to determine relative cell numbers in each well.
Cell Apoptosis Assay.
Cytoplasmic histone–associated DNA fragments were quantified using the Cell Death Detection ELISAPLUS kit (Roche Applied Science) according to the manufacturer’s protocol (Al-Azayzih et al., 2012). In brief, PC3 cells were plated in a 96-well plate at a density of 104 cells per well. After 24 hours, the cells were incubated in serum-free DMEM containing candesartan (0.5, 5, 10, 25, and 200 µM) for 24 hours. Control cells were incubated in DMEM alone. Cells were lysed and centrifuged at 200g for 10 minutes, and the collected supernatants were subjected to ELISA. The absorbance was measured at 405 nm (reference wavelength, 492 nm).
Immunohistochemistry.
Immunofluorescence staining of the tumor sections for laminin (blood vessels) was performed according to the standard protocol (Goc et al., 2012a). In brief, formalin-fixed, frozen prostate (PC3) xenograft tumor sections from nude mice were subjected to the standard xylene-ethanol dehydration process and permeabilized with 0.3% Triton X-100 in 1× PBS. The nonspecific staining was blocked with 5% goat serum for 1 hour at room temperature. The dehydrated, permeabilized, and blocked tissue sections were incubated with primary antibodies against laminin (dilution 1:750) overnight at 4°C followed by washing with 1× PBS (four times for 15 minutes each). Next, anti-mouse Alexa Fluor 488–labeled secondary antibodies (1:500) were applied for 1 hour at room temperature and washed four times for 15 minutes with 1× PBS. The slides were mounted with Vectashield (Vector Laboratories, Burlingame, CA), and the images were taken by a Zeiss fluorescent microscope (Zeiss Axiovert100M; Carl Zeiss, Jena, Germany). Analysis of the vascular lumen area and wall thickness was determined using NIH ImageJ software.
Statistical Analysis.
All data are presented as the mean ± S.D. of three to four independent experiments. To determine significant differences between treatment and control values, we used Student’s two-tailed t test. One-way analysis of variance was used for all of the concentration-dependent analyses in vitro. Significance was set at 0.05 levels (marked with symbols wherever data are statistically significant).
Results
Clinically Relevant Dose of Candesartan Inhibited Growth of Prostate Tumor Xenograft in Athymic Nude Mice.
Our data indicated that candesartan inhibited prostate cancer (PC3) tumor xenograft progression as detected by the size and weight of the tumors after treatment with the clinically relevant dose of candesartan (Fig. 1A). Although we observed a trend in inhibitory effect of candesartan on the growth of tumor xenograft starting from as early as 6 days post-treatment (12 days after PC3 cell administration), a significant antitumor effect was revealed from day 12 postcandesartan treatment (18 days after PC3 cell administration) (Fig. 1B). Additionally, candesartan significantly reduced the weight of prostate tumor xenografts measured after tumor xenograft extraction on day 24 (Fig. 1C). Furthermore, treatment with candesartan resulted in a significant reduction in the number of blood vessels approaching the tumor, suggesting an impairment of PC3 cell ability to attract host vasculature toward the tumor xenograft (Fig. 1D).
Fig. 1.
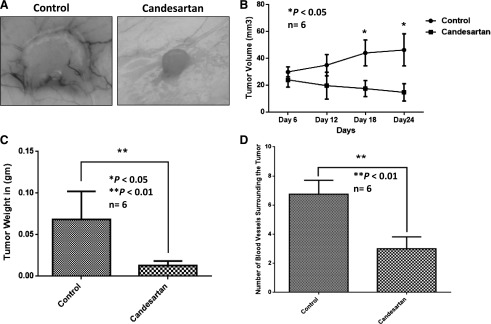
Candesartan inhibits the growth of prostate cancer xenograft in vivo at clinically relevant doses. (A) Representative pictures of PC3 cell prostate tumor xenografts showing reduced tumor size by treatment with clinically relevant doses of candesartan. (B) Graph showing the effect of clinical doses of candesartan on PC3 cell tumor xenograft volume as measured on days 12, 18, and 24 (6, 12, and 18 days post-treatment). (C) Bar graph showing the effect of clinical doses of candesartan on PC3 cell tumor xenograft weight on day 24, after 18-day treatment with candesartan. (D) Bar graph showing quantified data on the number of blood vessels surrounding the PC3 cell tumor xenografts in the presence and absence of clinical doses of candesartan. Data are presented as the mean ± S.D.
Candesartan Had a Modest Effect on the Migration and Apoptosis of PC3 Cells.
Since a clinical dose of candesartan was effective in inhibiting PC3 tumor growth and tumor neovascularization in vivo, we sought to determine if relevant concentrations of candesartan had any effect on PC3 tumor cell function in vitro. Our data showed that relevant concentrations of candesartan used in the in vivo tumor experiment induced a dose-dependent antiapoptotic effect in PC3 cells in vitro, which was observed even at supratherapeutic doses (Fig. 2A). Although modest, the effect of candesartan on tumor cell survival was significant and was in contrast with our data from in vivo analysis of tumor xenografts, which suggested that the effect of candesartan in vivo is much more complicated than its direct effect on tumor cells in vitro. Interestingly, although clinically relevant concentrations of candesartan did not elicit any effect on PC3 cell migration, supratherapeutic doses of candesartan had a paradoxical inhibitory effect, as reported by many other laboratories, which might be due to its toxic effect at high concentrations (Fig. 2B). Surprisingly, clinically relevant concentrations of candesartan did not have any effect on the proliferation or viability of PC3 cells in the short term (Fig. 2, C and D) or in the long term (72 hours) (Supplemental Fig. 1).
Fig. 2.

Candesartan elicits modest effects on prostate cancer cellular function in vitro. In vitro effects of candesartan on prostate cancer cell migration, viability, proliferation, and apoptosis were examined. (A) Bar graph showing the effect of clinical doses of candesartan on PC3 cell apoptosis determined by the levels of cytoplasmic histone–associated DNA fragments. (B) Bar graph showing the effect of clinical doses of candesartan on PC3 cell migration. (C) Bar graph showing the effect of clinical doses of candesartan on PC3 cell proliferation determined by BrdU incorporation assay. (D) Bar graph showing the effect of clinical doses of candesartan on PC3 cell viability determined by MTT assay. Except with high concentrations, candesartan did not have any appreciable effects on the progression of prostate cancer cells. Data are presented as the mean ± S.D.
Candesartan Did Not Affect the Major Survival Pathways in PC3 Cells.
In contrast to our in vivo data, in vitro data demonstrated a minimal effect of candesartan on the progression of cancer cells. To resolve this discrepancy, we analyzed the major survival pathways in PC3 cells in response to candesartan treatment. Our data demonstrated that therapeutic concentrations of candesartan did not alter the activity of either Akt or glycogen synthase kinase as determined by the changes in activity modulating phosphorylation (Fig. 3A). Consistent with previous reports (Uemura et al., 2003, 2005b, 2006), candesartan inhibited p38 MAPK signaling in a concentration-dependent manner (Fig. 3B), which might be responsible for the modest but significant decrease in prostate cancer cell apoptosis in vitro. A recent study indicated modulation of AT1 and AT2 receptor expression by candesartan in endothelial cells (Alhusban et al., 2013). Hence, we determined if treatment with candesartan will modulate AT1 and AT2 receptor expression in prostate cancer cells. Our study indicated that treatment with candesartan resulted in a significant and concentration-dependent decrease in AT1 receptor expression and increase in AT2 receptor expression in PC3 cells (Fig. 3, C and D, respectively), but not in DU145 cells (Supplemental Fig. 2, B and C), thus suggesting that effects of candesartan on AT1 and AT2 receptor expression, and probably the entire effect of candesartan on PC3 cells in vitro and tumor xenograft in vivo, may be purely limited to PC3 cells.
Fig. 3.
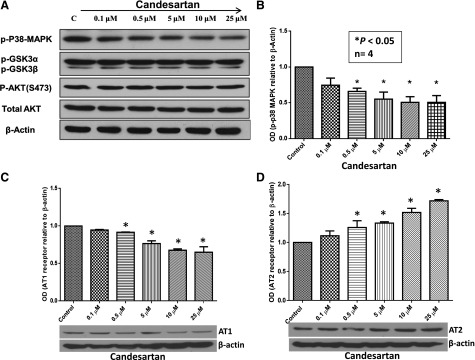
Candesartan inhibits p-p38 MAPK activation and modulates AT1/AT2 receptor expression in PC3 cells in vitro. PC3 cells were treated with a range of clinical doses of candesartan. (A) Western blot figures showing dose-dependent decrease in p38 MAPK phosphorylation in PC3 cell lysates with no changes in the activity levels of Akt and glycogen synthase kinase (GSK) 3. (B) Bar graph showing quantification of the optical densities (OD) of Western blot protein bands showing a significant and dose-dependent decrease in p38 MAPK phosphorylation in PC3 cell lysates. (C) Bar graph showing a significant dose-dependent inhibitory effect of candesartan on AT1 receptor expression in PC3 cells. (D) Bar graph showing a significant dose-dependent effect of candesartan on enhancing AT2 receptor expression in PC3 cells. Data are presented as the mean ± S.D.
Candesartan Inhibited Cancer-Induced Angiogenesis in Prostate Cancer Xenograft.
Since candesartan did not elicit a significant inhibitory effect on PC3 and DU145 prostate cancer cells in vitro, we postulated that the effect of candesartan in vivo could be a result of its ability to interfere with the tumor and stromal cell interactions. To assess the involvement of angiogenesis in candesartan-mediated inhibition of tumor growth in vivo, we determined the effect of a clinically relevant dose of candesartan on the expression of the proangiogenic growth factor, VEGF, and on tumor angiogenesis. Accordingly, PC3 tumor xenograft sections were stained with laminin, a marker of vascular density in xenografts, and tumor lysates were subjected to Western analysis for VEGF expression. Candesartan significantly inhibited tumor angiogenesis in PC3 tumor xenografts (Fig. 4, A and B), and this was corroborated with a significant inhibition of VEGF expression (Fig. 4, C and D). Furthermore, our in vitro analysis indicated that treatment with clinically relevant concentrations of candesartan inhibit VEGF expression in both PC3 cells (Fig. 4E) and DU145 cells (Supplemental Fig. 2A) in a concentration-dependent manner. Together, these data indicated that the effect of candesartan on tumor cell VEGF expression, tumor angiogenesis, and growth is indeed a global effect, not cell-specific, and may be via an AT1/AT2 receptor–independent mechanism.
Fig. 4.

Candesartan elicits an antiangiogenic effect in prostate cancer xenografts in mice. Treatment of nude mice with clinically relevant doses of candesartan reduced vascular density via inhibition of VEGF expression. (A) Representative fluorescent images of tumor xenograft sections showing laminin positive blood vessels as indicated by arrows. (B) Bar graph showing reduced vascular area in tumor xenografts treated with clinically relevant doses of candesartan as compared with saline-treated controls. Representative Western blot (C) and bar graph (D) showing reduced expression of VEGF in tumor xenografts treated with clinically relevant doses of candesartan as compared with saline-treated controls. (E) Representative Western blot (bottom) and bar graph (top) showing a dose-dependent inhibition of VEGF expression in PC3 tumor cells treated with clinically relevant doses of candesartan as compared with vehicle-treated controls. Data are presented as the mean ± S.D. OD, optical density.
Candesartan Inhibits VEGF mRNA Expression in Prostate Cancer Cells.
Next, we determined whether candesartan inhibits VEGF expression at the mRNA level or if its effect is limited to protein translation. Our study revealed that, in both PC3 and DU145 cells, candesartan treatment resulted in a significant reduction in VEGF mRNA levels at both the 0.5 and 25 µM candesartan concentrations (Fig. 5, A–C), thus indicating that the effect of candesartan on inhibition of VEGF expression in prostate cancer cells is at the transcriptional level.
Fig. 5.

Candesartan inhibits VEGF expression in PC3 and DU145 cells at the transcriptional level. (A) Representative image of PC3 and DU145 cell reverse-transcription PCR products for VEGF expression in the presence and absence of candesartan treatment as run on 2% agarose gels. (B and C) Bar graph showing densitometry analysis of VEGF mRNA levels in the presence and absence of candesartan treatment in PC3 (B) and DU145 cells (C). Data are presented as the mean ± S.D. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; OD, optical density.
Candesartan Induces Vascular Normalization in Tumor Xenografts In Vivo.
Since the effect of candesartan in vivo was predominantly on the tumor vasculature, we analyzed the tumor blood vessels to determine the effect of candesartan on tumor vascular normalization, an important feature that determines the efficacy of antiangiogenic and combinational therapies. According to our data, candesartan treatment resulted in significant reduction in the tumor vascular lumen perimeter (Fig. 6A) and lumen area (Fig. 6B) associated with a significant reduction in AT1 receptor (Fig. 6D) and increase in AT2 receptor (Fig. 6E) expression levels, indicating an overall vasodilatory effect of candesartan on tumor blood vessels. In addition, a significant increase in vascular wall thickness in candesartan-treated tumor blood vessels compared with saline-treated controls (Fig. 6C) suggested the ability of candesartan to enhance endothelial-barrier integrity and activation in vivo. Together, these data indicated the effect of candesartan on tumor vascular normalization, potentially via inhibition of tumor vascular permeability, an important feature essential to promote neovascularization in tumors.
Fig. 6.
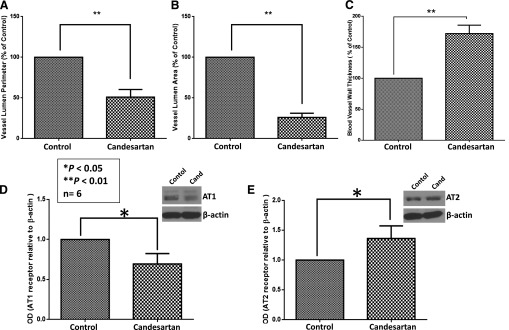
Candesartan treatment helps with vascular normalization in tumor xenografts in vivo. (A) Bar graph showing reduced lumen perimeter in tumor xenografts treated with clinically relevant doses of candesartan as compared with saline-treated controls. (B) Bar graph showing reduced vessel lumen area in tumor xenografts treated with clinically relevant doses of candesartan as compared with saline-treated controls. (C) Bar graph showing increased vessel wall thickness in tumor xenografts treated with clinically relevant doses of candesartan as compared with saline-treated controls. (D) Representative Western blot (insert) and bar graph showing significant inhibition of AT1 receptor expression in PC3 tumor xenografts with candesartan treatment. (E) Representative Western blot (insert) and bar graph showing significant increase in AT2 receptor expression in PC3 tumor xenografts with candesartan treatment. Data are presented as the mean ± S.D. Cand, candesartan; OD, optical density.
Conditioned Media from Candesartan-Treated PC3 Cells Did Not Compromise Human Microvascular Endothelial-Barrier Function.
The effect of candesartan on tumor vascular normalization prompted us to determine its effect on inhibition of vascular permeability by simulating the conditions in vitro using the ECIS technology. Our data showed that conditioned media collected from candesartan-treated PC3 cells preserved endothelial-barrier function in a concentration-dependent manner (Fig. 7A). This finding suggests that candesartan reduces the expression and release of mitogenic mediators such as VEGF by the prostate cancer cells, and that these mediators primarily produce a paracrine rather than an autocrine effect. On the other hand, candesartan-treated PC3 cells did not have a consistent effect on the endothelial-barrier function (Fig. 7B). PC3 cells treated with low concentrations of candesartan induced a trivial breakdown in endothelial-barrier function that was rapidly restored. Interestingly, PC3 cells treated with higher concentrations of candesartan failed to disrupt barrier function of endothelial cells.
Fig. 7.

Candesartan preserves endothelial-barrier integrity via inhibiting vascular permeability. (A) Graph showing real-time changes in endothelial-barrier resistance in response to conditioned media collected from PC3 cell cultures treated with various doses of candesartan. Data show that treatment with candesartan inhibits secretion of vascular permeability stimulating growth factors in a dose-dependent manner, thereby inhibiting the ability of the tumor cell–conditioned media to induce endothelial-barrier fenestrations. (B) Graph showing real-time changes in endothelial-barrier resistance in response to topically introduced PC3 cells pretreated with various doses of candesartan for 12 hours. Candesartan did not have an appreciable effect on the invasion potential of PC3 cells except at a higher concentration. (C) Bar graph showing quantification of the endothelial-barrier resistance at 4, 6, and 8 hours postaddition of control and candesartan-treated tumor cell culture conditioned medium. Data are presented as the mean ± S.D.
Effect of Candesartan on VEGF Expression by Tumor Cells Is Independent of AT2 Receptor Activation.
Finally, we determined whether the candesartan-mediated effect on VEGF expression by PC3 and DU145 cells was through activation of the AT2 receptor. To do this, we treated PC3 and DU145 cells with various concentrations of the AT2 receptor agonist CGP-121141A. Our data indicated that AT2 receptor activation had no effect on VEGF expression in either PC3 or DU145 cells (Supplemental Fig. 2).
Discussion
Our data demonstrate the ability of clinically relevant doses of candesartan to inhibit prostate cancer xenograft growth. Interestingly, candesartan-induced antitumorigenic effect was mediated indirectly through inhibition of cancer-induced angiogenesis. This finding was confirmed by the lack of significant effect of candesartan on the behavior of prostate cancer cells in vitro. Additionally, whereas conditioned media from candesartan-treated cells were able to maintain endothelial-barrier function in vitro, treatment with candesartan induced vascular normalization in tumor vasculature through inhibition of vascular permeability, thus impairing neovascularization and tumor perfusion.
Candesartan is an AT1 receptor antagonist and is widely prescribed for the management of hypertension and other cardiovascular disorders. It is extremely well tolerated, as are all of the other ARBs), and has an additional advantage of being dosed orally once daily. The pleiotropic properties of the ARBs, beyond blood pressure lowering, include prevention of pathologic remodeling after myocardial infarction, vascular protection, and the reduction of inflammation. The effects of ARBs on angiogenesis are debated and are likely to be dose-, tissue-, and context-dependent (Willis et al., 2011). Candesartan and other AT1 blockers have been demonstrated to inhibit the proliferation of cancer cells when used alone or in combination with other antineoplastic agents (Fujimoto et al., 2001; Miyajima et al., 2002; Kosugi et al., 2006; Kosaka et al., 2007; Chen et al., 2013). This antiproliferative effect was observed with high doses of AT1 blockers that are not clinically relevant and may have serious side effects, such as hypotension and kidney failure. Recently published meta-analyses had confusing results on the risk of cancer among AT1 blocker–treated patients (Sipahi et al., 2010; Mearns, 2011). Accordingly, we assessed the effect of clinically relevant doses of candesartan on the progression of prostate cancer using a xenograft model in nude mice. Similar to previously published data on the use of higher doses of candesartan (Uemura et al., 2006, 2008; Kosaka et al., 2007), our data also demonstrated the ability of candesartan to inhibit the progression of prostate cancer in vivo. This finding has a highly significant translational impact as it provides the first experimental evidence on the effect of candesartan on prostate tumor growth at doses comparable with that used in clinical practice, and can help to resolve the recently raised concerns about the safety of ARBs with regard to the risk of prostate cancer.
The antiproliferative effects of ARBs have been extensively reviewed (Uemura et al., 2006). A common theme among the majority of published literature is the concomitant treatment of prostate cancer cells with AngII (Uemura et al., 2003, 2005b, 2006). In this context, the observed antiproliferative effects of ARBs are basically a reflection of AT1-mediated signaling in cancer cells. This approach does not take into account the possible role of locally produced AngII, which has been demonstrated to be of major importance in other cell types (Reid et al., 2011; Angeli et al., 2013; Lu et al., 2013). In addition, this approach also ignores the possibility of ARB-induced AngII-independent effects which have been demonstrated in other cell types (Alhusban et al., 2013). Another source of complexity in understanding ARB-induced antiproliferative effects is the complex nature of the cancer microenvironment, which has been shown to play a major role in the progression of prostate cancer (Uemura et al., 2005a). To account for all of these variables, we were interested in assessing whether ARB-induced antiproliferative effects are due to the direct effects of the drug on prostate cancer cells. Accordingly, direct cytotoxicity of candesartan was assessed by treating PC3 cells with different concentrations of candesartan in the absence of exogenous AngII. Apoptosis, proliferation, viability, and migration of PC3 cells were assayed. Interestingly, the antiproliferative effect of candesartan was found to be unrelated to direct cytotoxicity against cancer cells. This finding was suggested by the relative lack of effect when candesartan was directly applied to prostate cancer cells. To our surprise, candesartan had an antiapoptotic effect on PC3 cells, and this was observed at all concentrations tested, including the supratherapeutic range. Candesartan-induced antiapoptotic effect was not associated with an increase in either migration or proliferation of PC3 cells. In contrast, higher concentrations of candesartan inhibited the rate of PC3 cell migration. Taken collectively, these data suggest that clinical doses of candesartan do not have an appreciable direct effect on PC3 cells. This finding supports previously published data on the inability of candesartan to affect the progression of prostate cancer cells in vitro (Matsuyama et al., 2010).
Interestingly, candesartan inhibited the phosphorylation of p38 MAPK in a concentration-dependent manner, and at all concentrations used. This finding reproduces previously published data on the effect of supratherapeutic doses of ARBs on prostate cancer in the presence of exogenously added AngII (Uemura et al., 2003). This similarity supports the hypothesis that observed ARB-induced antiproliferative effects are mediated by antagonizing the effects of locally produced AngII. AngII is expected to mediate its proangiogenic effects through AT2 receptors, as has recently been shown in our studies in brain endothelial cells (Alhusban et al., 2013). However, although candesartan treatment resulted in significantly decreased AT1 and increased AT2 receptor expression in PC3 cells and tumor xenografts, this effect was not observed in DU145 cells, suggesting that the common candesartan effects seen may be due to its direct inhibitory effects on AT1, as reported in melanoma (Akhavan et al., 2011). An alternative explanation is the possible AngII-independent effects of ARBs, which require more detailed, in-depth analyses that go beyond the scope of this investigation.
Cancer progression is a well orchestrated interplay between cancer cells and their microenvironment (Uemura et al., 2005a). This interplay can be best demonstrated by cancer-induced angiogenesis (Hanahan and Folkman, 1996). Cancer cells have high metabolic rates and require higher rates of nutrient delivery. To match their metabolic requirements, cancer cells induce angiogenesis, vascular permeability, and perfusion through the release of a variety of angiogenic mediators (Weidner et al., 1993). The lack of concordance between in vivo and in vitro effects of candesartan suggested possible involvement of other cell types in the observed antiproliferative effect of candesartan in vivo. VEGF plays a major role in prostate cancer–induced angiogenesis (Feng et al., 2008; Mahabeleshwar et al., 2008). Our in vivo data demonstrated elevated levels of VEGF in prostate tumor xenografts from saline-treated animals, and a significant inhibition of VEGF expression with a therapeutic dose of candesartan. The effect of candesartan on VEGF expression was also observed in both PC3 cells and DU145 cells. Interestingly, the AT2 agonist (CGP-121141A) had no significant effect on VEGF expression in either PC3 or DU145 cells, thus ruling out the role of AT2 receptor stimulation in candesartan-mediated inhibition of VEGF expression. Our previous studies in a stroke model indicated increased VEGF expression by candesartan (Guan et al., 2011), probably through endothelial cells (Alhusban et al., 2013; Soliman et al., 2014); in the current model, where angiogenesis is mainly driven by the tumor cells, the mechanism leading to inhibition of VEGF expression is not via AT2 stimulation. As reviewed previously, the effects of ARBs on angiogenesis are dose-, tissue-, and context-dependent (Willis et al., 2011)
Since no significant changes in survival pathways and tumor cell function with the clinical concentrations of candesartan were observed in the in vitro experiments, we wanted to rule out that the decrease in VEGF expression in candesartan-treated tumors is due to the paracrine effect from prostate cancer cells. To do this, and to confirm the antiangiogenic effect of candesartan, ECIS technique was used. In this system, endothelial cells cultured to confluence were challenged by either candesartan-treated PC3 cells or their conditioned media, and changes in electrical impedance were measured as a function of time to assess the changes in the endothelial-barrier resistance. ECIS findings demonstrated a protective effect of conditioned media from candesartan-treated PC3 cells on endothelial-barrier function compared with untreated control medium. This finding further supports the paracrine over the autocrine effect of VEGF in prostate cancer proliferation. In support of this, our immunohistochemistry analysis of tumor sections revealed that candesartan induced a vascular normalization effect on tumor vasculature as evidenced by reduced lumen size, increased vessel wall thickness, and more rounded appearance as compared with saline-treated controls. These characteristic features of tumor vasculature are associated with inhibition of tumor vascular permeability and tumor perfusion, thus inhibiting neovascularization and depriving the tumor cells of necessary nutrients to grow, rather than a direct antiproliferative effect of candesartan on tumor cells.
In conclusion, our data demonstrate the ability of candesartan to inhibit progression of prostate cancer through ablation of cancer-induced angiogenesis via inhibition of VEGF expression by prostate cancer cells. Although a role for AT1 and AT2 in candesartan-mediated inhibition of VEGF expression and/or prostate tumor growth is ruled out, existence of an AT2-independent pathway by AngII in candesartan-mediated inhibition of prostate tumor growth and neovascularization needs to be investigated. Nevertheless, our study conclusively demonstrated that clinically relevant doses of candesartan, alone or in combination with other chemotherapeutic drugs, can be developed into a potential therapeutic strategy for prostate cancer. Although monotherapy with candesartan induces vascular normalization and inhibits tumor angiogenesis in prostate tumors in our studies, it will be interesting to know the effect of candesartan in combination with other drugs on prostate tumors. Since vascular normalization is known to affect drug delivery in addition to its effects on tumor angiogenesis, the result of combining candesartan with other drugs can be interpreted in both ways. Additional research will be necessary to address this discrepancy.
Supplementary Material
Abbreviations
- Akt
protein kinase B
- ARB
angiotensin II type 1 receptor blocker
- AngII
angiotensin II
- AT1
angiotensin II type 1 receptor
- AT2
angiotensin II type 2 receptor
- BrdU
bromodeoxyuridine
- DMEM
Dulbecco’s modified Eagle’s medium
- ECIS
electric cell substrate impedance sensing
- ELISA
enzyme-linked immunosorbent assay
- MAPK
mitogen-activated protein kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- VEGF
vascular endothelial growth factor
Authorship Contributions
Participated in research design: Alhusban, Al-Azayzih, Fagan, Somanath.
Conducted experiments: Alhusban, Al-Azayzih, Goc, Gao.
Performed data analysis: Alhusban, Al-Azayzih, Goc, Gao, Fagan, Somanath.
Wrote or contributed to the writing of the manuscript: Alhusban, Al-Azayzih, Fagan, Somanath.
Footnotes
Funds were provided by the University of Georgia, Wilson Pharmacy Foundation, and in part by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant R01-HL103952] (to P.R.S.). A.A. and A.A.-A. were supported by predoctoral fellowships from Jordan University of Science and Technology. This material is the result of work supported with resources and the use of facilities at the Charlie Norwood VA Medical Center, Augusta, GA. The funders had no role in the study design, data collection, analysis, and decision to publish. Preparation of the manuscript and the contents do not represent the views of the Department of Veterans Affairs or the US Government.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Akhavan MM, Karimi M, Ghodrati M, Falahtpishe H. (2011) AT1 receptors activation enhances the expression of MMP-2, MMP-13 and VEGF but not MMP-9 in B16F10 melanoma cells. Pak J Biol Sci 14:821–830 [DOI] [PubMed] [Google Scholar]
- Al-Azayzih A, Gao F, Goc A, Somanath PR. (2012) TGFβ1 induces apoptosis in invasive prostate cancer and bladder cancer cells via Akt-independent, p38 MAPK and JNK/SAPK-mediated activation of caspases. Biochem Biophys Res Commun 427:165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhusban A, Kozak A, Ergul A, Fagan SC. (2013) AT1 receptor antagonism is proangiogenic in the brain: BDNF a novel mediator. J Pharmacol Exp Ther 344:348–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli JK, Cruz Pereira CA, de Oliveira Faria T, Stefanon I, Padilha AS, Vassallo DV. (2013) Cadmium exposure induces vascular injury due to endothelial oxidative stress: the role of local angiotensin II and COX-2. Free Radic Biol Med 65:838–848 [DOI] [PubMed] [Google Scholar]
- Chen X, Meng Q, Zhao Y, Liu M, Li D, Yang Y, Sun L, Sui G, Cai L, Dong X. (2013) Angiotensin II type 1 receptor antagonists inhibit cell proliferation and angiogenesis in breast cancer. Cancer Lett 328:318–324 [DOI] [PubMed] [Google Scholar]
- Euhus DM, Hudd C, LaRegina MC, Johnson FE. (1986) Tumor measurement in the nude mouse. J Surg Oncol 31:229–234 [DOI] [PubMed] [Google Scholar]
- Feng W, McCabe NP, Mahabeleshwar GH, Somanath PR, Phillips DR, Byzova TV. (2008) The angiogenic response is dictated by beta3 integrin on bone marrow-derived cells. J Cell Biol 183:1145–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto Y, Sasaki T, Tsuchida A, Chayama K. (2001) Angiotensin II type 1 receptor expression in human pancreatic cancer and growth inhibition by angiotensin II type 1 receptor antagonist. FEBS Lett 495:197–200 [DOI] [PubMed] [Google Scholar]
- Gleiter CH, Mörike KE. (2002) Clinical pharmacokinetics of candesartan. Clin Pharmacokinet 41:7–17 [DOI] [PubMed] [Google Scholar]
- Goc A, Abdalla M, Al-Azayzih A, Somanath PR. (2012a) Rac1 activation driven by 14-3-3ζ dimerization promotes prostate cancer cell-matrix interactions, motility and transendothelial migration. PLoS ONE 7:e40594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goc A, Al-Azayzih A, Abdalla M, Al-Husein B, Kavuri S, Lee J, Moses K, Somanath PR. (2013) P21 activated kinase-1 (Pak1) promotes prostate tumor growth and microinvasion via inhibition of transforming growth factor β expression and enhanced matrix metalloproteinase 9 secretion. J Biol Chem 288:3025–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goc A, Al-Husein B, Kochuparambil ST, Liu J, Heston WW, Somanath PR. (2011) PI3 kinase integrates Akt and MAP kinase signaling pathways in the regulation of prostate cancer. Int J Oncol 38:267–277 [PubMed] [Google Scholar]
- Goc A, Kochuparambil ST, Al-Husein B, Al-Azayzih A, Mohammad S, Somanath PR. (2012b) Simultaneous modulation of the intrinsic and extrinsic pathways by simvastatin in mediating prostate cancer cell apoptosis. BMC Cancer 12:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W, Somanath PR, Kozak A, Goc A, El-Remessy AB, Ergul A, Johnson MH, Alhusban A, Soliman S, Fagan SC. (2011) Vascular protection by angiotensin receptor antagonism involves differential VEGF expression in both hemispheres after experimental stroke. PLoS ONE 6:e24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86:353–364 [DOI] [PubMed] [Google Scholar]
- Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR. (2011) Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J Pharmacol Exp Ther 336:496–505 [DOI] [PubMed] [Google Scholar]
- Kosaka T, Miyajima A, Takayama E, Kikuchi E, Nakashima J, Ohigashi T, Asano T, Sakamoto M, Okita H, Murai M, et al. (2007) Angiotensin II type 1 receptor antagonist as an angiogenic inhibitor in prostate cancer. Prostate 67:41–49 [DOI] [PubMed] [Google Scholar]
- Kosugi M, Miyajima A, Kikuchi E, Horiguchi Y, Murai M. (2006) Angiotensin II type 1 receptor antagonist candesartan as an angiogenic inhibitor in a xenograft model of bladder cancer. Clin Cancer Res 12:2888–2893 [DOI] [PubMed] [Google Scholar]
- Lu X, Roksnoer LC, Danser AH. (2013) The intrarenal renin-angiotensin system: does it exist? Implications from a recent study in renal angiotensin-converting enzyme knockout mice. Nephrol Dial Transplant 28:2977–2982 [DOI] [PubMed] [Google Scholar]
- Mahabeleshwar GH, Chen J, Feng W, Somanath PR, Razorenova OV, Byzova TV. (2008) Integrin affinity modulation in angiogenesis. Cell Cycle 7:335–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama M, Funao K, Kuratsukuri K, Tanaka T, Kawahito Y, Sano H, Chargui J, Touraine JL, Yoshimura N, Yoshimura R. (2010) Telmisartan inhibits human urological cancer cell growth through early apoptosis. Exp Ther Med 1:301–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mearns BM. (2011) Hypertension: new meta-analysis shows no cancer risk with angiotensin-receptor blockers. Nat Rev Cardiol 8:243. [DOI] [PubMed] [Google Scholar]
- Miyajima A, Kosaka T, Asano T, Asano T, Seta K, Kawai T, Hayakawa M. (2002) Angiotensin II type I antagonist prevents pulmonary metastasis of murine renal cancer by inhibiting tumor angiogenesis. Cancer Res 62:4176–4179 [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. (2008) Dose translation from animal to human studies revisited. FASEB J 22:659–661 [DOI] [PubMed] [Google Scholar]
- Reid AC, Brazin JA, Morrey C, Silver RB, Levi R. (2011) Targeting cardiac mast cells: pharmacological modulation of the local renin-angiotensin system. Curr Pharm Des 17:3744–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2012) Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 125:e2–e220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. (2012) Cancer statistics, 2012. CA Cancer J Clin 62:10–29 [DOI] [PubMed] [Google Scholar]
- Sipahi I, Debanne SM, Rowland DY, Simon DI, Fang JC. (2010) Angiotensin-receptor blockade and risk of cancer: meta-analysis of randomised controlled trials. Lancet Oncol 11:627–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman S, Ishrat T, Pillai A, Somanath PR, Ergul A, El-Remessy AB, Fagan SC. (2014) Candesartan induces a prolonged proangiogenic effect and augments endothelium-mediated neuroprotection after oxygen and glucose deprivation: role of vascular endothelial growth factors a and B. J Pharmacol Exp Ther 349:444–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Uemura H, Seeni A, Tang M, Komiya M, Long N, Ishiguro H, Kubota Y, Shirai T. (2012) Therapeutic targeting of angiotensin II receptor type 1 to regulate androgen receptor in prostate cancer. Prostate 72:1559–1572 [DOI] [PubMed] [Google Scholar]
- Uemura H, Ishiguro H, Kubota Y. (2006) Angiotensin II receptor blocker: possibility of antitumor agent for prostate cancer. Mini Rev Med Chem 6:835–844 [DOI] [PubMed] [Google Scholar]
- Uemura H, Ishiguro H, Kubota Y. (2008) Pharmacology and new perspectives of angiotensin II receptor blocker in prostate cancer treatment. Int J Urol 15:19–26 [DOI] [PubMed] [Google Scholar]
- Uemura H, Ishiguro H, Nagashima Y, Sasaki T, Nakaigawa N, Hasumi H, Kato S, Kubota Y. (2005a) Antiproliferative activity of angiotensin II receptor blocker through cross-talk between stromal and epithelial prostate cancer cells. Mol Cancer Ther 4:1699–1709 [DOI] [PubMed] [Google Scholar]
- Uemura H, Ishiguro H, Nakaigawa N, Nagashima Y, Miyoshi Y, Fujinami K, Sakaguchi A, Kubota Y. (2003) Angiotensin II receptor blocker shows antiproliferative activity in prostate cancer cells: a possibility of tyrosine kinase inhibitor of growth factor. Mol Cancer Ther 2:1139–1147 [PubMed] [Google Scholar]
- Uemura H, Nakaigawa N, Ishiguro H, Kubota Y. (2005b) Antiproliferative efficacy of angiotensin II receptor blockers in prostate cancer. Curr Cancer Drug Targets 5:307–323 [DOI] [PubMed] [Google Scholar]
- Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J. (1993) Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol 143:401–409 [PMC free article] [PubMed] [Google Scholar]
- Willis LM, El-Remessy AB, Somanath PR, Deremer DL, Fagan SC. (2011) Angiotensin receptor blockers and angiogenesis: clinical and experimental evidence. Clin Sci (Lond) 120:307–319 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.