

SUMMARY
The nuclear envelope is a major barrier for nuclear uptake of plasmids and represents one of the most significant unsolved problems of non-viral gene delivery. We have previously shown that the nuclear entry of plasmid DNA is sequence-specific, requiring a 366 bp fragment containing the SV40 origin of replication and early promoter. In this report, we show that, although fragments throughout this region can support varying degrees of nuclear import, the 72 bp repeats of the SV40 enhancer facilitate maximal transport. The functions of the promoter and the origin of replication are not needed for nuclear localization of plasmid DNA. In contrast to the import activity of the SV40 enhancer, two other strong promoter and enhancer sequences, the human cytomegalovirus (CMV) immediate early promoter and the Rous sarcoma virus LTR, were unable to direct nuclear localization of plasmids. The inability of the CMV promoter to mediate plasmid nuclear import was confirmed by measurement of the CMV promoter-driven expression of green fluorescent protein (GFP) in microinjected cells. At times before cell division, as few as 3 to 10 copies per cell of cytoplasmically injected plasmids containing the SV40 enhancer gave significant GFP expression, while no expression was obtained with more than 1000 copies per cell of plasmids lacking the SV40 sequence. However, the levels of expression were the same for both plasmids after cell division in cytoplasmically injected cells and at all times in nuclear injected cells. Thus, the inclusion this SV40 sequence in non-viral vectors may greatly increase their ability to be transported into the nucleus, especially in non-dividing cells.
Keywords: nuclear import, gene expression, gene therapy, enhancer, plasmid delivery
INTRODUCTION
One of the major problems limiting the success of non-viral gene therapy vectors is their low efficiency of gene transfer to target cells. Many barriers exist for the efficient transfer of genes to cells, including the extracellular matrix, the endosomal/lysosomal environment, the endosomal membrane, and the nuclear envelope (1). Currently, extensive effort has been concentrated on the first three obstacles. DNA must enter the nucleus to be transcribed, replicated, and/or integrated, yet there has been relatively little attention directed toward discovering and exploiting the mechanisms by which plasmids enter the nucleus. These processes are especially relevant in post-mitotic and quiescent cells, since there is no breakdown of the nuclear envelope in the absence of mitosis. Because many cells that are targeted in gene therapies do not divide or divide very slowly, the entry of plasmids into the nucleus becomes the major limiting step in gene transfer.
We recently have begun to characterize transport of plasmids from the cytoplasm to the nucleus and have shown that plasmids are able to enter the nuclei of cells through the nuclear pore complex in the absence of cell division (2). Most importantly, and most intriguingly, we also have demonstrated that this nuclear entry is sequence-dependent. Although SV40 DNA was efficiently transported into the nucleus, bacterial plasmids such as pBR322 or pBluescript were excluded from the nucleus until mitosis occurred (2). The SV40 sequences necessary for nuclear import were localized to the origin of replication and early/late promoter region. When these sequences were incorporated into pBR322, they enabled hybrid plasmid transport into the nucleus. In the present report, we have further characterized the sequences involved in nuclear import and show their ability to achieve nuclear import of plasmids in non-dividing cells.
EXPERIMENTAL PROCEDURES
Construction and preparation of plasmids
SV40 DNA was purified from infected TC7 cells by Hirt lysis as described (3). The SV40 origin sequence (SV40 nts 5171 to 43) was excised from plasmid pOR(5171-43), a derivative of pBR322 (4), by digesting with EcoRI and HindIII and replaced with a multiple cloning site (HindIII-XbaI-NcoI-SmaI-KpnI-EcoRI) to create pDD201 and facilitate cloning (Fig. 1). The numbering of SV40 nucleotides is according to convention (5). The same multiple cloning site was inserted into the EcoRI and HindIII sites of the parent pBR322 plasmid to create pDD180. POR(5171-300) was constructed by subcloning the 372 bp HindIII-KpnI fragment from SV40 (nts 5171 to 300) containing the origin and promoter region into pDD201. The 372 bp SV40 origin fragment also was cloned into the TA cloning vector (Invitrogen, San Diego CA) to facilitate subsequent cloning steps. pOR(37-160) and pOR(128-200) contain the 123 bp NcoI-EcoRI (nts 37 to 160) and 72 bp SphI-SphI (nts 128 to 200) fragments from SV40 inserted into the corresponding restriction sites, respectively (Fig. 2). POR(37-300) was constructed by subcloning the 257 bp NcoI-KpnI (nts 37 to 300) fragment from SV40 into pDD201. pOR(200-300) was constructed by subcloning the 94 bp SphI-KpnI (nts 200 to 300) fragment into pDD201. pOR(5171-43), pOR(5171-160), pOR(5208-43)20C, and pOR(5208-43)5215G were kindly provided by Peter Tegtmeyer (SUNY Stony Brook) (4).
Figure 1. Constructs used in these studies.

Cartoons of the plasmids used in these studies are shown. Restriction endonuclease sites are abbreviated as follows: A, AflIII; Av, AvaI; B, BamHI; E, EcoRI; H, HindIII; K, KpnI; N, NcoI; S, SmaI; St, StuI; V, EcoRV; X, XbaI.
Figure 2. Functional organization of the SV40 origin/promoter region and map of DNA constructs.

The functional elements of the origin of replication, early and late promoters, and enhancer are shown (36). All SV40 DNA fragments shown were cloned into the pBR322 derived plasmid pOR(5171-43), replacing the original SV40 sequences present (4). pOR(5208-43)20C and pOR(5208-43)5215G are site-directed mutants of pOR(5208-43) that contain single point mutations in T antigen binding sites and are indicated by an “X” (9).
The 870 bp immediate early promoter/enhancer from the human cytomegalovirus (hCMViep) was removed from pBK-CMV (Stratagene, San Diego CA) by digestion with AflIII and EcoRI and cloned into pDD180 to create pBR-CMV. The 600 bp promoter contained within the Rous sarcoma virus long terminal repeat (RSV LTR) was excised from pRc/RSV (Invitrogen, San Diego CA) by digestion with BglII and HindIII and cloned into pDD180 to create pBR-RSV. The 372 bp SV40 origin and promoter region was removed from the TA coning vector by digestion with HindIII and XbaI and cloned into the same sites of pBR-CMV to produce pBR-CMV-SV40. The SV40 origin and promoter region were removed from the plasmid pRc/RSV by digestion with EcoRV and religation creating pRSVneo. pRSVneo-SV40 was made by removing a 398 bp StuI-EcoRV fragment containing the SV40 origin/promoter from the TA cloning vector and then re-introducing the SV40 sequence into the AvaI site of pRSVneo.
pGFPΔSV40 was derived from pEGFP-N1 (Clontech, Palo Alto, CA; referred to as pGFP in the text) by digestion with Stu I followed by partial digestion with Ssp I and ligation to remove the SV40 early promoter/enhancer region (nts 2218 to 2578). All manipulations were performed as described and all constructs were confirmed by double-stranded sequencing using Sequenase 2.0 (Amersham, Arlington Heights, IL) (6). Protein-free plasmid DNA was purified by alkaline lysis and Qiagen maxiprep columns (San Diego, CA). Agarose gel electrophoresis analysis of the purified plasmids revealed that all of the preparations contained predominantly supercoiled plasmid in the same relative amounts, with no RNA or low molecular weight contaminants (not shown).
Cell culture and microinjection
TC7 cells, a subline of African Green monkey kidney epithelium, were grown on coverslips in DMEM (Life Technologies, Gaithersburg MD) containing 10% fetal bovine serum and antibiotics in a humidified incubator at 37°C with 5% CO2. Purified protein-free plasmid was suspended in phosphate-buffered saline (PBS) at a concentration of 0.5 mg/ml and microinjected into the cytoplasm of cells grown on etched coverslips as described (7). A total volume of 3 × 10−10 ml was injected per cell, as determined by microinjection of a solution of 32P at known specific activity, and is in agreement with volumes delivered by others (8). At a plasmid concentration of 0.5 mg/ml, this volume contains approximately 20,000 molecules of plasmid. After microinjection, the cells were incubated in complete medium, unless otherwise indicated, for eight hours at 37°C. For GFP expression experiments, approximately 100 cells were injected for each DNA concentration, plasmid, and time point, and the experiments were repeated at least 4 times for each time point.
In situ hybridization
In situ hybridizations were performed as described (2). Briefly, after microinjection and incubation for 8 hours in the appropriate growth medium, the cells were rinsed in PBS, permeabilized with 0.5% Triton X-100 in PBS at 23°C for 45 seconds, fixed in acetone:methanol (1:1) at −20°C for 5 minutes, and incubated in 70% formamide in 2X SSC at 70°C for 2 minutes to denature the DNA. The cells were then hybridized overnight at 37°C with a fluorescently-labeled probe in hybridization buffer (2 mg/ml bovine serum albumin, 0.25 mg/ml sheared salmon sperm DNA, 0.25 mg/ml Torula yeast tRNA, 10% dextran sulfate, 2 X SSC, and 50% deionized formamide). All samples were treated with RNaseH (8U/ml) after hybridization. After the subsequent washing steps, the cells were mounted with 1 μg/ml DAPI and the anti-bleaching reagent DABCO according to the manufacturer’s instrcutions (Molecular Probes, Eugene, OR). Fluorescently-labeled probes were prepared by nick translation of pBR322, pRc/RSV, and SV40 DNA as described except that fluorescein-12-dUTP (Molecular Probes) were incorporated directly into the DNA. All photographs were taken with an Olympus BMAX50 epifluorescence microscope equipped with a PM20 photodocumentation system on 400 ASA Kodak Ektachrome or TriX-PAN film.
Quantification of GFP expression
Cells microinjected with pGFP or pGFPΔSV40 were observed by fluorescence microscopy after fixation in 4% paraformaldehyde in PBS for 15 min. at 4°C. Cells expressing GFP were recorded as showing very weak, weak, medium, strong, and very strong GFP fluorescence. These categories of intensity were assigned values of 1 (very weak) through 5 (very strong) and the relative expression is given as the total numerical value (cells × intensity) divided by the total number of injected cells. The validity of this quantitation method was confirmed by analyzing digital images of the GFP-expressing cells that were obtained with an Optronics cooled CCD camera and using the Image Pro-Plus software package. In all cases, the integrated optical density of the cells determined using the digital system gave results that were qualitatively indistinguishable from those obtained with our visual screening method.
Synchronization of cells
TC7 cells were plated onto etched coverslips in 6-well tissue culture plates and incubated in DMEM containing 10% fetal bovine serum until they reached 50% confluency. Lovastatin (Calbiochem, La Jolla, CA) was added to 50 μM and the cells were grown for an additional 24 hours. The medium was removed and the cells were washed with PBS. Fresh medium containing 5 mM mevalonic acid (Sigma, St. Louis, MO) was then added and the cells were grown for an additional 2 hours before microinjection. At the indicated times, the cells were washed with PBS, fixed for 15 min. in 4% paraformaldehyde in PBS, mounted with DAPI and DABCO, and observed for GFP expression by fluorescence microscopy.
RESULTS
Identification of DNA sequence elements needed for nuclear import
We have previously identified a 372 bp fragment of SV40 DNA containing the origin of replication and the early promoter and enhancer region as the sequence required for mediating nuclear entry of plasmids (2). To determine which sequences within this region are required for nuclear import and the possible function of these sequences in nuclear transport, we made a series of plasmids containing different segments of this region (Fig. 2) and tested them for their ability to localize in the nucleus (Table 1). As in our previous study, in situ hybridizations were performed to determine the subcellular localization of the cytoplasmically-injected DNA. The limit of detection of this method is about 300 copies of plasmid per nucleus. While the following experiments were performed in TC7 cells, we have shown previously that such sequence-specific nuclear import occurs in all cell types tested to date, including epithelial, endothelial, and smooth muscle cells from a variety of organisms (2).
Table 1.
Nuclear localization of plasmids.
| Subcellular Localization1 | |||
|---|---|---|---|
| Plasmid | Nucleus | Cytoplasm and Nucleus | Cytoplasm |
| SV40 (Complete Genome) | 82 ± 12 | 6 ± 2 | 12 ± 8 |
| pBR322 (No SV40) | 0 ± 0 | 0 ± 0 | 100 ± 0 |
| pOR-MCS (No SV40) | 0 ± 0 | 0 ± 0 | 100 ± 0 |
| pOR(5171-300) | 58 ± 4 | 22 ± 5 | 20 ± 9 |
| pOR(5171-43) | 8 ± 3 | 18 ± 1 | 74 ± 3 |
| pOR(5171-160) | 28 ± 11 | 27 ± 15 | 45 ± 5 |
| pOR(37-160) | 57 ± 3 | 21 ± 13 | 22 ± 10 |
| pOR(37-300) | 57 ± 6 | 20 ± 8 | 23 ± 2 |
| pOR(128-206) | 56 ± 3 | 16 ± 4 | 28 ± 5 |
| pOR(200-300) | 58 ± 7 | 20 ± 8 | 22 ± 15 |
| pOR(5208-43)20C | 8 ± 12 | 14 ± 8 | 77 ± 20 |
| pOR(5208-43)5215G | 6 ± 8 | 21 ± 11 | 73 ± 15 |
After cytoplasmic microinjection, cells were incubated for 8 hours, and scored for the subcellular location of the injected DNA by in situ hybridization. Cells were classified by either nuclear (≥80% nuclear signal), cytoplasmic and nuclear (≤80% nuclear signal), or cytoplasmic (0% nuclear) and tabulated as a percentage of cells with detectable signal. The results are the average (± st. dev.) of 4 experiments with between 50 and 100 cells visualized for each DNA.
Promoter function is not required for plasmid nuclear import
To determine whether a functional promoter was needed for import, we compared a series of plasmids made to dissect the SV40 origin of replication and promoter region. The plasmid pOR(5171-300) containing the entire 372 bp SV40 origin/promoter region localized to the nucleus to the same levels comparable to that of the entire SV40 genome (Table 1). The plasmid pOR(5171-43) contains 115 bp of SV40 sequence including the core region of the origin (4). While this plasmid supports DNA replication, it cannot drive transcription of a reporter gene since most of the promoter region is lacking. Eight percent of the cells injected with pOR(5171-43) showed complete nuclear localization of the plasmid while another 18% showed nuclear and cytoplasmic staining. Although this level of nuclear import is about 10% of that of the full length pOR(5171-300), the activity is greater than that of the backbone vector alone, pOR-MCS, which was completely unable to target to the nucleus in the absence of cell division. These results demonstrate that a functional promoter is not needed for the nuclear entry of plasmid DNA, but may be important for optimal import.
Origin of replication is not needed for nuclear import
To determine if the origin of replication was needed for nuclear import activity, several constructs were made that contained either (1) a non-functional origin, (2) distal flanking elements that are not sufficient to initiate DNA replication, or (3) none of the SV40 origin of replication. Two different point mutants, pOR(5208-43)20C and pOR(5208-43)5215G (9), which destroy origin activity in the pOR(5208-43) construct were also used. pOR(5208-43) is identical to pOR(5171-43), except for a 31 bp deletion between SV40 nucleotides 5178 and 5208, outside of the origin and promoter region (9). Both plasmids displayed identical levels of nuclear accumulation, which was also indistinguishable from the replication competent pOR(5208-43) and pOR(5171-43) wild type plasmids (Table 1). pOR(37-300), which contains nucleotides 37 to 300, lacks the core origin sequences. It does not support DNA replication, although it does contain the T-antigen binding region III (SV40 nts 30 to 72) which is needed for maximal DNA replication (4). pOR(37-300) was detected exclusively in the nuclei of 57% of the injected cells, a level similar to that seen with the plasmid containing the entire 372 bp region. Further, pOR(200-300), which contains only enhancer sequences and no origin sequences, was imported at similar levels. These results indicate that a functional origin of replication is not needed for plasmid nuclear localization.
Enhancer region provides majority of plasmid nuclear import activity
These results indicated that most of the plasmid nuclear import activity resided in regions outside of the origin of replication (SV40 nucleotides 5171 to 72). Indeed, inclusion of 117 bp in pOR(5171-43) extending into the GC boxes and much of the first 72 bp repeat of the enhancer, gave pOR(5171-160) which localized exclusively to the nucleus of 28% of injected cells and showed mixed nuclear and cytoplasmic localization in another 27% (Table 1). Removal of the core origin sequences produced pOR(37-160), which exhibited maximal transport activity. This result suggests that sequences around the origin may modulate or inhibit import. This result, coupled with the import activity of pOR(200-300) which contained one complete, contiguous copy of the 72 bp repeat, suggested that the majority of the import activity resided in portions of the 72 bp repeat. While pOR(128-206) contained an entire 72 bp repeat, it was not the intact, contiguous form: the 5′ 59 bp of the first repeat are followed by the 3′ 23 bp of the second repeat. Like pOR(200-300), this plasmid localized to the nucleus to the same degree as the entire 372 bp fragment. Neither pOR(200-300) or pOR(128-206) are able to function as promoters or origins. Since nuclear import of these plasmids occurs at maximal rates, this confirms that the function of neither the promoter or the origin is required for nuclear import of the DNA and that the majority of the import activity resides in DNA sequences within the enhancer domain.
Search for other promoters and enhancers that support plasmid nuclear entry
Because the most active SV40 sequence for nuclear import is the enhancer, it is possible that other enhancers and/or promoters may also give similar nuclear import activity. Alternatively, the SV40 sequence may be unique in its import properties. To test the hypothesis that plasmids with promoters and enhancers containing multiple transcription factor consensus binding sequences direct nuclear import, several other promoter-containing plasmids were tested for nuclear import. As likely candidates, two very strong in vitro and in vivo promoters were chosen: the immediate early promoter and enhancer from the human cytomegalovirus (hCMViep) and the LTR promoter from Rous sarcoma virus (RSV-LTR). Both promoters have been shown to be even stronger than the SV40 early promoter for gene expression in mouse muscles in vivo (10,11). As expected, when pOR(5171-300)was injected into the cytoplasm, it localized largely to the nucleus within eight hours as visualized by in situ hybridization (Fig. 3A). By contrast, when the 800 bp hCMViep was cloned into pBR322, the plasmid remained entirely in the cytoplasm of all cells 8 hours after injection (Fig. 3B). Similarly, two plasmids containing the RSV LTR promoter, pBR-RSV and pRSVneo, also failed to localize to the nucleus by eight hours (Fig. 3C and D). One explanation for the lack of nuclear localization is the cytoplasmic retention of these plasmids. However, when the entire 372 bp SV40 origin/enhancer region was cloned into either pBR-CMV (pBR-CMV-SV40; Fig. 3E) or pRSVneo (pRSVneo-SV40; Fig. 3F), the plasmids migrated to the nucleus as efficiently as did pOR(5171-300). Thus, it appears that the ability to promote nuclear import of plasmids is a unique property of the SV40 enhancer region.
Figure 3. Nuclear localization of plasmids containing viral promoters and enhancers.
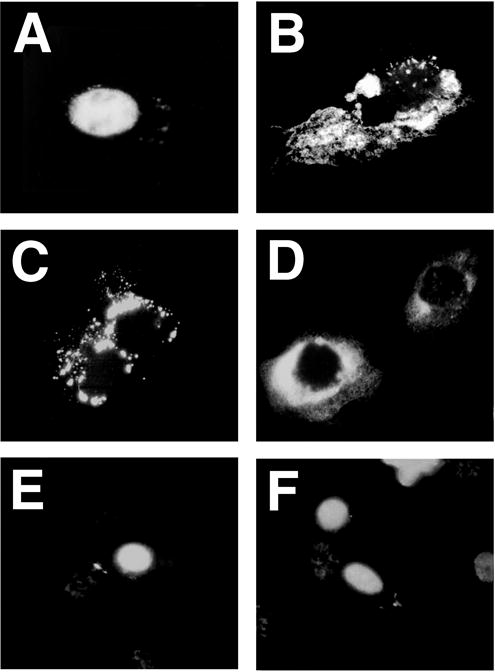
Several viral promoter/enhancers were cloned into luciferase or ß-galactosidase-expressing vectors. Plasmids were microinjected into the cytoplasm of TC7 cells and visualized 8 hours later by in situ hybridization. (A) pOR(5171-300) containing the 366 bp SV40 origin/promoter DNA fragment; (B) pBR-CMV; (C) pBR-RSV; (D) pRSVneo; (E) pBR-CMV-SV40 (pBR-CMV containing the 366 bp SV40 origin/promoter DNA fragment); and (F) pRSVneo-SV40 (pRSVneo containing the SV40 origin/promoter DNA fragment).
Plasmids carrying the SV40 promoter/enhancer express their products more rapidly and efficiently when injected into the cytoplasm
Although plasmids containing the hCMViep promoter and no SV40 sequences appeared to remain completely cytoplasmic when assayed by in situ hybridization, it is possible that a few copies of the plasmid (less than 300 copies per cell) were able to enter the nucleus but could not be detected by this assay. To exclude this possibility, a plasmid expressing GFP from the hCMViep promoter but containing no SV40 sequences (pGFPΔSV40) was injected into synchronized TC7 cells, as was the control plasmid which did contain the SV40 early promoter and enhancer (pGFP). In this synchronized system, cell division occurs between 12 and 16 hours. Expression of GFP was detected and quantified as the sum of the intensity of GFP signals in cells divided by the total number of cells injected. Both visual scoring of GFP intensities and digital analysis of images obtained with a cooled CCD camera and image analysis software yielded the same results. When either plasmid was injected into the nuclei of cells, expression of GFP could be detected within 2 hours. Both plasmids produced similar levels of GFP, as assayed by GFP fluorescence (Fig. 4A). Expression was detected in between 30% and 70% of nuclear injected cells at the highest DNA concentration, about 100 copies per cell.
Figure 4. Sequence-specific nuclear localization and GFP expression.
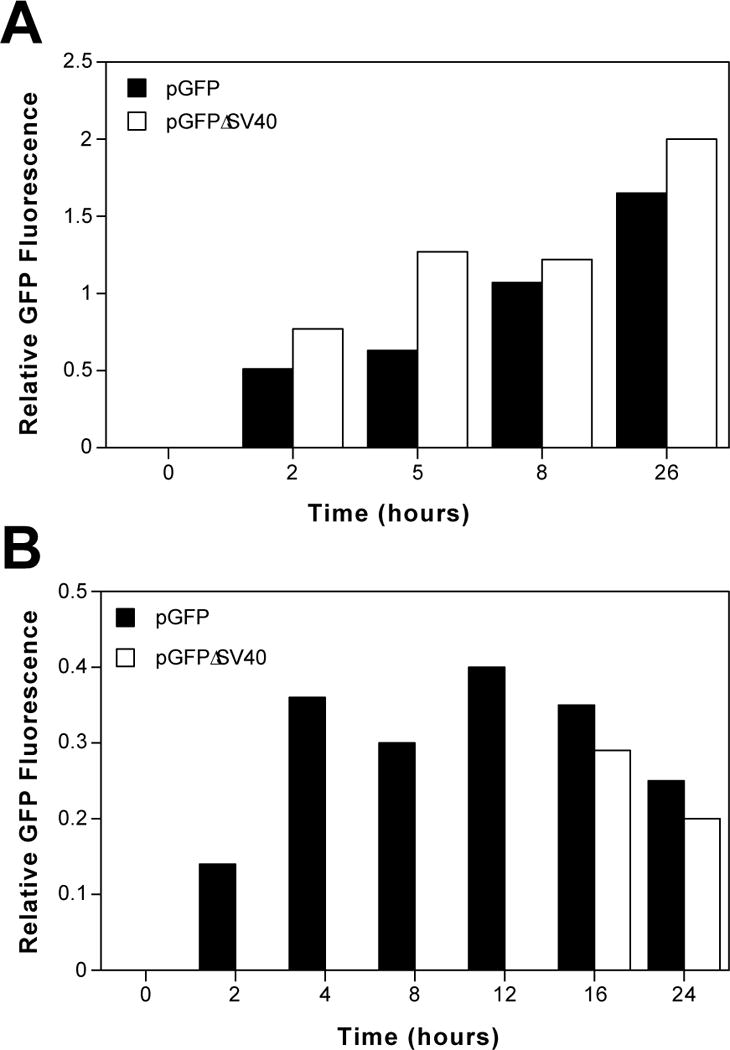
(A) Nuclear injected pGFP and pGFPΔSV40 display similar patterns and efficiencies of GFP expression. TC7 cells were synchronized by treatment with 60 μM lovastatin for 16 hours, washed and then incubated in medium containing serum and 5 mM mevalonic acid for two hours before microinjection. Thirty copies of either plasmid were injected into each nucleus of approximately 100–150 cells and assayed for GFP expression at later times. GFP expression which was recorded on an intensity scale of 1 to 5 and relative GFP expression is given as the total intensity divided by the total number of injected cells. (B) Plasmids with or without the SV40 enhancer differ greatly in their ability to express GFP when injected into the cytoplasm. Thirty copies of either pGFP or pGFPΔSV40 were microinjected into the cytoplasm of approximately 100 cells, synchronized as in (A), and relative GFP expression was measured. The results shown (A and B) are representative of 4 independent experiments.
However, when the plasmids were injected into the cytoplasm of synchronized cells, significant differences were observed between plasmids with and without the SV40 enhancer. When pGFP was injected into the cytoplasm of cells at 30 copies of plasmid per cell, GFP expression was detectable at 2 hours post-injection and increased at later times (Fig. 4B). By contrast, no GFP expression was detected in cells that had been cytoplasmically injected with the same number of pGFPΔSV40 plasmids for as long as 12 hours post-injection. However, at time points after cell division (16 and 24 hours), GFP expression was detected in cells cytoplasmically injected with both plasmids and the amount of GFP expression was roughly equivalent (Fig. 4B). This expression in cytoplasmically injected cells corresponded to between 25% and 50% of the injected cells producing a GFP signal.
Nuclear import of plasmid is rate-limiting for gene expression
GFP expression from pGFP increased with time in nuclear injected cells and the increase in expression was greatest in cells receiving low concentrations of plasmid (Fig. 5). For cells receiving 3 copies of plasmid per cell, expression increased from a relative value of 0.06 at 4 hours to 0.31 at 24 hours. Moreover, at the same concentration of plasmid, the absolute percentage of cells expressing GFP also increased accordingly from 2% to 20% at 4 and 24 hours, respectively. While increases were also seen at higher concentrations of plasmid, they were not as dramatic. At 100 copies per nucleus, the percentage of cells expressing GFP rose from 20% to 40% between 4 and 24 hours, and the relative expression also rose 2-fold over the same period. This time dependency of expression, typically 2-fold, was also seen in cytoplasmically injected cells (Fig. 5). Gene expression was also clearly dose-dependent at all times for both cytoplasmic and nuclear injected plasmid (Fig. 5). At 4, 8, and 24 hours, there was a 40-, 3-, and 7-fold increase, respectively, in relative gene expression in cells receiving 100 copies compared to 1 copy of pGFP per nucleus. Similarly, in cytoplasmically injected cells, the differences in relative gene expression were between 5- and 10-fold over a 100-fold concentration range. Further, a similar dose response was observed in nuclear injected cells for pGFPΔSV40. Although the absolute levels of gene expression seen in each experiment were variable with respect to each other, the same trends and results were obtained in 6 independent experiments, representing over 20,000 microinjected cells.
Figure 5. Nuclear import of plasmid DNA is rate limiting for gene expression at early times after microinjection.
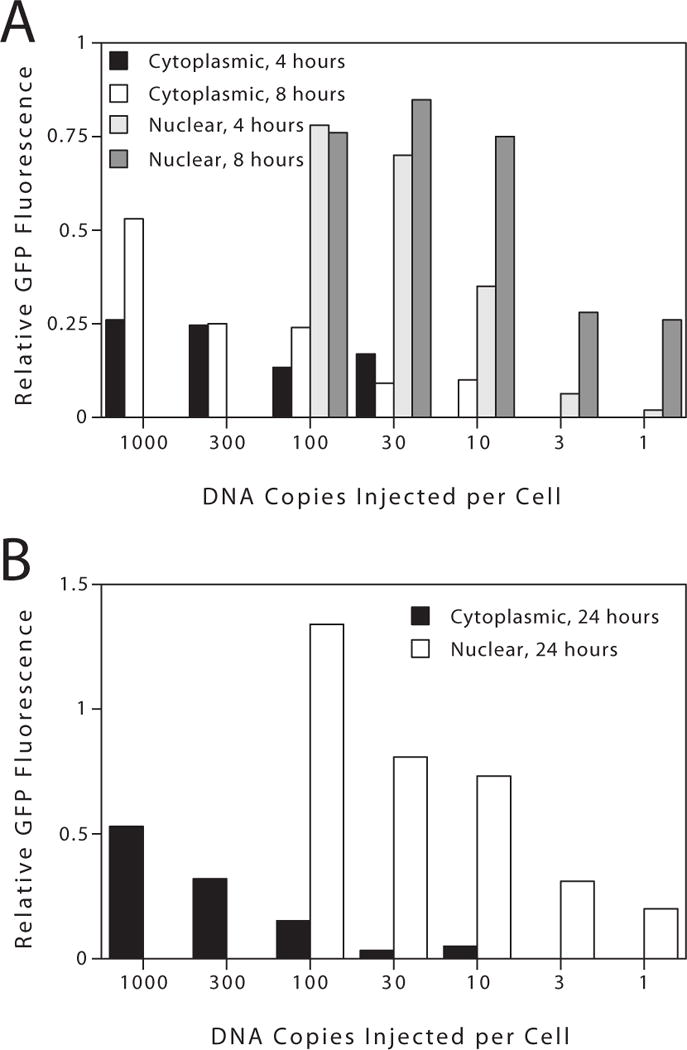
pGFP was microinjected into the nucleus or cytoplasm of TC7 cells at the indicated copy number per cell and assayed for gene expression at (A) 4 and 8 hours, and (B) 24 hours post-injection. Between 100 and 150 cells were injected for each concentration shown. The results shown (A and B) are representative of 4 similar experiments.
To determine to what extent nuclear import of plasmid DNA affects the level of gene expression, we compared the number of plasmids injected into each compartment needed to produce the same levels of GFP (Fig. 5A). At 4 hours, about 100 to 300 plasmids must be injected into the cytoplasm to give the same level of GFP as does 3 to 10 nuclear copies of the same plasmid. By 8 hours, much of the cytoplasmic DNA has been targeted to the nucleus, since as few as 10 copies of plasmid injected into the cytoplasm gives GFP expression (Fig. 5A). Compiling data from 6 similar experiments, we find that by 8 hours post-injection, roughly 20 times more of the SV40 DNA-containing plasmid must be injected into the cytoplasm, compared to the nucleus to yield equivalent gene expression. By contrast, amounts of the SV40-lacking plasmid as high as 1000 copies per cell gave no gene expression when injected into the cytoplasm until 16 to 24 hours after injection. Thus, as predicted, disassembly and reformation of the nuclear envelope during mitosis allows plasmids to have free access to the nucleus during cell division.
DISCUSSION
While many aspects of non-viral vector design are being addressed, one critical area that has received relatively little study is the nuclear import of plasmids. Plasmids must enter the nucleus to function, yet the entry mechanisms remain largely unexplored. In the present report, we have defined the sequences necessary for nuclear targeting of SV40 DNA in the absence of cell division. Although the origin of replication alone can support minimal nuclear import, the majority of the import activity resides in the promoter and enhancer region. Neither the function of the promoter nor the origin of replication is needed for efficient nuclear localization. Somewhat surprisingly, two other strong viral promoter and enhancer sequences, including the CMV immediate early promoter/enhancer and the RSV LTR, are unable to support nuclear import of plasmid in the absence of cell division. Using this observation, we were able to confirm the necessity of the SV40 sequences for nuclear import by following CMV promoter-driven GFP expression in plasmids with and without the SV40 promoter/enhancer. This approach allowed us to decrease the number of plasmids injected into the cells and to determine the contribution of nuclear transport of plasmids to gene expression. While not playing an crucial role in cells that divide during the course of the experiment, nuclear import of DNA plays a significant role in limiting gene expression in the absence of cell division.
All of our results, as well as those from many other researchers, clearly demonstrate that the nuclear envelope is a major barrier to gene transfer and expression (1,12–14). Others have shown that nuclear import is a rate-limiting step in gene expression (12,14). In his seminal microinjection experiments, Capecchi found that 50 to 100% of cells injected in the nucleus with plasmid expressed gene product as compared to only 0.01% of those receiving plasmid in the cytoplasm (12). More recently, another report has shown that at DNA copy numbers where 50% of nuclear injected cells display expression, only 1% of cytoplasmically injected cells do so (14). In these last experiments, significant gene expression was seen at 4 hours post-nuclear injection, but an additional 8 hours was needed to see any expression when the plasmids were introduced into the cytoplasm. Interestingly, both of these studies used plasmids containing no SV40 sequences. Our results are in complete agreement with these results. For plasmids without the SV40 sequences, more than 12 hours are needed to detect gene expression. Moreover, cytoplasmic injections are much less efficient than are nuclear injections (Figures 4 and 5). By contrast, when the SV40 enhancer sequence is present on the plasmid, gene expression is detected much more rapidly and requires fewer plasmids per cell to produce gene product (Figures 4 and 5 and ref. (15)). However, while the SV40 enhancer sequence must be present for a plasmid to enter the nucleus before cell division, at later time points, after the cells have undergone a round of cell division and the ensuing breakdown and reformation of the nuclear envelope, expression from plasmids without SV40 sequences would occur. During mitosis, the absence of a nuclear envelope negates the need for an NPC-mediated nuclear import signal. Indeed, this is evident from the fact that after division, pGFPΔSV40 that had been injected into the cytoplasm was capable of gene expression.
The ability of the SV40 enhancer to facilitate maximal nuclear localization of plasmid supports our previously proposed model for nuclear import, which is the following (Fig. 6). Newly synthesized transcription factors and DNA binding proteins bind to plasmid in the cytoplasm to create protein-DNA complexes. These DNA binding proteins are normally destined for the nucleus and contain nuclear localization sequences (NLSs) necessary for interaction with the nuclear import machinery (for a review see ref (16)). Thus, the complex is recognized through the NLS by the protein import machinery and is thereby targeted to the nucleus (2). Indeed, in experiments using digitonin-permeabilized HeLa cells, we have shown recently that nuclear import of plasmids can occur only when both nuclear extract and recombinant importinα, importinβ, and RAN are provided; neither nuclear extract alone nor the purified NLS-import machinery proteins alone can suffice (17). Of the 372 bp in the SV40 origin/promoter region, the enhancer contains the most number of consensus binding sites for such proteins. Thus, it is not surprising to find that the enhancer mediates maximal nuclear import, since the majority of transcription factors bind this region. Given the complexity of transcription, it seems unlikely that one specific transcription factor mediates DNA binding and nuclear import. More likely, a complex of proteins binds to the enhancer region and collectively mediates nuclear import of the plasmids.
Figure 6. Model for sequence specific nuclear import of plasmid DNA.
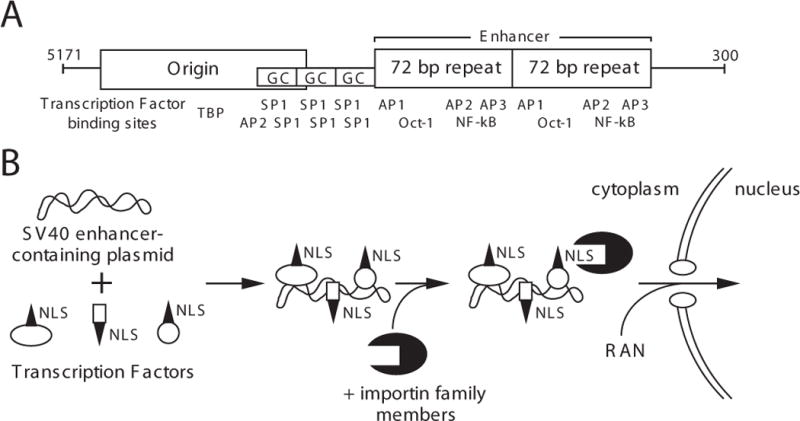
(A) Sequence elements and binding sites for transcription factors are shown for the 366 bp SV40 DNA nuclear targeting sequence. (B) Since these transcription factors are synthesized in the cytoplasm, once plasmid DNA has entered the cytoplasm by transfection, injection, or infection, the newly synthesized proteins can bind to the plasmid DNA to form a DNA-protein complex. Thus, the DNA is coated with NLSs from the transcription factors, allowing the NLS-mediated import machinery to recognize DNA as a substrate and target it into the nucleus.
Graessman has used microinjection to identify a helper activity of the SV40 enhancer in dividing cells that increased nuclear import based on subsequent gene expression (15). He reported that the AP1 binding site was absolutely required for nuclear import, since without it, the SV40 enhancer was unable to stimulate expression of a chloramphenicol acetyltransferase reporter gene when injected into the cytoplasm vs. the nucleus. However, our results demonstrate that AP1 is not essential for nuclear import. AP1 sites are not present in pOR(5171-43) (core origin fragment) although this fragment supports minimal nuclear transport. Moreover, pOR(200-300) which contains the majority of the second 72 bp enhancer repeat, but no AP1 site, also promotes maximal import.
Sequence analysis shows that AP2, a factor shown to bind to the SV40 enhancer (18,19) is the only factor that binds to pOR(5171-43), pOR(37-160) (GC boxes and the first 51 bp of the 72 bp repeat), pOR(128-206) (the first 72 bp repeat), and pOR(200-300) (the majority of the second 72 bp repeat). Further, the potential AP2 binding sites in the enhancer fragments are closer to the consensus than is the site at the end of the core origin fragment. That the hCMViep and the RSV LTR do not support nuclear import would imply that the putative transcription factor(s) do not bind to these promoters. Comparison of the transcription factor consensus binding sequences in these two promoters (20), and the sites identified experimentally (21), reveals that the binding sites for several transcription factors, including AP2 and AP3, are not present. To test the hypothesis that AP2 alone mediates the nuclear import of SV40 DNA, we tested the smooth muscle gamma actin promoter, which contains an AP2 site approximately 200 bp upstream of the transcriptional start site (22). Plasmids containing fragments of this promoter, ranging from 400 bp to 2.2 kbp, were unable to mediate nuclear import of the plasmids in TC7 and CV1 cells as analyzed by in situ hybridization (22). Most likely, AP2 is only one of a larger complex of proteins that forms on the SV40 promoter/enhancer region.
Several recent reports have proposed similar models for the NLS-dependent nuclear import of DNA. Längle-Rouault et al have shown that a plasmid containing the Epstein-Barr virus (EBV) oriP sequence gives almost 100-fold greater gene expression than a plasmid lacking the oriP repeats, either by transfection or cytoplasmic injection into cell lines expressing the oriP-binding nuclear EBNA-1 protein (23). However, this effect is limited to EBV-transformed cells.
Synthetic NLS peptides alone also can increase the nuclear localization of DNA when either covalently coupled to the DNA, or when complexed to the DNA by electrostatic interactions. When NLS peptides were covalently linked to plasmid DNA, plasmids localized to the nucleus rapidly in digitonin-permeabilized cells in a classical NLS-dependent manner (24). Unfortunately, these altered plasmids were unable to enter the nuclei of microinjected cells or give significant gene expression, calling into question the relevancy of this approach to increase in vivo gene transfer (24). By contrast, when synthetic NLS peptides were complexed with plasmid DNA and microinjected into the cytoplasm of zebrafish embryos, increased nuclear localization of plasmids was observed, as was increased integration and expression of the transgene (25–28). More recently, using an NLS peptide covalently linked to a hairpin oligonucleotide at one end of the gene, it was demonstrated that even a single NLS bound to a 3.3 kbp capped DNA molecule causes increased nuclear targeting and gene expression (29). Finally, several other reports have demonstrated that incorporation of DNA-binding proteins such as histone H1 or HMG proteins into liposomes can increase transfection efficiencies, presumably due to an increased nuclear targeting event (30–32).
By in situ hybridization experiments, there appears to be a quantal nuclear import response: the injected DNA is either entirely cytoplasmic or almost entirely nuclear (≥ 80%; Fig 3) (2). At first inspection, this finding is paradoxical, since this nuclear DNA has negligible effect on gene expression. By 8 hours post-injection, essentially all of the injected DNA accumulated within the nucleus (Fig. 3A), but the level of GFP expression was only 10 to 30% of that found in cells receiving the same number of plasmids injected directly into the nucleus (Figs. 4 and 5). The differences in the level of expression after nuclear and cytoplasmic injections may likely arise from the fact that gene expression is a time-dependent process. While the majority of the cytoplasmically injected DNA may have reached the nucleus by 8 hours post-injection, much of it has not yet begun to express. Indeed, even in the nuclear injections, GFP levels increase with time (Fig. 4 and 5). Furthermore, since GFP is a stable protein with a half-life of greater than 24 hours in cells, the levels of GFP should remain higher in nuclear injected cells than in cytoplasmically injected cells at all times because the DNA is available to the transcription machinery longer, and hence, more GFP will accumulate in the cells giving greater apparent gene expression.
It is somewhat surprising that the hCMViep and the RSV LTR sequences do not promote plasmid nuclear import, especially since they are two of the strongest promoters identified to date for use in cell culture and in muscle (10,11). In transfection experiments this is not surprising since most transfections are performed on subconfluent cells that are incubated for 24 to 48 hours, allowing at least one round of cell division and access to the nucleus during the transient breakdown of the nuclear envelope. However, both of these promoters generate very high levels of gene expression when injected into mouse quadricep muscles that are made up of post-mitotic, terminally differentiated myotubes (10,11). Thus, these plasmids gain access to the nuclei of these cells (33). One possible explanation for this paradox is that plasmids lacking the SV40 enhancer sequences are preferentially degraded in the cytoplasm, decreasing their numbers so that any entrance into the nucleus would be undetectable. However, it should be noted that good expression can be detected at 24 hours with as few as 10 copies of pGFPΔSV40 injected into the cytoplasm, a finding that suggests that very little degradation of the plasmid occurs. This conclusion is supported by others who have found little degradation of plasmid DNA in microinjected or transfected cells (15,34), although there is no consensus in the field regarding DNA degradation in transfections (35). An alternative explanation for our finding that the hCMViep and RSV LTR are incapable of targeting plasmids to the nucleus is that these plasmids are indeed capable of mediating nuclear import of plasmid DNA, but do so very slowly, requiring greater than 24 hours. In the experiments described here we would not detect such slow NPC-mediated nuclear import since our cells have undergone division by this time.
In summary, the inclusion this SV40 sequence in non-viral vectors greatly increases their ability to be transported into the nucleus and lead to increased levels of gene expression, especially in non-dividing cells.
Acknowledgments
We thank Peter Tegtmeyer (SUNY Stony Brook) for the gift of the SV40 origin plasmids, and Mike Sawdey (VICAL Inc.), Marston Manthorpe (VICAL Inc.), and Warren Zimmer (University of South Alabama) for helpful discussions. Supported in part by grants from the Alabama Affiliate of the American Heart Association (ALG 960006) and the National Heart, Lung, and Blood Institute (HL59956 and HL38639).
References
- 1.Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem. 1995;270:18997–19007. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]
- 2.Dean DA. Import of plasmid DNA into the nucleus is sequence specific. Exp Cell Res. 1997;230:293–302. doi: 10.1006/excr.1996.3427. [DOI] [PubMed] [Google Scholar]
- 3.Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 4.DeLucia AL, Deb S, Partin K, Tegtmeyer P. Functional interactions of the simian virus 40 core origin of replication with flanking regulatory sequences. J Virol. 1986;57:138–144. doi: 10.1128/jvi.57.1.138-144.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tooze J. Molecular biology of tumor viruses, 2nd edition. Cold Spring Harbor Laboratory, Cold Spring Harbor; NY: 1980. [Google Scholar]
- 6.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. John Wiley & Sons; New York: 1994. [Google Scholar]
- 7.Dean DA, Li PL, Lee LM, Kasamatsu H. Essential role of the Vp2 and Vp3 DNA-binding domain in SV40 morphogenesis. J Virol. 1995;69:1115–1121. doi: 10.1128/jvi.69.2.1115-1121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graessman M, Graessman A. Microinjection of tissue culture cells using glass microcapillaries: methods. In: Celis JE, Graessmann A, Loyter A, editors. Microinjection and organelle transplantation techniques: methods and applications. Academic Press; London: 1986. pp. 3–37. [Google Scholar]
- 9.Deb S, Tsui S, Koff A, DeLucia AL, Parsons R, Tegtmeyer P. The T-antigen-binding domain of the simian virus 40 core origin of replication. J Virol. 1987;61:2143–2149. doi: 10.1128/jvi.61.7.2143-2149.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartikka J, Sawdey M, Cornefert-Jensen F, Margalith M, Barnhart K, Nolasco M, Vahlsing HL, Meek J, Marquet M, Hobart P, Norman J, Manthorpe M. An improved plasmid DNA expression vector for direct injection into skeletal muscle. Hum Gene Ther. 1996;7:1205–1217. doi: 10.1089/hum.1996.7.10-1205. [DOI] [PubMed] [Google Scholar]
- 11.Manthorpe M, Cornefert-Jensen F, Hartikka J, Felgner J, Rundell A, Margalith M, Dwarki V. Gene therapy by intramuscular injection of plasmid DNA: studies on firefly luciferase gene expression in mice. Hum Gene Ther. 1993;4:419–431. doi: 10.1089/hum.1993.4.4-419. [DOI] [PubMed] [Google Scholar]
- 12.Capecchi MR. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell. 1980;22:479–88. doi: 10.1016/0092-8674(80)90358-x. [DOI] [PubMed] [Google Scholar]
- 13.Labat-Moleur F, Steffan AM, Brisson C, Perron H, Feugeas O, Furstenberger P, Oberling F, Brambilla E, Behr JP. An electron microscopy study into the mechanism of gene transfer with lipopolyamines. Gene Ther. 1996;3:1010–7. [PubMed] [Google Scholar]
- 14.Pollard H, Remy JS, Loussouarn G, Demolombe S, Behr JP, Escande D. Polyethylenimine but not cationic lipids promotes transgene delivery to the nucleus in mammalian cells. J Biol Chem. 1998;273:7507–11. doi: 10.1074/jbc.273.13.7507. [DOI] [PubMed] [Google Scholar]
- 15.Graessman M, Menne J, Liebler M, Graeber I, Graessman A. Helper activity for gene expression, a novel function of the SV40 enhancer. Nucleic Acids Res. 1989;17:6603–6612. doi: 10.1093/nar/17.16.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nigg EA. Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature. 1997;386:779–787. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- 17.Wilson GL, Dean BS, Wang G, Dean DA. Nuclear import of plasmid DNA in digitonin-permeabilized cells requires both cytoplasmic factors and specific DNA sequences. J Biol Chem. 1999;274:22025–22032. doi: 10.1074/jbc.274.31.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell PJ, Wang C, Tjian R. Positive and negative regulation of transcription in vivo: enhancer binding protein AP-2 is inhibited by SV40 T antigen. Cell. 1987;50:847–861. doi: 10.1016/0092-8674(87)90512-5. [DOI] [PubMed] [Google Scholar]
- 19.Dynan WS, Chervitz SA. Characterization of a minimal simian virus 40 late promoter: enhancer elements in the 72-base-pair repeat not required. J Virol. 1989;63:1420–1427. doi: 10.1128/jvi.63.3.1420-1427.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wingender E, Kel AE, Kel OV, Karas H, Heinemeyer T, Dietze P, Knueppel R, Romaschenko AG, Kolchanov NA. TRANSFAC, TRRD and COMPEL: Towards a federated database system on transcriptional regulation. Nucleic Acids Res. 1997;25:265–268. doi: 10.1093/nar/25.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson JA, Gnann JW, Jr, Ghazal P. Regulation and tissue-specific expression of human cytomegalovirus. Curr Top Microbiol Immunol. 1990;154:75–100. doi: 10.1007/978-3-642-74980-3_4. [DOI] [PubMed] [Google Scholar]
- 22.Vacik J, Dean BS, Zimmer WE, Dean DA. Cell-specific nuclear import of plasmid DNA. Gene Therapy. 1999;6:1006–1014. doi: 10.1038/sj.gt.3300924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langle-Rouault F, Patzel V, Benavente A, Taillez M, Silvestre N, Bompard A, Sczakiel G, Jacobs E, Rittner K. Up to 100-fold increase of apparent gene expression in the presence of Epstein-Barr virus oriP sequences and EBNA1: implications of the nuclear import of plasmids. J Virol. 1998;72:6181–5. doi: 10.1128/jvi.72.7.6181-6185.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sebestyén MG, Ludtke JL, Bassik MC, Zhang G, Budker V, Lukhtanov EA, Hagstrom JE, Wolff JA. DNA vector chemistry: the covalent attachment of signal peptides to plasmid DNA. Nature Biotech. 1998;16:80–85. doi: 10.1038/nbt0198-80. [DOI] [PubMed] [Google Scholar]
- 25.Collas P, Husebye H, Alestrom P. The nuclear localization sequence of the SV40 T antigen promotes transgene uptake and expression in zebrafish embryo nuclei. Transgenic Res. 1996;5:451–8. doi: 10.1007/BF01980210. [DOI] [PubMed] [Google Scholar]
- 26.Collas P, Alestrom P. Nuclear localization signal of SV40 T antigen directs import of plasmid DNA into sea urchin male pronuclei in vitro. Mol Reprod Dev. 1996;45:431–8. doi: 10.1002/(SICI)1098-2795(199612)45:4<431::AID-MRD4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 27.Collas P, Alestrom P. Rapid targeting of plasmid DNA to zebrafish embryo nuclei by the nuclear localization signal of SV40 T antigen. Mol Mar Biol Biotechnol. 1997;6:48–58. [PubMed] [Google Scholar]
- 28.Collas P, Alestrom P. Nuclear localization signals: a driving force for nuclear transport of plasmid DNA in zebrafish. Biochem Cell Biol. 1997;75:633–40. [PubMed] [Google Scholar]
- 29.Zanta MA, Belguise-Valladier P, Behr JP. Gene delivery: A single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc Natl Acad Sci U S A. 1999;96:91–96. doi: 10.1073/pnas.96.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaneda Y, Iwai K, Uchida T. Increased expression of DNA cointroduced with nuclear protein in adult rat liver. Science. 1989;243:375–8. doi: 10.1126/science.2911748. [DOI] [PubMed] [Google Scholar]
- 31.Bottger M, Zaitsev SV, Otto A, Haberland A, Vorob’ev VI. Acid nuclear extracts as mediators of gene transfer and expression. Biochim Biophys Acta. 1998;1395:78–87. doi: 10.1016/s0167-4781(97)00128-0. [DOI] [PubMed] [Google Scholar]
- 32.Hagstrom JE, Sebestyen MG, Budker V, Ludtke JJ, Fritz JD, Wolff JA. Complexes of non-cationic liposomes and histone H1 mediate efficient transfection of DNA without encapsulation. Biochim Biophys Acta. 1996;1284:47–55. doi: 10.1016/0005-2736(96)00106-x. [DOI] [PubMed] [Google Scholar]
- 33.Dowty ME, Williams P, Zhang G, Hagstrom JE, Wolff JA. Plasmid DNA entry into postmitotic nuclei of primary rat myotubes. Proc Natl Acad Sci USA. 1995;92:4572–4576. doi: 10.1073/pnas.92.10.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Escriou V, Ciolina C, Lacroix F, Byk G, Scherman D, Wils P. Cationic lipid-mediated gene transfer: effect of serum on cellular uptake and intracellular fate of lipopolyamine/DNA complexes. Biochim Biophys Acta. 1998;1368:276–88. doi: 10.1016/s0005-2736(97)00194-6. [DOI] [PubMed] [Google Scholar]
- 35.Lusky M, Botchan M. Inhibition of SV40 replication in simian cells by specific pBR322 DNA sequences. Nature. 1981;293:79–81. doi: 10.1038/293079a0. [DOI] [PubMed] [Google Scholar]
- 36.Cole CN. Polyomavirinae: the viruses and their replication. In: Fields BN, Knipe DM, Howley PM, Chanock RM, Melnick JL, Monath TP, Roizman B, Staus SE, editors. Virology. Raven Press; New York: 1996. pp. 1997–2043. [Google Scholar]