

Abstract
The purpose of the present study was to develop a rapid, reproducible method of non-viral gene transfer to the intact vasculature. Male Sprague-Dawley rats were anesthetized, a midline abdominal incision was made and segmental branches of the superior mesenteric artery were dissected free of surrounding mesentery. A specially designed electroporation probe was placed around the neurovascular bundle and the electroporation chamber filled with a solution containing the firefly luciferase expressing plasmid (pCMV-luxDTS) or the green fluorescent protein expressing plasmid (pEGFP-N1). Vessels were electroporated with eight 10 msec pulses of 200V/cm. Sixty seconds after electroporation, the DNA solution was removed, the intestine returned to the abdomen and the abdominal wall closed with suture and metal wound clips. Six hours to 5 days later, rats were sacrificed and electroporated vessels were recovered. Luciferase activity of the blood vessels was monitored. Gene expression was detected as early as 6 h post-electroporation, peaked at 1–3 days with levels up to 1 ng of reporter gene per vessel segment and returned towards baseline by day 5. Histological analysis of blood vessel segments revealed GFP positive cells throughout the thickness of the vessel wall (endothelial cells to adventitia). Responses of electroporated vessels to vasoconstricting stimuli were indistinguishable from those of control vessels at either 2 or 40 days post-treatment. The results of this study provide evidence that electroporation is an effective means for introducing naked DNA into the blood vessel wall and form the basis for future studies on targeted gene therapy to the intact vasculature.
Keywords: Gene transfer, gene therapy, electroporation, artery, in vivo, smooth muscle cells, endothelial cells
INTRODUCTION
Non-viral vectors show great potential for gene transfer and therapy in a variety of tissues. Unlike their viral counterparts, very little inflammation or pathology is associated with non-viral vectors and essentially no immune response is generated against DNA, either as naked plasmid or when complexed with liposomes or other polymer complexes such as polyethylenimine [1]. More importantly, high levels of plasmids can be produced and easily purified to contain no contaminants such as wild type or defective virus particles, a common problem of viral vectors. Thus, there are fewer safety concerns. Unfortunately, because of relatively poor efficiency of gene transfer by non-viral vectors, viral vectors have remained the choice for numerous groups pursuing in vivo gene transfer, especially to the vasculature.
Electroporation uses electrical fields to create transient pores in the cell membrane that allow the entry of normally impermeable macromolecules into the cytoplasm [2]. Although this technique is routinely used to transfer DNA to bacteria, yeast, and mammalian cells in culture [3], it has only recently been applied to intact tissues in living animals. The results achieved with electroporation are impressive; in most studies, the increase in gene expression to a tissue injected with DNA is on the order of 100 to 1000-fold compared to DNA in the absence of electroporation [4]. With this technique, yields of up to 100 ng per muscle of reporter or therapeutic gene product can be produced within 7 days [4, 5]. Such increases have been observed in intact skeletal muscle [4, 6, 7], liver [8, 9], cornea [10], and brain [11], as well as in explanted chicken embryo hearts [12]. When compared to other in vivo techniques in mouse testes, in vivo electroporation gave the same level of expression as the gene gun and almost 40-fold higher expression than liposomes [13].
Although electroporation protocols have been developed for and work well in solid tissues in which the DNA can be injected, few protocols for the vasculature have been developed. One approach to transfer plasmids to the vasculature has employed an electroporation electrode incorporated into a double-balloon catheter system [14]. Although well tolerated and efficient for transfer to the designated area of the arterial wall, a simpler approach was desired to obviate the need for specialized surgical skills and the tissue ischemia that can result from the use of a balloon occluder [15]. Furthermore, the balloon catheter would be unsuitable for small vessels in some experimental animals such as rats and mice. In this report, we demonstrate the efficient transfer of plasmid to the mesenteric arteries and subsequent high level gene expression in living animals using a novel electrode design that allows multiple vessels to be treated rapidly without disruption of blood flow and with the use of relatively low amounts of DNA. The present studies demonstrate the utility of this approach for the short-term expression and screening of genes for potential therapeutic use and the study of cellular physiology in the living animal.
MATERIALS AND METHODS
Plasmids
The plasmids pCMV-lux-DTS (5.8 kbp, [16]) and pEGFP-N1 (4.7 kbp, Clontech, Palo Alto, CA) express firefly luciferase and green flourescent protein (GFP), respectively, from the CMV immediate early promoter/enhancer. Plasmids were grown in Escherichia coli and purified using Qiagen Endotoxin-free maxi-prep kits, as described by the manufacturer (Qiagen, Chatsworth CA). The concentration of plasmid was adjusted to 2 mg/ml in 10 mM Tris, pH 8.0, 1 mM EDTA, and 140 mM NaCl. Agarose gel electrophoretic analysis demonstrated that greater than 80% of the purified DNA was present in the supercoiled form, and no RNA was detected.
Electrode design
To make electroporation a simple and reproducible procedure for gene transfer to the blood vessel wall, an electrode design was needed that (I) had constant and uniform separation of the wire electrodes, (II) isolated the electrodes from the surrounding tissue, (III) allowed the vessel to be in constant contact with the DNA solution, (IV) did not damage the vasculature, and (V) was simple and inexpensive to produce. The final design is shown in figure 1. Two nichrome wires were inserted parallel to each other into a 1 cm diameter rubber o-ring with a 3 mm gap between them. One end of each wire extended away from the o-ring for attachment to a BTX ECM2001 square wave electroporator. A small piece of plastic was cut to the size of the o-ring, cemented to its bottom with epoxy resin, and the entire electrode was sealed with epoxy. A channel into which the vessel sits was made by notching the o-ring with a hot soldering iron. The chamber was kept small such that when the vessel was placed into the chamber, it could be covered with 55 μl of DNA solution. The electrode was cleaned and sterilized with a 70% ethanol solution. This design satisfied all our criteria and gave uniform and reproducible results.
Figure 1. Cartoon of vascular electrode.
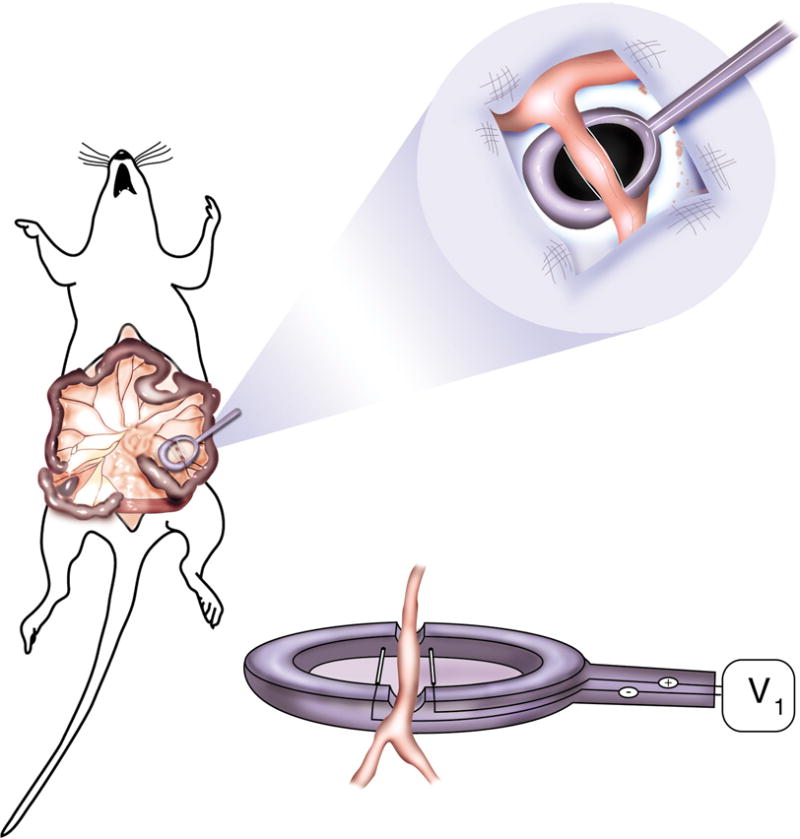
Nickel wires were set 3 mm apart from each other, flanking a channel cut into the top of the electrode in which the artery to be electroporated is placed. The total volume of the electrode’s chamber is 55 μl.
Animal surgery and electroporation
Male Sprague-Dawley rats (200–500 g, n = 25 rats) were anesthetized with isoflurane. A 5 cm midline incision was made and the small intestine was exposed to reveal the mesenteric arterial tree. A segment of distal small intestine and adjacent mesentery beginning at the ileocecal junction and extending proximally 7 – 10 cm was exteriorized and fanned out over sterile gauze pads. The mesentery surrounding segmental arteries supplying the small intestine was cut away from the neurovascular bundle. Individual vessels (200 to 400 μm diameter; 128 vessels in 25 animals) were placed into the electrode parallel to the conducting wires and 55 μl of the DNA solution was pipetted into the electrode, completely covering the vessel. Unless otherwise indicated, electroporations were carried out at a field strength of 200 V/cm, using eight 10 millisecond duration square wave pulses. Voltage pulses were delivered using a BTX ECM2001 electroporator (Genetronics, San Diego, CA). One minute after electroporation, the DNA solution was removed and used for the next vessel. Up to 10 vessels were electroporated in each animal. DNA transfer started with the distal most segmental branch of the superior mesenteric artery and proceeded proximally with respect to the ileocecal junction. Incisions were closed in layers with sutures and metal wound clips. Anesthetic was discontinued, the animals were allowed to recover and returned to the vivarium. Six hours to 5 days later, the rats were anesthetized, the abdomen reopened and the experimental vessels were ligated and removed. The animals were euthanized by thoracotomy and ventricular transection. Typically, 1 cm lengths of vessel were removed for analysis. All experiments were conducted in accordance with institutional guidelines in compliance with the recommendations of the Guide for Care and Use of Laboratory Animals. Animal protocols were approved by the University of South Alabama Institutional Animal Care and Use Committee.
Measurement of luciferase activity
Arterial segments were frozen in liquid nitrogen immediately after removal and extracts were prepared as described [17]. Briefly, the frozen vessels were placed onto 0.4 ml of frozen Promega lysis buffer in a 2.0 ml microfuge tube and the tissue was ground to a powder in a liquid nitrogen bath using a 5/16 inch drill bit run in reverse at a speed of 600 rpm. One hundred microliters of lysis buffer was added, the samples were thawed at room temperature and vortexed for 15 sec. The samples were refrozen in liquid nitrogen. Three freeze/thaw cycles were performed by alternating between liquid nitrogen and a room temperature water bath. Debris was removed by centrifugation. Luciferase activity was measured in duplicate using the Luciferase Assay System (Promega, Madison WI) in a Turner luminometer. Purified recombinant luciferase (Promega) was used to produce a standard curve for each experiment.
Histological analysis
In some experiments, vessels (25 vessels in 4 animals) were electroporated with a mixture of pCMV-Lux-DTS and pEGFP-N1 (2 mg/ml and 0.5 mg/ml, respectively). Upon removal from the animal, a 1 mm section of the electroporated vessel was excised from the rest of the vessel and embedded in OCT medium and frozen. Ten micron cryosections were cut, placed on polylysine-coated slides, and fixed in acetone for 30 seconds. GFP was localized directly by fluorescence microscopy using an Olympus BMAX-40 microscope and photographed with an Optronics CCD camera prior to staining with hemotoxylin and eosin.
Measurement of physiological responses to vasoconstricting stimuli
Rats were anesthetized with sodium pentobarbital (60mg/kg IP, supplemented as necessary) and placed on a surgical table where a tracheostomy was performed to ensure a clear airway. The abdomen was opened via a midline incision with a cauterizing iron to limit bleeding. The rat was then placed on his side on a Lucite stage where sections of the small intestine were drawn through the incision with moistened cotton-tipped applicators and placed within a groove in the stage to immobilize and orient the mesenteric arteries for viewing by intravital microscopy [18]. Throughout the procedure the mesentery was kept moist and warm by continuous addition of 37°C bicarbonate buffer, either lacking or containing phenylephrine. Arterial diameters were continuously monitored with a video caliper [18]. Mean arterial diameters were recorded during the 5th minute of drug administration.
RESULTS
Use of electroporation for gene transfer in the intact vasculature
Mesenteric arteries provided an ideal vascular tissue with which to perform these experiments. The ease of dissection and the ability to use anatomical location to identify the same vessels at a later date allowed up to 10 individual vessels to be treated in a given rat. Further, the design of the electrode and the rapid electroporation procedure allowed DNA to be transferred to up to 10 – 12 vessels within 30 minute time-frame. To follow gene expression, we chose two gene products, luciferase and GFP for quantitative and qualitative analysis, respectively. The fact that luciferase can be measured efficiently over a concentration range of 105 makes it an excellent choice for quantitative measurements. The stable expression of GFP coupled with the ability to detect directly as few as 300 molecules per cell with our microscope and CCD camera system make it useful for analyzing the distribution of gene expression in the tissues (unpublished observation).
When electric pulses similar to those used for muscle and liver (8 pulses of 10 millisecond duration at 200 V/cm;[4, 6–8]) were applied, significant luciferase activity was detected 2 days post-transfer (Figure 2 A). In contrast, when the vessels were incubated with the DNA solution without electroporation, no luciferase activity was seen 2 days post transfer. Slightly lower levels of luciferase expression were detected using a field strength of 100 V/cm when compared to the 200 V/cm field strength, but at 400 V/cm, gene expression was greatly diminished. Under all conditions, there appeared to be no trauma induced in the tissue, either at the time of electroporation or upon termination of the experiment. At the time of harvest vessels appeared healthy, as did the section of intestine that they were feeding. Further, at no time did there appear to be any bleeding, clotting, or intralumenal gas formed during or after the electroporation procedure. In cross section, the vessels appeared histologically normal. Because 200 V/cm worked sufficiently to induce luciferase expression, these settings were used throughout the remainder of the experiments.
Figure 2. Voltage-dependency and time course of gene transfer and expression.
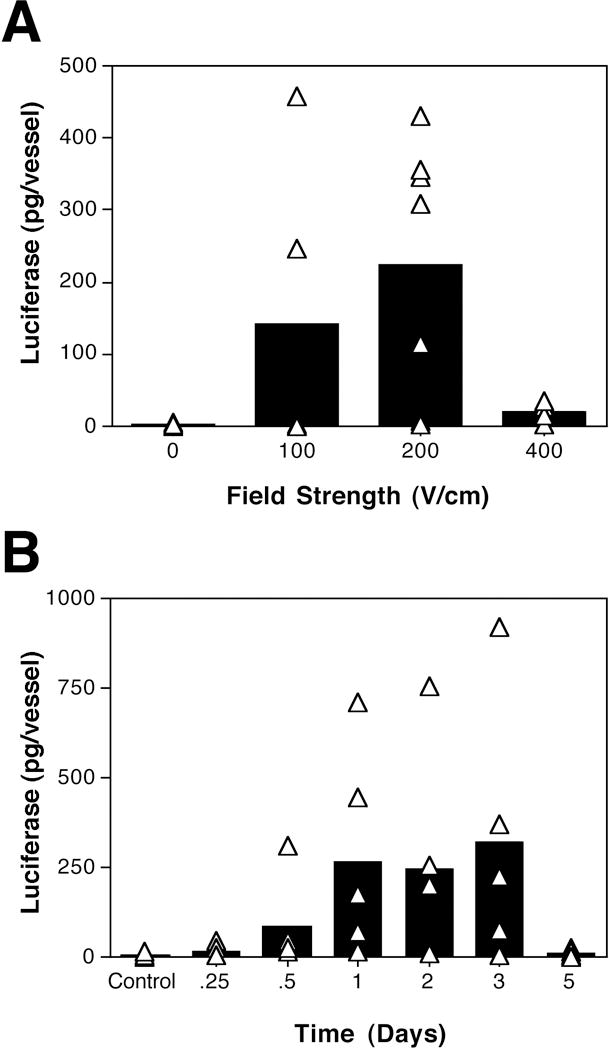
Rat mesenteric arteries were placed into the electrode and incubated with pCMV-Lux-DTS (2 mg/ml) for several seconds prior to electroporation. (A) Square wave electroporations were performed using a train of 8 pulses of 10 millisecond duration each at field strengths of 0 to 400 V/cm. The arteries were removed 2 days later and luciferase activity was measured as described in Materials and Methods. (B) Vessels were electroporated with plasmid pCMV-Lux-DTS (2 mg/ml; 200 V/cm, 8 pulses, 10 millisecond duration). Arteries were removed at the indicated times and luciferase activities were determined. The bars represent the average expression of all arteries and the triangles represent the individual arteries. Vessels receiving no DNA are shown as “control”.
To determine the duration of gene expression following electroporation, time course analyses were performed. Gene expression was detected very soon after electroporation, with luciferase activity appearing as early as 6 hours post-electroporation (Figure 2 B). Gene expression peaked at almost 1 ng of luciferase per vessel between days 1 and 3 post-transfer, and declined to levels just above background by day 5.
To ensure that this procedure was reproducible between multiple animals, the results from a series of experiments using vessels that were removed two days post-electroporation were compiled (Fig. 3). As can be seen, although there is variation of luciferase activities, the majority of vessels receiving DNA and electroporation displayed significant luciferase expression over background controls. Based on our results, electroporation proved to be a reproducible method for gene transfer to the vasculature, resulting in production of up to nanogram levels of gene product.
Figure 3. Luciferase expression in mesenteric arteries two days post-electroporation.
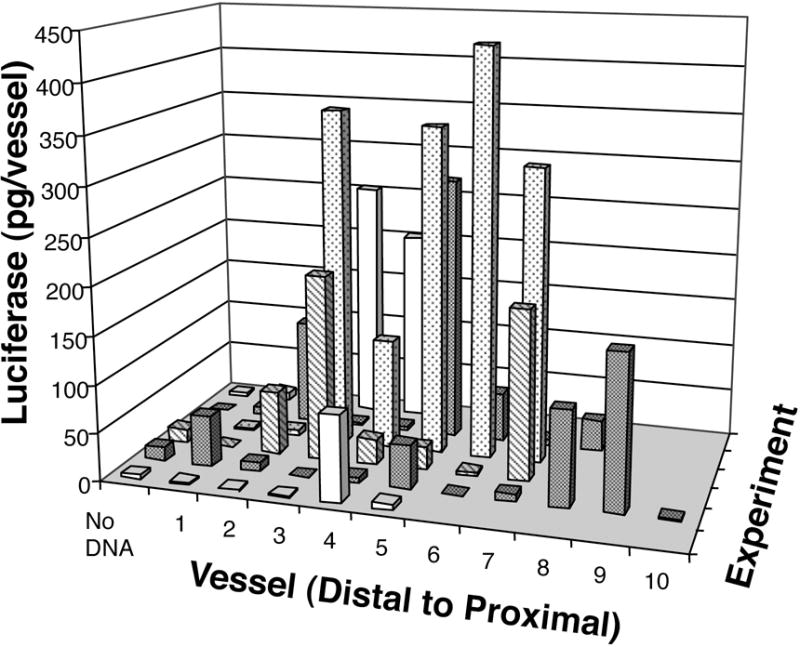
Electroporations of pCMV-Lux-DTS (2 mg/ml) were performed using the standard settings (200 V/cm, 8 pulses, 10 millisecond duration). Control values represent arteries that received no DNA. In all experiments, arteries were electroporated in order from distal to proximal with respect to the ileocecal junction. Two days post-electroporation, vessels were removed and luciferase activities were measured as described in Materials and Methods. Differing numbers of arteries were electroporated in each animal, depending on the anatomy of the animals. Results from independent experiments are shown along the y-axis.
Distribution of gene expression
To determine which cells in the vessel wall were expressing gene product, arteries were co-transfected with a mixture of luciferase-expressing and GFP-expressing plasmids. Upon removal from the animal, portions of each vessel were embedded and sectioned and the cell extract was made from the remainder of each vessel for quantitation of luciferase activity. Sections from those vessels expressing luciferase were analyzed for GFP expression with the assumption that arteries expressing one gene product would likely have received both plasmids and express both gene products. Figure 4A shows two representative cross sections of arteries that expressed luciferase. As can be seen, high levels of GFP fluorescence are detected in the tunica intima as well as in the tunica media. GFP was distributed throughout the vessel in adjacent sections. Figure 4C shows 11 serial sections of a representative artery that had been electroporated with pEGFP-N1 and pCMV-Lux-DTS. In this particular vessel, high levels of GFP expression were detected in over 30 serial sections. This suggests that the methods described in the present study are capable of producing uniform gene expression along the length of the blood vessel. In contrast, no GFP expression was detected in vessels that were bathed in DNA but not electroporated or in untreated vessels (Figure 4B).
Figure 4. Distribution of gene expression in electroporated arteries.

A mixture of pCMV-LUX-DTS and pEGFP (2mg/ml and 0.5 mg/ml, respectively) were transferred to mesenteric arteries with (A) or without (B) electroporation. After two days incubation, the arteries were removed and embedded for cryosectioning. Ten micron sections were prepared and GFP was visualized by fluorescence microscopy before the sections were stained with hemotoxylin and eosin. (C) GFP expression in successive 10 micron sections of an electroporated artery. All photographs were taken at an optical magnification of 25×, before computer analysis. Bars = 100 μm.
Physiological responses of electroporated vessels
Although the electroporated vessels appeared healthy upon visual inspection at the time of harvest, direct evidence was desired to demonstrate that they maintained unaltered vascular function after the procedure. Therefore, the responses of electroporated and control arteries to vasoconstricting stimuli were measured using intravital microscopy (Figure 5). Measurements were made on vessels harvested within the window of gene expression (day 2, n=3; Fig. 5A), or well after transgene expression had ceased (day 40, n=2; Fig. 5B). In both cases, control arteries constricted in a dose-dependent manner to increasing concentrations of phenylephrine. Similarly, the responses of the electroporated vessels were indistinguishable from those of the control vessels. Further, addition of either 0.1 mM adenosine or 50 μM isoproterenol to electroporated or control vessels constricted to closure resulted in nearly identical degrees of vessel dilation (not shown). These results demonstrate that vascular function, in terms of constriction and dilation, is unaltered by electroporation or the expression of the reporter genes in the vessels, for at least 40 days post-procedure.
Figure 5. Vasoconstriction of electroporated arteries.

Plasmid pCMV-LUX-DTS was transferred to rat mesenteric arteries by electroporation (8 pulses of 10 msec each; 200 V/cm). At 2 (A) or 40 (B) days post-transfer, the diameters of electroporated (filled bars) and untreated (“control”, open bars) vessels were determined by intravital microscopy using video calipers. Diameters are reported relative to baseline for the individual vessels, which ranged from 194 μm to 300 μm, n = 10 vessels. Average relative diameters ± standard deviations are shown for the vessels at each indicated concentration of phenylephrine. Vessels from 3 rats were used for measurements at day 2 and those from 2 rats were used for measurements at day 40.
DISCUSSION
In this study we have shown that delivery of naked DNA using electroporation is a simple and reproducible method of directing high level gene expression in the vasculature. Using our electrode design, we were able to transfer DNA to distinct regions of vessels of the mesenteric artery bundle with no apparent trauma or tissue damage. Maximal gene expression was obtained when 8 pulses of 10 millisecond duration each were applied using an electric field of around 200 V/cm. By contrast, essentially no gene expression was detected in the absence of electric pulses. The procedure is rapid and results in production of up to nanogram levels of gene product that are detectable as early as 6 to 12 hours and persist at high levels for 3 days. Further, expression of the transferred genes is seen in all cells of the arterial wall, including the endothelial cells, the medial smooth muscle layer, and the adventitial cells. Finally, electroporated arteries showed no differences from control vessels in terms of their ability to constrict or dilate to physiological stimuli up to 40 days after gene transfer. Thus, the present studies demonstrate that this technique can be used to study the short-term effects of exogenous gene expression in the vasculature of living animals and form the basis for further long-term studies of its efficacy. To our knowledge, this is the first time that this approach has been successfully applied to intact arteries without interruption of blood flow.
Within the past two years, electroporation has been used successfully to promote in vivo gene transfer and expression in a number of tissues, including skeletal muscle, liver, brain, and skin [4, 6–9, 11]. In all of these methods, the DNA is first injected into the tissue to allow for uniform distribution throughout the tissue and then electric pulses are applied to drive the DNA into the individual cells. It has been reported that up to 10% of the muscle fibers in the electroporated field take up and express plasmid [6, 7], a 10-fold increase over the amount that express in the absence of an electric field. By contrast, we saw no expression in the absence of an applied electric field but detected gene expression in up to 100% of cells in transverse sections of electroporated arteries. Thus, the electrode design chosen for this study is well suited to high level gene transfer in the vasculature. Further, the design of the electrode allowed us to bathe the vessels in a relatively dilute solution of DNA and to re-use the DNA solution with only loss of a few microliters per event. Because essentially no difference in the concentration of DNA was detected between successive uses of the same solution, it appears that very small amounts of DNA are being driven into the tissue. This is in contrast to the results seen in liver injections where the threshold for gene expression occurred with between 10 and 20 μg of DNA [9], and even to some extent in muscle where between 1.5 μg and 5 μg of plasmid are needed for expression of nanogram levels of luciferase per muscle [4].
The design of the electrode allowed us to transfer DNA to well defined regions of the vasculature with no contamination of the DNA or gene expression to regions of the vessel outside of the electrode and its induced electric field. Because of this, multiple transfers of different genes and vectors could in theory be transferred to distinct sites along the same vessel. Alternatively, untransformed segments of the same vessel could be used as controls in experiments. In terms of therapeutic applications for diseased vessels, such restricted gene transfer would prevent the applied gene from targeting to healthy tissue outside of the electrode’s chamber. Because of the fact that very little DNA appears to be transferred into the vessels during electroporation, it is unlikely that significant amounts of the DNA are reaching the bloodstream which could lead to gene transfer in distal sections of the vascular tree. Moreover, since the flow rate through the rat mesenteric arteries is on the order of 130 ml/min/100 g small intestine [19], any plasmid reaching the lumen would be quickly diluted in the blood supply and degraded over time, thereby minimizing any safety concerns.
Gene expression was detected as early as 6 to 12 hours after electroporation and reached maximal levels by 24 hours where it was maintained for three days. Gene expression then fell off rapidly to barely detectable levels by day 5. This pattern of expression is more transient than that found with adenovirus, which peaks between days 3 and 7 and falls off by day 14 [20, 21], and AAV, which peaks around day 10 and tapers off by day 30 in the vasculature [22]. Such transient expression has been observed repeatedly with plasmid vectors in vivo in a variety of tissues including the lung and cornea [23, 24]. This transient expression seen in the vasculature is in contrast to that seen in murine muscle using electroporation, where gene expression was detectable for out to 9 months [4]. However, this long term expression seen with plasmid in the muscle is more likely a function unique to skeletal muscle [25]. Thus, although electroporation of plasmid may not be a suitable approach when long-term vascular expression is desired, it is clearly the choice for short term expression desired very quickly after administration.
In these studies we have followed gene expression for up to one week post-administration. By this time, no trauma to the vessels or surrounding tissues was evident, and the treated arteries were patent and appeared to be as normal as surrounding untreated arteries. Moreover, treated vessels vasconstricted to phenylephrine in a dose-dependent manner, indistinguishable from control vessels at both 2 and 40 days post-treatment, suggesting the benign nature of the procedure. However, for this procedure to be considered for therapeutic use, the health and physiology of the treated vessels clearly must be followed for extended periods. The fact that no trauma or tissue damage has been found out to 6 to 9 months post-DNA injection and electroporation in murine muscle at the same filed strength supports our prediction that this procedure will not be hazardous to the vasculature [4, 5].
Whereas delivery of viral vectors via the lumen results largely in expression in the medial smooth muscle layer and occasionally in endothelial and adventitial cells, we found that when the DNA was added from the exterior of the vessel and electroporated into the vessel, gene expression was detected throughout all layers of the arterial wall. How the DNA is gaining access to the endothelial cells is unclear. It is well documented that electroporation induces pores on the order of 1 nm within cell membranes [2]. We would assume that all cells in the electroporated field are effected equally in this regard. However, little is known about the movement of DNA through the interstitial matrix. The fact that DNA introduced on the outside of the vessel wall gained access to the endothelial cells leads us to conclude that the interstitial matrix affords little restriction to DNA movement. Regardless of how the DNA is migrating into the interior of the vessel, its expression in all cell types at all areas within the vessel makes this technique highly attractive for delivery of DNA to endothelial as well as smooth muscle cells.
At present, we envision this technique to be highly suitable for two broad applications. First we believe that this approach will facilitate large-scale screening efforts to identify candidate genes for therapeutic use. With the advent of genomic informatics, large numbers of genes are being identified that must be assayed for biological effects. Electroporation offers a rapid screening tool to look at the immediate activities of these genes in vivo. Secondly, we believe that our approach is an ideal physiological tool for transferring genes into intact vasculature to examine biological pathways in the cell. For example, to study the role of a particular signaling pathway on a physiological process in the cell, such as smooth muscle contraction, the endogenous signaling pathway(s) can be knocked out by introduction of dominant negative mutant genes or enhanced by addition of constitutive mutants of a gene in the pathway. Although these approaches have been extensively used in cell culture systems, studies in intact tissue have been limited by the availability of rapid and uniform gene delivery systems. Data in the present study clearly shows that 1) effective gene delivery to intact blood vessels can be achieved via electroporation and 2) the vascular cells do express the gene product following electroporation. To this end, we believe that application of electroporation to the delivery of genes into the vascular cells represents a major advancement that sets the stage for functional studies with genetic rather that pharmacological manipulators of cell signaling pathways.
In summary, we have described a new approach for introduction of DNA into cells comprising the vessel wall. Our approach circumvents the need for vessel cannulation and occlusion as well as the use of viral vectors. We believe that the rapid expression of gene product, coupled with the ease, speed, and reproducibility of the procedure make this a strong addition to the areas of vascular gene therapy as well as a powerful tool for cell biologists and physiologists to dissect molecular mechanisms in an in vivo animal model.
Acknowledgments
We would like to thank Dr. Chris Abee of the Department of Comparative Medicine at the University of South Alabama for use of the BTX ECM2001 electroporator, Kathleen Blair-Parks, and Brenda Dean for technical assistance, and Matilde Tellaetxe-Issusi for the illustrations. This work was supported in part by grants HL59956 (DAD) and HL38639 (DAD and JNB) from the NIHLBI and DK51430 (JNB) from the NIDDK. We are also grateful to Dr. Aubrey Taylor and the Department of Physiology at the University of South Alabama for providing stipend support for JBM and the USA Summer Medical Student Research Program.
References
- 1.Parker SE, Vahlsing HL, Serfilippi LM, Franklin CL, Doh SG, Gromkowski SH, Lew D, Manthorpe M, Norman J. Cancer gene therapy using plasmid DNA: safety evaluation in rodents and non-human primates [see comments] Hum Gene Ther. 1995;6:575–590. doi: 10.1089/hum.1995.6.5-575. [DOI] [PubMed] [Google Scholar]
- 2.Mir LM, Banoun H, Paoletti C. Introduction of definite amounts of nonpermeant molecules into living cells after electropermeabilization: direct access to the cytosol. Exp Cell Res. 1988;175:15–25. doi: 10.1016/0014-4827(88)90251-0. [DOI] [PubMed] [Google Scholar]
- 3.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York: John Wiley & Sons; 1994. [Google Scholar]
- 4.Mir LM, Bureau MF, Gehl J, Rangara R, Rouy D, Caillaud J-M, Delaere P, Branellec D, Schwartz B, Scherman D. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc Natl Acad Sci USA. 1999;96:4262–4267. doi: 10.1073/pnas.96.8.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rizzuto G, Cappelletti M, Maione D, Savino R, Lazzaro D, Costa P, Mathiesen I, Cortese R, Ciliberto G, Laufer R, La Monica N, Fattori E. Efficient and regulated erythropoietin production by naked DNA injection and muscle electroporation. Proc Natl Acad Sci U S A. 1999;96:6417–6422. doi: 10.1073/pnas.96.11.6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathiesen I. Electropermeabilization of skeletal muscle enhances gene transfer in vivo. Gene Therapy. 1999;6:508–514. doi: 10.1038/sj.gt.3300847. [DOI] [PubMed] [Google Scholar]
- 7.Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998;16:867–870. doi: 10.1038/nbt0998-867. [DOI] [PubMed] [Google Scholar]
- 8.Heller R, Jaroszeski M, Atkin A, Moradpour D, Gilbert R, Wands J, Nicolau C. In vivo gene elctroinjection and expression in rat liver. FEBS Lett. 1996;389:225–228. doi: 10.1016/0014-5793(96)00590-x. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki T, Shin BC, Fujikura K, Matsuzaki T, Takata K. Direct gene transfer into rat liver cells by in vivo electroporation. FEBS Lett. 1998;425:436–440. doi: 10.1016/s0014-5793(98)00284-1. [DOI] [PubMed] [Google Scholar]
- 10.Oshima Y, Sakamoto T, Yamanaka I, Nishi T, Ishibashi T, Inomata H. Targeted gene transfer to corneal endothelium in vivo by electric pulse. Gene Ther. 1998;5:1347–1354. doi: 10.1038/sj.gt.3300725. [DOI] [PubMed] [Google Scholar]
- 11.Nishi T, Yoshizato K, Yamashiro S, Takeshima H, Sato K, Hamada K, Kitamura I, Yoshimura T, Saya H, Kuratsu J, Ushio Y. High-efficiency in vivo gene transfer using intraarterial plasmid DNA injection following in vivo electroporation. Cancer Res. 1996;56:1050–1055. [PubMed] [Google Scholar]
- 12.Harrison RL, Byrne BJ, Tung L. Electroporation-mediated gene transfer in cardiac tissue. FEBS Lett. 1998;435:1–5. doi: 10.1016/s0014-5793(98)00987-9. [DOI] [PubMed] [Google Scholar]
- 13.Muramatsu T, Nakamura A, Park H. In vivo electroporation: a powerful and convenient means of nonviral gene transfer to tissues of living animals (review) Int J Mol Med. 1998;1:55–62. doi: 10.3892/ijmm.1.1.55. [DOI] [PubMed] [Google Scholar]
- 14.Dev NB, Preminger TJ, Hofmann GA, Dev SB. Sustained local delivery of heparin to the rabbit arterial wall with an electroporation catheter. Cathet Cardiovasc Diagn. 1998;45:337–345. doi: 10.1002/(sici)1097-0304(199811)45:3<337::aid-ccd28>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 15.Liu MW, Roubin GS. Restenosis after coronary angioplasty: potential biologic determinants and role of intimal hyperplasia. Circulation. 1989;79:1374–1387. doi: 10.1161/01.cir.79.6.1374. [DOI] [PubMed] [Google Scholar]
- 16.Vacik J, Dean BS, Zimmer WE, Dean DA. Cell-specific nuclear import of plasmid DNA. Gene Therapy. 1999;6:1006–1014. doi: 10.1038/sj.gt.3300924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manthorpe M, Hartikka J, Vahlsing HL, Sawdey M. Quantification of plasmid DNA expression in vivo. In: Ferre F, editor. Gene Quantification. Burhaüser; Cambridge, MA: 1996. [Google Scholar]
- 18.Mesh CL, Joh T, Korthuis RJ, Granger DN, Benoit JN. Intestinal vascular sensitivity to vasopressin in portal hypertensive rats. Gastroenterology. 1991;100:916–921. doi: 10.1016/0016-5085(91)90264-l. [DOI] [PubMed] [Google Scholar]
- 19.Benoit JN, Womack WA, Korthuis RJ, Wilborn WH, Granger DN. Chronic portal hypertension: effects on gastrointestinal blood flow distribution. Am J Physiol. 1986;250:G535–539. doi: 10.1152/ajpgi.1986.250.4.G535. [DOI] [PubMed] [Google Scholar]
- 20.Schachtner SK, Rome JJ, Hoyt RF, Newman KD, Virmani R, Dichek DA. In vivo Adenovirus-mediated gene transfer via the pulmonary artery of rats. Circ Res. 1995;76:701–709. doi: 10.1161/01.res.76.5.701. [DOI] [PubMed] [Google Scholar]
- 21.Lee SW, Trapnell BC, Rade JJ, Virmani R, Dichek DA. In vivo adenoviral vector mediated gene transfer into balloon-injured rat carotid arteries. Circ Res. 1993;73:797–807. doi: 10.1161/01.res.73.5.797. [DOI] [PubMed] [Google Scholar]
- 22.Rolling F, Nong Z, Pisvin S, Collen D. Adeno-associated virus-mediated gene transfer into rat carotid arteries. Gene Ther. 1997;4:757–761. doi: 10.1038/sj.gt.3300465. [DOI] [PubMed] [Google Scholar]
- 23.Yew NS, Wysokenski DM, Wang KX, Ziegler RJ, Marshall J, McNeilly D, Cherry M, Osburn W, Cheng SH. Optimization of plasmid vectors for high-level expression in lung epithelial cells. Hum Gene Ther. 1997;8:575–584. doi: 10.1089/hum.1997.8.5-575. [DOI] [PubMed] [Google Scholar]
- 24.Daheshia M, Kuklin N, Kanangat S, Manickan E, Rouse BT. Suppression of ongoing ocular inflammatory disease by topical administration of plasmid DNA encoding IL-10. J Immunol. 1997;159:1945–1952. [PubMed] [Google Scholar]
- 25.Wolff JA, Ludtke JJ, Acsadi G, Williams P, Jani A. Long-term persistence of plasmid DNA and foreign gene expression in mouse muscle. Hum Mol Genetics. 1992;1:363–369. doi: 10.1093/hmg/1.6.363. [DOI] [PubMed] [Google Scholar]