

Abstract
Motivation
Genome-wide association (GWA) studies have reported susceptible regions in the human genome for many common diseases and traits, however, these loci only explain a minority of trait heritability. To boost the power of a GWA study, substantial research endeavors have been focused on integrating other available genomic information in the analysis. Advances in high through-put technologies have generated a wealth of genomic data, and made combining SNP and gene expression data become feasible.
Results
In this paper we propose a novel procedure to incorporate gene expression information into GWA analysis. This procedure utilizes weights constructed by gene expression measurements to adjust p values from a GWA analysis. Results from simulation analyses indicate that the proposed procedures may achieve substantial power gains while controlling family-wise type I error rate (FWER) at the nominal level. To demonstrate the implementation of our proposed approach, we apply the weight adjustment procedure to a GWA study for serum interferon-regulated chemokine levels in systemic lupus erythematosus (SLE) patients. The study results can provide valuable insights for the functional interpretation of GWA signals.
Availability
The R source code for implementing the proposed weighting procedure is available at http://www.biostat.umn.edu/~yho/research.html
Keywords: p value weighting, family-wise error rate, statistical power, integrative genomic analysis, SLE
1 Introduction
Over the past few years, genome-wide association (GWA) studies have been successful in localizing and identifying genetic regions that are related to common human diseases [1]. But studies have shown that the amount of genetic variation explained by GWA findings for any given disease is often significantly less than the estimated heritability of the disease [2]. One possible reason for the missing heritability is that GWA studies are under powered to detect genetic variants that possess small effects. Most common human diseases or traits have complex inheritance patterns with multiple underlying genes with small to moderate effects. Therefore, it requires a relatively large sample size for a GWA study to detect signals with such small effects. In order to boost statistical power of a GWA study, an important direction of recent research is to integrate available genomic information such as gene expression, single nucleotide polymorphism (SNP), copy number variation, transcription regulation, methylation, and protein abundance together in the analysis [3, 4]. With the advances of high through-put technologies, integrating gene expression with SNP information has drawn much attention in the past decade [5, 6, 7, 8, 9, 10, 11]. A recent study incorporating gene expression information into gene association mapping in mice showed assuring results in identifying functional networks that explain phenotypic alterations [12].
Genovese et al. [13] introduced the idea of utilizing prior knowledge to weight p values from a GWA study. Li et al. [14] adapted the p value weighting idea and proposed using gene expression information to formally derive weights and to apply the derived weights to p adjust the p values from a GWA study. They specified weights where peQTL is the p value for association between a SNP marker and gene expression (these SNP markers sometimes were referred to as eQTLs). They demonstrated power gain when incorporating information of association between SNP markers and gene expression profiles in their study. However, their approach did not utilize the information of association between gene expression profiles and phenotypic outcome of interest.
In this paper, we propose approaches that utilize both (SNP, gene expression) as well as (gene expression, phenotype) associations and are expected to achieve greater power gains than that of Li et al. [14] under some situations. A study-wise threshold for the weight adjusted p values can then be used to determine genome-wide significance. In addition to the expected power gain, the weights calculated based on gene expression can provide useful information for prioritizing SNPs for further functional validation experiments.
To assess the performance of our proposed approach, we conducted simulation analyses under various scenarios to evaluate the family-wise type-I error rate (FWER) and statistical power. We also compared the performance of our proposed weighting approaches to that of Li et al. [14] in the simulation analysis.
We apply the proposed approach to a study related to lupus activity in Systemic lupus erythematosus (SLE) patients. Through experimental data analysis, our primary focus is to demonstrate the implementation of the proposed weight adjustment approach. SLE is a chronic, inflammatory autoimmune disease that can cause damage to organs and tissues throughout the body. The exact cause of SLE is unknown, but a combination of genetic and environmental factors are thought to trigger the disease. More than 45 genetic variants are known to be associated with SLE, and over half of these can be linked to the type I interferon (IFN) pathway [15]. The type I IFNs are a family of antiviral cytokines that are implicated in the pathogenesis of lupus, and IFN-inducible transcripts and proteins are candidate biomarkers for this disease [16, 17, 18, 19]. We previously evaluated a panel of 3 IFN-inducible serum chemokines (IP-10, MCP-1, MIP-3b) as predictors of lupus flare [20]. Identification of genetic variants associated with elevated chemokine levels could improve the treatment of SLE patients and may assist in identifying additional disease susceptibility loci.
The structure of the paper is as follows: we describe the study population and the weighting approach in detail in Section 2, the simulation analysis in Section 3. The implementation of the proposed approach in the SLE GWA study is described in Section 4, with a discussion and a final conclusion in Section 5.
2 Research Design and Methods
Phenotype and Study Population
In this study, our primary outcome of interest is lupus activity in SLE patients. Three interferon (IFN) regulated serum chemokines: CXCL10 (IP-10), CCL2 (MCP-1) and CCL19 (MIP-3β) were measured using SearchLight (Pierce, Woburn, MA) chemiluminescence sandwich-based immunoassays. A single normalized composite chemokine score on a 100-points scale was calculated using the three chemokine measurements as described in [20]. The chemokine score could serve as a biomarker for lupus activity and predict future disease flares in patient cohort [20].
The data we used was collected from 309 SLE patients with consent from the Hopkins Lupus Cohort [21] enrolled through Autoimmune Biomarkers Collaborative Network (ABCoN) described in Bauer et al. [20]. All patients were Caucasian and the majority were females. They received treatments for lupus during the study period including hydroxychloroquine, cyclophosphamide, mycophenolate mofetil, azathioprine, methotrexate, chlorambucil, and oral prednisone.
SNP Genotyping and Gene Expression Profiling
Whole genome genotyping for 555,352 SNP markers were carried out using Illumina 550K SNP array version 1 (HJ550v1; Illumina Inc., San Diego, CA, USA). Gene expression of 24,849 genes were measured using Illumina Human-6 Expression BeadChip; data files were analyzed with Illumina’s GenomeStudio gene expression module to report quantile-normalized, background-corrected gene expression signal levels.
Method
In this study, we propose using weights [22, 13, 23] computed based on gene expression measurements to adjust p values from GWA analysis. Roeder et al. [24] suggested to use a weight (wi > 0) to adjust a p value (pi) and reject a null hypothesis Hi if the adjusted p value is smaller than the Bonferroni corrected threshold , where α is the significance level and m is the total number of hypotheses. Hence the set of rejected hypotheses (Hi) is defined as:
In Roeder et al. [25], they provided theoretical proof that the rejection set R described above controls FWER at level α, as long as wi > 0 and .
Based on their theoretical results, we utilize the p value weighting approach to gain power in detecting signal while controlling FWER at the nominal level. After weight adjustment, SNP loci that have strong contributions to phenotype-associated gene expression will have smaller p values. The weighting mechanism is as follows. We assign a weight, to each SNP marker i such that , and the average of weights ( ) is 1. The weight for a given SNP (Li) is a product of two parts: wLiEj and wEjP. wLiEj indicates the effect of SNP locus Li on the j-th gene expression measurement, Ej. The second term wEjP describes whether gene expression measurement (Ej) is associated with the phenotypic outcome (P).
To ensure correct control of FWER, we use the model below to calculate wLiEj:
| (1) |
and . The estimate from the above regression model is equivalent to that of βr in the following model:
where r1 and r2 are residuals from the following two models respectively:
In regression model (1), the reason for adjusting for P is to remove the effect of P to prevent the correlation between the weight wLiEj and the GWA p value. In an extreme situation, for example, consider SNP marker Li is not associated with Ej while Ej and P are highly correlated. The p value for βLiEj in the model Ej = β0+βLiEj Li+ε will be highly correlated with the GWA p value for β in the GWA model P = β0+β Li+ε, due to the high correlation between Ej and P. Hence, after adjusting for the effect of P in model (1), the p value for βLiEj will be less likely to be significant, yielding a derived weight not highly correlated with the GWA p value.
Similarly, we adjust for Li when calculating weight wEjP using the model: P = β0 + βEjPEj + γLi + ε, and assign . Then the product of the two weights is used to describe the effect of SNP locus Li on P through Ej. The useful benefit of taking the product of two weights is that if either wLiEj or wEjP is zero, then the resulting product will be zero. On the other hand, if both wLiEj and wEjP are reasonably large, then taking the product of the two parts will result in an amplified overall weight. A crude weight for SNP locus Li is determined by the maximum of the products among all gene expression measurements:
Finally, we divide crude weights (wMP) by their average ( ) so that as required by Roeder et al. [25]:
With the , adjusted p value for the ith SNP can then be calculated as:
In the weighting approach described above, we assume that the effect of SNP genotype on gene expression is linear. More generally, we may code two dummy variables to indicate Li = 1 versus 0, and Li = 2 versus 0 respectively, then the χ2 statistic derived from the likelihood ratio test can be used in replacement of the squared t statistic. Furthermore, in the case-control study design, a similar approach can be applied by deriving the weights through logistic regression.
Because the weights are constructed as products of two parts, to ensure both the associations between Li & Ej and also Ej & P are substantial, we propose a trimming method for wLiEj and wEjP as follows. We set wLiEj × wEjP = 0 if or . In the analysis below, we set . Then can be calculated as described above.
When only a subset of individuals have gene expression profiles available in a dataset, the p values can be derived from the full dataset, and weights can be calculated from the partial data set. Then the same weight adjusting procedure can be implemented as described above. The R source code for implementing the proposed weighting procedure is available at http://www.biostat.umn.edu/~yho/research.html.
3 Simulation
To mimic data from a GWA study, we constructed simulated data using the marginal distribution parameters of the genotype scores of the SNP markers, the gene expression measurements and the phenotype obtained from the SLE study described in the previous section. We selected one SNP marker to be the true underlying SNP (SNPtrue) and simulated a single gene expression level according to this model: E1 = β0E + βE × SNPtrue + ε, . In the described model, β0E and were obtained from the mean and the variance of a randomly selected gene expression. The value of the phenotypic outcome was simulated based on the model: . The β0P, and σP were determined by the mean and variance of the chemokine score in the SLE dataset. We used standardized E1 in the above equation for the ease of interpreting βP.
To investigate family-wise error control, we randomly selected another 9 non-phenotype-associated SNP markers and 1,000 gene expression measurement from the experimental data in the SLE study. Hence, the simulated dataset consisted of 10 SNP markers and 1,001 gene expression measures, and one continuous phenotypic outcome.
Scenario 1
In the first simulation, we assumed βE = βP = 0 to examine the type-I error rate and the results are presented in Table 1. In Table 1, we recorded the fraction of times that a SNP was declared significant at for the four approaches: the conventional GWA analysis, the weight adjusted approach with ( ) and without (WMP) trimming and Li’s approach [14]. The overall FWERs from the 10,000 simulations were estimated by counting the percent of times when any significant results were reported. In Table 1, we observed the estimated FWERs were close to 0.05 for all four approaches. In addition, we observed similar results when assuming βE = 10, and βP = 0.
Table 1.
FWER of the conventional GWA analysis and three weight adjusting approaches in scenario 1 and 2. Results are from 10,000 simulation iterations. WMP: Weight adjustment approach without trimming. : Weight adjustment approach with trimming.
| GWA | WMP |
|
Li | ||
|---|---|---|---|---|---|
| Scenario 1 | 0.053 | 0.055 | 0.053 | 0.051 | |
| Scenario 2 | 0.046 | 0.048 | 0.047 | 0.078 | |
| Scenario 3 | 0.051 | 0.051 | 0.051 | 0.059 |
Scenario 2
A second simulation was conducted by assuming βE = 0 and βP = 10. The results are shown in Table 1. According to the simulation results shown in Table 1, the FWER remained controlled at the 0.05 level for GWA, WMP, and .
Scenario 3
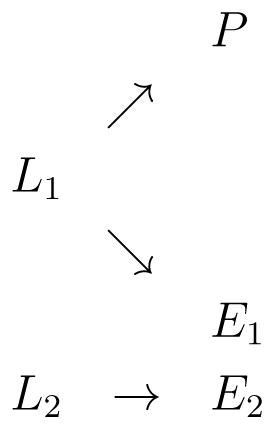
In this simulation scenario, the underlying SNP marker L was not associated with outcome P and gene expression E. However, two unobserved latent variables (F1, and F2) were in association with the observed L, E and P as as illustrated in Figure 1. This latent model can account population stratification, or admixture. In this simulation scenario, we are interested in examine whether the latent variables could create a spurious dependency for the proposed analyses. According to the result shown in Table 1, the FWER remained controlled at the 0.05 level for all four approaches in this scenario.
Figure 1.

A diagram illustrates simulation Scenario 3.
Scenario 4
To assess the power of the proposed weighting approaches, we conducted simulations assuming and the results are shown in Table 2. The result suggested substantial power gain by the weighting approaches when compared to the conventional GWA analysis. By incorporating gene expression information, in the moderate effect size setting ( ), power increased by 56% (from 42.5% to 66.1%) when was compared to the standard GWA analysis.
Table 2.
Power of the conventional GWA analysis and three weight adjusting approaches in scenario 3, 4, and 5. Results are from 10,000 simulation iterations. WMP: Weight adjustment approach without trimming. : Weight adjustment approach with trimming.
| GWA | WMP |
|
Li | ||
|---|---|---|---|---|---|
| Scenario 4 | 0.425 | 0.653 | 0.661 | 0.602 | |
| Scenario 5 | 0.421 | 0.617 | 0.640 | 0.184 | |
| Scenario 6 | 0.419 | 0.594 | 0.601 | 0.474 | |
| Scenario 7 | 0.810 | 0.736 | 0.418 | 0.736 | |
| Scenario 8 | 0.795 | 0.814 | 0.433 | 0.861 |
Furthermore, the results also indicated that Li’s approach had less power gain than our proposed weight approaches due to the fact that the former’s weight does not incorporate information of association between gene expression and phenotype.
Scenario 5
In this scenario, we assumed ; in addition, we assumed another SNP maker (SNPadd) had an effect on a gene expression with effect size , but this gene expression was not associated with phenotype as illustrated by the diagram below.
The results from this simulation scenario are shown in Table 2. In Table 2, WMP and demonstrated considerable power gains over the conventional GWA. However, Li’s weighting approach suffered dramatic power loss compared to the GWA approach in this scenario. Li’s weighting approach only accounted for the association between SNP and gene expression, which resulted in incorrectly assigning higher weights to SNPadd.
Scenario 6
In this scenario, we assumed ; only a subset of the individuals had expression measurements available but more samples had SNP genotype and phenotype information. In the simulation, we assumed that only 1/3 of individuals had expression profiles available. In this setting, the proposed weighting approach still demonstrated considerable power gain (with power increased from 41.9% to 60.1%) compared to the conventional GWA, although the power gain was less prominent than that of scenario 4 when the expression profiles for the whole study population were available.
Scenario 7
This scenario assumes a reactive model [26] as illustrated by the diagram below. We assumed a SNP marker L had an effect on outcome P, and the alteration of gene expression E was the result of change in outcome P. We assumed in the simulation. The result shown in Table 2 indicates that all three weight adjustment approaches (WMP, , Li’s approach) assigned lower weights for SNP markers with gene expression in reactive model compared to the original GWS p value. In the proposed weighting approaches, the model constructed for calculating wLE (equation 1) adjusted for outcome P. As a result, wLE was less likely to be large and more likely be reduced to 0 in the trimming approach. Hence we observed that assigned the lowest weights for the reactive model.
Scenario 8
This scenario was modified from Scenario 5, where an independence model was assumed as illustrated in the diagram below. A SNP marker L1 had an effect on outcome P and also had an effect on gene expression E1. But P and E1 were independent. We assumed and in the simulation.
The results from this simulation scenario shown in Table 2 indicates that assigned lowest weights for SNP markers that arose from independence model due to small wEP values. On the contrary, according to the simulation result, Li’s approach undesirably assigned larger weights for SNP markers that were in the independence model.
In summary, the proposed weighting procedures control FWER at the nominal level when L is not associated with E or E is not associated with P or both. Our proposed methods aim to find genes that undergo L → E → P mechanism, where gene expression is in the middle of the pathway. Hence our methods exhibit largest power gain in identifying genes described in Scenario 4.
4 Data Analysis Results
4.1 Results
In the SLE dataset described in Section 2, the obtained chemokine score is considered a biomarker for lupus activity for SLE patients. To demonstrate the implementation of the proposed approach, we applied the chemokine score as the primary outcome and performed weight adjustment analysis for SNP loci with allele frequency > 0.1. The simulation results suggest that the trimming approach could allocate more weights on relevant SNP loci. Therefore, we applied with trimming in the following analysis.
The 13 SNPs with GWA p value < 10−5 are listed in Table 3. In addition to the GWA p values indicated in the third column of Table 3, weights and weight adjusted p values are also presented. In the last two columns of Table 3, we recorded the annotation of SNP markers, and the gene expression that reported the maximum weight for the SNP.
Table 3.
SNP loci with p value < 10−5 according to the GWA analysis or the weight adjustment procedure. chr: chromosome. Adjusted p value is calculated as .
| SNP | chr | GWA p value |
|
Li | Adjusted p value | SNP Annotation | Expression | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 | rs17415112 | 10 | 1.20e-07 | 1.28 | 1.10 | 9.37e-08 | EXOSC1 | LOC441019 | |
| 2 | rs7744944 | 6 | 1.48e-07 | 1.38 | 1.37 | 1.07e-07 | SP140 | ||
| 3 | rs6536732 | 4 | 5.57e-07 | 0.98 | 1.54 | 5.69e-07 | RMI2 | ||
| 4 | rs17218497 | 4 | 9.89e-07 | 1.04 | 1.06 | 9.53e-07 | CARD17 | ||
| 5 | rs931481 | 4 | 2.96e-06 | 0.71 | 1.11 | 4.18e-06 | |||
| 6 | rs4892151 | 18 | 4.30e-06 | 0.74 | 1.03 | 5.79e-06 | CARD17 | ||
| 7 | rs767528 | 20 | 4.34e-06 | 1.89 | 1.01 | 2.29e-06 | IFI35 | ||
| 8 | rs12778761 | 10 | 4.49e-06 | 0.58 | 1.01 | 7.79e-06 | ARHGAP19-SLIT1 | TNFSF10 | |
| 9 | rs11247300 | 15 | 5.53e-06 | 0.51 | 1.44 | 1.07e-05 | PCSK6 | LGALS9 | |
| 10 | rs306254 | 5 | 5.83e-06 | 0.68 | 0.90 | 8.54e-06 | SP140 | ||
| 11 | rs2810693 | 13 | 6.90e-06 | 0.63 | 1.17 | 1.10e-05 | GBP5 | ||
| 12 | rs173577 | 5 | 6.99e-06 | 0.57 | 0.92 | 1.23e-05 | RNASE2 | ||
| 13 | rs4919096 | 10 | 8.09e-06 | 0.66 | 0.80 | 1.23e-05 | TIMM10 | ||
| 14 | rs596346 | 11 | 1.22e-05 | 1.54 | 1.30 | 7.95e-06 | BC041900 | FAM212B | |
| 15 | rs624676 | 11 | 1.40e-05 | 1.46 | 1.39 | 9.55e-06 | BC041900 | FAM212B | |
| 16 | rs514604 | 11 | 1.40e-05 | 1.46 | 1.39 | 9.55e-06 | BC041900 | FAM212B |
The weights obtained from the proposed and Li’s approach have similar trend for 10 out of 16 in the top SNP markers shown in this table. Six SNP markers (rs2810693, rs11247300, rs931481, rs6536732, rs4892151, rs122778761) were down-weighted ( ) in our approach while having w > 1 using Li’s approach. One possible explanation is that these SNP markers might undergo the independence model.
The top SNP hit (rs17415112) is located within the EXOSC1 gene. EXOSC1 encodes a core component of the exosome and may be involved in Ig class switch recombination and degradation of mRNA transcripts for histone proteins, which are implicated in SLE [27]. Our results suggest a possible interaction of EXOSC1 and LOC441019, a locus related to immune system, cytokine and interferon gamma signaling. In addition, several transcripts reported in Table 3 are involved in the immune response, inflammation, and cytokine pathways that are known to be associated with active SLE [28]. These include the inflammatory caspase-related CARD17 [29], the interferon-inducible IFI35, the apoptosis-inducing ligand TNFSF10 [30], and the galectin LGALS9 [31].
Furthermore, according to Table 3 we noticed that there were several SNP markers at the top of the list having lowered p values after the weight adjustment. Using the threshold of 10−5, three additional SNPs (rs596346, rs624676 and rs514604) became significant. These three SNP markers are in linkage disequilibrium and mapped to a noncoding mRNA transcript BC041900. This region is about 500K upstream of gene clusters on chromosome 11 where FAM181B, PRCP, SNORA70E, C11orf82, LOC100506233, AK311356, ANKRD42, RAB30 locate.
To investigate SNP loci with moderate p values but large weights, we ranked SNP loci with unadjusted p value < 10−3 and . The top 40 ranked loci are listed in Table 4. The complete list of SNP loci with unadjusted p value < 10−3 and is available in Supplementary Data. Interestingly, in some cases multiple SNPs on different chromosomes appear to interact with a single transcript. Among these transcripts are BST2, PARP12, SP140, TIMM10, UBQLNL, and XAF1. This suggests that distinct genetic variants may lead to altered expression of a single gene and provide divergent ways to trigger elevated chemokine levels.
Table 4.
Top 40 ranked SNP loci with unadjusted p value < 10−3 and . chr: chromosome. Adjusted p value is calculated as .
| SNP | chr | GWA p value |
|
Li | Adjusted p value | GWA Rank | SNP Annotation | Expression | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs6495528 | 15 | 9.74e-04 | 6.24 | 0.94 | 1.56e-04 | 580 | KIAA1199 | XAF1 | |
| 2 | rs2869580 | 17 | 4.50e-04 | 4.12 | 1.58 | 1.09e-04 | 285 | RGS9 | UBQLNL | |
| 3 | rs12374087 | 3 | 7.05e-04 | 3.55 | 0.96 | 1.99e-04 | 421 | TIMM10 | ||
| 4 | rs12681426 | 8 | 4.66e-04 | 3.30 | 1.00 | 1.41e-04 | 299 | PARP12 | ||
| 5 | rs13100138 | 3 | 9.93e-04 | 3.05 | 1.02 | 3.26e-04 | 596 | TIMM10 | ||
| 6 | rs2222255 | 3 | 8.15e-04 | 3.05 | 0.96 | 2.67e-04 | 483 | BST2 | ||
| 7 | rs2086369 | 8 | 9.50e-04 | 3.01 | 1.38 | 3.16e-04 | 566 | NKAIN3 | BATF2 | |
| 8 | rs1021012 | 8 | 8.81e-04 | 2.97 | 1.36 | 2.97e-04 | 527 | NKAIN3 | BATF2 | |
| 9 | rs7015211 | 8 | 8.81e-04 | 2.97 | 1.36 | 2.97e-04 | 527 | NKAIN3 | BATF2 | |
| 10 | rs4721478 | 7 | 8.41e-04 | 2.86 | 0.77 | 2.94e-04 | 506 | ISPD | MDK | |
| 11 | rs4809388 | 20 | 4.94e-04 | 2.83 | 1.60 | 1.74e-04 | 313 | PRPF6 | SP140 | |
| 12 | rs2071570 | 17 | 2.61e-04 | 2.83 | 0.96 | 9.22e-05 | 178 | NR1D1 | PARP12 | |
| 13 | rs433074 | 21 | 4.12e-04 | 2.78 | 1.33 | 1.48e-04 | 264 | ISG15 | ||
| 14 | rs12497437 | 3 | 5.36e-04 | 2.68 | 1.01 | 2.00e-04 | 328 | PSME2 | ||
| 15 | rs478420 | 8 | 1.24e-04 | 2.30 | 1.25 | 5.38e-05 | 89 | PCM1 | SP140 | |
| 16 | rs4737609 | 8 | 9.86e-04 | 2.28 | 1.19 | 4.33e-04 | 591 | NKAIN3 | UBQLNL | |
| 17 | rs12986829 | 2 | 6.12e-05 | 2.18 | 1.52 | 2.81e-05 | 56 | TIMM10 | ||
| 18 | rs1400030 | 2 | 1.21e-04 | 2.18 | 1.47 | 5.55e-05 | 84 | TIMM10 | ||
| 19 | rs12536527 | 7 | 9.26e-04 | 2.13 | 1.19 | 4.34e-04 | 556 | SDK1 | GBP5 | |
| 20 | rs13426554 | 2 | 3.45e-05 | 2.12 | 1.39 | 1.63e-05 | 38 | DDX58 | ||
| 21 | rs2345102 | 2 | 3.45e-05 | 2.12 | 1.39 | 1.63e-05 | 38 | DDX58 | ||
| 22 | rs11203933 | 8 | 2.73e-04 | 2.10 | 0.95 | 1.30e-04 | 187 | SP140 | ||
| 23 | rs7912066 | 10 | 6.61e-04 | 2.02 | 0.86 | 3.27e-04 | 397 | CTNNA3 | TIMM10 | |
| 24 | rs4402422 | 13 | 2.19e-04 | 2.02 | 1.21 | 1.09e-04 | 149 | UBQLNL | ||
| 25 | rs12329145 | 2 | 1.01e-04 | 2.02 | 1.41 | 4.99e-05 | 76 | TIMM10 | ||
| 26 | rs1041530 | 10 | 1.24e-04 | 1.99 | 1.21 | 6.21e-05 | 88 | SP140 | ||
| 27 | rs4739003 | 8 | 2.54e-04 | 1.99 | 1.48 | 1.28e-04 | 173 | NKAIN3 | UBQLNL | |
| 28 | rs7003189 | 8 | 3.79e-04 | 1.96 | 1.43 | 1.93e-04 | 245 | NKAIN3 | UBQLNL | |
| 29 | rs2239540 | 9 | 2.92e-04 | 1.94 | 1.12 | 1.50e-04 | 200 | ZBTB6 | SAMD9L | |
| 30 | rs938716 | 4 | 5.66e-05 | 1.90 | 0.77 | 2.97e-05 | 50 | SP140 | ||
| 31 | rs1530433 | 17 | 1.93e-04 | 1.90 | 1.39 | 1.01e-04 | 133 | DNAH17 | FLJ11286 | |
| 32 | rs11226922 | 11 | 7.26e-04 | 1.89 | 1.28 | 3.83e-04 | 429 | HERC6 | ||
| 33 | rs767528 | 20 | 4.34e-06 | 1.89 | 1.01 | 2.29e-06 | 7 | IFI35 | ||
| 34 | rs2127559 | 8 | 7.20e-04 | 1.89 | 1.47 | 3.81e-04 | 426 | NKAIN3 | UBQLNL | |
| 35 | rs12710667 | 2 | 6.26e-04 | 1.88 | 0.79 | 3.34e-04 | 378 | PARP12 | ||
| 36 | rs988667 | 11 | 2.47e-04 | 1.87 | 1.18 | 1.32e-04 | 164 | DKK3 | LOC400759 | |
| 37 | rs2161210 | 19 | 6.77e-04 | 1.86 | 1.23 | 3.65e-04 | 404 | MT2A | ||
| 38 | rs647619 | 1 | 8.13e-04 | 1.84 | 1.35 | 4.42e-04 | 481 | TNFSF10 | ||
| 39 | rs761523 | 14 | 3.86e-04 | 1.84 | 0.94 | 2.11e-04 | 250 | BST2 | ||
| 40 | rs12882238 | 14 | 5.86e-05 | 1.82 | 0.97 | 3.22e-05 | 52 | XAF1 |
In Table 4, there are 11 SNP markers were up-weighted ( ) using our approach but down-weighted by Li’s approach. These differences in weights could be due to the mechanism described in simulation scenario 2.
4.2 Functional Annotation
Many of the transcripts identified in our study are known to be regulated by type I IFN [16]. Furthermore, several of the transcript abundance are also known to be altered in blood cells of patients with SLE compared to healthy controls [16]. Among those transcript abundance that are both altered in SLE and IFN-inducible are IFI35, TNFSF10, XAF1, PARP12, BST2, ISG15, DDX58, HERC6, and MT2A. Only a few transcripts in our results: SP140, PSME2, and LGALS9 are IFN-inducible but were not observed to be changed in our studies of SLE patients.
While the current findings require validation to confirm the association of these variants to gene expression and serum chemokine levels, several of the identified loci harbor genes with known functions related to the immune system. These include DKK3, which plays a role in peripheral CD8 T-cell tolerance [32]; the transcription factor NR1D1, which regulates the production of inflammatory cytokines [33]; RGS9, which may have a role in chemokine-induced lymphocyte migration [34]; and SDK1, which is associated with combined variable immunodeficiency [35]. In addition, several expressed genes listed in the ninth column of Table 4, that interact with the SNP variants described above, participate in cellular apoptosis such as: XAF1, UBQLNL, TIMM10, BST2, HERC6, ISG15, PSME2 [36, 37].
5 Discussion
Through experimental data analysis, our primary focus is to demonstrate the implementation of the proposed weight adjustment approach. In the analysis of SLE GWA data, we identified three SNP loci with large weights that became significant after weight adjustment as shown in Table 3. Several genes listed in Tables 3 and 4 have pivotal roles in immune functions. Our results identified several genetic interactions among immune responses and cellular apoptosis pathways and seemed to suggest the importance of their interactions in active SLE symptoms. These SNP loci and corresponding associated gene expression provided valuable information for further functional evaluation. Replication of these findings in other cohorts is necessary to demonstrate the biological significance of the additional loci identified by our method.
Yet we also observed SNP loci with small p values that showed no evidence of gene expression association when analyzing SLE GWA data. This could be due to that phenotype contribution mechanisms were not measured in the current study, such as through unmeasured gene expression transcripts, DNA structures, or through other mechanisms. These SNPs will not be favored in the proposed weighting method since they will be down-weighted due to lack of evidence of association with gene expression. These unmeasured mechanisms might explain the moderate differences in p values we observed in the SLE data analysis results described in Section 4. Hence in practice, we suggest to pursue both (1) SNPs with tiny GWA p values without weighting, (2) SNPs with small weight adjusted-p values. The proposed weighting approach assists researchers to prioritize GWA SNP findings by integrating available genomic information.
In this paper, we demonstrated our method in a paired gene expression and GWA study data from the same cohort. To date, relatively few published studies have utilized gene expression data from the same patients studied by GWA study [38, 39, 40, 41, 42]. However, as appreciation grows for the power of eQTL analysis, efforts such as the Genotype-Tissue Expression project will come to fruition [43], and methods such as ours will facilitate the analysis of such datasets, we believe the collection of RNA for expression profiling from GWA study participants will be useful.
In addition, if eQTL (SNP, gene expression) data, and (gene expression, phenotype) data are available from two different sets of cohorts, instead of paired gene expression and GWA study data from the same cohort, our proposed approaches can be easily modified and applied.
Our proposal provides a formalized procedure to incorporate additional genomic information into GWA analyses. In additional to gene expression measurements as demonstrated in this paper, with the advent of a wealth of genetic data generated through high-throughput technologies, the proposed method is extendable to integrate other sources of information such as DNA methylation status, transcription regulation, and protein abundance.
The weighted hypothesis testing concept appeared to be first introduced by Holm [22], and since then theoretical developments have been advanced to form the basis for p value weighting in order to increase power while controlling FWER in a multiple hypothesis testing setting. Roeder et al. [25] provided an applicable theory for constructing weights which control FWER at the nominal level. Roeder et al. [24] applied p value weighting procedures to GWA analysis and demonstrated power gain compared to conventional analysis.
In the spirit of integrating genomic information from multiple sources for power gain, we proposed novel weighting procedures based on the theory by Roeder et al. [25] to incorporate gene expression into GWA analyses. Our simulation results confirmed that the proposed weighting procedure dramatically improved the statistical power of GWA studies while controlling FWER at the nominal level, when gene expression is in the middle of the etiological pathway. Under this mechanism, our methods demonstrated greater power gain compared to that of Li et al. [14]. It also provides ways to draw valuable information from massive data to assist functional interpretations of GWA signals.
Acknowledgments
Y.-Y. Ho is supported by NIH grant P30 CA77598, P50CA101955, UL1TR000114 andU54-MD008620. W. Pan is supported by NIH grants R01GM081535, R01HL65462 and R01HL105397.
Funding: The data collection are supported by the Alliance for Lupus Research, and the Arthritis Foundation.
Footnotes
Conflict of Interest: Ward Ortmann, Timothy W. Behrens, Robert R. Graham, Tushar R. Bhangale are employees of Genentech Inc.
References
- 1.Hindorff Lucia A, Sethupathy Praveen, Junkins Heather A, Ramos Erin M, Mehta Jayashri P, Collins Francis S, Manolio Teri A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009 Jun;106(23):9362–9367. doi: 10.1073/pnas.0903103106. URL http://dx.doi.org/10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manolio Teri A, Collins Francis S, Cox Nancy J, Goldstein David B, Hindorff Lucia A, Hunter David J, McCarthy Mark I, Ramos Erin M, Cardon Lon R, Chakravarti Aravinda, Cho Judy H, Guttmacher Alan E, Kong Augustine, Kruglyak Leonid, Mardis Elaine, Rotimi Charles N, Slatkin Montgomery, Valle David, Whittemore Alice S, Boehnke Michael, Clark Andrew G, Eichler Evan E, Gibson Greg, Haines Jonathan L, Mackay Trudy FC, McCarroll Steven A, Visscher Peter M. Finding the missing heritability of complex diseases. Nature. 2009 Oct;461(7265):747–753. doi: 10.1038/nature08494. URL http://dx.doi.org/10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conlon Erin M, Liu X Shirley, Lieb Jason D, Liu Jun S. Integrating regulatory motif discovery and genome-wide expression analysis. Proc Natl Acad Sci U S A. 2003 Mar;100 (6):3339–3344. doi: 10.1073/pnas.0630591100. URL http://dx.doi.org/10.1073/pnas.0630591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Zuowei, Sahin Orhan, Shen Zhangqi, Liu Peng, Miller William G, Zhang Qijing. Multi-omics approaches to deciphering a hypervirulent strain of campylobacter jejuni. Genome Biol Evol. 2013 Nov; doi: 10.1093/gbe/evt172. URL http://dx.doi.org/10.1093/gbe/evt172. [DOI] [PMC free article] [PubMed]
- 5.Nicolae Dan L, Gamazon Eric, Zhang Wei, Duan Shiwei, Dolan M Eileen, Cox Nancy J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010 Apr;6(4):e1000888. doi: 10.1371/journal.pgen.1000888. URL http://dx.doi.org/10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Shaoyu, Lu Qing, Cui Yuehua. A systems biology approach for identifying novel pathway regulators in eQTL mapping. J Biopharm Stat. 2010 Mar;20(2):373–400. doi: 10.1080/10543400903572803. URL http://dx.doi.org/10.1080/10543400903572803. [DOI] [PubMed] [Google Scholar]
- 7.Li Shaoyu, Williams Barry L, Cui Yuehua. A combined p-value approach to infer pathway regulations in eQTL mapping. Statistics and Its Interface. 2011;4:389–402. [Google Scholar]
- 8.Cusanovich Darren A, Billstrand Christine, Zhou Xiang, Chavarria Claudia, De Leon Sherryl, Michelini Katelyn, Pai Athma A, Ober Carole, Gilad Yoav. The combination of a genome-wide association study of lymphocyte count and analysis of gene expression data reveals novel asthma candidate genes. Hum Mol Genet. 2012 May;21(9):2111–2123. doi: 10.1093/hmg/dds021. URL http://dx.doi.org/10.1093/hmg/dds021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Can, Wang Lin, Zhang Shuqin, Zhao Hongyu. Accounting for non-genetic factors by low-rank representation and sparse regression for eQTL mapping. Bioinformatics. 2013 Apr;29(8):1026–1034. doi: 10.1093/bioinformatics/btt075. URL http://dx.doi.org/10.1093/bioinformatics/btt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kendziorski CM, Chen M, Yuan M, Lan H, Attie AD. Statistical methods for expression quantitative trait loci (eQTL) mapping. Biometrics. 2006 Mar;62(1):19–27. doi: 10.1111/j.1541-0420.2005.00437.x. URL http://dx.doi.org/10.1111/j.1541-0420.2005.00437.x. [DOI] [PubMed] [Google Scholar]
- 11.Sun Wei. A statistical framework for eQTL mapping using RNA-seq data. Biometrics. 2012 Mar;68(1):1–11. doi: 10.1111/j.1541-0420.2011.01654.x. URL http://dx.doi.org/10.1111/j.1541-0420.2011.01654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Yanqing, Zhu Jun, Lum Pek Yee, Yang Xia, Pinto Shirly, MacNeil Douglas J, Zhang Chunsheng, Lamb John, Edwards Stephen, Sieberts Solveig K, Leonardson Amy, Castellini Lawrence W, Wang Susanna, Champy Marie-France, Zhang Bin, Emilsson Valur, Doss Sudheer, Ghazalpour Anatole, Horvath Steve, Drake Thomas A, Lusis Aldons J, Schadt Eric E. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008 Mar;452(7186):429–435. doi: 10.1038/nature06757. URL http://dx.doi.org/10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genovese Christopher R, Roeder Kathryn, Wasserman Larry. False discovery control with p-value weighting. Biometrika. 2006;93(3):509–524. URL http://EconPapers.repec.org/RePEc:oup:biomet:v:93:y:2006:i:3:p:509-524. [Google Scholar]
- 14.Li Lin, Kabesch Michael, Bouzigon Emmanuelle, Demenais Florence, Farrall Martin, Moffatt Miriam F, Lin Xihong, Liang Liming. Using eQTL weights to improve power for genome-wide association studies: a genetic study of childhood asthma. Front Genet. 2013;4:103. doi: 10.3389/fgene.2013.00103. URL http://dx.doi.org/10.3389/fgene.2013.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bronson Paola G, Chaivorapol Christina, Ortmann Ward, Behrens Timothy W, Graham Robert R. The genetics of type I interferon in systemic lupus erythematosus. Curr Opin Immunol. 2012 Oct;24(5):530–537. doi: 10.1016/j.coi.2012.07.008. URL http://dx.doi.org/10.1016/j.coi.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Baechler Emily C, Batliwalla Franak M, Karypis George, Gaffney Patrick M, Ortmann Ward A, Espe Karl J, Shark Katherine B, Grande William J, Hughes Karis M, Kapur Vivek, Gregersen Peter K, Behrens Timothy W. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003 Mar;100(5):2610–2615. doi: 10.1073/pnas.0337679100. URL http://dx.doi.org/10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett Lynda, Palucka A Karolina, Arce Edsel, Cantrell Victoria, Borvak Josef, Banchereau Jacques, Pascual Virginia. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003 Mar;197(6):711–723. doi: 10.1084/jem.20021553. URL http://dx.doi.org/10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crow Mary K, Kirou Kyriakos A, Wohlgemuth Jay. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003 Dec;36(8):481–490. doi: 10.1080/08916930310001625952. [DOI] [PubMed] [Google Scholar]
- 19.Bauer Jason W, Baechler Emily C, Petri Michelle, Batliwalla Franak M, Crawford Dianna, Ortmann Ward A, Espe Karl J, Li Wentian, Patel Dhavalkumar D, Gregersen Peter K, Behrens Timothy W. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006 Dec;3(12):e491. doi: 10.1371/journal.pmed.0030491. URL http://dx.doi.org/10.1371/journal.pmed.0030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bauer Jason W, Petri Michelle, Batliwalla Franak M, Koeuth Thearith, Wilson Joseph, Slattery Catherine, Panoskaltsis-Mortari Angela, Gregersen Peter K, Behrens Timothy W, Baechler Emily C. Interferon-regulated chemokines as biomarkers of systemic lupus erythematosus disease activity: a validation study. Arthritis Rheum. 2009 Oct;60(10):3098–3107. doi: 10.1002/art.24803. URL http://dx.doi.org/10.1002/art.24803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petri M. Hopkins lupus cohort. 1999 update. Rheum Dis Clin North Am. 2000 May;26(2):199–213. v. doi: 10.1016/s0889-857x(05)70135-6. [DOI] [PubMed] [Google Scholar]
- 22.Holm S. A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics. 1979;6:65–70. [Google Scholar]
- 23.Roeder Kathryn, Devlin B, Wasserman Larry. Improving power in genome-wide association studies: weights tip the scale. Genet Epidemiol. 2007 Nov;31(7):741–747. doi: 10.1002/gepi.20237. URL http://dx.doi.org/10.1002/gepi.20237. [DOI] [PubMed] [Google Scholar]
- 24.Roeder Kathryn, Bacanu Silvi-Alin, Wasserman Larry, Devlin B. Using linkage genome scans to improve power of association in genome scans. Am J Hum Genet. 2006 Feb;78(2): 243–252. doi: 10.1086/500026. URL http://dx.doi.org/10.1086/500026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roeder Kathryn, Wasserman Larry. Genome-wide significance levels and weighted hypothesis testing. Stat Sci. 2009 Nov;24(4):398–413. doi: 10.1214/09-STS289. URL http://dx.doi.org/10.1214/09-STS289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schadt Eric E, Lamb John, Yang Xia, Zhu Jun, Edwards Steve, Guhathakurta Debraj, Sieberts Solveig K, Monks Stephanie, Reitman Marc, Zhang Chunsheng, Lum Pek Yee, Leonardson Amy, Thieringer Rolf, Metzger Joseph M, Yang Liming, Castle John, Zhu Haoyuan, Kash Shera F, Drake Thomas A, Sachs Alan, Lusis Aldons J. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005 Jul;37(7):710–717. doi: 10.1038/ng1589. URL http://dx.doi.org/10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fishbein E, Alarcon-Segovia D, Vega JM. Antibodies to histones in systemic lupus erythematosus. Clin Exp Immunol. 1979 Apr;36(1):145–150. [PMC free article] [PubMed] [Google Scholar]
- 28.Rahman Anisur, Isenberg David A. Systemic lupus erythematosus. N Engl J Med. 2008 Feb;358(9):929–939. doi: 10.1056/NEJMra071297. URL http://dx.doi.org/10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- 29.Lamkanfi Mohamed, Denecker Geertrui, Kalai Michael, D’hondt Kathleen, Meeus Ann, Declercq Wim, Saelens Xavier, Vandenabeele Peter. INCA, a novel human caspase recruitment domain protein that inhibits interleukin-1β generation. J Biol Chem. 2004 Dec;279 (50):51729–51738. doi: 10.1074/jbc.M407891200. URL http://dx.doi.org/10.1074/jbc.M407891200. [DOI] [PubMed] [Google Scholar]
- 30.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996 May;271(22):12687–12690. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 31.Zhu Chen, Anderson Ana C, Schubart Anna, Xiong Huabao, Imitola Jaime, Khoury Samia J, Zheng Xin Xiao, Strom Terry B, Kuchroo Vijay K. The tim-3 ligand galectin-9 negatively regulates t helper type 1 immunity. Nat Immunol. 2005 Dec;6(12):1245–1252. doi: 10.1038/ni1271. URL http://dx.doi.org/10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 32.Papatriantafyllou Maria, Moldenhauer Gerhard, Ludwig Julia, Tafuri Anna, Garbi Natalio, Hollmann Gorana, Küblbeck Günter, Klevenz Alexandra, Schmitt Sabine, Pougialis Georg, Niehrs Christof, Gröne Hermann-Josef, Hämmerling Gunter J, Arnold Bernd, Oelert Thilo. Dickkopf-3, an immune modulator in peripheral cd8 t-cell tolerance. Proc Natl Acad Sci U S A. 2012 Jan;109(5):1631–1636. doi: 10.1073/pnas.1115980109. URL http://dx.doi.org/10.1073/pnas.1115980109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibbs Julie E, Blaikley John, Beesley Stephen, Matthews Laura, Simpson Karen D, Boyce Susan H, Farrow Stuart N, Else Kathryn J, Singh Dave, Ray David W, Loudon Andrew SI. The nuclear receptor rev-erbα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc Natl Acad Sci U S A. 2012 Jan;109(2):582–587. doi: 10.1073/pnas.1106750109. URL http://dx.doi.org/10.1073/pnas.1106750109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agenès Fabien, Bosco Nabil, Mascarell Laurent, Fritah Sabrina, Ceredig Rod. Differential expression of regulator of G-protein signalling transcripts and in vivo migration of cd4+ naïve and regulatory t cells. Immunology. 2005 Jun;115(2):179–188. doi: 10.1111/j.1365-2567.2005.02146.x. URL http://dx.doi.org/10.1111/j.1365-2567.2005.02146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keller Michael D, Jyonouchi Soma. Chipping away at a mountain: Genomic studies in common variable immunodeficiency. Autoimmun Rev. 2012 Nov; doi: 10.1016/j.autrev.2012.10.017. URL http://dx.doi.org/10.1016/j.autrev.2012.10.017. [DOI] [PMC free article] [PubMed]
- 36.Straszewski-Chavez Shawn L, Visintin Irene P, Karassina Natasha, Los Georgyi, Liston Peter, Halaban Ruth, Fadiel Ahmed, Mor Gil. XAF1 mediates tumor necrosis factor-α-induced apoptosis and X-linked inhibitor of apoptosis cleavage by acting through the mitochondrial pathway. J Biol Chem. 2007 Apr;282(17):13059–13072. doi: 10.1074/jbc.M609038200. URL http://dx.doi.org/10.1074/jbc.M609038200. [DOI] [PubMed] [Google Scholar]
- 37.Sijts EJAM, Kloetzel PM. The role of the proteasome in the generation of mhc class I ligands and immune responses. Cell Mol Life Sci. 2011 May;68(9):1491–1502. doi: 10.1007/s00018-011-0657-y. URL http://dx.doi.org/10.1007/s00018-011-0657-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schadt Eric E, Molony Cliona, Chudin Eugene, Hao Ke, Yang Xia, Lum Pek Y, Kasarskis Andrew, Zhang Bin, Wang Susanna, Suver Christine, Zhu Jun, Millstein Joshua, Sieberts Solveig, Lamb John, GuhaThakurta Debraj, Derry Jonathan, Storey John D, Avila-Campillo Iliana, Kruger Mark J, Johnson Jason M, Rohl Carol A, van Nas Atila, Mehrabian Margarete, Drake Thomas A, Lusis Aldons J, Smith Ryan C, Guengerich F Peter, Strom Stephen C, Schuetz Erin, Rushmore Thomas H, Ulrich Roger. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008 May;6(5):e107. doi: 10.1371/journal.pbio.0060107. URL http://dx.doi.org/10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emilsson Valur, Thorleifsson Gudmar, Zhang Bin, Leonardson Amy S, Zink Florian, Zhu Jun, Carlson Sonia, Helgason Agnar, Walters G Bragi, Gunnarsdottir Steinunn, Mouy Magali, Steinthorsdottir Valgerdur, Eiriksdottir Gudrun H, Bjornsdottir Gyda, Reynisdottir Inga, Gudbjartsson Daniel, Helgadottir Anna, Jonasdottir Aslaug, Jonasdottir Adalbjorg, Styrkarsdottir Unnur, Gretarsdottir Solveig, Magnusson Kristinn P, Stefansson Hreinn, Fossdal Ragnheidur, Kristjansson Kristleifur, Gislason Hjortur G, Stefansson Tryggvi, Leifsson Bjorn G, Thorsteinsdottir Unnur, Lamb John R, Gulcher Jeffrey R, Reitman Marc L, Kong Augustine, Schadt Eric E, Stefansson Kari. Genetics of gene expression and its effect on disease. Nature. 2008 Mar;452(7186): 423–428. doi: 10.1038/nature06758. URL http://dx.doi.org/10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 40.Borel Christelle, Deutsch Samuel, Letourneau Audrey, Migliavacca Eugenia, Montgomery Stephen B, Dimas Antigone S, Vejnar Charles E, Attar Homa, Gagnebin Maryline, Gehrig Corinne, Falconnet Emilie, Dupré Yann, Dermitzakis Emmanouil T, Antonarakis Stylianos E. Identification of cis- and trans-regulatory variation modulating microrna expression levels in human fibroblasts. Genome Res. 2011 Jan;21(1):68–73. doi: 10.1101/gr.109371.110. URL http://dx.doi.org/10.1101/gr.109371.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hao Ke, Bossé Yohan, Nickle David C, Paré Peter D, Postma Dirkje S, Laviolette Michel, Sandford Andrew, Hackett Tillie L, Daley Denise, Hogg James C, Elliott W Mark, Couture Christian, Lamontagne Maxime, Brandsma Corry-Anke, van den Berge Maarten, Koppelman Gerard, Reicin Alise S, Nicholson Donald W, Malkov Vladislav, Derry Jonathan M, Suver Christine, Tsou Jeffrey A, Kulkarni Amit, Zhang Chunsheng, Vessey Rupert, Opiteck Greg J, Curtis Sean P, Timens Wim, Sin Don D. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet. 2012;8(11):e1003029. doi: 10.1371/journal.pgen.1003029. URL http://dx.doi.org/10.1371/journal.pgen.1003029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almlöf Jonas Carlsson, Lundmark Per, Lundmark Anders, Ge Bing, Maouche Seraya, Göring Harald HH, Liljedahl Ulrika, Enström Camilla, Brocheton Jessy, Proust Carole, Godefroy Tiphaine, Sambrook Jennifer G, Jolley Jennifer, Crisp-Hihn Abigail, Foad Nicola, Lloyd-Jones Heather, Stephens Jonathan, Gwilliam Rhian, Rice Catherine M, Hengstenberg Christian, Samani Nilesh J, Erdmann Jeanette, Schunkert Heribert, Pastinen Tomi, Deloukas Panos, Goodall Alison H, Ouwehand Willem H, Cambien François, Syvänen Ann-Christine. Powerful identification of Cis-regulatory SNPs in human primary monocytes using allele-specific gene expression. PLoS One. 2012;7(12):e52260. doi: 10.1371/journal.pone.0052260. URL http://dx.doi.org/10.1371/journal.pone.0052260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lonsdale John, Thomas Jeffrey, Salvatore Mike, Phillips Rebecca, Lo Edmund, Shad Saboor, Hasz Richard, Walters Gary, Garcia Fernando, Young Nancy, Foster Barbara, Moser Mike, Karasik Ellen, Gillard Bryan, Ramsey Kimberley, Sullivan Susan, Bridge Jason, Magazine Harold, Syron John, Fleming Johnelle, Siminoff Laura, Traino Heather, Mosavel Maghboeba, Barker Laura, Jewell Scott, Rohrer Dan, Maxim Dan, Filkins Dana, Harbach Philip, Cortadillo Eddie, Berghuis Bree, Turner Lisa, Hudson Eric, Feenstra Kristin, Sobin Leslie, Robb James, Branton Phillip, Korzeniewski Greg, Shive Charles, Tabor David, Qi Liqun, Groch Kevin, Nampally Sreenath, Buia Steve, Zimmerman Angela, Smith Anna, Burges Robin, Robinson Karna, Valentino Kim, Bradbury Deborah, Cosentino Mark, Diaz-Mayoral Norma, Kennedy Mary, Engel Theresa, Williams Penelope, Erickson Kenyon, Ardlie Kristin, Winckler Wendy, Getz Gad, DeLuca David, MacArthur Daniel, Kellis Manolis, Thomson Alexander, Young Taylor, Gelfand Ellen, Donovan Molly, Meng Yan, Grant George, Mash Deborah, Marcus Yvonne, Basile Margaret, Liu Jun, Zhu Jun, Tu Zhidong, Cox Nancy J, Nicolae Dan L, Gamazon Eric R, Im Hae K, Konkashbaev Anuar, Pritchard Jonathan, Stevens Matthew, Flutre Timothee, Wen Xiaoquan, Dermitzakis Emmanouil T, Lappalainen Tuuli, Guigo Roderic, Monlong Jean, Sammeth Michael, Koller Daphne, Battle Alexis, Mostafavi Sara, McCarthy Mark, Rivas Manual, Maller Julian, Rusyn Ivan, Nobel Andrew, Wright Fred, Shabalin Andrey, Feolo Mike, Sharopova Nataliya, Sturcke Anne, Paschal Justin, Anderson James M, Wilder Elizabeth L, Derr Leslie K, Green Eric D, Struewing Jeffery P, Temple Gary, Volpi Simona, Boyer Joy T, Thomson Elizabeth J, Guyer Mark S, Ng Cathy, Abdallah Assya, Colantuoni Deborah, Insel Thomas R, Koester Susan E, Roger Little A, Bender Patrick K, Lehner Thomas, Yao Yin, Compton Carolyn C, Vaught Jimmie B, Sawyer Sherilyn, Lockhart Nicole C, Demchok Joanne, Moore Helen F. The Genotype-Tissue Expression (GTEx) project. Nature Genetics. 2013 May;45(6):580–585. doi: 10.1038/ng.2653. URL http://dx.doi.org/10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]