

In the simplest terms, diabetes mellitus results when pancreatic beta cells are unable to maintain adequate insulin secretion to prevent hyperglycemia. A combination of genetic and environmental factors causes the underlying beta-cell failure. In type 1 diabetes, a T-cell–mediated autoimmune response against beta cells appears to be the main disease mechanism, whereas insulin resistance is the key metabolic abnormality in type 2 diabetes. Yet the way in which insulin resistance triggers beta-cell failure remains obscure. The report by Larsen et al. in this issue of the Journal (pages 1517– 1526) provides insight into a shared mechanism of beta-cell dysfunction in type 1 diabetes and type 2 diabetes and suggests a new therapeutic approach to type 2 diabetes. The authors report that anakinra, a receptor antagonist of the inflammatory cytokine interleukin-1, which has long been implicated in beta-cell damage in type 1 diabetes, improved blood glucose control and insulin secretion in patients with type 2 diabetes. This agent also reduced markers of systemic inflammation.
For many years, attempts to distinguish different types of diabetes have been associated with changing definitions that reflect the state of our knowledge. Terms have changed from juvenile and adult diabetes to insulin-dependent and non-insulin-dependent diabetes and finally to type 1 and type 2 diabetes. In addition, there are several intermediate forms, such as latent autoimmune diabetes in adults, or “type 1.5.” The genetic determinants of the two main forms of diabetes are distinct. Type 1 diabetes is strongly associated with certain HLA haplotypes, a link that supports the notion that this condition is primarily an autoimmune disease. In type 2 diabetes, examples of candidate genes are KCNJ11 (potassium inwardly rectifying channel, subfamily J, member 11), which encodes the islet ATP-sensitive potassium channel Kir6.2, and TCF7L2 (transcription factor 7–like 2), which regulates proglucagon gene expression and thus the production of glucagon-like peptide 1.
It has become clear that the effectors of beta-cell failure are similar in the two main types of diabetes, regardless of the inciting event (see illustration). Glucotoxicity and lipotoxicity induce oxidative stress and up-regulate inflammatory cytokines, thereby leading to cellular damage and promoting apoptosis in all beta cells, regardless of the type of diabetes. Recently, abnormal islet innervation has been described as another common contributing element in animal models of type 1 and type 2 diabetes. Thus, alleviating beta-cell stress opens the door to therapeutic approaches that would probably be useful in all types of diabetes.
An example is treatment with anakinra, a recombinant version of the naturally occurring human interleukin-1–receptor antagonist that blocks the effects of interleukin-1α and β.1 Both forms of interleukin-1, which are produced by many cells (e.g., lymphocytes, endothelial cells, adipocytes, and beta cells), play important roles in host defense but also can induce fever, anorexia, hypotension, cartilage destruction, and beta-cell apoptosis. Treatment with interleukin-1–receptor antagonist was approved by the Food and Drug Administration for rheumatoid arthritis in 2001. The drug has also been shown to alleviate symptoms in neonatal-onset multisystem inflammatory disease. Generally, few side effects have been observed, but one death from cardiac causes was recently reported in a patient with Still's disease (systemic-onset juvenile rheumatoid arthritis) shortly after the initiation of anakinra therapy2.
The value of the observations reported by Larsen et al. goes beyond the specific role of the interleukin-receptor antagonist. An important consideration is that although the role of “smoldering” inflammation in the pathogenesis of insulin resistance is well established, its relevance to mechanisms of beta-cell dysfunction and death is poorly understood.3 In this respect, the study by Larsen et al. raises an interesting point. The authors report a modest improvement in insulin secretion, along with a sharp drop in the levels of C-reactive protein and interleukin-6. Although the authors do not favor this interpretation, the response to anakinra might also be attributable to mildly improved insulin sensitivity resulting from reduced systemic inflammation. Such a conclusion is consistent with the demonstration that certain cytokines impair insulin signaling and supports the concept that treating the underlying inflammation improves diabetes control, which is probably an aspect of the beneficial effects of statins and glitazones.
Anakinra is not alone in bridging the divide between disease mechanisms and appropriate treatments for the different types of diabetes. For example, some oral agents that are administered to improve insulin sensitivity in type 2 diabetes are also antiapoptotic (metformin) or have immunemodulatory effects (glitazones). Thus, these or similar drugs may find a place in the treatment of type 1 diabetes. There is growing interest in developing therapies that not only improve beta-cell function in the short term (controlling postprandial hyperglycemia, for example) but also preserve beta-cell mass or promote beta-cell regeneration. Two recently approved drugs (exenatide and sitagliptin) not only are insulinotropic but also promote beta-cell proliferation in rodents. It remains to be seen whether these effects occur in humans with diabetes as well. The basis of these new therapeutic approaches is the identification of various molecular mechanisms of beta-cell failure and the recognition that beta cells have an innate ability to replicate, albeit very slowly4.
A note of caution, however: anakinra treatment led to a modest improvement in the control of glycemia, with a maximal effect at 4 weeks but with an upward trend in the glycated hemoglobin level at 13 weeks. Since the patients varied with respect to baseline blood-glucose control and type of concomitant treatment, it is difficult to deduce the effectiveness of anakinra therapy in various stages of disease. Moreover, none of the currently available medications (including the aforementioned oral agents) are successful as long-term monotherapy, and none of them have effectively halted the continuous decline in beta-cell mass. Although the findings regarding anakinra break new ground by demonstrating that inhibition of cytokine function can restore insulin secretion, the ultimate goal is the improvement of blood-glucose control without the need for triple- and quadruple-combination therapies, as well as the prevention of beta-cell death.
In the first century B.C., Publilius Syrus declared, “Better use medicines at the outset than at the last moment.” His recommendation may serve as a motto for our continued search for an optimal remedy for diabetes.
Development of Diabetes.
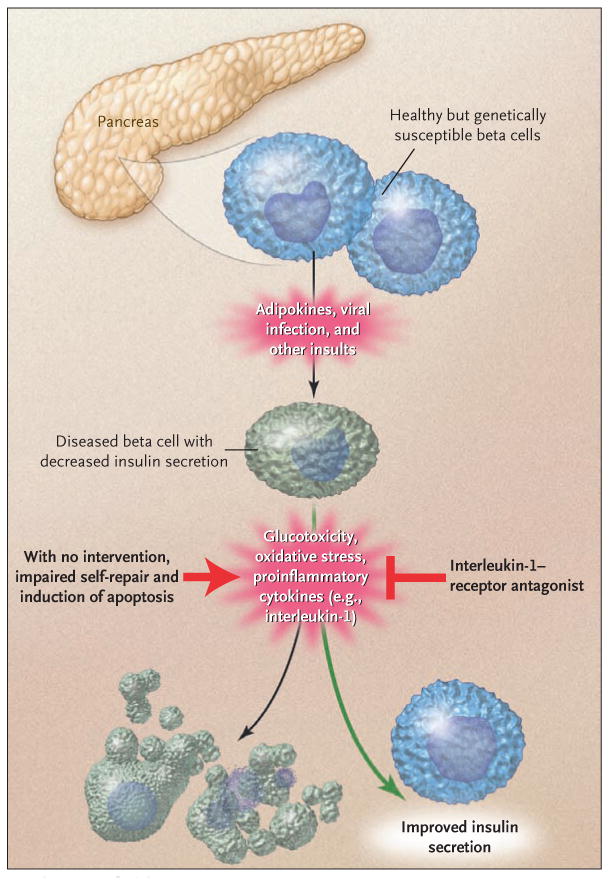
In the upper part of the diagram, a beta cell receives a primary insult. It is not known whether this insult is identical in type 1 diabetes and type 2 diabetes but leads to different responses because of variations in genetic susceptibility. The currently accepted view is that an immune-mediated insult leads to the primary beta-cell dysfunction in type 1 diabetes, whereas a different, unknown insult causes type 2 diabetes. Once injured, beta cells have a similar fate in the two types of diabetes. Glucotoxicity, oxidative stress, and cytotoxic cytokines lead to further damage, which eventually results in beta-cell death if the process is not countered by effective self-repair and therapeutic interventions.
References
- 1.Perrier S, Darakhshan F, Hajduch E. IL-1 receptor antagonist in metabolic diseases: Dr Jekyll or Mr Hyde? FEBS Lett. 2006;580:6289–94. doi: 10.1016/j.febslet.2006.10.061. [DOI] [PubMed] [Google Scholar]
- 2.Ruiz PJ, Masliah E, Doherty TA, Quach A, Firestein GS. Cardiac death in a patient with adult-onset Still's disease treated with the interleukin 1 receptor inhibitor anakinra. Ann Rheum Dis. 2007;66:422–3. doi: 10.1136/ard.2006.060541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 4.Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48:2221–8. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]