

Abstract
Study Objectives:
Sleep deprivation is common in patients with neuropathic pain, but the effect of sleep deprivation on pathological pain remains uncertain. This study investigated whether sleep deprivation aggravates neuropathic symptoms and enhances microglial activation in the cuneate nucleus (CN) in a median nerve chronic constriction injury (CCI) model. Also, we assessed if melatonin supplements during the sleep deprived period attenuates these effects.
Design:
Rats were subjected to sleep deprivation for 3 days by the disc-on-water method either before or after CCI. In the melatonin treatment group, CCI rats received melatonin supplements at doses of 37.5, 75, 150, or 300 mg/kg during sleep deprivation. Melatonin was administered at 23:00 once a day.
Participants:
Male Sprague-Dawley rats, weighing 180-250 g (n = 190), were used.
Measurements:
Seven days after CCI, behavioral testing was conducted, and immunohistochemistry, immunoblotting, and enzyme-linked immunosorbent assay were used for qualitative and quantitative analyses of microglial activation and measurements of proinflammatory cytokines.
Results:
In rats who underwent post-CCI sleep deprivation, microglia were more profoundly activated and neuropathic pain was worse than those receiving pre-CCI sleep deprivation. During the sleep deprived period, serum melatonin levels were low over the 24-h period. Administration of melatonin to CCI rats with sleep deprivation significantly attenuated activation of microglia and development of neuropathic pain, and markedly decreased concentrations of proinflammatory cytokines.
Conclusions:
Sleep deprivation makes rats more vulnerable to nerve injury-induced neuropathic pain, probably because of associated lower melatonin levels. Melatonin supplements to restore a circadian variation in melatonin concentrations during the sleep deprived period could alleviate nerve injury-induced behavioral hypersensitivity.
Citation:
Huang CT, Chiang RP, Chen CL, Tsai YJ. Sleep deprivation aggravates median nerve injury-induced neuropathic pain and enhances microglial activation by suppressing melatonin secretion. SLEEP 2014;37(9):1513-1523.
Keywords: median nerve, melatonin, microglia, neuropathic pain, sleep deprivation
INTRODUCTION
Peripheral nerve injury arising from disease or trauma induces long-lasting pathological pain, namely neuropathic pain,1 which manifests as spontaneous pain, allodynia (pain evoked by a normally innocuous stimulus), or hyperalgesia (heightened pain evoked by a noxious stimulus). Tactile allodynia in particular is a cardinal symptom of neuropathic pain.2 Recently, neuropathic pain has been thought to arise primarily from neuronal dysfunction. Several lines of evidence demonstrate that activation of glial cells in the central nervous system is associated with the posttraumatic neuronal plasticity and sensitization, and contributes to the development of neuropathic pain.3–5 In our previous work, activation of microglia in the cuneate nucleus (CN) was observed in a median nerve chronic constriction injury (CCI) model and inhibition of microglial activation by minocycline prevented the development of tactile allodynia.6
Disturbed sleep and short sleep duration are not uncommon in patients with various chronic pain disorders.7–9 Rats with peripheral neuropathy induced by sciatic nerve constriction injury also have a poor quality of sleep with reduced sleep efficiency,10 although there are reports of studies using the Bennett model of sciatic nerve ligation that show no untoward effects on sleep.11 Accumulating evidence suggests that insufficient sleep and poor sleep quality are risk factors for inflammation-related conditions such as cardiovascular disease,12 immune dysfunction,13,14 and metabolic disorders.12,15 In healthy human subjects, sleep deprivation increases pain sensitivity.16 Pain-related behavioral responses of male rats subjected to 72 h of sleep deprivation were significantly greater when noxious mechanical, thermal, or electrical stimuli were applied.17 Certainly, a vicious cycle develops in that pain interferes with sleep and inadequate sleep has an influence on pain perception.18–20 Thus, it is of paramount importance to better understand the interplay between pain and sleep, and its mechanism. Specifically, we are wondering if sleep deprivation aggravates neuropathic pain. Further, if this is the case, what role does activation of microglia play in this phenomenon?
Melatonin (N-acetyl-5-methoxytryptamine) is an indoleamine synthesized in the mammalian pineal gland21 and secreted into the bloodstream.22 Melatonin is involved in numerous biological functions, including circadian rhythm regulation,23 sleep,24,25 analgesia,26–28 and antioxidant effect.29 There is evidence that systemic or central administration of melatonin effectively prevents neuronal damage on the sleep deprived animals.30,31 However, it is unknown whether melatonin supplements during the sleep deprived period counteracts the effect of sleep deprivation on the presence and severity of neuropathic pain. For clinicians taking care of nerve-injured patients experiencing neuropathic pain, it is always helpful to understand the influence of sleep deprivation on neuropathic symptoms and the potential new remedies.
In the current study, we aimed to investigate the effects of sleep deprivation on neuropathic pain symptoms and microglial activation in the rat CN after median nerve CCI. In addition, we used melatonin supplementation to assess its effects on the sleep deprived rats with regard to neuropathic pain symptoms and microglial activation following CCI of the median nerve.
METHODS
Animal Preparation
The experimental protocol was approved by the National Science Council Committee and the Institutional Animal Care and Use Committee (IACUC) of Fu-Jen Catholic University. Ethical guidelines of the International Association for the Study of Pain,32 regarding the animal experimentation, were followed. All efforts were made to minimize the animal suffering and reduce the number of experimental animals. Animals used in this study were male Sprague-Dawley rats weighing 180-250 g, and they were housed under approved conditions with a 12/12 h light/dark cycle and with food and water available ad libitum.
Nerve Injury Surgery
The experimental model of median nerve CCI was established based on the methods described by Bennett and Xie.33 Briefly, anesthesia was induced by an intraperitoneal (i.p.) injection of 7% chloral hydrate (0.45 mL/100 g body weight). Under a dissecting microscope, the right median nerve was separated from the surrounding tissue at the elbow level, immediately proximal to the entry between two heads of the pronator teres muscle. Four loose ligatures (4.0 chromic gut) were made around the nerve,6,34–36 and then the incision was sutured. For the sham surgery, the median nerve of the right forelimb was manipulated similarly without ligation.
Sleep Deprivation Procedure
The disc-on-water method was slightly modified to achieve total sleep deprivation (TSD) in this study.37 The apparatus comprised two rectangular clear plastic chambers (60 × 20 × 60 cm each) placed side by side for housing two rats simultaneously. A single plastic disc (40 cm in diameter), serving as the rat-carrying platform, was built into the lower quarter of the two chambers. Beneath the disc, and extending to the chamber walls, was a rectangular tray filled with water to a depth of 5 cm. This disc was controlled by a motor set to rotate it for 8 sec at a speed of 3.5 rpm and then stop for 15 sec. The rats placed on the disc had to keep awake and walk against the direction of disc rotation to avoid falling into the water. Sleep deprivation depended on the rats' aversion to water, as rats rarely entered water spontaneously. Before the experiment, rats scheduled to undergo sleep deprivation were placed in the apparatus for environmental adaptation for at least 7 days. During the adaptive period and throughout the experiment, food and water were made available through grids placed on the top of the chambers. In the current study, the sleep of the rats was not recorded and the disc-on-water method designed for use with sleep deprived rats yoked to control rats was not implemented.
The experimental animals were divided into four groups (Figure 1). Rats in the first group (preinjury TSD group) were subjected to TSD for 3 days. Then, the rats were anesthetized immediately and underwent CCI of the median nerve (n = 10) or sham operation (n = 10). Animals in the second group were housed in the same apparatus for 3 days but were allowed to sleep (control for total sleep deprivation, TSDC). Subsequently, the rats (preinjury TSDC group) had median nerve CCI (n = 10) or sham operation (n = 10). In the third group (postinjury TSD group), animals underwent median nerve CCI (n = 10) or sham operation (n = 10). Immediately thereafter, the rats were subjected to TSD for 3 days. In the fourth group (postinjury TSDC group), animals underwent CCI of the median nerve (n = 10) or sham operation (n = 10). Then, the rats were housed in the same apparatus for 3 days but were permitted to sleep.
Figure 1.
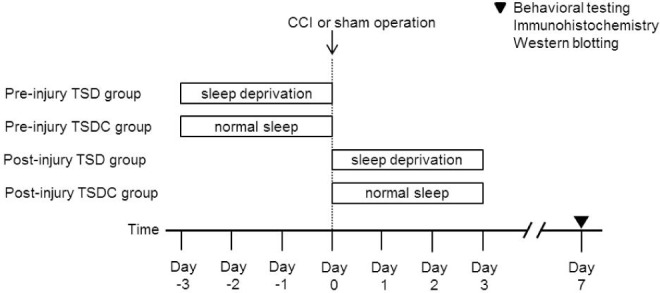
Study design. Rats were randomized to undergo either chronic constriction injury (CCI) or sham operation of the median nerve. In addition, the animals were randomized to receive total sleep deprivation (TSD) or normal sleep as controls for total sleep deprivation (TSDC) either preinjury or postinjury.
The animals in all the four groups survived 7 days after median nerve injury or sham operation, and then the behavioral testing was conducted on these animals. Afterward, the rats were randomly assigned to one of the two experiments. In one experiment, the animals were processed for immunohisto-chemistry; in the other experiment, animals were processed for Western blotting.
Melatonin Treatment
In a pilot study, we found that serum melatonin levels were consistently low during the sleep deprived period. The experimental animals were assigned to two study groups. Rats in one group underwent median nerve CCI under chloral hydrate anesthesia and then were subjected to TSD for 3 days. During the sleep deprived period, the animals were given vehicle (n = 10) or melatonin (N-acetyl-5-methoxytryptamine; Sigma-Aldrich, St. Louis, MO, USA) at doses of 37.5 (n = 10), 75 (n = 10), 150 (n = 10), or 300 mg/kg (n = 10). Rats in the other group underwent CCI of the median nerve but were allowed to sleep (TSDC) for 3 days. During the TSDC period, the rats were given vehicle (n = 10) or melatonin at doses of 37.5 (n = 10), 75 (n = 10), 150 (n = 10), or 300 mg/kg (n = 10). Melatonin was administered at 23:00 once a day taking into consideration the diurnal melatonin production in rats. Melatonin and vehicle were fed through an orogastric tube. Melatonin was dissolved in absolute alcohol and diluted in Ringer solution to achieve an ethanol concentration of < 1%. Animals in the control group (n = 10) underwent sham operation, but were not subjected to TSD. The animals were allowed to survive 7 days after median nerve injury or sham operation, and then the behavioral testing was conducted on them. Afterward, the rats were randomly assigned to one of the two experiments. In one experiment, the animals were processed for immunohistochemistry, and in the other, they were processed for Western blotting and enzyme-linked immunosorbent assay (ELISA).
Behavioral Testing
All studies were performed in a randomized, blinded manner to avoid expectation bias. In short, all animals were selected in a random order for behavioral testing, and another investigator blinded to the experimental status of the rats made the observations. The behavioral testing was accomplished during the light phase of the diurnal cycle.
Mechanical Allodynia
Rats were placed in individual Plexiglass chambers (25 × 40 × 18 cm) with wire mesh bottoms and were allowed to acclimatize to the environment for 30 min. The mechanical withdrawal threshold of the rat forepaws was determined using a series of von Frey filaments (Semmes-Weinstein Monofilaments, North Coast Medical, Inc., Gilroy, CA, USA; bending force: 0.16, 0.4, 0.6, 1.0, 1.4, 2.0, 4.0, 6.0, 8.0, 10.0, 15.0, and 26.0 g). The measurement was performed as previously described.38 Quantitative mechanical stimuli were applied to the medial plantar surface of each forepaw in an ascending order to evaluate the withdrawal threshold. Each von Frey filament was applied five times and the duration of each stimulus was 5 sec. The minimum time interval between two successive stimuli was 10 min. When rats displayed two or more withdrawal responses to a given filament, the bending force of the filament was defined as the withdrawal threshold.
Thermal Hyperalgesia
To measure the thermal withdrawal latency of the forepaws, the plantar test (Ugo Basile, Comerio, Italy) was used. In brief, rats were individually placed in one of three Plexiglass containers (22 × 17 × 14 cm) located on the elevated floor of a clear glass plate (3 mm thick) and were habituated to the apparatus for 30 min. A radiant heat source was positioned under the glass plate and directly beneath the plantar surface of the forepaw. The withdrawal latency was automatically measured as the time elapsed from the onset of radiant heat stimulation to the withdrawal of the forepaw. To avoid tissue damage, the maximum thermal stimulus duration was 20 sec. The use of the apparatus has been described in detail previously.39 Each forepaw was alternately tested five times with a minimal interval of 10 min between measurements, and readings were recorded to the nearest 0.1 sec. Five latency values per side were averaged. Animals were tested at least 5 h after von Frey filament testing.
Immunohistochemistry
The rats underwent perfusion with 4% paraformaldehyde/0.1 M phosphate buffer (PB), pH 7.4, after deep anesthesia (7% chloral hydrate, 0.45 mL/100 g body weight, i.p.). Tissue blocks of the medulla containing the CN were obtained and stored in PB/30% sucrose overnight, and were transversely cut into 30-μm slices with a cryostat (Leica, Nussloch, Germany).
The floating sections were retrieved and treated with 1% H2O2, blocked with 2% normal goat serum (NGS; GibcoBRL, Grand Island, NY, USA) in PB for 1 h, and then incubated in the mouse monoclonal anti-OX-42 antibody (CD11b, 1:200; Serotec, Indianapolis, IN) at 4°C for 48 h. The primary antibody was diluted in 0.01 M phosphate buffered saline (PBS, pH 7.4), containing 0.1% Triton X-100 and 5% NGS. After rinsing with PBS, sections were incubated in 1:200 biotinylated antimouse immunoglobulin G (IgG) (Vector, Burlingame, CA, USA) for 2 h and processed with avidin-biotin-horseradish peroxidase complex (Vector) at room temperature for 1 h. Peroxidase activity was subsequently visualized using the Vector® SG Substrate Kit. Finally, floating sections were mounted onto gelatinized slides and observed light microscopically (Zeiss Axiophot, Jena, Germany). No immunolabeling was observed in sections when normal mouse serum was substituted for the aforementioned primary antibody or the same antibody was omitted as controls.
Sample Preparation and Western Blot Analysis
Rats were sacrificed by decapitation after i.p. anesthesia with chloral hydrate. The brainstem including the CN was removed and cut into 150-μm-thick slices with a vibratome (TPI, Series 1000, Portland, OR, USA). Sections were stained with a 0.05% toluidine blue solution (Wako, Osaka, Japan) for 1 min and washed three times in PBS. Sections were then mounted onto glass slides and air-dried for 10 min. The CN was microdissected using a blunt-ended microdissection needle under a dissecting stereomicroscope (Leica).40 The retrieved tissue was homogenized in 100 μl lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 1% NP-40, 0.5% deoxycholic acid, 0.1% sodium docecyl sulfate (SDS), 1 mM Na3VO4, 20 μg/mL aprotinin, 20 μg/mL leupeptin, 20 μg/mL pepstatin A) with a grinder on ice. The homogenate was centrifuged at 10,000 g at 4°C for 20 min. The supernatant was obtained and its protein concentration was determined by a detergent-compatible protein assay with a bovine serum albumin standard (BioRad, Hercules, CA, USA). Proteins (20 μg) were separated on 10% sodium docecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to the nitrocellulose membrane (GE Healthcare, Piscataway, NJ, USA). Equal protein loading was confirmed by Ponceau S staining. The membrane was blocked in the 5% nonfat dry milk in PBS/0.1% Tween-20 (PBS-T) for 1 h and then incubated in mouse monoclonal anti-OX-42 antibody (CD11b, 1:1000; BD Biosciences, San Jose, CA, USA) at 4°C for 18 h. The membrane was washed three times in PBS-T and incubated with horseradish peroxidase (HRP)-conjugated sheep antimouse IgG (1:5000; GE Healthcare) at room temperature for 1 h. Peroxidase activity was measured by incubating the membrane with the enhanced chemiluminescence (ECL) Western blotting detection reagents (GE Healthcare) for 1 min and exposing it to the hyper-film (GE Healthcare) for 5 min. The blots were then incubated in stripping buffer (67.5 mM Tris, pH 6.8, 2% SDS, and 0.7% β-mercaptoethanol) and reprobed with mouse anti-β-actin monoclonal antibody (1:5000; Sigma-Aldrich) as loading controls. The optical density of specific OX-42 bands was measured with a computer-assisted image analysis system (Gel-Pro Analyzer software, Media Cybernetics, Inc., Bethesda, MD, USA).
Enzyme-Linked Immunosorbent Assay
Concentrations of proinflammatory cytokines in the CN were determined by ELISA. The samples were prepared as described previously. The supernatant was collected and tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and inter-leukin-6 (IL-6) levels were quantified using TNF-α Immuno-assay Kit (R&D Systems, Inc., Minneapolis, MN, USA), IL-1β Immunoassay Kit (R&D Systems, Inc.), and IL-6 Immunoassay Kit (R&D Systems, Inc.), respectively. The microplates were read using a microplate reader (Molecular Devices Corporation, Sunnyvale, CA, USA). All procedures were performed following manufacturer's instructions. Detection limits of the assays were 5 (TNF-α), 5 (IL-1β), and 21 (IL-6) pg/mL with intra-assay and interassay coefficients of variation of < 9% and < 10%, respectively.
Melatonin Assay
Serum melatonin concentrations were measured in rats subjected to TSD, either preinjury or postinjury, by the commercial immunoassay kit (GenWay Biotech, Inc., San Diego, CA, USA) according to the manufacturer's instructions. Blood samples were collected from the orbital sinus using the standard procedure described by Parasuraman et al.41 The rats were anesthetized with 10% ether using a vaporizer (Buxco Electronics, Inc., Wilmington, NC, USA). The inlet in the exposure chamber was placed at the bottom to ensure a homogeneous concentration. Animals were placed in the chamber for 2 min and then a topical ophthalmic anesthetic agent was applied to their eyes prior to blood sample collection. The animal was fixed with the thumb and forefinger of the nondominant hand, and the skin around the eye was pulled taut. A capillary tube was inserted into the medial canthus of the eye. Slight thumb pressure was enough to puncture the tissue and enter the orbital sinus. After blood sampling, the capillary tube was gently removed and wiped with sterile cotton. Bleeding could be stopped by applying gentle finger pressure over the puncture site. The rats were checked for postoperative and periorbital lesions 30 min after blood collection. We performed blood sampling twice a day at 11:00 and 23:00 from 2 days before to 2 days after TSD. At each time point, 5 rats were randomly selected for blood collection; however, at most two bleedings, one for each eye, were allowed for each animal during the study period. The sample collection tubes were centrifuged at 4,000 g at 4°C for 30 min and serum aliquots were frozen at -20°C. The melatonin assay had a detection limit of 4 pg/mL, an intra-assay coefficient of variation of 7–10%, and an interassay coefficient of variation of 10–13%.
Data Presentation and Statistical Analysis
All results were presented as mean ± standard deviation (SD). A factorial analysis of variance (ANOVA) was computed to test the main effects of different factors and their interactions. If there were significant main effects or interactions, post hoc pairwise comparisons were carried out with the Bonferroni correction. A P value of < 0.05 denoted statistical significance. All statistical analyses were performed using the SPSS software (version 19.0, SPSS, Inc., Chicago, IL, USA).
RESULTS
Effect of TSD on the Behavioral Testing After Median Nerve CCI
A factorial ANOVA with behavioral measures of either mechanical or thermal sensitivity as the dependent variable and sleep deprivation (TSD versus TSDC), CCI (CCI versus sham operation) and timing of intervention (preinjury versus postinjury) as fixed factors revealed a significant main effect of CCI (both P < 0.05). The sham-operated rats in all the four groups had similar mechanical withdrawal thresholds (Figure 2A) and thermal withdrawal latencies (Figure 2B). A marked decrease of the mechanical withdrawal threshold and thermal withdrawal latency was observed in CCI rats in each of the study groups as compared to the corresponding sham-operated rats. The subsequent 2 × 2 factorial design analysis including only CCI rats on behavioral measures of mechanical or thermal sensitivity showed significant main effects of both sleep deprivation and timing of intervention, and a significant sleep deprivation*timing of intervention interaction (all P < 0.05). The mechanical withdrawal threshold and thermal withdrawal latency of CCI rats in the postinjury TSDC group did not differ from those of CCI rats in preinjury TSD or TSDC groups; further, the latter two groups displayed similar behavioral testing results. Of note, a significantly decreased mechanical withdrawal threshold and thermal withdrawal latency was discernible in CCI rats of the postinjury TSD group than in those of the other three study groups.
Figure 2.
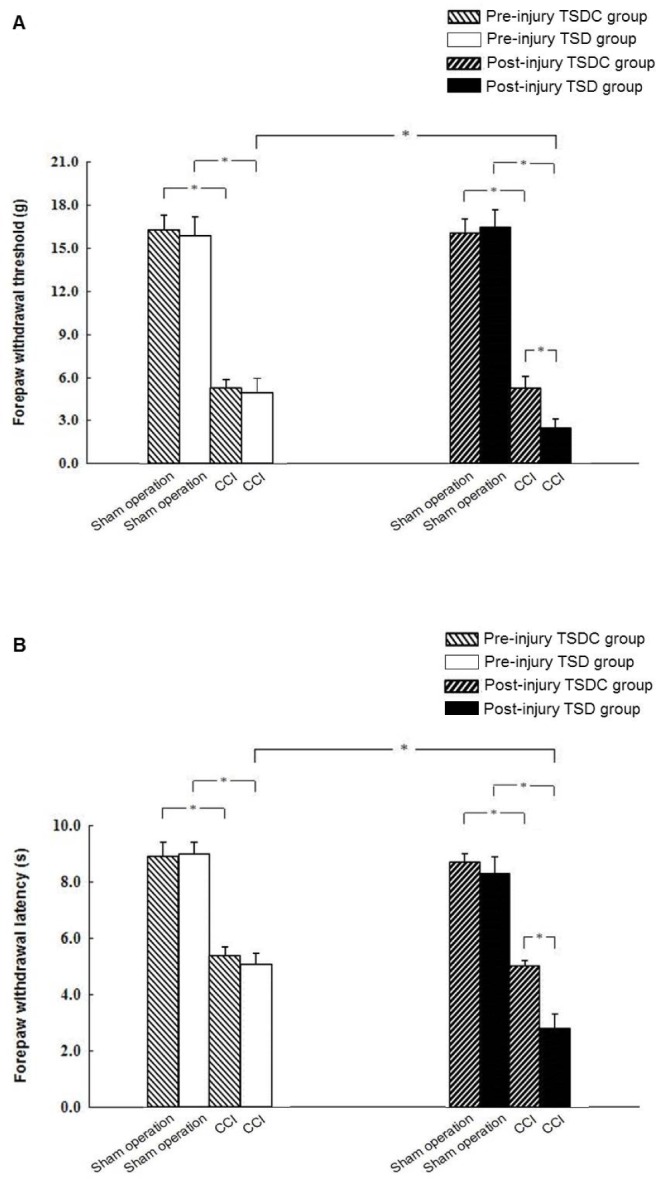
Effect of preinjury or postinjury total sleep deprivation (TSD) on nerve injury-induced mechanical allodynia and thermal hyperalgesia. The chronic constriction injury (CCI) and sham-operated rats were subjected to TSD or control for total sleep deprivation (TSDC) for 3 days either preinjury or postinjury. The mechanical withdrawal thresholds (A) and thermal withdrawal latencies (B) were assessed 7 days after CCI or sham operation. Data are expressed as mean ± standard deviation (error bars); n = 10 rats per group; * Bonferroni-adjusted P < 0.05.
Effect of TSD on Microglial Activation After Median Nerve CCI
A significant main effect of CCI (P < 0.05) on OX-42 expression was identified by using a factorial ANOVA in which the three factors in the design were sleep deprivation, CCI, and timing of intervention. Immunohistochemistry (Figures 3A, 3B, 3E, 3F) and immunoblotting (Figure 4) showed little expression of OX-42 in the CN of sham-operated rats in the four study groups. When compared to sham operation, CCI resulted in a significant increase in OX-42 expression in rat CN within each study group (Figures 3C, 3D, 3G, 3H, 4). The 2 × 2 factorial design analysis in CCI rats on OX-42 immunoreactivity showed significant main effects of sleep deprivation and timing of intervention, and a significant interaction between them (all P < 0.05). Among CCI rats, OX-42 was more profoundly expressed in the CN in the postinjury TSD group than the other three groups. There was similar expression of OX-42 in the CN of CCI rat between preinjury TSD and TSDC groups.
Figure 3.
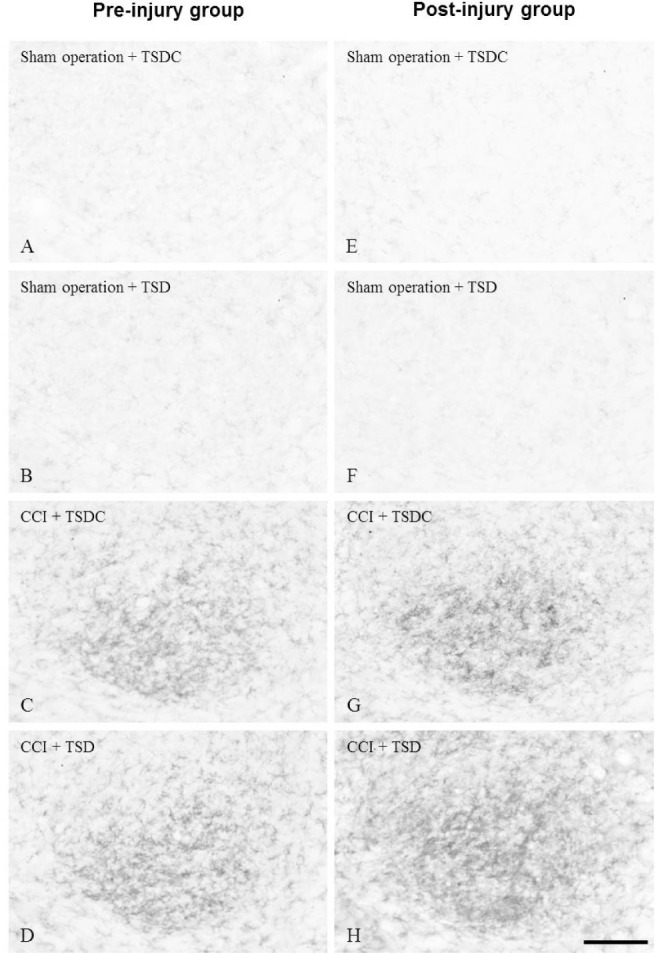
Photomicrographs showing OX-42 immunoreactivity in the ipsilateral cuneate nucleus (CN) of chronic constriction injury (CCI) or sham-operated rats. The rats were treated with total sleep deprivation (TSD) (B,D,F,H) or control for total sleep deprivation (TSDC) (A,C,E,G) for 3 days either preinjury (left panels) or postinjury (right panels). Each animal was sacrificed 7 days after CCI (C-D,G-H) or sham operation (A-B,E-F). The CN was harvested and processed for immunohistochemistry using the OX-42 antibody. Bar = 100 μm.
Figure 4.
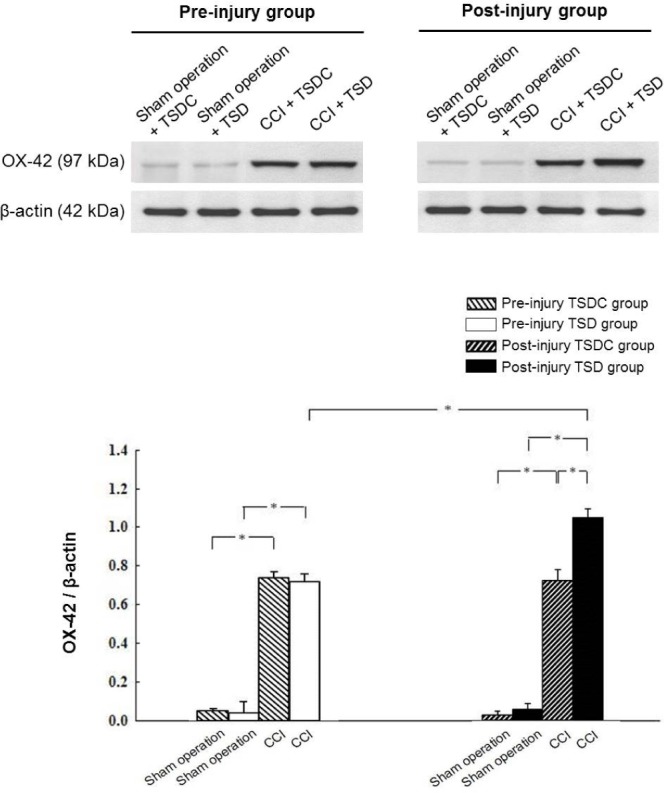
Western blot analysis showing the levels of OX-42 in the ipsilateral cuneate nucleus (CN) of chronic constriction injury (CCI) and sham-operated rats. The CN was homogenized and 20 μg of protein was analyzed by Western blot using anti-OX-42 antibody. β-actin was used as a loading control. Data are expressed as mean ± standard deviation (error bars); n = 7 rats per group; * Bonferroni-adjusted P < 0.05. TSD, total sleep deprivation; TSDC, control for total sleep deprivation.
Influence of Melatonin Treatment on Behavioral Testing, Microglial Activation, and Proinflammatory Cytokine Expression in CCI Rats
Serum melatonin concentrations displayed a circadian variation with low daytime and high nighttime values before and after the period of TSD (Figure 5). During the sleep deprived period, serum melatonin levels were low over the 24-h period.
Figure 5.
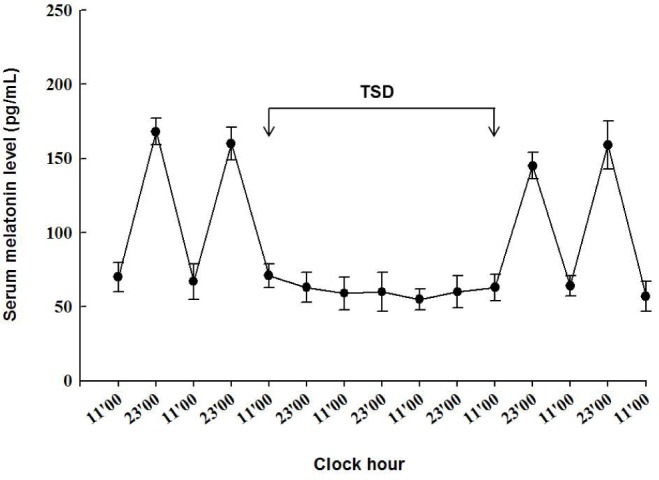
The diagram showing serum melatonin levels during the study period. Serum melatonin concentrations were measured twice a day at 11:00 and 23:00. Data are expressed as mean ± standard deviation (error bars); n = 5 rats at each time point. TSD, total sleep deprivation.
A 2 × 2 factorial ANOVA with each of measurement of mechanical and thermal sensitivity, OX-42 immunoreactivity, and cytokine levels as the dependent variable and sleep deprivation and melatonin treatment (melatonin versus vehicle) as fixed factors revealed significant main effects of sleep deprivation and melatonin treatment (all P < 0.05), but not their interactions. Behavioral testing demonstrated that the mechanical withdrawal threshold (Figure 6A) and thermal withdrawal latency (Figure 6B) were significantly decreased in vehicle-treated CCI rats that were subjected to TSD or TSDC as compared to sham-operated control rats. Administration of 75, 150, or 300 mg/kg melatonin to TSD-treated CCI rats dose-dependently increased mechanical withdrawal threshold and thermal withdrawal latency. Similar effects of melatonin on behavioral testing in TSDC-treated CCI rats were also observed. Generally speaking, a more profoundly decreased mechanical withdrawal threshold and thermal withdrawal latency was measured in TSD-treated CCI rats than TSDC-treated ones at various doses of melatonin.
Figure 6.
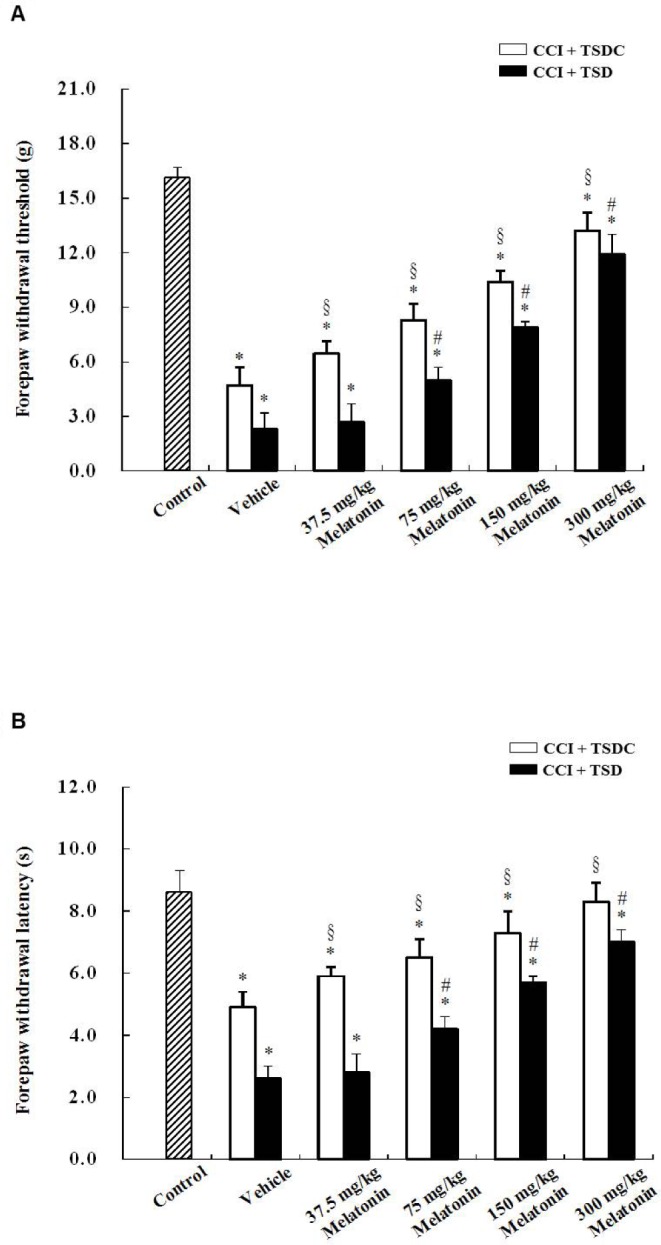
Effect of melatonin treatment on nerve injury-induced mechanical allodynia and thermal hyperalgesia in chronic constriction injury (CCI) and sham-operated control rats. The rats received CCI of the median nerve and then were treated with total sleep deprivation (TSD) or were allowed to sleep (control for total sleep deprivation, TSDC) for 3 days. During the 3-day period, the animals were administered with either vehicle or 37.5, 75, 150, or 300 mg/kg melatonin at 23:00 once a day. Sham-operated control rats were not subjected to TSD. Mechanical allodynia (A) and thermal hyperalgesia (B) were evaluated 7 days after CCI or sham operation. Data are expressed as mean ± standard deviation (error bars); n = 10 rats per group. * Bonferroni-adjusted P < 0.05 as compared to sham-operated control rats; # Bonferroni-adjusted P < 0.05 as compared to TSD- and vehicle-treated CCI rats; § Bonferroni-adjusted P < 0.05 as compared to TSDC- and vehicle-treated CCI rats.
Little OX-42 immunoreactivity was present in the CN of sham-operated control rats (figure not shown). There was intense OX-42 immunoreactivity in the ipsilateral CN of vehicle-treated CCI rats subjected to TSD or TSDC (Figures 7A, 7F); however, TSD led to more profoundly increased OX-42 immunoreactivity than TSDC. After administration of different doses of melatonin, OX-42 immunoreactivity was decreased in the CN of TSD- or TSDC-treated CCI rats in a dose-dependent manner (Figures 7B–7E, 7G–7J). Western blot analysis (Figure 8) further confirmed the findings of immunohistochemistry.
Figure 7.
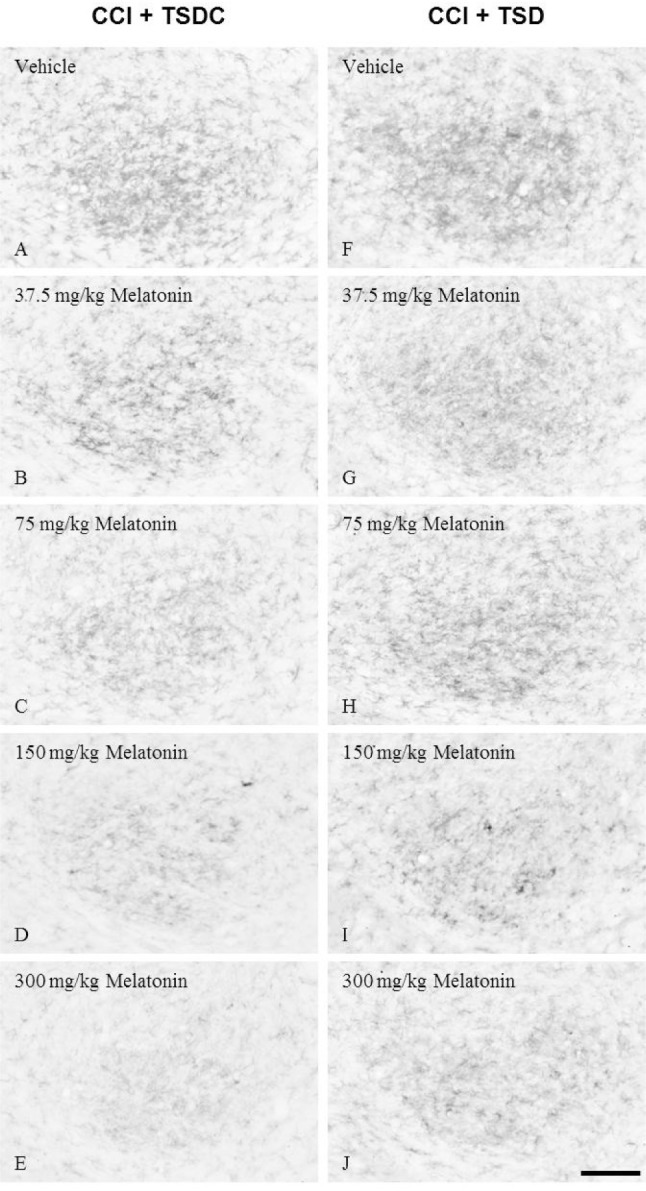
Photomicrographs showing OX-42 immunoreactivity in the ipsilateral cuneate nucleus (CN) of rats with chronic constriction injury (CCI) that received melatonin supplements during the sleep deprived or sleep period. The CCI rats underwent total sleep deprivation (TSD) (F-J) or were allowed to sleep (control for total sleep deprivation, TSDC) (A-E) for 3 days after surgery. In the meantime, vehicle or melatonin at a dose of 37.5, 75, 150, or 300 mg/kg was given to these animals at 23:00 once a day. Bar = 100 μm.
Figure 8.
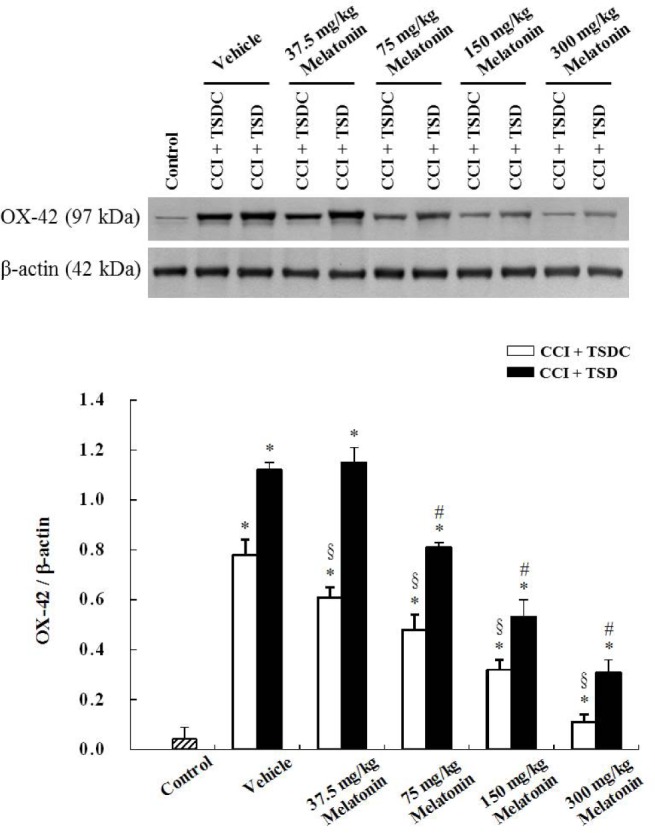
Western blot analysis showing the OX-42 levels in the ipsilateral cuneate nucleus (CN) of rats with chronic constriction injury (CCI) that were given melatonin treatment during the sleep deprived or sleep period. The rats were subjected to total sleep deprivation (TSD) or were allowed to sleep (control for total sleep deprivation, TSDC) for 3 days after CCI of the median nerve. During the same period of TSD or TSDC, the animals also received vehicle or different doses of melatonin at 23:00 once a day. The sham-operated rats were used as controls. β-actin was used as a loading control. Data are expressed as mean ± standard deviation (error bars); n = 7 rats per group. * Bonferroni-adjusted P < 0.05 when compared with sham-operated rats; # Bonferroni-adjusted P < 0.05 when compared with CCI rats treated with TSD and vehicle; § Bonferroni-adjusted P < 0.05 when compared with CCI rats treated with TSDC and vehicle.
Low levels of TNF-α, IL-1β, and IL-6 were detected in the CN of sham-operated control rats; however, these cytokine levels were markedly increased in the ipsilateral CN of vehicle-treated CCI rats that were subjected to TSD or TSDC (Figure 9). Among TSD-treated CCI rats, melatonin treatment at various doses dose-dependently decreased the levels of TNF-α, IL-1β, and IL-6 in the ipsilateral CN as compared to vehicle treatment. Similar effects of melatonin on cytokine expression in TSDC-treated CCI rats were also observed. In general, a more evident production of TNF-α, IL-1β, and IL-6 was found in CCI rats subjected to TSD than in those subjected to TSDC at a given dose of melatonin.
Figure 9.
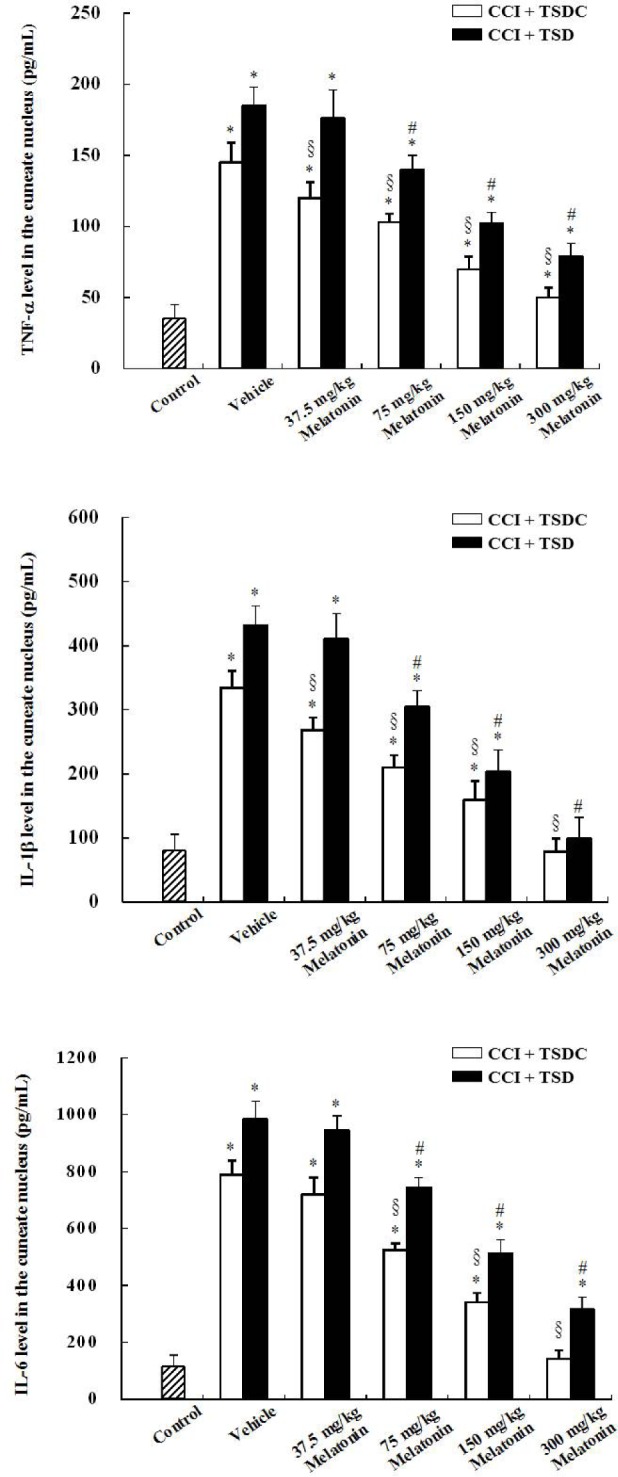
Histograms showing the proinflammatory cytokine expression in the ipsilateral cuneate nucleus (CN) of rats with chronic constriction injury (CCI) that received melatonin treatment during the sleep deprived or sleep period. The rats with CCI were subjected to total sleep deprivation (TSD) or were allowed to sleep (control for total sleep deprivation, TSDC) for 3 days after surgery. During the period, they were given vehicle or melatonin at a dose of 37.5, 75, 150, or 300 mg/kg at 23:00 once a day. Proinflammatory cytokines, tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6 levels in the CN were measured at 7 days after CCI. Data are expressed as mean ± standard deviation (error bars); n = 7 rats per group. * Bonferroni-adjusted P < 0.05 as compared with sham-operated rats; # Bonferroni-adjusted P < 0.05 as compared with TSD- and vehicle-treated CCI rats; § Bonferroni-adjusted P < 0.05 as compared with TSDC- and vehicle-treated rats with CCI.
DISCUSSION
This study demonstrates that 3-day TSD applied immediately after but not before CCI of the median nerve enhanced OX-42 immunoreactivity in the ipsilateral CN and development of neuropathic pain behavior in rats that were measured 7 days following nerve injury. During the sleep deprived period, serum melatonin levels were low throughout the day; in the meantime, melatonin treatment effectively reduced OX-42 immunoreactivity and the release of proinflammatory cytokines, and successfully ameliorated neuropathic pain in CCI rats.
Microglial activation in the central nervous system is a common phenomenon following peripheral nerve injury42,43 and is considered to play a key role in the development of neuropathic pain.44 The activation of microglia is accompanied with upregulation of the surface antigen, complement receptor 3 (CR3); herein, we assessed the microglial activation response in the CN following median nerve CCI using immunocyto-chemical detection of OX-42 monoclonal antibody, which recognizes CR3.45 Previous work showed that the activation of microglia reaches a peak and mechanical hypersensitivity is most pronounced on day 7 after median nerve injury.6 Therefore, in the current study, we chose day 7 as the time point for experimental observations.
Peripheral nerve injury evokes a barrage of injury discharges from the injured nerve fibers and their dorsal root ganglia. Then, these ectopic discharges prompt the release of excitatory amino acids, such as glutamate and aspartate, from damaged primary afferents, resulting in microglial activation and the development of neuropathic pain.33,46,47 In the CN, glutamate is one of the major neurotransmitters in the primary afferent terminals.48,49 Several studies have shown that sleep deprivation leads to a rise in glutamate concentrations50 and has an effect on the endogenous opioid system51 in the rat brain that may facilitate nociceptive transmission. Further, the electroencephalographic study found that in patients with head injury, 24-h sleep deprivation causes an increase in the neuronal discharges in regions of brain contusion.52 In the curreent study, post-CCI total sleep deprivation increased microglial activation in the CN and aggravated nerve injury-induced neuropathic pain. Based on these observations, it is thought that postinjury sleep deprivation could exacerbate injury-induced ectopic discharges from the median nerve and enhance glutamate release that in turn aggravates the activation of microglia and the development of neuropathic pain. Additionally, in our previous study, we examined the microglial activation responses in the CN after median nerve CCI at different time points and found that the levels of OX-42 were evidently increased as early as 1 day after nerve injury.6 When the rats were subjected to TSD for 3 days after CCI, microglia in the CN had already been activated. On the contrary, microglia were inactive in rats treated with TSD prior to CCI. It is speculated that TSD has influence only on activated microglia; accordingly, more profoundly activated microglia and more severe neuropathic pain were observed in rats treated with post-CCI TSD than those given pre-CCI TSD.
Pharmacological studies showed that in cultured microglia, glutamate leads to activation of the N-methyl-d-aspartate (NMDA) receptor, which depolarizes the cell membrane and thereby opens voltage-gated Ca2+ channels.53,54 A large influx of Ca2+ ions results in activation of microglia. Recent work demonstrated that activated microglia release signal mediators, such as proinflammatory cytokines (IL-1β, IL-6, and TNF-α), cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase (iNOS), and these mediators appear contributory to different features of pathological pain.3,55,56 During the period of sleep deprivation, systemic administration of melatonin reduced inflammatory mediators in an animal model of lipopolysaccharide-induced lung injury.57 The current study found that oral administration of melatonin in CCI rats treated with TSD significantly attenuated OX-42 immunoreactivity and proinflammatory cytokine levels in the CN and ameliorated nerve injury-induced neuropathic pain. Several studies demonstrated that melatonin blocks the opening of voltage-gated Ca2+ channels by directly crossing the lipid bilayer of cell membranes or indirectly binding to two G-protein-coupled receptors (MT1 and MT2).31,58,59 These two melatonin receptors have recently been identified in the cell membranes of microglia.60 In addition, an electrophysiological experiment showed that melatonin depresses activation of the NMDA receptor and its associated activity in an in vitro model.61 It is suggested that melatonin attenuates the release of proinflammatory cytokines from microglia and the development of neuropathic pain through the inhibition of Ca2+ influx-mediated microglial activation. Our study also showed that during the sleep deprived period, serum melatonin levels were low throughout the day. Moreover, microglial activation was aggravated and neuropathic pain worsened in CCI rats subjected to postinjury TSD as compared to those allowed to sleep after the injury. Prior studies demonstrated that reduced endogenous blood melatonin levels by pinealectomy increase contusion area after traumatic brain injury and abolish the analgesic action of melatonin.62,63 Thus, these results suggest that melatonin is an important neuroprotectant and a paucity of melatonin makes the rats more vulnerable to nerve injury-induced neuropathy.
A couple of studies demonstrated that healthy rats would display mechanical hypersensitivity immediately after a period of sleep deprivation16,64; however, we did not find a similar result in the current study. Compared to prior work, the behavioral testing was conducted in our sham-operated rats a number of days after TSD. This suggests that neuropathic pain behavior associated with the application of sleep deprivation could be resolved after normal sleep for a few days. Nonetheless, aggravated mechanical allodynia and thermal hyperalgesia were still present after the same period of rest in our CCI rats subjected to postinjury TSD. This may be explained by insufficient melatonin secretion during the sleep deprivation in that it worsens nerve injury-induced neuropathic pain; thus, it is speculated that a longer period of time is required for the neuropathy to subside. In addition, we found that melatonin supplements to restore its circadian characteristics ameliorated not only sleep deprivation-aggravated pain behavior but nerve injury-induced neuropathic pain.
Although inconsistent findings of experimental human studies on the effects of sleep deprivation on pain perception exist, there is a tendency of results indicating that sleep deprivation produces hyperalgesic changes in healthy subjects.64,65 Moreover, patients with primary insomnia have lower pain thresholds, and sleep deprivation exacerbates pain in patients with rheumatoid arthritis.66,67 However, clinical observations found that restorative sleep is independently associated with the resolution of chronic widespread pain in a population-based prospective study,68 and measures to improve sleep, such as an extended bedtime and continuous positive airway pressure, have been shown to reduce pain sensitivity in mildly sleepy healthy subjects and patients with obstructive sleep apnea, respectively.69,70 However, for ethical reasons, it is hardly possible to conduct human studies to precisely evaluate the reciprocal time relationship between a pain state and sleep. Our animal study is advantageous in this regard and shows that prenerve injury sleep deprivation had little, if any, effects on neuropathic pain; instead, postnerve injury sleep deprivation significantly intensified neuropathic pain. In addition, there remains an issue about identifying sleep related mediators of the decrease in pain sensitivity.71 Sleep deprivation in healthy subjects is associated with an increase in proinflammatory cytokines, such as IL-6, and TNF-α,65 that in turn have been shown to sensitize sensory neurons and facilitate pain.72 Although we demonstrated herein the beneficial effects of melatonin on reduction of inflamma-tory mediators and neuropathic pain in sleep deprived rats, the application of melatonin in sleep management and pain in various clinical situations needs further investigation in human studies.
One of the limitations of this study is that the sleep was not recorded in the rats. Based on the study by Rechtschaffen et al.,37 the disc carrying the rats rotated only 23% of the total recording time, and total sleep was reduced by 87% in sleep deprived rats and 31% in control rats from baseline to experiment. In the current study, the disc was rotated on a regular basis to occupy approximately one third of the sleep deprived period. Thus, it is anticipated that the methodology used here would be as effective to achieve sleep deprivation as that applied by Rechtschaffen et al.37 However, we should acknowledge that without electroencephalogram recording, the exact role of different stages of sleep deprivation in pain modulation could not be in depth explored in this study.
In summary, inadequate sleep prior to nerve injury does not aggravate postoperative neuropathic pain; however, increased neuropathic pain behavior is observed when sleep deprivation happens after nerve injury. Therefore, when patients with nerve injury encounter sleep problems, it should be seriously managed to prevent aggravation of neuropathic pain. Melatonin treatment to restore a circadian variation is a potential remedy in such a situation. Our study provides further knowledge about the interplay between neuropathy and sleep, and paves a way for the development of treatment modality.
DISCLOSURE STATEMENT
This was not an industry supported study. This study was supported by research grants from the National Science Council of Taiwan (NSC 98-2320-B-030-002-MY3; NSC 101-2320-B-030-001). The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
Chun-Ta Huang and Rayleigh Ping-Ying Chiang contributed equally to this study. The authors are grateful to Ms. Yi-Tien Li for the technical assistance. We also thank Dr. Jou-Wei Lin for his assistance with the review of the statistical analysis.
Footnotes
A commentary on this article appears in this issue on page 1405.
REFERENCES
- 1.Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]
- 2.Dworkin RH, Backonja M, Rowbotham MC, et al. Advances in neuropathic pain: diagnosis, mechanisms, and treatment recommendations. Arch Neurol. 2003;60:1524–34. doi: 10.1001/archneur.60.11.1524. [DOI] [PubMed] [Google Scholar]
- 3.Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–5. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 4.Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- 5.Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–7. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Lin SC, Yeh JH, Chen CL, Chou SH, Tsai YJ. Effects of local lidocaine treatment before and after median nerve injury on mechanical hypersensitivity and microglia activation in rat cuneate nucleus. Eur J Pain. 2011;15:359–67. doi: 10.1016/j.ejpain.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Morin CM, Gibson D, Wade J. Self-reported sleep and mood disturbance in chronic pain patients. Clin J Pain. 1998;14:311–4. doi: 10.1097/00002508-199812000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Ohayon MM. Relationship between chronic painful condition and insomnia. J Psychiatr Res. 2005;39:151–9. doi: 10.1016/j.jpsychires.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Smith MT, Haythornthwaite JA. How do sleep disturbance and chronic pain inter-relate? Insights from longitudinal and cognitive-behavioral clinical trials literature. Sleep Med Rev. 2004;8:119–32. doi: 10.1016/S1087-0792(03)00044-3. [DOI] [PubMed] [Google Scholar]
- 10.Andersen ML, Tufik S. Sleep patterns over 21-day period in rats with chronic constriction of sciatic nerve. Brain Res. 2003;984:84–92. doi: 10.1016/s0006-8993(03)03095-6. [DOI] [PubMed] [Google Scholar]
- 11.Kontinen VK, Ahnaou A, Drinkenburg WH, Meert TF. Sleep and EEG patterns in the chronic constriction injury model of neuropathic pain. Physiol Behav. 2003;78:241–6. doi: 10.1016/s0031-9384(02)00966-6. [DOI] [PubMed] [Google Scholar]
- 12.Mullington JM, Haack M, Toth M, Serrador JM, Meier-Ewert HK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog Cardiovasc Dis. 2009;51:294–302. doi: 10.1016/j.pcad.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci. 2009;10:199–210. doi: 10.1038/nrn2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lange T, Dimitrov S, Fehm HL, Westermann J, Born J. Shift of monocyte function toward cellular immunity during sleep. Arch Intern Med. 2006;166:1695–700. doi: 10.1001/archinte.166.16.1695. [DOI] [PubMed] [Google Scholar]
- 15.Knutson KL, Spiegel K, Penev P, Van Cauter E. The metabolic consequences of sleep deprivation. Sleep Med Rev. 2007;11:163–78. doi: 10.1016/j.smrv.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kundermann B, Krieg JC, Schreiber W, Lautenbacher S. The effect of sleep deprivation on pain. Pain Res Manag. 2004;9:25–32. doi: 10.1155/2004/949187. [DOI] [PubMed] [Google Scholar]
- 17.Onen SH, Alloui A, Jourdan D, Eschalier A, Dubray C. Effects of rapid eye movement (REM) sleep deprivation on pain sensitivity in the rat. Brain Res. 2001;900:261–7. doi: 10.1016/s0006-8993(01)02320-4. [DOI] [PubMed] [Google Scholar]
- 18.Carli G, Montesano A, Rapezzi S, Paluffi G. Differential effects of persistent nociceptive stimulation on sleep stages. Behav Brain Res. 1987;26:89–98. doi: 10.1016/0166-4328(87)90158-6. [DOI] [PubMed] [Google Scholar]
- 19.Landis CA, Robinson CR, Levine JD. Sleep fragmentation in the arthritic rat. Pain. 1988;34:93–9. doi: 10.1016/0304-3959(88)90186-8. [DOI] [PubMed] [Google Scholar]
- 20.Onen SH, Onen F, Courpron P, Dubray C. How pain and analgesics disturb sleep. Clin J Pain. 2005;21:422–31. doi: 10.1097/01.ajp.0000129757.31856.f7. [DOI] [PubMed] [Google Scholar]
- 21.Stehle JH, Saade A, Rawashdeh O, et al. A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J Pineal Res. 2011;51:17–43. doi: 10.1111/j.1600-079X.2011.00856.x. [DOI] [PubMed] [Google Scholar]
- 22.Vanecek J. Cellular mechanisms of melatonin action. Physiol Rev. 1998;78:687–721. doi: 10.1152/physrev.1998.78.3.687. [DOI] [PubMed] [Google Scholar]
- 23.Hardeland R, Madrid JA, Tan DX, Reiter RJ. Melatonin, the circadian multioscillator system and health: the need for detailed analyses of peripheral melatonin signaling. J Pineal Res. 2012;52:139–66. doi: 10.1111/j.1600-079X.2011.00934.x. [DOI] [PubMed] [Google Scholar]
- 24.Cardinali DP, Srinivasan V, Brzezinski A, Brown GM. Melatonin and its analogs in insomnia and depression. J Pineal Res. 2012;52:365–75. doi: 10.1111/j.1600-079X.2011.00962.x. [DOI] [PubMed] [Google Scholar]
- 25.Santhi N, Thorne HC, van der Veen DR, et al. The spectral composition of evening light and individual differences in the suppression of melatonin and delay of sleep in humans. J Pineal Res. 2012;53:47–59. doi: 10.1111/j.1600-079X.2011.00970.x. [DOI] [PubMed] [Google Scholar]
- 26.Zahn PK, Lansmann T, Berger E, Speckmann EJ, Musshoff U. Gene expression and functional characterization of melatonin receptors in the spinal cord of the rat: implications for pain modulation. J Pineal Res. 2003;35:24–31. doi: 10.1034/j.1600-079x.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 27.Gitto E, Aversa S, Salpietro CD, et al. Pain in neonatal intensive care: role of melatonin as an analgesic antioxidant. J Pineal Res. 2012;52:291–5. doi: 10.1111/j.1600-079X.2011.00941.x. [DOI] [PubMed] [Google Scholar]
- 28.Wilhelmsen M, Amirian I, Reiter RJ, Rosenberg J, Gögenur I. Analgesic effects of melatonin: a review of current evidence from experimental and clinical studies. J Pineal Res. 2011;51:270–7. doi: 10.1111/j.1600-079X.2011.00895.x. [DOI] [PubMed] [Google Scholar]
- 29.Galano A, Tan DX, Reiter RJ. Melatonin as a natural ally against oxidative stress: a physiochemical examination. J Pineal Res. 2011;51:1–16. doi: 10.1111/j.1600-079X.2011.00916.x. [DOI] [PubMed] [Google Scholar]
- 30.Chang HM, Wu UI, Lan CT. Melatonin preserves longevity protein (sirtuin 1) expression in the hippocampus of total sleep-deprived rats. J Pineal Res. 2009;47:211–20. doi: 10.1111/j.1600-079X.2009.00704.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Zhang HQ, Liang XY, Zhang HF, Zhang T, Liu FE. Melatonin ameliorates cognitive impairment induced by sleep deprivation in rats: role of oxidative stress, BDNF and CaMKII. Behav Brain Res. 2013;256:72–81. doi: 10.1016/j.bbr.2013.07.051. [DOI] [PubMed] [Google Scholar]
- 32.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–10. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 33.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 34.Tsai YJ, Lin CT, Huang CT, et al. Neuropeptide Y modulates c-Fos protein expression in the cuneate nucleus and contributes to mechanical hypersensitivity following rat median nerve injury. J Neurotrauma. 2009;26:1609–21. doi: 10.1089/neu.2008.0642. [DOI] [PubMed] [Google Scholar]
- 35.Chen JJ, Lue JH, Lin LH, et al. Effects of pre-emptive drug treatment on astrocyte activation in the cuneate nucleus following rat median nerve injury. Pain. 2010;148:158–66. doi: 10.1016/j.pain.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Tsai YJ, Huang CT, Lin SC, Yeh JH. Effects of regional and whole-body hypothermic treatment before and after median nerve injury on neuropathic pain and glial activation in rat cuneate nucleus. Anesthesiology. 2012;116:415–31. doi: 10.1097/ALN.0b013e318242a801. [DOI] [PubMed] [Google Scholar]
- 37.Rechtschaffen A, Gilliland MA, Bergmann BM, Winter JB. Physiological correlates of prolonged sleep deprivation in rats. Science. 1983;221:182–4. doi: 10.1126/science.6857280. [DOI] [PubMed] [Google Scholar]
- 38.Tal M, Bennett GJ. Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain. 1994;57:375–82. doi: 10.1016/0304-3959(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 39.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 40.Palkovits M, Brownstein MJ. Maps and guide to microdissection of the rat brain. New York: Elsevier; 1988. pp. 199–204. [Google Scholar]
- 41.Parasuraman S, Raveendran R, Kesavan R. Blood sample collection in small laboratory animals. J Pharmacol Pharmacother. 2010;1:87–93. doi: 10.4103/0976-500X.72350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aldskogius H, Kozlova EN. Central neuron-glial and glial-glial interactions following axon injury. Prog Neurobiol. 1998;55:1–26. doi: 10.1016/s0301-0082(97)00093-2. [DOI] [PubMed] [Google Scholar]
- 43.Eriksson NP, Persson JK, Svensson M, Arvidsson J, Molander C, Aldskogius H. A quantitative analysis of the microglial cell reaction in central primary sensory projection territories following peripheral nerve injury in the adult rat. Exp Brain Res. 1993;96:19–27. doi: 10.1007/BF00230435. [DOI] [PubMed] [Google Scholar]
- 44.Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126:56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Obata H, Eisenach JC, Hussain H, Bynum T, Vincler M. Spinal glial activation contributes to postoperative mechanical hypersensitivity in the rat. J Pain. 2006;7:816–22. doi: 10.1016/j.jpain.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 46.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 47.Wall PD, Gutnick M. Ongoing activity in peripheral nerves: the physiology and pharmacology of impulses originating from a neuroma. Exp Neurol. 1974;43:580–93. doi: 10.1016/0014-4886(74)90197-6. [DOI] [PubMed] [Google Scholar]
- 48.Lue JH, Jiang-Shieh YF, Shieh JY, Ling EA, Wen CY. Multiple inputs of GABA-immunoreactive neurons in the cuneate nucleus of the rat. Neurosci Res. 1997;27:123–32. doi: 10.1016/s0168-0102(96)01139-x. [DOI] [PubMed] [Google Scholar]
- 49.Lue JH, Chen SH, Shieh JY, Wen CY. Afferent synaptic contacts on glycine-immunoreactive neurons in the rat cuneate nucleus. Synapse. 2001;41:139–49. doi: 10.1002/syn.1068. [DOI] [PubMed] [Google Scholar]
- 50.Bettendorff L, Sallanon-Moulin M, Touret M, Wins P, Margineanu I, Schoffeniels E. Paradoxical sleep deprivation increases the content of glutamate and glutamine in rat cerebral cortex. Sleep. 1996;19:65–71. doi: 10.1093/sleep/19.1.65. [DOI] [PubMed] [Google Scholar]
- 51.Fadda P, Tortorella W, Fratta W. Sleep deprivation decreases mu and delta opioid receptor binding in the rat limbic system. Neurosci Lett. 1991;129:315–7. doi: 10.1016/0304-3940(91)90489-g. [DOI] [PubMed] [Google Scholar]
- 52.Thomaides TN, Kerezoudi EP, Chaudhuri KR, Cheropoulos C. Study of EEGs following 24-hour sleep deprivation in patients with posttraumatic epilepsy. Eur Neurol. 1992;32:79–82. doi: 10.1159/000116796. [DOI] [PubMed] [Google Scholar]
- 53.Kim WK, Ko KH. Potentiation of N-methyl-D-aspartate-mediated neurotoxicity by immunostimulated murine microglia. J Neurosci Res. 1998;54:17–26. doi: 10.1002/(SICI)1097-4547(19981001)54:1<17::AID-JNR3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 54.Lohr C, Deitmer JW. Calcium signaling in invertebrate glial cells. Glia. 2006;54:642–9. doi: 10.1002/glia.20368. [DOI] [PubMed] [Google Scholar]
- 55.Deleo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 56.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–55. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 57.Kim JY, Lee YD, Kim BJ, et al. Melatonin improves inflammatory cytokine profiles in lung inflammation associated with sleep deprivation. Mol Med Rep. 2012;5:1281–4. doi: 10.3892/mmr.2012.814. [DOI] [PubMed] [Google Scholar]
- 58.Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res. 2007;42:28–42. doi: 10.1111/j.1600-079X.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 59.Ambriz-Tututi M, Rocha-González HI, Cruz SL, Granados-Soto V. Melatonin: a hormone that modulates pain. Life Sci. 2009;84:489–98. doi: 10.1016/j.lfs.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 60.Olivier P, Fontaine RH, Loron G, et al. Melatonin promotes oligodendroglial maturation of injured white matter in neonatal rats. PLoS One. 2009;4:e7128. doi: 10.1371/journal.pone.0007128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Escames G, León J, López LC, Acuña-Castroviejo D. Mechanisms of N-methyl-D-aspartate receptor inhibition by melatonin in the rat striatum. J Neuroendocrinol. 2004;16:929–35. doi: 10.1111/j.1365-2826.2004.01250.x. [DOI] [PubMed] [Google Scholar]
- 62.Ebadi M, Govitrapong P, Phansuwan-Pujito P, Nelson F, Reiter RJ. Pineal opioid receptors and analgesic action of melatonin. J Pineal Res. 1998;24:193–200. doi: 10.1111/j.1600-079x.1998.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 63.Kilic E, Ozdemir YG, Bolay H, Keleştimur H, Dalkara T. Pinealectomy aggravates and melatonin administration attenuates brain damage in focal ischemia. J Cereb Blood Flow Metab. 1999;19:511–6. doi: 10.1097/00004647-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 64.Lautenbacher S, Kundermann B, Krieg JC. Sleep deprivation and pain perception. Sleep Med Rev. 2006;10:357–69. doi: 10.1016/j.smrv.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 65.Haack M, Sanchez E, Mullington JM. Elevated inflammatory markers in response to prolonged sleep restriction are associated with increased pain experience in healthy volunteers. Sleep. 2007;30:1145–52. doi: 10.1093/sleep/30.9.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Irwin MR, Olmstead R, Carrillo C, et al. Sleep loss exacerbates fatigue, depression, and pain in rheumatoid arthritis. Sleep. 2012;35:537–43. doi: 10.5665/sleep.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haack M, Scott-Sutherland J, Santangelo G, Simpson NS, Sethna N, Mullington JM. Pain sensitivity and modulation in primary insomnia. Eur J Pain. 2012;16:522–33. doi: 10.1016/j.ejpain.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davies KA, Macfarlane GJ, Nicholl BI, et al. Restorative sleep predicts the resolution of chronic widespread pain: results from the EPIFUND study. Rheumatology (Oxford) 2008;47:1809–13. doi: 10.1093/rheumatology/ken389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roehrs TA, Harris E, Randall S, Roth T. Pain sensitivity and recovery from mild chronic sleep loss. Sleep. 2012;35:1667–72. doi: 10.5665/sleep.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Khalid I, Roehrs TA, Hudgel DW, Roth T. Continuous positive airway pressure in severe obstructive sleep apnea reduces pain sensitivity. Sleep. 2011;34:1687–91. doi: 10.5665/sleep.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Doghramji K. Sleep extension in sleepy individuals reduces pain sensitivity: new evidence regarding the complex, reciprocal relationship between sleep and pain. Sleep. 2012;35:1587–8. doi: 10.5665/sleep.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp Neurol. 2005;192:444–62. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]