

Abstract
Study Objectives:
To determine whether sleep disturbances are found in the valproic acid model of autism spectrum disorders (ASD).
Design:
Comparative study for sleep behavior, sleep architecture, electroencephalogram (EEG) spectral analysis, and glutamic acid decarboxylase (GAD) 65/67 protein expression in juvenile rats exposed to valproic acid (VPA), sodium salt, or saline in utero.
Setting:
N/A.
Participants:
Juvenile (postnatal day 32) male and female Sprague-Dawley rats.
Interventions:
In utero exposure to either saline or 400 mg/kg VPA administered intraperitoneally to the dams on gestational day 12.5. On postnatal days 22-24, all rats were implanted with transmitters to record EEG and electromyogram (EMG) activity.
Measurements and Results:
During the light phase, when nocturnal animals are typically quiescent, the VPA-exposed animals spent significantly more time in wake (∼35 min) and significantly less time in non-rapid eye movement (NREM) sleep (∼26 min) compared to the saline controls. Furthermore, spectral analysis of the EEG reveled that VPA-exposed animals exhibited increased high-frequency activity during wake and rapid eye movement (REM) sleep and reduced theta power across all vigilance states. Interestingly, the gamma-aminobutyric acid (GABA)-ergic system, which modulates the induction and maintenance of sleep states, was also disrupted, with reduced levels of both GAD 65 and GAD67 in the cortical tissue of VPA-exposed animals compared to saline controls.
Conclusions:
To date, the current animal models of ASD have been underutilized in the investigation of associated sleep disturbances. The VPA animal model recapitulates aspects of sleep disruptions reported clinically, providing a tool to investigate cellular and molecular dysregulation contributing to sleep disruptions in ASD.
Citation:
Cusmano DM, Mong JA. In utero exposure to valproic acid changes sleep in juvenile rats: a model for sleep disturbances in autism. SLEEP 2014;37(9):1489-1499.
Keywords: autism spectrum disorders, sleep, valproic acid
INTRODUCTION
Autism spectrum disorders (ASD) are an array of developmental disorders that primarily disrupt social interactions, communication, and behavioral patterns. Clinical observations have also indicated a high prevalence of sleep disturbances in children with ASD.1–6 Recent clinical studies report that the prevalence of sleep problems in children with ASD is approximately 44% to 83% of diagnosed cases.2,7–10 These sleep dysfunctions typically manifest as difficulties initiating and maintaining sleep, sleep fragmentation, insomnia, and parasomnias.8,11–21 Quality sleep is imperative for the maintenance of good health, and sleep loss can lead to or exacerbate existing behavioral problems associated with ASD.14,22–24 Alterations in sleep architecture have been linked with cognitive and behavioral deficits as well as stress and anxiety.3,22,23,25,26 Because children in whom ASD has been diagnosed share symptoms similar to those experiencing sleep loss, it is plausible to suspect that sleep disturbances in ASD may contribute to its symptomology. However, it appears that sleep problems in ASD are not influenced by the severity of cognitive deficits or ASD subtypes.16,27,28
Although these sleep disruptions have been characterized, their cause remains unknown and relatively unexplored. A question that remains unanswered is whether the neurocircuitry underlying sleep pathways is developmentally changed in ASD or whether disruptions in sleep are a consequence of changes in daily anxiety levels. Distinguishing between the two possibilities in a clinical setting would be challenging. The rodent's neurocircuitry and neurochemistry of sleep share similarities with humans, suggesting that rats would be a good model system for basic investigations.29 To date, the current animal models of ASD have been underutilized in the investigation of ASD associated sleep disturbances. Here we present data supporting the use of a rodent model in elucidating the etiology of sleep disruptions in ASD.
One animal model of ASD is prenatal exposure to valproic acid (VPA) or the salt, sodium valproate. VPA is an antiepileptic drug used to treat seizure disorders. Although VPA can prevent the induction of seizures during pregnancies, it has been classified as a teratogen and linked to a clinical phenotype known as fetal valproate syndrome (FVS), which includes various types of craniofacial malformations and developmental and cognitive delays.30–32 Many studies have also shown that FVS is associated with autistic behaviors and it is now thought that VPA exposure during a critical period of development significantly increases the risk of ASD.30,33–35 In a rodent model, animals exposed to VPA prenatally show similar alterations in brain architecture and sexspecific behavioral deficits seen in children with ASD.36–45 Therefore, it is thought to be a valid animal model for studying ASD.
To our knowledge, only one study has indirectly evaluated sleep/wake behavior in an animal model of ASD using locomotor and feeding behaviors.46 With the VPA model, Tsujino and colleagues46 have shown abnormal circadian rhythms in their animals, marked by increased locomotor activity and feeding behaviors during their sleep phase. Additionally, using micro-dialysis, they found that basal levels of serotonin (5-HT) are higher in VPA-exposed animals and that there is a caudal shift of 5-HT positive neurons in the dorsal raphe nucleus. Interestingly, the dorsal raphe supplies the serotinergic input involved in the ascending arousal system of the sleep/wake circuitry.
In this study, we investigated sleep/wake behaviors, including the sleep patterning, and electroencephalogram (EEG) characteristics related to the individual vigilance states in juvenile rats. Most studies investigating sleep in ASD report findings from children and adolescents with ASD; therefore, we recorded from juvenile/prepubertal rats to better model brain maturation and sleep in children and young adults. Because sleep/wake cycles are controlled by a complex system of reciprocal connections between sleep-promoting nuclei and arousal centers, we investigated whether VPA exposure alters some aspect of the sleep-wake neurocircuitry. Specifically, we investigated whether VPA alters the gamma-aminobutyric acid (GABA)ergic system, which plays a significant role in sleep onset and maintenance,47 through quantification of the expression of glutamic acid decarboxylase (GAD), the rate-limiting enzyme in GABA synthesis. Here, we measured GAD expression in the cortex and basal forebrain (BF), which is involved in sleep/wake and has been shown to regulate activity of cortical neurons.48–50
We found that VPA-exposed animals have more consolidated bouts of wake and non-rapid eye movement (NREM) sleep, resulting in a disruption of the normal sleep architecture. Further, we see changes in the EEG spectrum related to rapid eye movement (REM) sleep and arousal, with decreased theta power and increased gamma power. As implicated in children with autism, the GABAergic system is also affected in these animals. Expression levels of both isoforms (65 and 67) of GAD are significantly decreased in the cortex, but not the BF. Overall, these data suggest that in utero exposure to VPA induces sleep disruptions that correlate with the clinical ASD phenotype and that it provides a valid model for studying the underlying mechanisms regulating these changes in sleep.
METHODS
Animals
All experimental procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments were approved by and were in accordance with the guidelines of the University of Maryland Institutional Animal Care and Use Committee.
Female Sprague-Dawley rats were mated overnight (12-16-h) and the next day was designated day 1 of gestation. Pregnancy was determined by percent weight gain by gestational day 12. VPA sodium salt (Sigma-Aldrich, St. Louis, MO) was dissolved in 0.9% sterile saline, pH 7.4. Pregnant dams received a single intraperitoneal injection of 400 mg/kg VPA (VPA-exposed) or sterile saline (SAL-exposed) on gestational day 12.5. Each female was housed individually and raised her own litter. For this study, two SAL-exposed and two VPA-exposed litters were used. Pups were weaned on postnatal day 21.
The following experiments were performed in SAL- (n = 5) and VPA-exposed (n = 6) juvenile male and female rats (∼70 -80 g). Tail malformations that manifest as a distal bend or a kink have been reported as a marker of VPA toxicity during development.39,51,52 Only VPA-exposed rats with tail malformations were used in this study (see next paragraphs). All animals were housed with a littermate under a 12:12 h, reverse light:dark cycle with free access to food and water for the duration of the experiment.
Surgery
Surgical implantation of the telemeter and EEG/electro-myographic (EMG) electrodes were according to previously published protocols53–57 with the following modifications. Animals were anesthetized using isofluorane and placed in a stereotaxic frame to secure the head. A bi-potential-lead transmitter (TL11M2-F20-EET, Data Sciences International (DSI), New Brighton, MN) was implanted subcutaneously through a dorsal incision of the abdominal region. Another incision was made along the midline of the head and neck to expose the skull and neck muscle. Two burr holes (0.5 mm diameter) were drilled asymmetrically and two dental screws (Plastics One, Roanoke, VA) were implanted into the skull at 2.0 mm anterior/+1.5 mm lateral and 6.0 mm posterior/-1.5 mm lateral from bregma. The EEG leads were attached to the screws and secured with dental cement. The two EMG leads were implanted in the dorsal cervical neck muscle approximately 1.0 mm apart and sutured into place. The skin along the head was sutured and the dorsal incision was stapled closed with wound clips. Animals were treated with antibiotic ointment and topical lidocaine, as well as 0.1-cc buprenorphine, postoperatively, and allowed 7 days to recover before being reunited with their non-experimental cagemate.
Data Acquisition and Analysis
All recordings took place in a designated room shielded from noise or other background disturbances. EEG and EMG waveform data for postnatal days 31–34 were collected using Dataquest ART 4.0 software (DSI) set to a continuous sampling mode. Sleep is typically measured as a function of EEG and EMG activity. For each of the animals, the 24-h (12:12 light:dark cycle) period on postnatal day 32 was scored using NeuroScore software (DSI). EEG/EMG waveforms were scored according to visual inspection of 5-sec epochs into wake (low-amplitude, high-frequency EEG combined with high-amplitude EMG), NREM (high-amplitude, low-frequency EEG combined with low EMG tone) or REM (low-amplitude, high-frequency EEG combined with muscle atonia and occasional muscle twitches). Transitions were scored when four or more epochs of the new state were noted within another stage. This rule was followed unless there was a period punctuated by a new state, which then persisted; this was considered a transition period so these epochs were scored as the new state.
The scored epochs were summed over the 12-h light phase, 12-h dark phase, and 24-h period and reported as the total time (in min) spent in each state. Within each 12-h phase, the average bout duration (in sec) and the number of each bout and transition were also determined for each vigilance state.
The DSI module for periodogram powerbands in NeuroScore was used to generate the mean power of frequencies present in the three vigilance states (wake, NREM sleep, and REM sleep) across the light and dark. The periodograms estimate the power for each defined frequency band (delta: 0.5-4 Hz, theta: 4-8 Hz, alpha: 8-12 Hz, sigma: 12-16 Hz, beta: 16-24, and gamma: 30-80 Hz) on the fast Fourier transform (FFT) designed for continuous data. The relative power band value (percentage; desired band over total power in the signal) was determined in 1-h bins and then averaged across each 12-h phase for each vigilance state.
Western Blotting
Following the recording period, on postnatal day 40, brains were collected and frozen on dry ice. Sections (180 μm) were cut on a cryostat and micropunches were collected from the somatosensory cortex and BF of each animal, as well as three extra age-matched SAL-controls (total SAL n = 7). The tissue was then homogenized via sonication in a cell lysis buffer containing a protease inhibitor cocktail. The protein concentration for each sample was determined using a bicinchoninic acid (BCA) assay kit (Pierce, Rockford, IL). Protein (1 μg) of each sample (SAL = 7, VPA = 6) was loaded into a 10% Tris-glycine sodium dodecyl sulfate polyacrylamide gel electrophoresis gel (Invitrogen, Carlsbad, CA), and then the electrophoresed proteins were blotted onto a polyvinyl difluoride membrane (Invitrogen). The membrane was washed in 20 mM Tris-buffered saline solution with 0.05% Tween 20 (T-TBS). The membrane was blocked overnight in 5% powdered milk at 4 oC and incubated the next day in the anti-GAD 65/67 primary antibody solution (1:30,000 in T-TBS; AB1511, Millipore, Billerica, MA) for 2 h at room temperature. Following the primary antibody incubation, the membrane was washed three times in T-TBS, and incubated for 1 h at room temperature in an anti-rabbit immunoglobulin G, horseradish peroxidase (HRP)-linked secondary antibody solution (1:2000 in T-TBS; #7074, Cell Signaling Technologies, Danvers, MA). The Phototype-HRP chemiluminescent system (Cell Signaling Technology) was used for detection of the protein recognized by the antisera. To correct for errors in sample loading, the membrane was also probed with an antibody to the housekeeping enzyme glyceraldehyde- 3-phosphate dehydrogenase (GAPDH; 1:1,000,000; MAB374, Millipore) as previously described.54,58,59 Membrane was exposed to Hyperfilm-ECL (Kodak, Rochester, NY) for varying exposure times. The films were then scanned into a computer at 1,200 dpi and analyzed using ImageJ64 software (http://rsbweb.nih.gov/ij/download.html). The optical densities (o.d.) were measured for each individual band and normalized to the o.d. of the GAPDH bands.
Statistical Analysis
Statistical differences between SAL- and VPA-exposed animals were determined with Student t-tests for each sleep parameter studied. Differences in the power spectrum densities were determined with a Mann-Whitney U test for nonpara-metric data. A two-way repeated measures analysis of variance (ANOVA) was used to analyze wake gamma power across the 24-h period. Two-way ANOVAs were used to analyze NREM sleep and REM sleep gamma power across the 24-h period because these sleep states were not represented in every hour. Student t-tests were used comparing o.d. for SAL- and VPA-exposed tissue. One-way ANOVA followed by a Newman-Keuls post hoc test was used to compare the o.d. values for SAL- and VPA-exposed GAD expression by tail malformation severity.
RESULTS
VPA Exposure Increases Wakefulness at the Expense of Sleep in Juvenile Rats
Nocturnal adult rodents, like the Sprague-Dawley rat, cycle through bouts of sleep (NREM and REM) and wake in both the dark (active) and light (quiescent) phases. Typically, a higher percentage of accumulated sleep occurs in the light phase with consolidated bouts of wake occurring in the dark phase. Visual inspection of hypnograms generated from the scored EEG traces from SAL- and VPA-exposed animals suggested the juvenile controls (SAL-exposed) followed a normal adult-like pattern with consolidated bouts of wake primarily present in the dark phase. In contrast, the VPA-exposed rats appeared to experience consolidated bouts of wake in both the light and dark phases (Figure 1).
Figure 1.

Representative hypnograms of juvenile rats exposed to saline (SAL, top) and valproic acid (VPA, bottom) in utero across a 24-h period. W, wake; N, NREM sleep; R, REM sleep. The solid black bar is highlighting consolidated wake bouts during the light phase of VPA-exposed rats, which are absent in the SAL-exposed controls. Gray area is lights off.
Quantitative analysis of the scored traces for the total time spent in NREM, REM, and wake revealed significant differences between the SAL- and VPA-exposed groups. Across the 12-h of the light phase, the VPA-exposed animals spent significantly more time in wake (∼35 min; t9 = 2.741, P = 0.023; Figure 2A) with no significant differences in the dark phase (t9 = 0.788, P > 0.05). Moreover, across the 24-h light:dark period, VPA-exposed rats again spent significantly more time in wake with the difference increasing to approximately 48 min compared to the SAL-exposed controls (t9 = 2.382, P = 0.041; Figure 2A).
Figure 2.
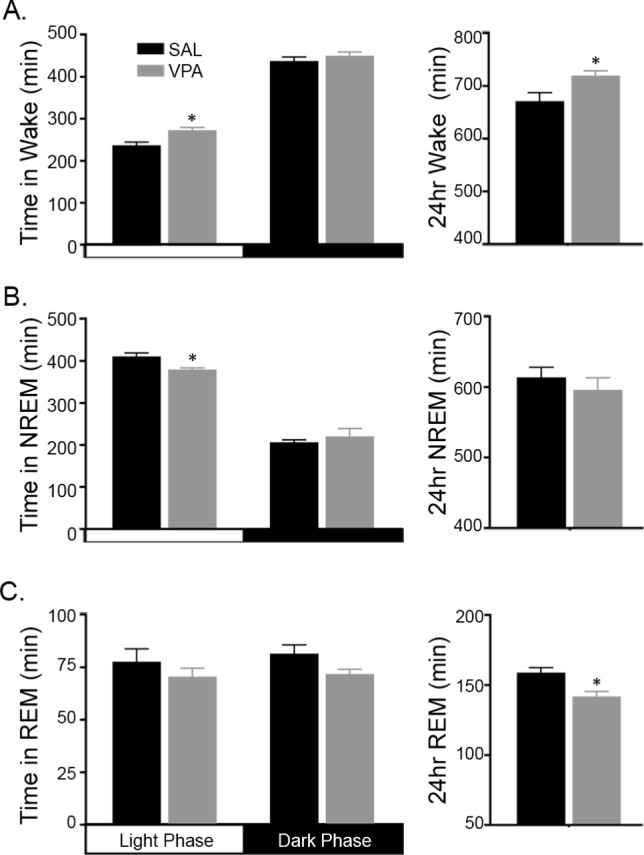
Juvenile rats exposure to valproic acid (VPA) spend more time awake than asleep compared to saline (SAL)-exposed controls. (A) During the light phase, VPA-exposed rats spend more time in wake (∼35 min) compared to SAL-exposed rats. Across the 24-h period, VPA-exposed rats spend ∼48 min more in wake than SAL-exposed controls. (B) VPA exposure reduced non-rapid eye movement (NREM) sleep time by ∼30 min during the light phase only. (C) Rapid eye movement (REM) sleep during the individual light and dark phases was not affected by VPA exposure, however; the 24-h REM sleep total is reduced by ∼17 min in the VPA-exposed juveniles compared to SAL-exposed controls. * P < 0.05 versus SAL-exposed controls. Data are represented as mean ± standard error of the mean.
As a consequence of the increased wake time in the light phase, total sleep time (TST; sum of the time spent in NREM and REM sleep) decreased significantly in the VPA-exposed animals by approximately 38 min compared to controls (t9 = 3.126, P = 0.012; data not shown). Changes in NREM sleep accounted for the overall difference in TST. In the light phase, the VPA-exposed animals spent significantly less time in NREM sleep (∼30 min; t9 = 2.621, P = 0.028; Figure 2B) with no significant differences in the dark phase (t9 = 0.553, P = 0.594), whereas REM sleep was unaffected by VPA-exposure in either the light or dark phase (Figure 2C; t9 = 0.920, P = 0.382, light phase and t9 = 1.905, P = 0.09, dark phase). In contrast to individual 12-h phases, the 24-h total of REM sleep revealed that VPA-exposed animals spent significant less time in REM sleep (∼17 min) compared to the controls (t9 = 2.799, P = 0.021; Figure 2C).
VPA Exposure Disrupts the Pattern of Juvenile Sleep/Wake Cycles
In utero exposure to VPA markedly changed the sleep/wake architecture (mean bout duration, bout number, and number of transitions into Wake, NREM, or REM sleep) compared to controls. VPA exposure significantly increased the mean duration of a wake bout in the light and dark phases by ∼117% and ∼70%, respectively, compared to the SAL-exposed animals (t9 = 6.966, P < 0.0001, light phase and t9 = 2.552, P < 0.031, dark phase; Figure 3A). The increase in bout duration was associated with a significant decrease in the number of wake bouts in the light phase only (t9 = 6.532, P = 0.0001, Figure 3A).
Figure 3.
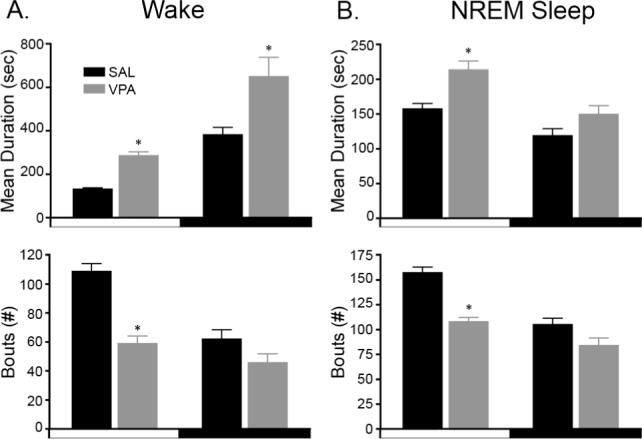
Valproic acid (VPA)-exposed juvenile rats have more consolidated sleep and wake behaviors. (A-top) During both the light and dark phases, VPA-exposed rats have, on average, longer bouts of wakefulness. (A-bottom) The increased duration of wake bouts is associated with a decrease in the number of wake bouts during the light phase only. (B-top) Non-rapid eye movement (NREM) sleep bouts, on average, are substantially increased in VPA-exposed rats as well during the light phase. (B-bottom) This increase in NREM bout duration is also associated with a decrease in the number of NREM bouts during the light phase. * P < 0.05 versus SAL-exposed controls. Data are represented as mean ± standard error of the mean.
For sleep, only NREM sleep demonstrated significant changes in architecture. Curiously, in the light phase, VPA exposure significantly increased the average duration of a NREM sleep bout by ∼36% (t9 = 3.517, P = 0.007; Figure 3B) whereas the number of NREM bouts was significantly reduced by ∼32% compared to SAL-exposed animals (t9 = 6.646, P < 0.0001, Figure 3B). For REM sleep, there were no significant differences in the mean bout duration or bout number in either phase (data not shown).
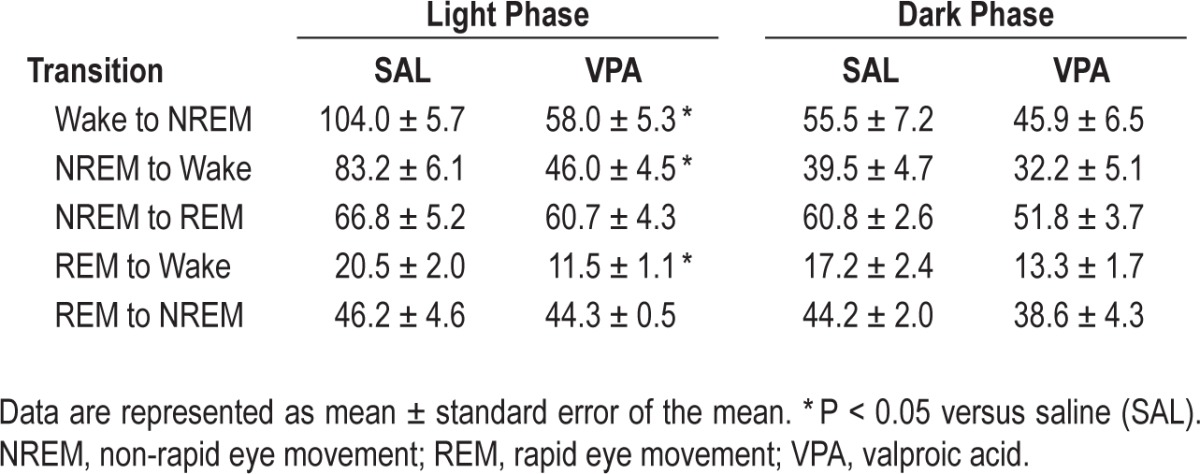
To assess whether VPA exposure affects the stability of maintaining a wake or sleep state, the number of transitions into and out of wake, NREM sleep, and REM sleep were quantified across the light and dark phases. In the light phase, the number of transitions to and from wake was significantly reduced by approximately 50% in VPA-exposed animals compared to the SAL- exposed animals (Table 1; wake to NREM: t9 = 6.460, P = 0.0001, NREM to wake: t9 = 6.509, P = 0.0001, and REM to wake: t9 = 3.484, P = 0.007). No significant differences were observed in the dark phase or when transitioning between sleep states (Table 1).
Table 1.
Number of transitions to and from each vigilance state during the light and dark phase
VPA Exposure Results in Changes to the Power Spectra
Power spectral analysis was used to study the cortical EEG of the juvenile rats exposed to VPA or SAL in utero during the light and dark phase. We also investigated differences in power in wake, NREM sleep, and REM sleep. In the spectral analysis of all frequencies in the continuous EEG, VPA-exposure reduced the theta power in both the 12-h light and dark phases (Table 2, P < 0.05). When we calculated the mean power for each frequency band in for the individual vigilance states (wake, NREM sleep, and REM sleep) we found that VPA exposure reduced midrange frequencies while increasing higher range frequencies, primarily in wake and REM sleep; the two states characterized by EEG desynchrony (Figure 4A, 4C, and Table 3). In all vigilance states, VPA exposure decreased theta band densities (4-8 Hz) in both the light phase (wake: U = 0.0, P = 0.004; NREM: U = 2.0, P = 0.017; REM: U = 0.0, P = 0.004; Figure 4A-C) and the dark phase (wake: U = 2.0, P = 0.017; NREM: U = 3.0, P = 0.03; REM: U = 0.0, P = 0.004); Figure 4A-C). Most notable was the approximate 20% decrease in theta band density during REM sleep regardless of phase (Figure 4C). Alpha band densities (8-12 Hz) were also significantly decreased during wake in the light (10%) and dark (13%) phases in VPA-exposed rats compared to SAL-exposed controls (Table 3; light: U = 1.0, P = 0.009; dark: U = 0.0, P = 0.004).
Table 2.
Averaged power spectra (% total power) in the light and dark phase
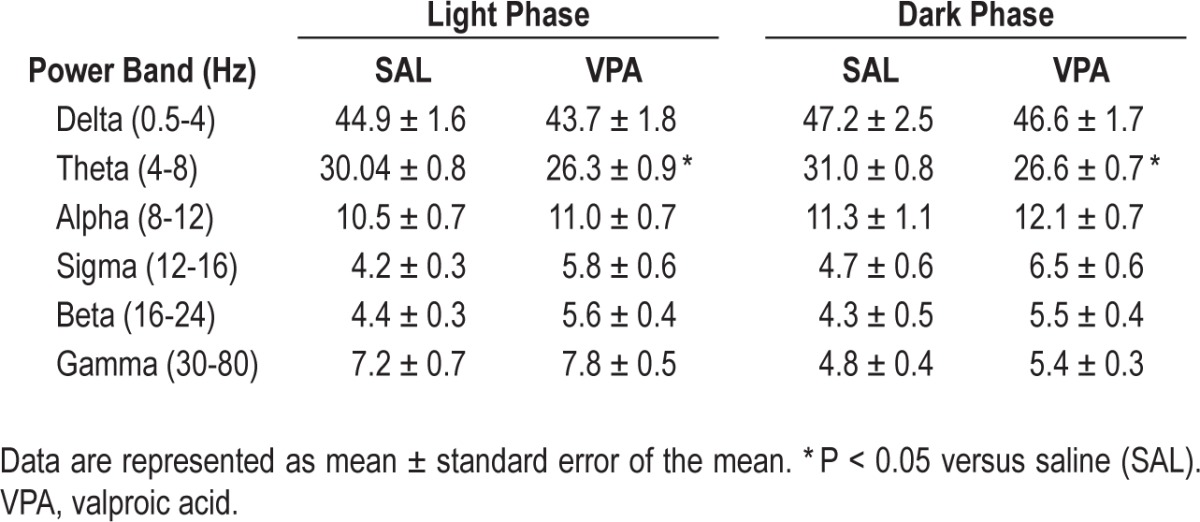
Figure 4.

Valproic acid (VPA) exposure in utero reduced theta power but increased gamma power compared to saline (SAL). The average theta power in (A) wake, (B) non-rapid eye movement (NREM) sleep, and (C) rapid eye movement (REM) sleep is reduced during both the light and dark phases of VPA-exposed rats. In both the light and dark phases, gamma power is increased during (A) wake and (C) REM sleep in VPA-exposed rats compared to SAL-exposed controls. Gamma power (1-h bins) across the 24-h analysis period is significantly increased in VPA-exposed animals during (D) wake and (F) REM sleep bouts but not (E) NREM sleep bouts. * P < 0.05 versus SAL-exposed controls. Data are represented as mean ± standard error of the mean.
Table 3.
Mean power (% total power) for alpha, sigma, and beta frequency bands
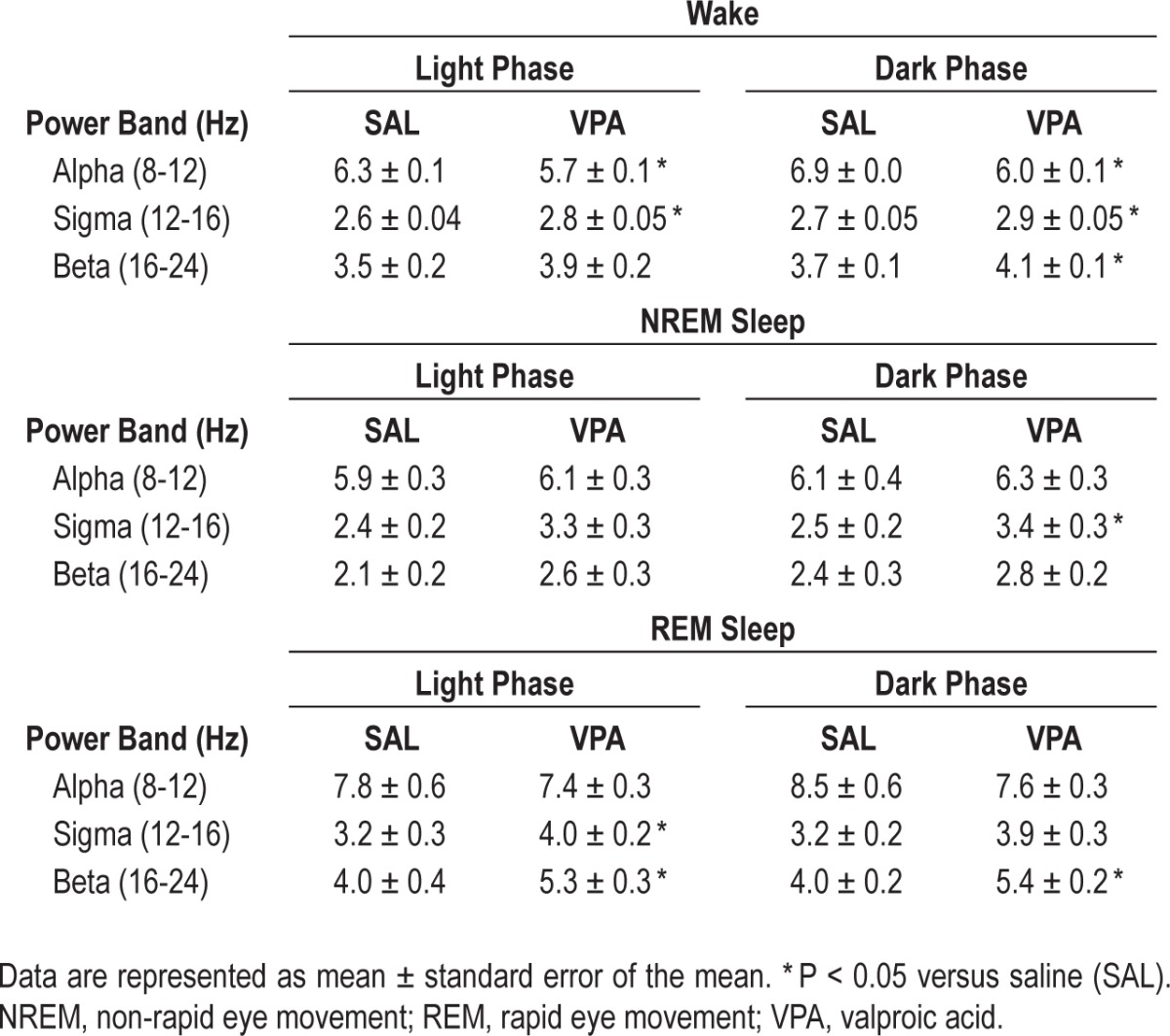
Conversely, VPA exposure increased high frequency gamma band densities (30-80 Hz) during wake and REM sleep in the light phase (Figure 4A and 4C; wake: U = 1.0, P = 0.009; REM: U = 0.0, P = 0.004) and dark phase (wake: U = 0.0, P = 0.004; REM: U = 0.0, P = 0.004) compared to the SAL-exposed controls. Interestingly, the greatest magnitude of change in the frequency spectrum occurred in REM sleep gamma power in the light (∼50%) and dark (∼37%) phases. Sigma and beta also increased, albeit less reliably, during wake and sleep, primarily REM sleep, in the light phase (Table 3; wake: sigma, U = 0.0, P = 0.004; beta, U = 4.0, P = 0.052, and REM: sigma, U = 4.0, P = 0.052; beta: U = 3.0, P = 0.03) and dark phase (Table 3; wake: sigma, U = 3.0, P = 0.03; beta, U = 0.0, P = 0.004; NREM: sigma. U = 4.0, P = 0.052; and REM: beta, U = 3.0, P = 0.004) in VPA-exposed animals. Again, the most prominent changes were during REM sleep; there was an approximate 30% change in beta and 22% change in sigma.
Because spectral analysis removes time as a factor, it is difficult to address whether changes in the mean spectral powers are caused by the power itself or longer/shorter durations of the vigilance states. To address this, we specifically analyzed the mean gamma power by hour -across the 24-h period. There was a main effect (Figure 4D; F1,23 = 14.50; P = 0.004) of in utero exposure on wake gamma power across the 24-h period. Similarly, there was a main effect (Figure 4F; F1,23 = 147.9; P < 0.0001) of in utero exposure on REM sleep gamma power across the 24-h period. VPA-exposed animals had elevated gamma power across the 24-h period during wake and REM sleep bouts compared to SAL-exposed animals. There was no significant difference in NREM sleep gamma power across the 24-h period (Figure 4E).
VPA Exposure Results in Changes to the Cortical GABAergic System
In the current study, we observed varying degrees of tail malformations in the VPA-exposed animals that included barely discernable changes (Figure 5A; arrows) or kinks and bends (Figure 5A; asterisks). Western blot analysis of protein isolated from primary somatosensory cortex micropunches from all animals revealed no differences in the cortical GABAergic system in VPA-treated animals compared to SAL-treated controls (Figure 5B and 5C; GAD67: t11 = 0.5943, P > 0.05; GAD65: t11 = 0.9328; P > 0.05). Interestingly, the o.d. values for the two VPA-exposed animals with the barely discernable tail malformations (n = 2, circled in Figure 5C) appeared more similar to the controls. Thus, if the VPA-exposed animals were regrouped according to the severity of the tail malformations (minor, M and severe, S), then both isoforms of GAD (65 and 67) were significantly reduced in VPA-exposed animals with severe tail malformations (GAD 67: F2,10 = 6.294, P = 0.02; GAD 65: F2,10 = 4.616, P = 0.04) compared to SAL (Figure 5C; Newman-Keuls post hoc GAD 67: P = 0.04; GAD 65: P = 0.04) and to VPA-exposed animals with minor tail malformations (Figure 5C; Newman-Keuls post hoc GAD 67: P = 0.006; GAD 65: P = 0.02). There were no changes in GAD expression in the BF (data not shown; GAD 65: SAL-1.76 ± 0.67, VPA-2.07 ± 0.14, t9 = 1.204, P > 0.05; GAD 67: SAL-0.61 ± 0.07, VPA-0.69 ± 0.05, t9 = 0.870, P > 0.05).
Figure 5.
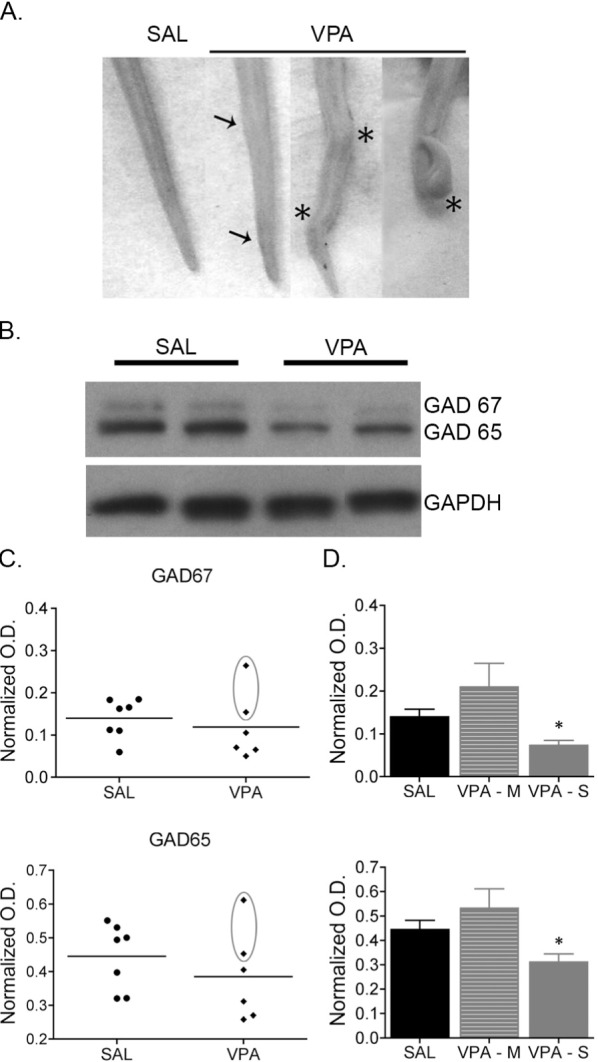
Cortical expression of GAD65 and GAD67 is reduced in valproic acid (VPA)-exposed juvenile rats. (A) Images of the tails of juvenile rats exposed to either saline (SAL) or VPA. The tails of VPA-exposed rats had varying degrees of malformations, including barely discernable changes (arrows; referred to as minor) to more severe kinks and bends (asterisks; referred to as severe). (B) Representative immunoblots from micropunches of the primary somatosensory cortex of SAL- and VPA-exposed rats. (C) Plots of the individual optical density (o.d.) values from SAL and VPA exposed animals. The circled points are values from VPA-exposed animals with minor tail malformations. (D) GAD65 and GAD67 protein expression is reduced in VPA-exposed rats with severe tail malformations compared to SAL-exposed controls and VPA-exposed animals with minor tail malformations. * P < 0.05 versus SAL-exposed controls and VPA-exposed animals with minor tail malformations. Data are represented as mean ± standard error of the mean.
DISCUSSION
In this study, sleep and wake behaviors as well as cortical EEG power spectral frequencies were analyzed in an animal model of ASD. We found that animals exposed to VPA in utero displayed consolidated bouts of arousal during the expected sleep phase compared to SAL-exposed controls. Perhaps equally exciting is our finding that changes in the cortical spectral densities and cortical GABAergic system correlate with this enhanced behavioral arousal. Together, our data suggest the occurrence of developmental changes in the neurocircuitry and/ or neurochemistry intimately involved in sleep-wake behavior in a teratogenic model of ASD.
Sleep in ASD
Studies investigating sleep in children with ASD have ranged from parent-reported and sleep diary studies to more objective studies including actigraphy, and polysomnographic (PSG) studies. Commonly, these studies indicate that children with ASD suffer from insomnia, particularly difficulties initiating and maintaining sleep, and parasomnias.8,11–21 Parents report that bedtime routines are often challenging, which may lead to delayed sleep onset and increase sleep anxiety.8,13,15,16,18,19,21,60 Both questionnaire and PSG studies show that children with ASD acquire less total sleep compared to typically developing children, awaken frequently during the night and remain awake for periods of time, and overall experience less efficient sleep.8,13,18,21,61 More specifically, sleep spindles62 and the time spent in stage 3 NREM63 and REM13,64 sleep are decreased in ASD subjects compared to control subjects.
Parental reports indicate that children's sleep problems negatively affect both the children's and the families' daily functioning.16 Sleep disruptions in children with ASD are correlated with their daytime behavior, and the quality of their sleep may predict the severity of their behaviors.14,24 In fact, treating the sleep problems has been shown to alleviate the severity of some of the characteristic behaviors associated with ASD.65–67
Our findings are consistent with the clinical findings of sleep disruptions and patterns reported in the ASD phenotypes. Sprague-Dawley rats are nocturnal and acquire most sleep during their quiescent, or light, phase. In our model, in utero VPA exposure increases wakefulness at the expense of sleep, specifically NREM sleep, during the light phase. Once awake, these VPA-exposed animals maintain consolidated wake bouts compared to SAL-exposed controls. These consolidated, lengthy periods of wake during sleep mirrors the behavior seen in ASD.
GABAergic Dysfunction in ASD and Potential Effect on Sleep
Sleep is a complex behavior that requires cooperative actions of multiple brain regions. The GABAergic system is involved in the onset and regulation of sleep.47 There are reciprocal GABAergic connections between major sleep-promoting regions, such as preoptic area, and wake-promoting areas, such as the lateral hypothalamus, which control the transitioning between vigilance states.68 There is also a strong GABAergic innervation of the cortex by neurons in the BF, which influence cortical output and behavior.48,69 Together, these GABAergic networks orchestrate the balance between sleep and wake states.
Like our behavioral findings, the decreases in GAD 65 and GAD 67 protein expression in the VPA-exposed rats compared to SAL are consistent with the clinical findings. Surprisingly, these differences in GAD65 and GAD67 protein expression were specific to the cortex and not found in the BF. Although we anticipated decreases in GAD 65 and GAD 67 in the BF, because of its role in the control of vigilance states, it is possible that our micropunches limited our analysis of GAD expression in this region. Micropunches only contain the components within the local circuit. GAD 65 is localized mainly to synaptic terminals, whereas GAD 67 is found both in cell bodies and in terminals.70,71 Thus, the origin of the GAD in the current cortical punch technique cannot distinguish between GAD in terminal regions of BF projection neurons or cortical somas. Nevertheless, reduced cortical GAD 65/67 will result in decreased GABA production and ultimately decreased inhibitory tone, which will have a direct effect on EEG oscillations and may affect behavioral outputs. Ongoing laboratory studies are investigating GAD 65/67 protein in critical sleep nuclei to determine whether mechanisms controlling the onset and maintenance of sleep are disrupted in this model.
In ASD, there is clear evidence that the GABAergic system is substantially disrupted. GABAergic receptor expression and binding in various brain regions are substantially reduced in ASD.72–75 Furthermore, GAD 65/67 messenger RNA and protein levels are reduced in parietal cortex and cerebellum postmortem tissue from individuals who had an ASD.76–78 Because GAD is the enzyme responsible for the synthesis of GABA, it is likely that there is also a reduction in inhibitory signals. A recent in vivo study of children with ASD reported that GABA concentrations are reduced in the frontal cortex, leading to a lower [GABA]/[glutamate] ratio as measured by 3-T MRI.79 Such findings are indicative of cortical disinhibition, which contribute to a more aroused state.
Excitatory/Inhibitory Imbalance in ASD
Imbalance in the excitatory/inhibitory (E/I) ratio has been implicated in the etiology of ASD.80–82 Because balanced excitation and inhibition among local neural circuits is critical for the stable functioning of these circuits, it is plausible that this E/I imbalance can disrupt the delicate interplay between brain regions involved in complex behaviors, such as communication, sensory processing, social behaviors, and even sleep. Many studies support this notion that an imbalance, particularly an elevation, in the E/I ratio may underlie behavioral deficits associated with ASD. The abundance of evidence implicating an aberrant GABAergic system in ASD, described previously, could suggest that the inhibitory tone in ASD brains is reduced. Furthermore, various studies have linked ASD83–86 and ASD-like behaviors in animal models87–90 to a hyperexcited brain.
Spectral Analysis of EEG in ASD
Clinically, spectral analysis of EEG in ASD children has consistently revealed abnormal EEG characteristics. Recently in preclinical models, it has been shown that increasing the E/I ratio via optogenetic manipulations in the medial prefrontal cortex increases high-frequency activity, specifically in the gamma range.85 Higher frequency bands, especially gamma (30-80 Hz) frequencies, are associated with cognition, attention, and sensory processing.86,87 Elevated E/I ratio also impairs social behavior and conditioning,85 behavioral deficits observed in ASD, and animal models of ASD. In our study, VPA exposure increases high-frequency EEG bands in the sigma, beta, and gamma range during wake and REM sleep. The decreases in GAD 65/67 protein expression in the cortex may influence the EEG frequency bands such that lower inhibitory tone (GABA) would lead to increased higher frequency oscillations.
Additionally, several clinical findings consistently demonstrate a reduction in the alpha power in children with ASD compared to controls.88–91 In humans, the alpha bands have been associated with suppression of nonsalient inputs allowing for selective attention.92,93 Our current data demonstrate that VPA-exposed rats also have significantly less alpha power during wake. This finding further supports the use of this model for future investigations, not only into the mechanisms underlying changes in sleep but also changes in EEG oscillations in ASD.
Circadian Disruptions in ASD
Sleep disruptions, especially those related to the timing of sleep, may be caused by disruptions in circadian rhythm. Many studies indicate that children with ASD may also suffer from circadian rhythm disorders.1,93 The pineal gland, whose activity is regulated by the master pacemaker, secretes melatonin during the night. The rhythm of melatonin secretion as well as the amount is a proxy for circadian rhythmicity and duration of sleep acquired. In ASD, many studies report that melatonin levels, as well as its metabolite, are reduced.94–99 Studies also show that melatonin is effective in treating some sleep disruptions in ASD.65,67,100,101 Our data and that of others suggest there are also circadian disruptions in the VPA model of ASD. Here, we show that prenatal VPA exposure decreases the amount of time animals spend asleep and increases wakefulness during the animals' sleep phase. These data support the findings by Tsujino and colleagues46 that VPA exposure increases animals' locomotor and feeding behaviors during the quiescent phase compared to saline controls. Although melatonin levels have not been measured in the VPA model, chronic melatonin treatment has recently been shown to be effective at restoring social interaction, phosphorylation of calcium/calmodulin-dependent protein kinase II, N-methyl-d-aspartate receptor 1, and protein kinase A, as well as long-term potentiation deficits in the hippo-campus of VPA-treated rats.102
Consequence to Sleep Loss
Sleep loss can have a significant effect on a child's overall health and quality of daily life.22,23 As in adults, sleep loss in children and adolescents not only leads to fatigue but can also negatively impact physiological functions, mood, cognition, and behavior.22,103,104 Insufficient sleep can cause poor academic performance,104,105 increase irritability and aggressiveness, impair attention, and lead to hyperactivity.22,106 In fact, sleep problems are often associated with behavioral problems seen in attention deficit hyperactivity disorders (ADHD).107,108 Insufficient sleep is also associated with depression and anxiety.25,26,109 Children with a diagnosis of ASD share similar symptoms110 and disrupted sleep may lead to these symptoms or exacerbate ASD symptomology.14,24 Loss of REM sleep during critical periods during development can have profound effects on the developing brain, particularly the development on the sensory systems.111 Adolescence is a time of brain maturation and reorganization112; therefore, it is possible that insufficient REM sleep during this period can alter further brain maturation in children with ASD.
CONCLUSION
Surprisingly, animal models of ASD have been underutilized in advancing our understanding of the etiology of sleep disruptions in ASD. Our findings suggest that juvenile rats exposed to VPA in utero display sleep behavior similar to that seen in children with ASD. These similarities validate the use of this model to further investigate whether there are biological mechanisms underlying these changes in sleep or if they are consequences ASD symptomology.
DISCLOSURE STATEMENT
This was not an industry supported study. Support was provided by a DOD Research Program AR080087 awarded to Dr. Mong. The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors would like to thank Shaun S. Viechweg and Michael A. Castello for their technical assistance.
REFERENCES
- 1.Cortesi F, Giannotti F, Ivanenko A, Johnson K. Sleep in children with autistic spectrum disorder. Sleep Med. 2010;11:659–64. doi: 10.1016/j.sleep.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Johnson K, Giannotti F, Cortesi F. Sleep patterns in autism spectrum disorders. Child Adolesc Psychiatr Clin N Am. 2009;18:917–28. doi: 10.1016/j.chc.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Johnson KP, Malow BA. Sleep in children with autism spectrum disorders. Curr Neurol Neurosci Rep. 2008;8:155–61. doi: 10.1007/s11910-008-0025-y. [DOI] [PubMed] [Google Scholar]
- 4.Malow BA. Sleep disorders, epilepsy, and autism. Ment Retard Dev Disabil Res Rev. 2004;10:122–5. doi: 10.1002/mrdd.20023. [DOI] [PubMed] [Google Scholar]
- 5.Reynolds AM, Malow BA. Sleep and autism spectrum disorders. Pediatr Clin North Am. 2011;58:685–98. doi: 10.1016/j.pcl.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Kotagal S, Broomall E. Sleep in children with autism spectrum disorder. Pediatr Neurol. 2012;47:242–51. doi: 10.1016/j.pediatrneurol.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Johnson CR. Sleep problems in children with mental retardation and autism. Child Adolesc Psychiatr Clin N Am. 1996;5:673–683. [Google Scholar]
- 8.Miano S, Bruni O, Elia M, et al. Sleep in children with autistic spectrum disorder: a questionnaire and polysomnographic study. Sleep Med. 2007;9:64–70. doi: 10.1016/j.sleep.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Richdale AL, Prior MR. The sleep/wake rhythm in children with autism. Eur Child Adolesc Psychiatry. 1995;4:175–86. doi: 10.1007/BF01980456. [DOI] [PubMed] [Google Scholar]
- 10.Schreck KA, Mulick JA. Parental report of sleep problems in children with autism. J Autism Dev Disord. 2000;30:127–35. doi: 10.1023/a:1005407622050. [DOI] [PubMed] [Google Scholar]
- 11.Elia M, Ferri R, Musumeci SA, et al. Sleep in subjects with autistic disorder: a neurophysiological and psychological study. Brain Dev. 2000;22:88–92. doi: 10.1016/s0387-7604(99)00119-9. [DOI] [PubMed] [Google Scholar]
- 12.Gail Williams P, Sears LL, Allard A. Sleep problems in children with autism. J Sleep Res. 2004;13:265–8. doi: 10.1111/j.1365-2869.2004.00405.x. [DOI] [PubMed] [Google Scholar]
- 13.Giannotti F, Cortesi F, Cerquiglini A, Vagnoni C, Valente D. Sleep in children with autism with and without autistic regression. J Sleep Res. 2011;20:338–47. doi: 10.1111/j.1365-2869.2010.00882.x. [DOI] [PubMed] [Google Scholar]
- 14.Hoffman CD, Sweeney DP, Gilliam JE, Apodaca DD, Lopez-Wagner MC, Castillo MM. Sleep problems and symptomology in children with autism. Focus on Autism and Other Developmental Disabilities. 2005;20:194–200. [Google Scholar]
- 15.Hoffman CD, Sweeney DP, Gilliam JE, Lopez-Wagner MC. Sleep problems in children with autism and in typically developing children. Focus on Autism and Other Developmental Disabilities. 2006;21:146–52. [Google Scholar]
- 16.Krakowiak P, Goodlin-Jones B, Hertz-Picciotto I, Croen LA, Hansen RL. Sleep problems in children with autism spectrum disorders, developmental delays, and typical development: a population-based study. J Sleep Res. 2008;17:197–206. doi: 10.1111/j.1365-2869.2008.00650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Limoges E, Mottron L, Bolduc C, Berthiaume C, Godbout R. Atypical sleep architecture and the autism phenotype. Brain. 2005;128:1049–61. doi: 10.1093/brain/awh425. [DOI] [PubMed] [Google Scholar]
- 18.Malow BA, Marzec ML, McGrew SG, Wang L, Henderson LM, Stone WL. Characterizing sleep in children with autism spectrum disorders: a multidimensional approach. Sleep. 2006;29:1563–71. doi: 10.1093/sleep/29.12.1563. [DOI] [PubMed] [Google Scholar]
- 19.Patzold LM, Richdale AL, Tonge BJ. An investigation into sleep characteristics of children with autism and Asperger's Disorder. J Paediatr Child Health. 1998;34:528–33. doi: 10.1046/j.1440-1754.1998.00291.x. [DOI] [PubMed] [Google Scholar]
- 20.Wiggs L, Stores G. Sleep patterns and sleep disorders in children with autistic spectrum disorders: insights using parent report and actigraphy. Dev Med Child Neurol. 2004;46:372–80. doi: 10.1017/s0012162204000611. [DOI] [PubMed] [Google Scholar]
- 21.Souders MC, Mason TB, Valladares O, et al. Sleep behaviors and sleep quality in children with autism spectrum disorders. Sleep. 2009;32:1566–78. doi: 10.1093/sleep/32.12.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dahl RE. The impact of inadequate sleep on children's daytime cognitive function. Semin Pediatr Neurol. 1996;3:44–50. doi: 10.1016/s1071-9091(96)80028-3. [DOI] [PubMed] [Google Scholar]
- 23.Maski KP, Kothare SV. Sleep deprivation and neurobehavioral functioning in children. Int J Psychophysiol. 2013;89:259–64. doi: 10.1016/j.ijpsycho.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 24.Schreck KA, Mulick JA, Smith AF. Sleep problems as possible predictors of intensified symptoms of autism. Res Dev Disabil. 2004;25:57–66. doi: 10.1016/j.ridd.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Chorney DB, Detweiler MF, Morris TL, Kuhn BR. The interplay of sleep disturbance, anxiety, and depression in children. J Pediatr Psychol. 2008;33:339–48. doi: 10.1093/jpepsy/jsm105. [DOI] [PubMed] [Google Scholar]
- 26.Gregory AM, Caspi A, Eley TC, Moffitt TE, O'Connor TG, Poulton R. Prospective longitudinal associations between persistent sleep problems in childhood and anxiety and depression disorders in adulthood. J Abnorm Child Psychol. 2005;33:157–63. doi: 10.1007/s10802-005-1824-0. [DOI] [PubMed] [Google Scholar]
- 27.Polimeni MA, Richdale AL, Francis AJ. A survey of sleep problems in autism, Asperger's disorder and typically developing children. J Intellect Disabil Res. 2005;49:260–8. doi: 10.1111/j.1365-2788.2005.00642.x. [DOI] [PubMed] [Google Scholar]
- 28.Richdale AL. Sleep problems in autism: prevalence, cause, and intervention. Dev Med Child Neurol. 1999;41:60–6. doi: 10.1017/s0012162299000122. [DOI] [PubMed] [Google Scholar]
- 29.Datta S, Hobson JA. The rat as an experimental model for sleep neurophysiology. Behav Neurosci. 2000;114:1239–44. doi: 10.1037//0735-7044.114.6.1239. [DOI] [PubMed] [Google Scholar]
- 30.Christianson AL, Chesler N, Kromberg JG. Fetal valproate syndrome: clinical and neuro-developmental features in two sibling pairs. Dev Med Child Neurol. 1994;36:361–9. doi: 10.1111/j.1469-8749.1994.tb11858.x. [DOI] [PubMed] [Google Scholar]
- 31.Kini U. Fetal valproate syndrome: a review. Paediatr Perinat Drug Ther. 2006;7:123–30. [Google Scholar]
- 32.Nicolai J, Vles JSH, Aldenkamp AP. Neurodevelopmental delay in children exposed to antiepileptic drugs in utero: A critical review directed at structural study-bias. J Neurol Sci. 2008;271:1–14. doi: 10.1016/j.jns.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Asher O. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod Toxicol. 2009;28:1–10. doi: 10.1016/j.reprotox.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 34.Rasalam AD, Hailey H, Williams JHG, et al. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev Med Child Neurol. 2005;47:551–5. doi: 10.1017/s0012162205001076. [DOI] [PubMed] [Google Scholar]
- 35.Williams G, King J, Cunningham M, Stephan M, Kerr B, Hersh JH. Fetal valproate syndrome and autism: additional evidence of an association. Dev Med Child Neurol. 2001;43:202–6. [PubMed] [Google Scholar]
- 36.Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci. 2005;23:183–7. doi: 10.1016/j.ijdevneu.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 37.Binkerd PE, Rowland JM, Nau H, Hendrickx AG. Evaluation of valproic acid (VPA) developmental toxicity and pharmacokinetics in Sprague-Dawley rats. Fundam Appl Toxicol. 1988;11:485–93. doi: 10.1016/0272-0590(88)90112-1. [DOI] [PubMed] [Google Scholar]
- 38.Ingram JL, Peckham SM, Tisdale B, Rodier PM. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol Teratol. 2000;22:319–24. doi: 10.1016/s0892-0362(99)00083-5. [DOI] [PubMed] [Google Scholar]
- 39.Kim KC, Kim P, Go HS, et al. The critical period of valproate exposure to induce autistic symptoms in Sprague-Dawley rats. Toxicol Lett. 2011;201:137–42. doi: 10.1016/j.toxlet.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 40.Miyazaki K, Narita N, Narita M. Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: implication for pathogenesis of autism. Int J Dev Neurosci. 2005;23:287–97. doi: 10.1016/j.ijdevneu.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 41.Rodier PM, Ingram JL, Tisdale B, Croog VJ. Linking etiologies in humans and animal models: studies of autism. Reprod Toxicol. 1997;11:417–22. doi: 10.1016/s0890-6238(97)80001-u. [DOI] [PubMed] [Google Scholar]
- 42.Rodier PM, Ingram JL, Tisdale B, Nelson S, Romano J. Embryological origin for autism: developmental anomalies of the cranial nerve motor nuclei. J Comp Neurol. 1996;370:247–61. doi: 10.1002/(SICI)1096-9861(19960624)370:2<247::AID-CNE8>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 43.Schneider T, Przewlocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology. 2005;30:80–9. doi: 10.1038/sj.npp.1300518. [DOI] [PubMed] [Google Scholar]
- 44.Schneider T, Roman A, Basta-Kaim A, et al. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology. 2008;33:728–40. doi: 10.1016/j.psyneuen.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 45.Schneider T, Ziolkowska B, Gieryk A, Tyminska A, Przewlocki R. Prenatal exposure to valproic acid disturbs the enkephalinergic system functioning, basal hedonic tone, and emotional responses in an animal model of autism. Psychopharmacology. 2007;193:547–55. doi: 10.1007/s00213-007-0795-y. [DOI] [PubMed] [Google Scholar]
- 46.Tsujino N, Nakatani Y, Seki Y, et al. Abnormality of circadian rhythm accompanied by an increase in frontal cortex serotonin in animal model of autism. Neurosci Res. 2007;57:289–95. doi: 10.1016/j.neures.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 47.Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–42. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gritti I, Mainville L, Mancia M, Jones BE. GABAergic and other noncholinergic basal forebrain neurons, together with cholinergic neurons, project to the mesocortex and isocortex in the rat. J Comp Neurol. 1997;383:163–77. [PubMed] [Google Scholar]
- 49.Henny P, Jones BE. Projections from basal forebrain to prefrontal cortex comprise cholinergic, GABAergic and glutamatergic inputs to pyramidal cells or interneurons. Eur J Neurosci. 2008;27:654–70. doi: 10.1111/j.1460-9568.2008.06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones BE. From waking to sleeping: neuronal and chemical substrates. Trends Pharmacol Sci. 2005;26:578–86. doi: 10.1016/j.tips.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 51.Ehlers K, Sturje H, Merker HJ, Nau H. Valproic acid-induced spina bifida: a mouse model. Teratology. 1992;45:145–54. doi: 10.1002/tera.1420450208. [DOI] [PubMed] [Google Scholar]
- 52.Gogolla N, Leblanc JJ, Quast KB, Sudhof TC, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord. 2009;1:172–81. doi: 10.1007/s11689-009-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cusmano DM, Hadjimarkou MM, Mong JA. Gonadal steroid modulation of sleep and wakefulness in male and female rats is sexually differentiated and neonatally organized by steroid exposure. Endocrinology. 2014;155:204–14. doi: 10.1210/en.2013-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hadjimarkou MM, Benham R, Schwarz JM, Holder MK, Mong JA. Estradiol suppresses rapid eye movement sleep and activation of sleep-active neurons in the ventrolateral preoptic area. Eur J Neurosci. 2008;27:1780–92. doi: 10.1111/j.1460-9568.2008.06142.x. [DOI] [PubMed] [Google Scholar]
- 55.Schwartz MD, Mong JA. Estradiol suppresses recovery of REM sleep following sleep deprivation in ovariectomized female rats. Physiol Behav. 2011;104:962–71. doi: 10.1016/j.physbeh.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwartz MD, Mong JA. Estradiol modulates recovery of REM sleep in a time-of-day-dependent manner. Am J Physiol Regul Integr Comp Physiol. 2013;305:R271–80. doi: 10.1152/ajpregu.00474.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McDowell KA, Hadjimarkou MM, Viechweg S, et al. Sleep alterations in an environmental neurotoxin-induced model of parkinsonism. Exp Neurol. 2010;226:84–9. doi: 10.1016/j.expneurol.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blutstein T, Baab PJ, Zielke HR, Mong JA. Hormonal modulation of amino acid neurotransmitter metabolism in the arcuate nucleus of the adult female rat: a novel action of estradiol. Endocrinology. 2009;150:3237–44. doi: 10.1210/en.2008-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blutstein T, Devidze N, Choleris E, Jasnow AM, Pfaff DW, Mong JA. Oestradiol up-regulates glutamine synthetase mRNA and protein expression in the hypothalamus and hippocampus: implications for a role of hormonally responsive glia in amino acid neurotransmission. J Neuroendocrinol. 2006;18:692–702. doi: 10.1111/j.1365-2826.2006.01466.x. [DOI] [PubMed] [Google Scholar]
- 60.Giannotti F, Cortesi F, Cerquiglini A, et al. An investigation of sleep characteristics, EEG abnormalities and epilepsy in developmentally regressed and non-regressed children with autism. J Autism Dev Disord. 2008;38:1888–97. doi: 10.1007/s10803-008-0584-4. [DOI] [PubMed] [Google Scholar]
- 61.Malow B, McGrew S. Sleep disturbances and autism. Sleep Med Clin. 2008;3:479–88. [Google Scholar]
- 62.Vriend JL, Corkum PV, Moon EC, Smith IM. Behavioral interventions for sleep problems in children with autism spectrum disorders: current findings and future directions. J Pediatr Psychol. 2011;36:1017–29. doi: 10.1093/jpepsy/jsr044. [DOI] [PubMed] [Google Scholar]
- 63.Maes JHR, Eling PATM, Wezenberg E, Vissers CTWM, Kan CC. Attentional set shifting in autism spectrum disorder: Differentiating between the role of perseveration, learned irrelevance, and novelty processing. J Clin Exper Neuropsychol. 2011;33:210–7. doi: 10.1080/13803395.2010.501327. [DOI] [PubMed] [Google Scholar]
- 64.Buckley AW, Rodriguez AJ, Jennison K, et al. Rapid eye movement sleep percentage in children with autism compared with children with developmental delay and typical development. Arch Pediatr Adolesc Med. 2010;164:1032–7. doi: 10.1001/archpediatrics.2010.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malow B, Adkins KW, McGrew SG, et al. Melatonin for sleep in children with autism: a controlled trial examining dose, tolerability, and outcomes. J Autism Dev Disord. 2012;42:1729–37. doi: 10.1007/s10803-011-1418-3. author reply 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malow BA, McGrew SG, Harvey M, Henderson LM, Stone WL. Impact of treating sleep apnea in a child with autism spectrum disorder. Pediatr Neurol. 2006;34:325–8. doi: 10.1016/j.pediatrneurol.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 67.Paavonen EJ, Nieminen-von Wendt T, Vanhala R, Aronen ET, von Wendt L. Effectiveness of melatonin in the treatment of sleep disturbances in children with Asperger disorder. J Child Adolesc Psychopharmacol. 2003;13:83–95. doi: 10.1089/104454603321666225. [DOI] [PubMed] [Google Scholar]
- 68.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 69.Gritti I, Manns ID, Mainville L, Jones BE. Parvalbumin, calbindin, or calretinin in cortically projecting and GABAergic, cholinergic, or glutamatergic basal forebrain neurons of the rat. J Comp Neurol. 2003;458:11–31. doi: 10.1002/cne.10505. [DOI] [PubMed] [Google Scholar]
- 70.Kaufman D, Houser C, Tobin A. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J Neurochem. 1991;56:720–3. doi: 10.1111/j.1471-4159.1991.tb08211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Esclapez M, Tillakaratne N, Kaufman D, Tobin A, Houser C. Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J Neurosci. 1994;14:1834–55. doi: 10.1523/JNEUROSCI.14-03-01834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fatemi SH, Folsom TD, Reutiman TJ, Thuras PD. Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum. 2009;8:64–9. doi: 10.1007/s12311-008-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD. GABA(A) Receptor downregulation in brains of subjects with autism. J Autism Dev Disord. 2009;39:223–30. doi: 10.1007/s10803-008-0646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord. 2001;31:537–43. doi: 10.1023/a:1013238809666. [DOI] [PubMed] [Google Scholar]
- 75.Collins AL, Ma D, Whitehead PL, et al. Investigation of autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics. 2006;7:167–74. doi: 10.1007/s10048-006-0045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry. 2002;52:805–10. doi: 10.1016/s0006-3223(02)01430-0. [DOI] [PubMed] [Google Scholar]
- 77.Yip J, Soghomonian JJ, Blatt GJ. Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol. 2007;113:559–68. doi: 10.1007/s00401-006-0176-3. [DOI] [PubMed] [Google Scholar]
- 78.Yip J, Soghomonian JJ, Blatt GJ. Decreased GAD65 mRNA levels in select subpopulations of neurons in the cerebellar dentate nuclei in autism: an in situ hybridization study. Autism Res. 2009;2:50–9. doi: 10.1002/aur.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harada M, Taki MM, Nose A, et al. Non-Invasive evaluation of the GABAergic/glutamatergic system in autistic patients observed by MEGA-editing proton MR spectroscopy using a clinical 3 Tesla instrument. J Autism Dev Disord. 2011;41:447–54. doi: 10.1007/s10803-010-1065-0. [DOI] [PubMed] [Google Scholar]
- 80.Hussman JP. Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J Autism Dev Disord. 2001;31:247–8. doi: 10.1023/a:1010715619091. [DOI] [PubMed] [Google Scholar]
- 81.Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–67. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pizzarelli R, Cherubini E. Alterations of GABAergic Signaling in autism spectrum disorders. Neural Plasticity. 2011;2011 doi: 10.1155/2011/297153. 297153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gomot M, Belmonte MK, Bullmore ET, Bernard FA, Baron-Cohen S. Brain hyper-reactivity to auditory novel targets in children with high-functioning autism. Brain. 2008;131:2479–88. doi: 10.1093/brain/awn172. [DOI] [PubMed] [Google Scholar]
- 84.Dichter GS, Felder JN, Bodfish JW. Autism is characterized by dorsal anterior cingulate hyperactivation during social target detection. Soc Cogn Affect Neurosci. 2009;4:215–26. doi: 10.1093/scan/nsp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Orekhova EV, Stroganova TA, Nygren G, et al. Excess of high frequency electroencephalogram oscillations in boys with autism. Biol Psychiatry. 2007;62:1022–9. doi: 10.1016/j.biopsych.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 86.Rojas DC, Maharajh K, Teale P, Rogers SJ. Reduced neural synchronization of gamma-band MEG oscillations in first-degree relatives of children with autism. BMC Psychiatry. 2008;8:66. doi: 10.1186/1471-244X-8-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Han S, Tai C, Westenbroek RE, et al. Autistic-like behaviour in Scn1a+/−mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489:385–90. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin HC, Gean PW, Wang CC, Chan YH, Chen PS. The amygdala excitatory/inhibitory balance in a valproate-induced rat autism model. PLoS One. 2013;8:e55248. doi: 10.1371/journal.pone.0055248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rinaldi T, Kulangara K, Antoniello K, Markram H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc Natl Acad Sci U S A. 2007;104:13501–6. doi: 10.1073/pnas.0704391104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yizhar O, Fenno LE, Prigge M, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–8. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Engel AK, Fries P, Singer W. Dynamic predictions: oscillations and synchrony in top-down processing. Nat Rev Neurosci. 2001;2:704–16. doi: 10.1038/35094565. [DOI] [PubMed] [Google Scholar]
- 92.Wang XJ. Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev. 2010;90:1195–268. doi: 10.1152/physrev.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Glickman G. Circadian rhythms and sleep in children with autism. Neurosci Biobehav Rev. 2010;34:755–68. doi: 10.1016/j.neubiorev.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 94.Kulman G, Lissoni P, Rovelli F, Roselli MG, Brivio F, Sequeri P. Evidence of pineal endocrine hypofunction in autistic children. Neuroendocrinol Lett. 2000;21:31–4. [PubMed] [Google Scholar]
- 95.Tordjman S, Anderson GM, Pichard N, Charbuy H, Touitou Y. Nocturnal excretion of 6-sulphatoxymelatonin in children and adolescents with autistic disorder. Biol Psychiatry. 2005;57:134–8. doi: 10.1016/j.biopsych.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 96.Nir I, Meir D, Zilber N, Knobler H, Hadjez J, Lerner Y. Brief report: circadian melatonin, thyroid-stimulating hormone, prolactin, and cortisol levels in serum of young adults with autism. J Autism Dev Disord. 1995;25:641–54. doi: 10.1007/BF02178193. [DOI] [PubMed] [Google Scholar]
- 97.Melke J, Goubran Botros H, Chaste P, et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol Psychiatry. 2008;13:90–8. doi: 10.1038/sj.mp.4002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leu RM, Beyderman L, Botzolakis EJ, Surdyka K, Wang L, Malow BA. Relation of melatonin to sleep architecture in children with autism. J Autism Dev Disord. 2011;41:427–33. doi: 10.1007/s10803-010-1072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tordjman S, Anderson GM, Bellissant E, et al. Day and nighttime excretion of 6-sulphatoxymelatonin in adolescents and young adults with autistic disorder. Psychoneuroendocrinology. 2012;37:1990–7. doi: 10.1016/j.psyneuen.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 100.Giannotti F, Cortesi F, Cerquiglini A, Bernabei P. An open-label study of controlled-release melatonin in treatment of sleep disorders in children with autism. J Autism Dev Disord. 2006;36:741–52. doi: 10.1007/s10803-006-0116-z. [DOI] [PubMed] [Google Scholar]
- 101.Cortesi F, Giannotti F, Sebastiani T, Panunzi S, Valente D. Controlled-release melatonin, singly and combined with cognitive behavioural therapy, for persistent insomnia in children with autism spectrum disorders: a randomized placebo-controlled trial. J Sleep Res. 2012;21:700–9. doi: 10.1111/j.1365-2869.2012.01021.x. [DOI] [PubMed] [Google Scholar]
- 102.Tian Y, Yabuki Y, Moriguchi S, et al. Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J Pineal Res. 2014;56:1–11. doi: 10.1111/jpi.12081. [DOI] [PubMed] [Google Scholar]
- 103.O'Brien LM. The neurocognitive effects of sleep disruption in children and adolescents. Child Adolesc Psychiatr Clin N Am. 2009;18:813–23. doi: 10.1016/j.chc.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 104.Dewald JF, Meijer AM, Oort FJ, Kerkhof GA, Bogels SM. The influence of sleep quality, sleep duration and sleepiness on school performance in children and adolescents: A meta-analytic review. Sleep Med Rev. 2010;14:179–89. doi: 10.1016/j.smrv.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 105.Wolfson AR, Carskadon MA. Understanding adolescents' sleep patterns and school performance: a critical appraisal. Sleep Med Rev. 2003;7:491–506. doi: 10.1016/s1087-0792(03)90003-7. [DOI] [PubMed] [Google Scholar]
- 106.Fallone G, Owens JA, Deane J. Sleepiness in children and adolescents: clinical implications. Sleep Med Rev. 2002;6:287–306. doi: 10.1053/smrv.2001.0192. [DOI] [PubMed] [Google Scholar]
- 107.Perfect MM, Archbold K, Goodwin JL, Levine-Donnerstein D, Quan SF. Risk of behavioral and adaptive functioning difficulties in youth with previous and current sleep disordered breathing. Sleep. 2013;36:517–25B. doi: 10.5665/sleep.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bonuck K, Freeman K, Chervin RD, Xu LZ. Sleep-disordered breathing in a population-based cohort: behavioral outcomes at 4 and 7 years. Pediatrics. 2012;129:E857–E65. doi: 10.1542/peds.2011-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gregory AM, Rijsdijk FV, Dahl RE, McGuffin P, Eley TC. Associations between sleep problems, anxiety, and depression in twins at 8 years of age. Pediatrics. 2006;118:1124–32. doi: 10.1542/peds.2005-3118. [DOI] [PubMed] [Google Scholar]
- 110.Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383:896–910. doi: 10.1016/S0140-6736(13)61539-1. [DOI] [PubMed] [Google Scholar]
- 111.Graven S. Sleep and brain development. Clin Perinatol. 2006;33:693–706. vii. doi: 10.1016/j.clp.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 112.Arain M, Haque M, Johal L, et al. Maturation of the adolescent brain. Neuropsychiatr Dis Treat. 2013;9:449–61. doi: 10.2147/NDT.S39776. [DOI] [PMC free article] [PubMed] [Google Scholar]