

Abstract
Plasmid DNA serves as a simple and easily modifiable form of antigen delivery for vaccines. The USDA approval of DNA vaccines for several non-human diseases underscores the potential of this type of antigen delivery method as a cost-effective approach for the treatment or prevention of human diseases, including cancer. However, while DNA vaccines have demonstrated safety and immunological effect in early phase clinical trials, they have not consistently elicited robust anti-tumor responses. Hence many recent efforts have sought to increase the immunological efficacy of DNA vaccines, and we have specifically evaluated several target antigens encoded by DNA vaccine as treatments for human prostate cancer. In particular, we have focused on SSX2 as one potential target antigen, given its frequent expression in metastatic prostate cancer. We have previously identified two peptides, p41–49 and p103–111, as HLA-A2-restricted SSX2-specific epitopes. In the present study we sought to determine whether the efficacy of a DNA vaccine could be enhanced by an altered peptide ligand (APL) strategy wherein modifications were made to anchor residues of these epitopes to enhance or ablate their binding to HLA-A2. A DNA vaccine encoding APL modified to increase epitope binding elicited robust peptide-specific CD8+ T cells producing Th1 cytokines specific for each epitope. Ablation of one epitope in a DNA vaccine did not enhance immune responses to the other epitope. These results demonstrate that APL encoded by a DNA vaccine can be used to elicit increased numbers of antigen-specific T cells specific for multiple epitopes simultaneously, and suggest this could be a general approach to improve the immunogenicity of DNA vaccines encoding tumor antigens.
Keywords: SSX, CTA, Prostate cancer, DNA vaccine, APL
1. Introduction
Plasmid DNA delivered by direct injection was demonstrated to be an effective means of gene transfer in rodent studies over two decades ago [1]. Subsequently, it was demonstrated that this could elicit antigen-specific CD8+ T cells in multiple model systems due to the presentation of the encoded antigen, expressed by a eukaryotic promoter, via the endogenous antigen presentation pathway [2]. In fact, plasmid DNA vaccines have been approved by the USDA for the treatment of West Nile virus in horses, and infectious hematopoietic necrosis virus in salmon [3,4]. The first anti-tumor DNA vaccine was approved in the U.S. in 2010 for the treatment of canine melanoma based on results from non-randomized clinical trials demonstrating safety and improved survival compared to historical controls [5,6]. With the demonstration that immune responses to an encoded antigen, and CD8+ cytolytic T cells in particular, could be elicited in larger mammals, DNA vaccines as a therapeutic treatment for cancer have entered human clinical trials. Despite many phase I trials that have demonstrated safety and immunological efficacy, few have demonstrated robust immune responses with a limited number of vaccinations, few have demonstrated marked anti-tumor responses, and hence few have progressed to phase II trials [7]. This has been largely attributed to the low efficiency of plasmid DNA transfer to antigen-presenting cells, the low immunogenicity of this approach relative to other methods of antigen transfer such as by viral immunization, and potentially due to the antigens targeted. Consequently, many recent studies have sought to identify methods able to increase the efficiency of plasmid DNA gene transfer and/or immunogenicity of the antigen.
We have been specifically interested in developing vaccines as treatments for human prostate cancer. Recently the Food and Drug Administration (FDA) approved the first anti-tumor vaccine for castrate-resistant prostate cancer, Sipuleucel-T (Provenge®, Dendreon Corp.). This autologous cellular therapy, loaded ex vivo with a prostate tumor antigen (prostatic acid phosphatase, PAP) fused to GM-CSF, was found to confer a survival benefit in patients with castrate-resistant metastatic disease over placebo [8]. Unfortunately, the high cost and cumbersome production of this autologous cellular therapy have limited its widespread implementation. We have demonstrated that a DNA vaccine encoding this same PAP antigen was safe and immunologically effective in eliciting PAP-specific T cells in patients with recurrent prostate cancer [9,10]. However, immune responses were not detected in all individuals, suggesting that modifications to improve the immunogenicity of DNA vaccines might be advantageous.
Several other antigens have been evaluated as targets encoded by DNA vaccines in clinical trials for patients with prostate cancer [11,12]. To date, however, the optimal targets for vaccines remain undefined and controversial. As other potential targets for anti-tumor vaccines, we have been evaluating a family of cancer-testis antigens (CTA), the synovial sarcoma chromosome X breakpoint (SSX) proteins [13–18]. We have specifically focused on SSX2, which we found is expressed in ~25% of metastatic prostate cancer lesions [18], and immune responses to which we have observed in patients with prostate cancer [14,17]. As a CTA this protein likely has limited central tolerance since its expression in normal tissues is restricted to immune-privileged testis tissue [19]. We previously identified two HLA-A2-restricted SSX2 epitopes (p41–49 and p103–111), T cells specific for which can lyse SSX2-expressing prostate cancer cells, and we demonstrated that HLA-A2 transgenic mice immunized with a DNA vaccine encoding SSX2 develop p41–49- and p103–111-specific T cells [17,18].
In the current study we sought to determine whether modifications to a plasmid DNA vaccine targeting SSX2, encoding altered MHC class I epitopes, could augment the frequency and efficacy of antigen-specific CD8+ T cells with cytolytic activity. We first identified altered peptide ligands (APL) for SSX2 with greater or reduced HLA-A2 binding. We then sought to establish whether immunization directly with these peptides resulted in cross-reactive CTL specific for the native epitopes, and whether DNA vaccines encoding these altered peptides could be used to elicit these responses in vivo. Finally, we sought to determine whether multiple modifications, encoding epitopes with increased or decreased MHC binding, would result in broad epitope-specific CD8+ T cells with effector function. We found that modification to both epitopes, to increase MHC class I binding, elicited the greatest number of antigen-specific CD8+ T cells to each epitope, with a Th1-biased cytokine phenotype. These findings suggest that this approach could be generally applied as a means to increase the immunological efficacy of plasmid DNA vaccines.
2. Materials and methods
2.1. Mice
HLA-A2.01/HLA-DR1-expressing, murine MHC class I/II knockout transgenic mice (HHDII-DR1) on C57Bl/6 background were obtained from Charles River Labs (France) courtesy of Dr. François Lemonnier [20]. Mice were maintained in microisolater cages under aseptic conditions and all experimental procedures were conducted under an Institutional Animal Care and Use Committee-approved protocol.
2.2. HLA-A2 T2 binding affinity assays
Modified peptides were designed from the amino acid sequence of SSX2 to have either reduced or enhanced affinity for HLA-A2 in the SYFPEITHI and BIMAS prediction algorithms [21,22]. These peptides were synthesized, and the purity and identity of each peptide was confirmed by mass spectrometry and gas chromatography (United Biochemical Research, Seattle, WA) [23]. Peptide binding affinities were assessed with TAP-deficient/HLA-A2+ T2 cells as described previously [17,23].
2.3. Plasmid DNA vaccine constructs
A DNA vaccine encoding SSX2 (pTVG-SSX2) was described previously [17]. Modifications to the DNA vaccine were made by site-directed mutagenesis using the Phusion™ Site-Directed Mutagenesis Kit (New England BioLabs (Ipswich, MA, USA)). Amplified products were ligated, cloned, and sequence verified using standard molecular biology techniques. pTVG4, pTVG-SSX2, and modified pTVG-SSX2 plasmids were purified from E. coli using the Endo-free Plasmid Giga Kit (Qiagen, Valencia, CA).
2.4. Peptide and DNA immunization of HHDII-DR1 mice
For peptide vaccinations, 4–6 week-old HHDII-DR1 mice were immunized subcutaneously with 100 μg of an individual peptide in complete Freund’s adjuvant (Sigma, St. Louis, MO). Mice were euthanized seven days later and spleens were collected, processed through a mesh screen, and splenocytes were isolated by centrifugation after red blood cell osmotic lysis with ammonium chloride/potassium chloride lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) [17]. For DNA vaccinations, 4–6 week-old HHDII-DR1 mice were immunized intradermally in the ear pinna with 100 μg of DNA plasmid. Mice were immunized at 14-day intervals. Mice were immunized two to six times, six times having been previously found to elicit robust immune responses with the native pTVG-SSX2 DNA vaccine [17]. Fewer immunizations were given, in a purposefully suboptimal fashion, to detect differences in magnitude of immune response to the subdominant epitope (using four immunizations) or to the dominant epitope (using two immunizations). Two weeks after the last immunization, mice were euthanized and spleens were collected and processed as described above.
2.5. IFNγ ELISPOT
Interferon (IFN)-γ enzyme-linked immunosorbent spot (ELISPOT) was performed according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN) as previously described [9,23]. Dried plates were counted with an automated plate reader (Autoimmun Diagnostika). The number of spots was corrected for media alone negative control, and reported as the mean number of peptide-specific IFNγ spot-forming units (SFU) per 106 splenocytes. All assays were conducted in triplicate, and were conducted with splenocytes without prior in vitro stimulation.
2.6. Cytotoxicity assay – lactate dehydrogenase release
Splenocytes from immunized animals were evaluated for cytolytic activity toward peptide-pulsed T2 cells and LNCaP human prostate cancer cells transfected to constitutively express HLA-A2 (gift of Dr. Lawrence Fong) as previously described [23].
2.7. Intracellular cytokine staining
Splenocytes from immunized animals were pooled by treatment group, enriched for CD8+ T cells (Stemcell Technologies, Vancouver, BC, Canada), and stimulated in vitro with 2 μg/mL of individual SSX2 peptide (or 40 ng/mL phorbol 12-myristate 13-acetate (PMA, Sigma) and 2.6 μg/mL ionomycin (MP Biomedicals, Santa Ana, CA) as a positive control) in RPMI medium, supplemented as above, for 6 (PMA/Ionomycin) or 24 h (peptide stimulation). Following 6 h of treatment with 1.5 μM monensin, intracellular cytokine staining was performed as per the manufacturer’s protocol (Cytofix/Cytoperm Kit, BD Biosciences). Antibodies included anti-CD3-CV510, anti-CD4-AF700, anti-CD8-PECy5, anti-IFNγ-PerCP-Cy5.5, anti-TNFα-PECy7, anti-IL-2-APC, anti-IL-4-BV421, anti-IL-10-FITC, anti-IL-17-APC-Cy7, or corresponding fluorescently labeled IgG controls (eBiocience, San Diego, CA).
3. Results
3.1. Modified SSX2 peptides designed to have enhanced affinity for HLA-A2 have increased affinity compared to native peptides
We have reported previously that SSX2 peptides p41–49, p57–65, p99–107, and p103–111 have affinity for HLA-A2 [17]. But while all four of these peptides were found to bind HLA-A2 in vitro, studies in HHDII-DR1 transgenic mice (expressing HLA-A2) revealed that only p41–49 and p103–111 were HLA-A2-restricted epitopes, T cells specific for which were able to lyse an HLA-A2-expressing LNCaP prostate cancer cell line [17]. We wished to evaluate whether the T-cell responses to any or all four of the previously identified HLA-A2-binding SSX2 peptides could be enhanced by modifying the anchor residues of the peptides for HLA-A2 binding, while maintaining recognition of the native peptide residues by T cells, and whether the immunological efficacy of a DNA vaccine could be improved by encoding these modified epitopes. Using two MHC-peptide binding algorithms, BIMAS [21], and SYFPEITHI [22], we designed homologous peptides for p41–49, p57–65, p99–107, and p103–111 with predicted increased or ablated HLA-A2 binding affinity (Table 1). Using T2 binding assays, we found that all peptides designed to have enhanced binding to HLA-A2 were able to stabilize the surface expression of HLA-A2 on T2 cells to a higher degree than the native peptides (Fig. 1), while peptides designed to ablate HLA-A2 affinity showed no HLA-A2 stabilization above vehicle control.
Table 1.
Predicted HLA-A2 affinity of modified SSX2 peptides.
| Name | Sequence | BIMAS (t1/2 min) | SYFPEITHI (arbitrary units) | Purpose |
|---|---|---|---|---|
| p41 | KASEKIFYV | 1017 | 22 | Native peptide |
| p41-AV | KVSEKIFYV | 6407 | 22 | Enhances |
| p41-AL | KLSEKIFYV | 73,228 | 28 | Enhances |
| p41-VP | KASEKIFYP | 0.218 | 12 | Reduces |
| p57 | AMTKLGFKA | 20 | 16 | Native peptide |
| p57-AV | AMTKLGFKV | 291 | 22 | Enhances |
| p57-AP | AMTKLGFKP | 0.06 | 12 | Reduces |
| p99 | MTFGRLQGI | 2 | 20 | Native peptide |
| p99-TI | MIFGRLQGI | 20 | 24 | Enhances |
| p99-TP | MPFGRLQGI | 0.9 | 16 | Reduces |
| p103 | RLQGISPKI | 10 | 23 | Native peptide |
| p103-RF | FLQGISPKI | 48 | 24 | Enhances |
| p103-IV | RLQGISPKV | 69 | 25 | Enhances |
| p103-IP | RLQGISPKP | 0.01 | 15 | Reduces |
Shown are the peptide names, amino acid sequences, and predicted HLA-A2 affinity scores of modified SSX2 peptides. Highlighted are the amino acid changes to the anchor residues of the native peptide designed to enhance or reduce binding to HLA-A2. Predicted HLA-A2 peptide affinities were determined using two different peptide prediction algorithms (BIMAS and SYFPEITHI) [21,22].
Fig. 1.

Modified SSX2 peptides have enhanced or reduced HLA-A2 affinity in vitro compared to native peptides. Shown is the relative HLA-A2 binding affinity of each modified SSX2 peptide as determined by mean fluorescence intensity (MFI) of HLA-A2 stabilization on T2 cells. Values were measured in triplicate, ± SD, for each peptide and normalized to the MFI of a no-antigen vehicle control. Results are representative of two independent experiments. A known HLA-A2-binding peptide derived from the influenza A matrix protein (GILGFVFTL, Flu) was used as a positive control for comparison.
3.2. HHDII-DR1 mice immunized with modified SSX2 peptides develop enhanced peptide-specific immune responses and CTL capable of lysing prostate cancer cells
To determine whether modified SSX2 peptides could elicit enhanced, cross-reactive peptide-specific immune responses, immunization studies were carried out in HHDII-DR1 mice. Animals were immunized once with either a native peptide (p41–49, p57–65, p99–107, or p103–111) or a modified peptide. Splenocytes were collected from these animals and tested for the frequency of peptide-specific immune responses by IFNγ ELISPOT. As shown in Fig. 2, animals immunized with the peptides designed to enhance HLA-A2 binding for p41–49 (Fig. 2A) and p103–111 (Fig. 2D) developed higher frequencies of peptide-specific T cells compared to animals immunized with the native peptides. These T cells recognized the modified immunizing peptide, but could also cross-recognize the native epitopes as assessed by IFNγ release following peptide stimulation detected by ELISPOT (Fig. 2A and D). Animals immunized with modified p57–65 (Fig. 2B) or p99–107 (Fig. 2C) peptides did not develop peptide-specific T cells, suggesting that these peptides are simply not immunogenic despite their affinity for HLA-A2.
Fig. 2.

HHDII-DR1 mice immunized with modified peptides p41–49 and p103–111 develop increased peptide-specific immune responses. Splenocytes from individual mice immunized once with either a native SSX2 peptide, an HLA-A2 enhanced-binding peptide, or an HLA-A2 reduced-binding peptide were analyzed for the frequency of peptide-specific immune responses by IFNγ ELISPOT. Splenocytes were evaluated for responses to each of the immunizing peptides or concanavalin A (ConA) positive control. The influenza A matrix protein HLA-A2 epitope (GILGFVFTL, Flu) was evaluated as a negative control. Each colored symbol represents the frequency of peptide-specific IFNγ-secreting responses from an individual mouse for the modified p41–49 group (panel A), modified p57–65 group (panel B), modified p99–107 group (panel C), and modified p103–111 group (panel D), with n = 4–8 mice per group.
Splenocytes isolated from peptide-immunized animals were tested for their ability to recognize and lyse peptide-pulsed target cells and SSX2-expressing cancer cells. We found that animals immunized with p41–49 were unable to lyse prostate cancer cell lines, however splenocytes from animals immunized with the modified peptide p41–49-AL or p41–49-AV (not shown) were able to lyse peptide-pulsed T2 target cells as well as the SSX2-expressing HLA-A2+ LNCaP prostate cancer cell line (Fig. 3). Lysis was abrogated in the presence of an HLA-A2 blocking antibody, demonstrating that peptide p41–49 is an HLA-A2-restricted epitope presented by LNCaP cells. Splenocytes from animals immunized with the native p103–111 peptide or the modified p103–111-RF peptide were similarly able to lyse both peptide-pulsed target cells and the LNCaP cell line in an HLA-A2-restricted fashion. Of note, we have previously demonstrated that SSX2 is expressed in LNCaP cells, albeit at low levels [14,18]. Hence, differences in % lysis observed with the p103- and p41-specific T cells may be related to differences in presentation of each native epitope. Not unexpectedly, no CTL lysis was observed using splenocytes isolated from animals immunized with native or enhanced-binding p57–99 or p99–107 peptides (data not shown). These results suggest that peptides p41–49 and p103–111 are the only SSX2-specific HLA-A2-restricted epitopes, CTL specific for these epitopes are capable of lysing an SSX2-expressing tumor cell line in an HLA-A2-restricted manner, and p103–111 is a dominant epitope. Additionally, in the case of peptide p41–49, it was advantageous to immunize with the APL, potentially due to increasing the number, avidity and/or effector function of p41–49-specific cytolytic T cells with this APL immunization.
Fig. 3.

HHDII-DR1 mice immunized with modified SSX2 peptides develop CTL capable of lysing peptide-pulsed target cells and the LNCaP cancer cell line. Splenocytes from HHDII-DR1 mice vaccinated with native SSX2 peptides p41–49 (panel A), p103–111 (panel C) or modified SSX2 peptides p41-AL (panel B) or p103-RF (panel D), were stimulated with the native peptide for five days and tested for specific lysis of peptide-pulsed T2 target cells (left panels), the HLA-A2+ LNCaP prostate cancer cell line, or the LNCaP cell line pre-incubated with an HLA-A2 blocking antibody (right panels). Shown are the means and standard deviations of percent specific lysis at three effector-to-target ratios. Data is from individual mice and representative of multiple independent experiments.
3.3. HHDII-DR1 mice immunized with SSX2 plasmid DNA vaccines encoding APL with increased HLA-A2 binding develop epitope-specific immune responses at higher frequency than animals immunized with the native vaccine
Using single peptides as vaccines there is no competition at the level of the antigen-presenting cell among epitopes for presentation or T-cell recognition. However, other groups have demonstrated that epitope competition can result in the development of dominant and subdominant immune responses to multiple peptides delivered directly or encoded by a genetic vaccine [24–26]. We hypothesized that dominant epitope-specific immune responses generated by a DNA vaccine encoding SSX2 might mask the development of responses to subdominant epitopes by competition for MHC presentation and/or T-cell activation. Therefore, we tested DNA vaccines that both ablated HLA-A2 binding of a dominant epitope (p103–111) as well as vaccines that were designed to enhance binding of a subdominant epitope (p41–49) in an effort to identify an optimal vaccine to elicit simultaneous immune responses to both SSX2 epitopes. Modified plasmid vaccines were generated by utilizing site-directed mutagenesis to alter the native pTVG-SSX2 plasmid to encode the APL identified above. Mice were immunized two to six times; six times having been previously found to elicit robust immune responses with the native pTVG-SSX2 DNA vaccine [17]. Two to four immunizations were given to detect differences in magnitude of immune response to the dominant or subdominant epitopes in a purposefully suboptimal fashion.
We first wished to determine whether the only HLA-A2-specific immune responses are conferred by the dominant (p103–111) and subdominant (p41–49) SSX2 epitopes, and whether responses to these epitopes might mask responses to other potentially weaker “cryptic” epitopes (e.g. p57–65 and p99–107). Animals were immunized six times biweekly with the native SSX2 vaccine or a modified SSX2 vaccine encoding peptides p41–49 and p103–111 with mutations to ablate HLA-A2 binding. As expected, ablation of peptides p41–49 and p103–111 by introduction of proline mutations abrogated immune responses to both of these epitopes, however, animals also failed to elicit immune responses to other HLA-A2-binding peptides p57–65 or p99–107 (Fig. 4A). These results confirm that peptides p57–65 and p99–107 are not HLA-A2 epitopes in this system, and do not represent “cryptic” epitopes that might be recognized following ablation of the dominant epitopes.
Fig. 4.

HHDII-DR1 mice immunized with SSX2 plasmid DNA vaccines encoding APL develop epitope-specific immune responses at higher frequency than animals immunized with the native vaccine. (Panel A) Splenocytes from individual mice immunized with either native pTVG-SSX2 plasmid vaccine (n = 6) or pTVG-SSX2 p41-VP/p103-IP (encoding epitopes with ablated HLA-A2 binding, n = 6) were analyzed for the frequency of peptide-specific immune responses by IFNγ ELISPOT. (Panel B) Splenocytes from individual mice immunized with either native pTVG-SSX2, a plasmid encoding p41-AL (enhanced binding), a plasmid ablating p103–111 HLA-A2 binding (p103-IP), or a plasmid encoding both mutations (p41-AL/p103-IP), were analyzed for the frequency of peptide-specific immune responses by IFNγ ELISPOT (n = 5 for each treatment group). (Panel C) Splenocytes from individual mice immunized with either native pTVG-SSX2, a plasmid encoding p103-RF, or a plasmid ablating p103–111 HLA-A2 binding (p103-IP), were analyzed for the frequency of peptide-specific immune responses by IFNγ ELISPOT (n = 11 for each treatment group). (Panel D) Splenocytes from individual mice immunized with native pTVG-SSX2, a plasmid encoding p41-AL, or a plasmid encoding both p41-AL and p103-RF (p41-AL/p103-RF) were analyzed for the frequency of peptide-specific immune responses by IFNγ ELISPOT (n = 6 for each vaccine). Splenocytes were stimulated for 48 h with each of the stimulator peptides indicated or a concanavalin A positive control prior to IFNγ detection. Flu peptide was evaluated as a negative control. Each colored symbol represents the frequency of peptide-specific IFNγ-secreting responses from an individual mouse. * Indicates a significant (P < 0.05) difference in the mean number of IFNγ spot-forming units (SFU) between the 2 groups (2-tailed t test).
We next evaluated whether immunization with a DNA vaccine encoding modifications designed to increase HLA-A2 binding of the epitopes could enhance peptide-specific immune responses, and whether ablating binding of peptide p103–111 to HLA-A2 could increase immune responses to the subdominant p41–49 epitope. Epitope alterations to the DNA plasmid were based on the results from the direct peptide vaccination studies. Mice were immunized four times with a vaccine that encoded p41-AL (pTVG-SSX2 p41-AL), a vaccine that encoded an ablated peptide p103–111 binding to HLA-A2 (pTVG-SSX2 p103-IP), or a vaccine that encoded both mutations (pTVG-SSX2 p41-AL/p103-IP). Interestingly, we found that ablating binding of peptide p103–111 to HLA-A2 did not increase the number of p41–49 peptide-specific immune responses as we had predicted (Fig. 4B), however both constructs encoding peptide p41-AL elicited statistically significantly higher frequencies of native p41–49 peptide-specific T cells in the immunized animals.
We next sought to determine whether immunization with DNA encoding modifications to the dominant p103–111 epitope could elicit more robust immune responses to that epitope, and/or whether this might lead to diminished responses to the subdominant p41–49 epitopes. Mice were immunized two times with the native vaccine, a vaccine encoding peptide p103–111 with enhanced HLA-A2 affinity (pTVG-SSX2 p103-RF), and a vaccine that ablated peptide p103–111 binding to HLA-A2 (pTVG-SSX2 p103-IP). As expected, we found that a greater number of animals had higher frequencies of peptide p103–111-specific immune responses when immunized with p103–111-RF compared to the native vaccine (Fig. 4C). While few responses to peptide p41–49 were detected with two immunizations only, this frequency was not significantly affected by vaccines encoding increased (p103-RF) or ablated (p103-IP) binding to the p103–111 epitope.
Finally, we conducted studies to determine whether modifications to both epitopes could elicit a higher frequency of T cells specific for both p41–49 and p103–111 simultaneously. HHDII-DR1 mice were immunized four times with the native vaccine or vaccines encoding p41-AL alone (pTVG-SSX2 p41–49-AL) or encoding both p41-AL and p103-RF (pTVG-SSX2 p41-AL/p103-RF). We found that the vaccine encoding both enhancing modifications was able to elicit the highest frequency of peptide specific immune responses to both of the native p41–49 and p103–111 epitopes (Fig. 4D). Moreover, as shown in Fig. 5, epitope-specific CD8+ T cells of a Th1-biased phenotype (secreting IFNγ, TNFα, and/or IL-2) were elicited with both the native and epitope-modified vaccine, but a higher frequency of epitope-specific CD8+ T cells was identified following immunization with the epitope-modified DNA vaccine. In addition, IL-17-expressing CD8+ T cells were also observed following immunization with the epitope-modified DNA vaccine, suggesting a multifunctional phenotype resulted following immunization with this epitope-modified vaccine.
Fig. 5.
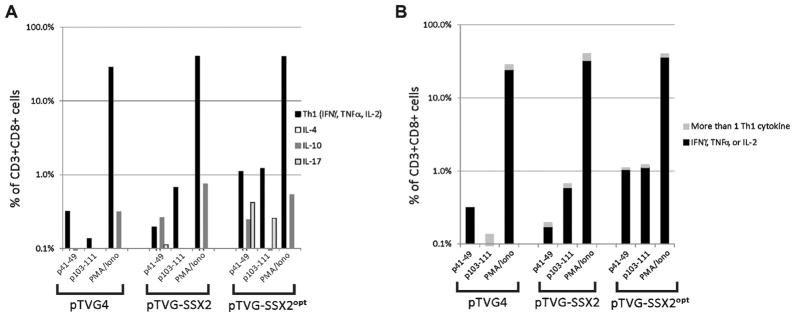
HHDII-DR1 mice immunized with SSX2 plasmid DNA vaccines develop epitope-specific immune responses with a multi-functional phenotype. (Panel A) Splenocytes from animals (n = 3–4 animals per group) immunized with pTVG4 (control vector), pTVG-SSX2, or pTVG-SSX2 p41-AL/p103-RF (pTVG-SSX2opt) were pooled, enriched for CD8+ T cells, and stimulated with p41–49 or p103–111 peptide overnight. The frequency of CD3+ CD8+ T cells secreting Th1 (IFNγ, TNFα, or IL-2), IL-4, IL-10, or IL-17 under each antigen-stimulating condition and for each experimental group was determined by intracellular cytokine staining. (Panel B) The CD3+ CD8+ T cells secreting Th1 cytokines (IFNγ, TNFα, or IL-2) were subdivided among those concurrently expressing one or more of these cytokines.
4. Discussion
Since the concept of using APL to augment T-cell immune responses was first introduced two decades ago, this strategy has been used to investigate multiple aspects of antigen-specific immunity [27,28]. APL have been used by many groups to both up-regulate and down-regulate immune responses to peptide antigens expressed by pathogens, tumor cells, and somatic cells in applications targeting infectious diseases, cancer, and autoimmune diseases [28]. Tumor-associated antigens (TAA) are usually weakly immunogenic, containing epitopes that have low binding affinity for HLA molecules. Strong immune responses have been generated to several TAA in both pre-clinical mouse studies and human clinical trials using APL strategies [29]. For example, Lazoura et al. demonstrated that immune responses to mucin 1 could be enhanced in mice after in vivo vaccination with MUC1-8, an altered peptide with 100-fold increased binding to H-2Kb due to an anchor residue modification [30]. To date, APL have demonstrated efficacy in eliciting enhanced immune responses to many different tumor and viral antigens that are otherwise weakly immunogenic or tolerized [24–26,31–33].
In the present study we sought to enhance epitope-specific immune responses to the SSX2 tumor antigen using an APL strategy to boost responses to multiple SSX2 epitopes simultaneously by means of a DNA vaccine. We evaluated whether APL-encoding DNA vaccines could elicit enhanced frequencies of SSX2 peptide-specific T cells, and specifically CD8+ T cell responses with effector function. It should be noted that we specifically made amino acid substitutions to peptide anchor residues to enhance HLA-A2 binding while potentially maintaining interaction with the TCR of lymphocytes specific to these peptide/MHC complexes.
We had previously identified that peptide p103–111 is the immunodominant HLA-A2-restricted SSX2 epitope based on the frequency of peptide-specific T cells from immunized animals, and that p41–49 is subdominant [17]. We found that APL for peptides p41–49 and p103–111, modified at MHC/peptide anchor residues, had enhanced binding affinity to HLA-A2 in vitro, and immunization with these APL could elicit greater numbers of peptide-specific T cells in HHDII-DR1 mice after peptide immunization. T cells from these animals were cytolytic and cross-reactive to the native epitopes. By modifying a SSX2 DNA plasmid vaccine to incorporate these APL we were able to generate increased frequencies of peptide p41–49- and p103–111-specific immune responses simultaneously, and with a Th1-biased multifunctional phenotype. Our goal was to elicit multiple, epitope-specific immune responses to potentially enhance the breadth of immune responses against epitopes that may be presented by tumor cells, as we had previously observed that the most robust lysis of prostate cancer cell lines in vitro by cytolytic T cells from occurred when animals developed IFNγ-secreting immune responses to both peptides p41–49 and p103–111 [17]. The development of tumor models expressing SSX2, to be able to test the anti-tumor efficacy of these modified DNA vaccines, will be an aim of future studies.
Based on previous findings of T-cell competition among dominant and subdominant viral epitopes [25,26], we had hypothesized that it may be possible to elicit enhanced immune responses to a subdominant SSX2 epitope using an APL strategy to ablate binding of a dominant epitope encoded by a DNA vaccine. By encoding both epitopes within a plasmid DNA, individual APC would process both epitopes and could possibly exhibit competition for MHC presentation. However, we found that ablating HLA-A2 binding of the dominant epitope (p103–111) did not elicit increased frequency of T cells specific for the subdominant epitope (p41–49). And, as in Fig. 4A, encoding modifications to ablate binding of both epitopes did not result in recognition of other HLA-A2-binding peptides as potential “cryptic” epitopes. These results suggest that epitope competition is not a major mechanism to “mask” subdominant epitopes, at least in this model system. These results differ from what other groups have found by ablating HLA binding of dominant HBV antigen epitopes in a DNA plasmid vaccine [26]. Riedl et al. observed uncovering of immune responses to cryptic epitopes in mice that were immunized with a vaccine that ablated HLA affinity of dominant epitopes. However, this un-masking was only observed in a setting of tolerance to the dominant epitope. Thus, this difference in findings may be due to an antigen-dependent effect, the mouse model and HLA haplotype used, or more likely, the presence or absence of regulatory mechanisms already in place to dampen immune responses to the dominant epitopes.
Curiously, while the studies demonstrated that immune responses elicited were Th1 biased, IL-10-secreting p41–49-specific T cells were also elicited. IL-10 has traditionally been associated with regulatory or tolerant immune responses, and with suppression of IFNγ production by CD8+ T cells [34]. However, more recent murine studies have also demonstrated a role for IL-10 in receptor-mediated CD8+ T-cell proliferation and cytolytic activity [35]. IL-10 may also play a direct antagonistic role in tumor growth and metastasis [36]. The function of these antigen-specific IL-10-secreting CD8+ T cells is currently unknown, however a prior study found that mice immunized concurrently with plasmid DNA and the protein antigen itself developed antigen-specific CD4+ T cells with immune suppressive activity and expressing IFNγ and IL-10 [37]. Thus, it is conceivable that the IL-10-expressing population does represent cells with decreased effector function, and may have accounted for the decreased cytolytic activity of the cells that we observed (Fig. 2). Of note, we have previously found that IL-10-secreting antigen-specific T cells can be amplified with high plasmid DNA dose and repetitive immunization in rats [38]. It is further conceivable that the p41–49 epitope shares homology with a native murine epitope. While we have not identified any murine proteins known to share this amino acid sequence, this could potentially explain the “subdominance” of this particular epitope and why pre-existing tolerant IL-10-type responses might be amplified with immunization. Future studies will be needed to determine whether these IL-10-secreting T cells have immune effector or regulatory function, and whether they might share the same CDR3 repertoire as purely Th1-biased cells.
An approach similar to encoding APL within a genetic vaccine is the strategy of using xenoantigen targets. In principle, shared epitopes between two species’ proteins may be dissimilar by a few amino acids, and in some instances this has been shown to elicit higher frequency epitope-specific immune responses than immunizing with the autologous protein [39–43]. In fact, the only USDA-approved anti-tumor vaccine is a DNA plasmid encoding the human tyrosinase xenoantigen for the treatment of canine melanoma [44]. We have similarly evaluated a xenoantigen approach to DNA vaccination by immunizing Lewis strain rats with DNA encoding human PAP [45]. However, we found that immune responses developed to uniquely human epitopes without cross-reactivity to rat PAP [46]. Thus, while this strategy can work to elicit cross-reactive immunity, it is equally likely that encoded foreign epitopes may weaken MHC binding of epitopes that are otherwise immunogenic, thereby dampening the epitope/MHC:TCR interaction. Hence, we believe that immunization with epitope-specific modifications with defined MHC binding affinity, as in the current study, and potentially targeting multiple MHC class I types, offers a more rational approach for future DNA vaccination strategies to increase the number and function of tumor antigen-specific CD8+ T cells.
Acknowledgments
The HLA-A2 transgenic HHDII-DR1 mice are the property of the Institut Pasteur, 25–28 rue du Docteur Roux, Paris France 75015 and were generously provided by Dr. François Lemonnier. The HLA-A2-expressing LNCaP cell line was provided by Dr. Larry Fong. We would also like to thank Dr. Brian Olson and Jordan Bloom for technical assistance.
Abbreviations
- SSX
synovial sarcoma chromosome X breakpoint
- CTA
cancer testis antigen
- TAA
tumor-associated antigen
- APL
altered peptide ligand
- MHC
major histocompatibility complex
- ELISPOT
enzyme-linked immunosorbent spot
- HLA
human leukocyte antigen
- CTL
cytotoxic T lymphocyte
Footnotes
This work was supported by the Department of Defense Prostate Cancer Research Program W81XWH-10-1-0495 and W81XWH-08-1-0341, and by the National Institutes of Health R01-CA142608.
References
- 1.Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, et al. Direct gene transfer into mouse muscle in vivo. Science. 1990;247:1465–8. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 2.Iwasaki A, Torres CA, Ohashi PS, Robinson HL, Barber BH. The dominant role of bone marrow-derived cells in CTL induction following plasmid DNA immunization at different sites. J Immunol. 1997;159:11–4. [PubMed] [Google Scholar]
- 3.Alonso M, Leong JA. Licensed DNA vaccines against infectious hematopoietic necrosis virus (IHNV) Recent Pat DNA Gene Seq. 2013;7:62–5. doi: 10.2174/1872215611307010009. [DOI] [PubMed] [Google Scholar]
- 4.Hall RA, Khromykh AA. West Nile virus vaccines. Expert Opin Biol Ther. 2004;4:1295–305. doi: 10.1517/14712598.4.8.1295. [DOI] [PubMed] [Google Scholar]
- 5.Grosenbaugh DA, Leard AT, Bergman PJ, Klein MK, Meleo K, Susaneck S, et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am J Vet Res. 2011;72:1631–8. doi: 10.2460/ajvr.72.12.1631. [DOI] [PubMed] [Google Scholar]
- 6.Bergman PJ, McKnight J, Novosad A, Charney S, Farrelly J, Craft D, et al. Long-term survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: a phase I trial. Clin Cancer Res. 2003;9:1284–90. [PubMed] [Google Scholar]
- 7.Colluru VT, Johnson LE, Olson BM, McNeel DG. Preclinical and clinical development of DNA vaccines for prostate cancer. Urol Oncol. 2013 doi: 10.1016/j.urolonc.2013.09.014. http://dx.doi.org/10.1016/j.urolonc.2013.09.014 [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 8.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 9.McNeel DG, Dunphy EJ, Davies JG, Frye TP, Johnson LE, Staab MJ, et al. Safety and immunological efficacy of a DNA vaccine encoding prostatic acid phosphatase in patients with stage D0 prostate cancer. J Clin Oncol. 2009;27:4047–54. doi: 10.1200/JCO.2008.19.9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Becker JT, Olson BM, Johnson LE, Davies JG, Dunphy EJ, McNeel DG. DNA vaccine encoding prostatic acid phosphatase (PAP) elicits long-term T-cell responses in patients with recurrent prostate cancer. J Immunother. 2010;33:639–47. doi: 10.1097/CJI.0b013e3181dda23e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavlenko M, Roos AK, Lundqvist A, Palmborg A, Miller AM, Ozenci V, et al. A phase I trial of DNA vaccination with a plasmid expressing prostate-specific antigen in patients with hormone-refractory prostate cancer. Br J Cancer. 2004;91:688–94. doi: 10.1038/sj.bjc.6602019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Low L, Mander A, McCann K, Dearnaley D, Tjelle T, Mathiesen I, et al. DNA vaccination with electroporation induces increased antibody responses in patients with prostate cancer. Hum Gene Ther. 2009;20:1269–78. doi: 10.1089/hum.2009.067. [DOI] [PubMed] [Google Scholar]
- 13.Smith HA, McNeel DG. The SSX family of cancer-testis antigens as target proteins for tumor therapy. Clin Dev Immunol. 2010;1505:91. doi: 10.1155/2010/150591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubovsky JA, McNeel DG. Inducible expression of a prostate cancer-testis antigen, SSX-2, following treatment with a DNA methylation inhibitor. Prostate. 2007;67:1781–90. doi: 10.1002/pros.20665. [DOI] [PubMed] [Google Scholar]
- 15.dos Santos NR, Torensma R, de Vries TJ, Schreurs MW, de Bruijn DR, Kater-Baats E, et al. Heterogeneous expression of the SSX cancer/testis antigens in human melanoma lesions and cell lines. Cancer Res. 2000;60:1654–62. [PubMed] [Google Scholar]
- 16.Cronwright G, Le Blanc K, Gotherstrom C, Darcy P, Ehnman M, Brodin B. Cancer/testis antigen expression in human mesenchymal stem cells: down-regulation of SSX impairs cell migration and matrix metalloproteinase 2 expression. Cancer Res. 2005;65:2207–15. doi: 10.1158/0008-5472.CAN-04-1882. [DOI] [PubMed] [Google Scholar]
- 17.Smith HA, McNeel DG. Vaccines targeting the cancer-testis antigen SSX-2 elicit HLA-A2 epitope-specific cytolytic T cells. J Immunother. 2011;34:569–80. doi: 10.1097/CJI.0b013e31822b5b1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith HA, Cronk RJ, Lang JM, McNeel DG. Expression and immunotherapeutic targeting of the SSX family of cancer-testis antigens in prostate cancer. Cancer Res. 2011;71:6785–95. doi: 10.1158/0008-5472.CAN-11-2127. [DOI] [PubMed] [Google Scholar]
- 19.Huijbers IJ, Soudja SM, Uyttenhove C, Buferne M, Inderberg-Suso EM, Colau D, et al. Minimal tolerance to a tumor antigen encoded by a cancer-germline gene. J Immunol. 2012;188:111–21. doi: 10.4049/jimmunol.1002612. [DOI] [PubMed] [Google Scholar]
- 20.Pajot A, Michel ML, Fazilleau N, Pancre V, Auriault C, Ojcius DM, et al. A mouse model of human adaptive immune functions: HLA-A2. 1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur J Immunol. 2004;34:3060–9. doi: 10.1002/eji.200425463. [DOI] [PubMed] [Google Scholar]
- 21.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152:163–75. [PubMed] [Google Scholar]
- 22.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–9. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 23.Olson BM, Frye TP, Johnson LE, Fong L, Knutson KL, Disis ML, et al. HLA-A2-restricted T-cell epitopes specific for prostatic acid phosphatase. Cancer Immunol Immunother. 2010;59:943–53. doi: 10.1007/s00262-010-0820-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alves PM, Viatte S, Fagerberg T, Michielin O, Bricard G, Bouzourene H, et al. Immunogenicity of the carcinoembryonic antigen derived peptide 694 in HLA-A2 healthy donors and colorectal carcinoma patients. Cancer Immunol Immunother. 2007;56:1795–805. doi: 10.1007/s00262-007-0323-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kloverpris H, Karlsson I, Bonde J, Thorn M, Vinner L, Pedersen AE, et al. Induction of novel CD8+ T-cell responses during chronic untreated HIV-1 infection by immunization with subdominant cytotoxic T-lymphocyte epitopes. Aids. 2009;23:1329–40. doi: 10.1097/QAD.0b013e32832d9b00. [DOI] [PubMed] [Google Scholar]
- 26.Riedl P, Wieland A, Lamberth K, Buus S, Lemonnier F, Reifenberg K, et al. Elimination of immunodominant epitopes from multispecific DNA-based vaccines allows induction of CD8+ T cells that have a striking antiviral potential. J Immunol. 2009;183:370–80. doi: 10.4049/jimmunol.0900505. [DOI] [PubMed] [Google Scholar]
- 27.Evavold BD, Allen PM. Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand. Science. 1991;252:1308–10. doi: 10.1126/science.1833816. [DOI] [PubMed] [Google Scholar]
- 28.Unanue ER. Altered peptide ligands make their entry. J Immunol. 2011;186:7–8. doi: 10.4049/jimmunol.1090118. [DOI] [PubMed] [Google Scholar]
- 29.Katsara M, Minigo G, Plebanski M, Apostolopoulos V. The good, the bad and the ugly: how altered peptide ligands modulate immunity. Expert Opin Biol Ther. 2008;8:1873–84. doi: 10.1517/14712590802494501. [DOI] [PubMed] [Google Scholar]
- 30.Lazoura E, Lodding J, Farrugia W, Ramsland PA, Stevens J, Wilson IA, et al. Enhanced major histocompatibility complex class I binding and immune responses through anchor modification of the non-canonical tumour-associated mucin 1–8 peptide. Immunology. 2006;119:306–16. doi: 10.1111/j.1365-2567.2006.02434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gritzapis AD, Mahaira LG, Perez SA, Cacoullos NT, Papamichail M, Baxevanis CN. Vaccination with human HER-2/neu (435–443) CTL peptide induces effective antitumor immunity against HER-2/neu-expressing tumor cells in vivo. Cancer Res. 2006;66:5452–60. doi: 10.1158/0008-5472.CAN-05-4018. [DOI] [PubMed] [Google Scholar]
- 32.van Stipdonk MJ, Badia-Martinez D, Sluijter M, Offringa R, van Hall T, Achour A. Design of agonistic altered peptides for the robust induction of CTL directed towards H-2Db in complex with the melanoma-associated epitope gp100. Cancer Res. 2009;69:7784–92. doi: 10.1158/0008-5472.CAN-09-1724. [DOI] [PubMed] [Google Scholar]
- 33.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of self-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–86. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Endharti AT, Rifa’i M, Shi Z, Fukuoka Y, Nakahara Y, Kawamoto Y, et al. Cutting edge: CD8+ CD122+ regulatory T cells produce IL-10 to suppress IFN-gamma production and proliferation of CD8+ T cells. J Immunol. 2005;175:7093–7. doi: 10.4049/jimmunol.175.11.7093. [DOI] [PubMed] [Google Scholar]
- 35.Emmerich J, Mumm JB, Chan IH, Laface D, Truong H, McClanahan T, et al. IL-10 directly activates and expands tumor-resident CD8+ T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. 2012;72:3570–81. doi: 10.1158/0008-5472.CAN-12-0721. [DOI] [PubMed] [Google Scholar]
- 36.Tanikawa T, Wilke CM, Kryczek I, Chen GY, Kao J, Nunez G, et al. Interleukin-10 ablation promotes tumor development, growth, and metastasis. Cancer Res. 2012;72:420–9. doi: 10.1158/0008-5472.CAN-10-4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang Y, Jin H, Zheng G, Du X, Xiao C, Zhang X, et al. Co-inoculation of DNA and protein vaccines induces antigen-specific T cell suppression. Biochem Biophys Res Commun. 2007;353:1034–9. doi: 10.1016/j.bbrc.2006.12.124. [DOI] [PubMed] [Google Scholar]
- 38.Johnson LE, Frye TP, Arnot AR, Marquette C, Couture LA, Gendron-Fitzpatrick A, et al. Safety and immunological efficacy of a prostate cancer plasmid DNA vaccine encoding prostatic acid phosphatase (PAP) Vaccine. 2006;24:293–303. doi: 10.1016/j.vaccine.2005.07.074. [DOI] [PubMed] [Google Scholar]
- 39.Kianizad K, Marshall LA, Grinshtein N, Bernard D, Margl R, Cheng S, et al. Elevated frequencies of self-reactive CD8+ T cells following immunization with a xenoantigen are due to the presence of a heteroclitic CD4+ T-cell helper epitope. Cancer Res. 2007;67:6459–67. doi: 10.1158/0008-5472.CAN-06-4336. [DOI] [PubMed] [Google Scholar]
- 40.Fong L, Brockstedt D, Benike C, Breen JK, Strang G, Ruegg CL, et al. Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol. 2001;167:7150–6. doi: 10.4049/jimmunol.167.12.7150. [DOI] [PubMed] [Google Scholar]
- 41.Hu B, Wei YQ, Tian L, Zhao X, Lu Y, Wu Y, et al. Human T lymphocyte responses against lung cancer induced by recombinant truncated mouse EGFR. Cancer Immunol Immunother. 2006;55:386–93. doi: 10.1007/s00262-005-0028-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gyorffy S, Rodriguez-Lecompte JC, Woods JP, Foley R, Kruth S, Liaw PC, et al. Bone marrow-derived dendritic cell vaccination of dogs with naturally occurring melanoma by using human gp100 antigen. J Vet Intern Med. 2005;19:56–63. doi: 10.1892/0891-6640(2005)19<56:bmdcvo>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 43.Kievits F, Boerenkamp WJ, Lokhorst W, Ivanyi P. Specificity and frequency of primary anti-HLA cytotoxic T lymphocytes in normal and HLA-B27.2-, HLA-B27.5-, and HLA-Cw3-transgenic mice. A transgenic model for MHC xenoantigen recognition. J Immunol. 1990;144:4513–9. [PubMed] [Google Scholar]
- 44.USDA licenses DNA vaccine for treatment of melanoma in dogs. J Am Vet Med Assoc. 2010;236:495. doi: 10.2460/javma.236.5.488. [DOI] [PubMed] [Google Scholar]
- 45.Johnson LE, Frye TP, Chinnasamy N, Chinnasamy D, McNeel DG. Plasmid DNA vaccine encoding prostatic acid phosphatase is effective in eliciting autologous antigen-specific CD8+ T cells. Cancer Immunol Immunother. 2007;56:885–95. doi: 10.1007/s00262-006-0241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson LE, Frye TP, McNeel DG. Immunization with a prostate cancer xenoantigen elicits a xenoantigen epitope-specific T-cell response. Oncoimmunology. 2012;1:1546–56. doi: 10.4161/onci.22564. [DOI] [PMC free article] [PubMed] [Google Scholar]