

Abstract
Unactivated α-branched primary and secondary aliphatic alcohols have been successfully transformed into their corresponding alkyl chlorides in high yields upon treatment with a mixture of triphosgene and pyridine in dichloromethane at reflux. These mild chlorination conditions are high yielding, stereospecific, and well tolerated by numerous sensitive functionalities. Furthermore, no nuisance waste products are generated in the course of the reactions.
INTRODUCTION
The preparation of alkyl chlorides continues to be an area of significant interest in organic synthesis. While several new methods have been developed in recent years, many of these have limited variability of substrate tolerance and efficiency.1-6 Our involvement in the development of new chlorination methodology originated from our synthetic interests in chlorine-containing natural products, such as the chlorosulfolipids (Figure 1). The chlorosulfolipid class of natural products is essentially highlighted by multiple chlorine and sulfate sub-stitutions along the long hydrocarbon backbone. These note-worthy features are accompanied by multiple stereogenic centers of chlorine atoms, which present significant challenges for structure elucidation and total synthesis.7,8
Figure 1.

Examples of chlorosulfolipid natural products.
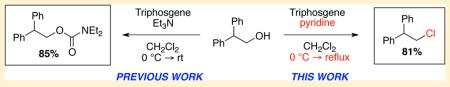
Intrigued by the unusual structural features of this class of natural products, we initiated research to develop a mild method for chemoselective chlorination of aliphatic alcohols, particularly in the presence of nearby sensitive functionalities. In fact, we recently reported a new method for the chemoselective chlorination of primary aliphatic alcohols using a mixture of triphosgene and triethylamine in dichloromethane.9 As shown in Scheme 1, these conditions were effective in producing primary alkyl chlorides in high yields while being compatible with various acid- and base-sensitive functionalities that would be problematic under classical conditions. Furthermore, these conditions were operationally simple, due to the fact that triphosgene exists as a stable nonhygroscopic crystalline material at room temperature, and this permits easy and safe handling.10-12
Scheme 1.

Previous Work on Triphosgene–Triethylamine-Promoted Chlorination
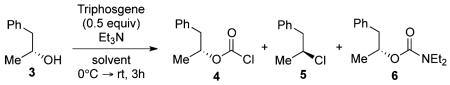
During the course of substrate studies, we observed that sterically hindered alcohols were not suitable for our methodology. For example, activation of 2,2-diphenyl-1-ethanol (1) with a triphosgene–triethylamine mixture yielded only diethylcarbamate adduct 2 in 85% yield, while the chlorination product was not detected. Furthermore, exposure of secondary alcohol 3 under identical conditions produced a mixture of alkyl chloride 5 and diethylcarbamate 6 in 45% and 27% yields, respectively. It appeared that the selectivity between formation of alkyl chloride and diethylcarbamate was driven by sterics via an intermediacy of acylammonium ion species 7, where the ensuing nucleophilic attack by chloride ions could competitively occur at two possible electrophilic carbon centers.13-15.
Our mechanistic investigation also concluded that alcohols remain unreactive toward triphosgene in the absence of triethylamine, which serves as a base and nucleophilic catalyst to promote chlorination.9 The broader applicability of our method to sterically congested substrates is clearly limited by these selectivity problems, and therefore, it is imperative that we address these crucial issues.
RESULTS AND DISCUSSION
As shown in Table 1, we proceeded with a comprehensive optimization study by initially varying the amount of triethylamine while maintaining 0.5 equiv of triphosgene. Secondary alcohol 3 was used as a model substrate for these studies. We hypothesized that perhaps reducing the amount of triethylamine would inhibit formation of the undesired diethylcarbamate functionality. As shown in entry 1, the use of 1.0 equiv of triethylamine predominantly produced chloroformate 4 along with minimal alkyl chloride 5 and diethylcarbamate 6.16 According to our expectation, an incremental increase of triethylamine from 1.0 to 2.0 equiv resulted in the disappearance of 4. In these cases, however, formation of 5 and 6 was found to be equally facile. As indicated in entries 2–5, the two competing products were produced in a nearly 1:1 ratio regardless of the quantity of triethylamine. Interestingly, entries 6–10 indicated the resistance of chloroformate 4 to further transformation to either products 5 or 6 when an identical series of optimization reactions were executed in toluene.
Table 1. Optimization Study Varying the Amount of Triethylamine.
| ||||||
|---|---|---|---|---|---|---|
| yield, %a |
||||||
| entry | amt of Et3N, equiv | solvent | 3 | 4 | 5 | 6 |
| 1 | 1.00 | CH2Cl2 | 6 | 73 | 14 | 7 |
| 2 | 1.25 | CH2Cl2 | 3 | 75 | 13 | 9 |
| 3 | 1.50 | CH2Cl2 | 2 | 45 | 27 | 27 |
| 4 | 1.75 | CH2Cl2 | 1 | 16 | 44 | 39 |
| 5 | 2.00 | CH2Cl2 | 0 | 3 | 51 | 46 |
| 6 | 1.00 | toluene | 11 | 86 | 3 | 0 |
| 7 | 1.25 | toluene | 3 | 87 | 5 | 5 |
| 8 | 1.50 | toluene | 2 | 87 | 6 | 5 |
| 9 | 1.75 | toluene | 1 | 54 | 18 | 27 |
| 10 | 2.00 | toluene | 2 | 65 | 21 | 13 |
Yields were determined by GC-MS analysis of the crude mixtures, assuming that these compounds elicited identical GC responses.
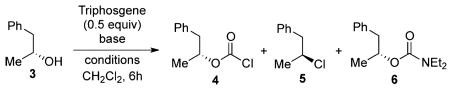
As a result of these initial studies, we concluded that triethylamine alone, as a base/activator, would not be effective for our intended chlorination. Optimization studies then shifted to the exploration of a mixture of amine bases, such as pyridine and triethylamine. On the basis of the proposed N-acylammonium ion intermediate 7,9 our chlorination reaction could employ a stoichiometric amount of pyridine, which would serve as a base in the chloroformylation step,16 while maintaining a substoichiometric amount of triethylamine to promote chlorination. We realized that formation of triethylammonium ion species would be favored in the proton transfer equilibrium between protonated pyridine and triethylamine upon chloroformylation of the alcohol starting material, due to the difference in the pKa values of their conjugate acids.17 However, a low concentration of unprotonated triethylamine is expected to remain in the equilibrium and should promote nucleophilic substitution by chloride ions by forming acylammonium ion intermediate 7.
As shown in Table 2, entries 1–8, activation of secondary alcohol 3 with triphosgene in the presence of 1.2 equiv of pyridine with varying substoichiometric amounts of triethylamine successfully suppressed the formation of diethylcarbamate 6. These reactions primarily afforded chloroformate 4 at room temperature (entries 1–4), but heating the reaction mixtures to reflux (entries 5–8) led to an increased production of alkyl chloride 5. In fact, entry 7 details the GC-MS analysis of the crude materials, revealing that the use of 1.2 equiv of pyridine and 0.75 equiv of triethylamine in dichloromethane at reflux quantitatively converted secondary alcohol 3 to alkyl chloride 5. Much to our surprise, an attempt to further investigate the role of pyridine in this reaction yielded unanticipated results, as studies in entries 9–12 indicated that the use of pyridine by itself readily generated the target alkyl chloride. In fact, secondary alcohol 3 was completely consumed and converted to alkyl chloride 5 when the reaction was performed using excess pyridine in refluxing dichloromethane (entries 13–16). These conditions completely eliminated the problematic formation of diethylcarbamate byproduct. Interestingly, while the use of 1.0 equiv of pyridine led to a mixture of 2:1 mixture of starting material 3 and alkyl chloride 5 (entry 13), increasing the amount of pyridine to 1.2 equiv fully consumed alcohol 3 and yielded a 1:1 mixture of chloroformate 4 and alkyl chloride 5 (entry 14). These observations again strongly suggested an intermediacy of the chloroformate species in our chlorination reaction.
Table 2. Optimization Study with Mixed Amine Base Systems.
| |||||||
|---|---|---|---|---|---|---|---|
| yield, %b |
|||||||
| entry | amt of Py, equiv |
amt of Et3N, equiv |
conditionsa | 3 | 4 | 5 | 6 |
| 1 | 1.2 | 0.25 | 0 °C → room temp | 0 | 98 | 1 | 1 |
| 2 | 1.2 | 0.50 | 0 °C → room temp | 0 | 75 | 25 | 0 |
| 3 | 1.2 | 0.75 | 0 °C → room temp | 0 | 60 | 39 | 0 |
| 4 | 1.2 | 1.00 | 0 °C → room temp | 0 | 50 | 50 | 0 |
| 5 | 1.2 | 0.25 | 0 °C → reflux | 0 | 34 | 66 | 0 |
| 6 | 1.2 | 0.50 | 0 °C → reflux | 0 | 9 | 90 | 1 |
| 7 | 1.2 | 0.75 | 0 °C → reflux | 0 | 0 | 100 | 0 |
| 8 | 1.2 | 1.00 | 0 °C → reflux | 0 | 1 | 90 | 9 |
| 9 | 1.0 | 0 | 0 °C → room temp | 93 | 0 | 7 | 0 |
| 10 | 1.2 | 0 | 0 °C → room temp | 75 | 0 | 25 | 0 |
| 11 | 1.7 | 0 | 0 °C → room temp | 35 | 0 | 65 | 0 |
| 12 | 2.2 | 0 | 0 °C → room temp | 28 | 0 | 72 | 0 |
| 13 | 1.0 | 0 | 0 °C → reflux | 69 | 0 | 31 | 0 |
| 14 | 1.2 | 0 | 0 °C → reflux | 0 | 44 | 56 | 0 |
| 15 | 1.7 | 0 | 0 °C → reflux | 2 | 0 | 98 | 0 |
| 16 | 2.2 | 0 | 0 °C → reflux | 0 | 0 | 100 | 0 |
Reagents were added at 0 °C, and then the reaction mixture was warmed to room temperature or reflux.
Yields were determined by GC-MS analysis of the crude mixtures, assuming that these compounds elicited identical GC responses.
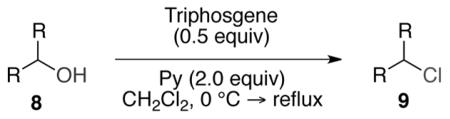
These results were intriguing, as there are precedents which demonstrate that a triphosgene–pyridine mixture readily chlorinates activated alcohols, such as those in benzylic, allylic, or propargylic systems.18 There are also reports that demonstrate the use of this mixture to convert aliphatic alcohols to their corresponding chloroformates.19-22 However, to the best of our knowledge, chlorination of unreactive aliphatic secondary alcohols using this mixture has remained unexplored. With these preliminary results in hand, we then examined the generality of these chlorination conditions by screening a series of secondary alcohols containing various common functional and protecting groups. A typical reaction protocol involved addition of 0.5 equiv of triphosgene and 2.0 equiv of pyridine to a solution of 1.0 equiv of secondary alcohol in dichloromethane at 0 °C. The reaction mixture was then warmed to gentle reflux overnight, followed by aqueous workup with a dilute HCl solution and flash chromatography. It is crucial to note that unlike the classical chlorination methods using SOCl2 or PPh3–NCS activation, our reaction does not produce any nuisance waste products. The typical crude reaction mixture upon workup cleanly contains the desired alkyl chloride, although in most cases, a minor elimination product (<10%) was detected by crude GC-MS or 1H NMR analyses.
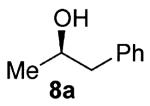
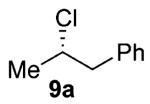
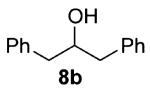
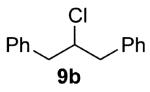
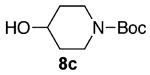
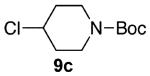
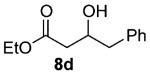
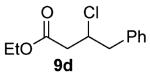
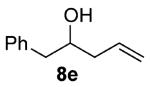
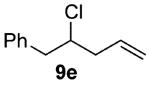
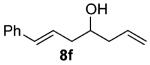
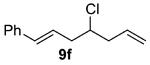
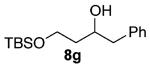
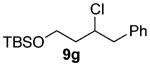
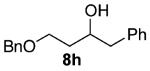
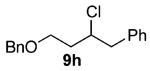
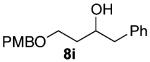
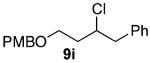
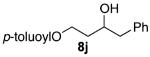
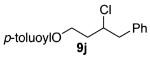
As shown in Table 3 entries 1–3, simple secondary alcohols in both acyclic and cyclic forms 8a–c were readily converted to their corresponding secondary alkyl chlorides 9a–c in excellent yields. The use of enantiomerically pure alcohol 8a produced optically active 9a in high yield. The absolute configuration of this secondary chloride and its enantiomeric purity were determined by comparing its optical rotation to the literature value.23 These results confirmed that our chlorination reaction is stereospecific and proceeds via an inversion of stereo-chemistry. Our chlorination conditions proved to be mild, as β-hydroxy ester 8d readily afforded β-chloro ester 9d in 82% yield without suffering from any substantial elimination. Olefins were also found to be a suitable functionality. Secondary alcohols 8e,f, containing internal and terminal olefins, provided their corresponding chlorides 9e,f in good yields. Common protecting groups were also compatible under the reaction conditions. Entries 7–10 show that tert-butyldimethylsilyl ether, benzyl ether, p-methoxybenzyl ether, and p-toluoyl containing starting materials 8g–j proceeded to chlorination without complications to give the secondary chlorides 9g–j in excellent yields.
Table 3. Chlorination of Secondary Alcohols Containing Various Functionalities and Protecting Groups.
| entry | starting materiala | product | yield(b,c) |
|---|---|---|---|
| 1 |
|
|
89% |
| 2 |
|
|
87% |
| 3 |
|
|
88% |
| 4 |
|
|
82% |
| 5 |
|
|
94% |
| 6 |
|
|
82% |
| 7 |
|
|
90% |
| 8 |
|
|
84% |
| 9 |
|
|
82% |
| 10 |
|
|
85% |
Alcohols 8d–j are racemic.
Yield based on product isolated by flash chromatography.
In most cases, a minor (<10%) elimination product was detected in the crude mixture by either GC-MS or 1H NMR.
Our previous report also revealed the incompatibility of α-branched primary alcohols with our triphosgene–triethylamine chlorination conditions. Such substrates were readily transformed to the diethylcarbamate functionality.9 This problem was readily rectified with the new triphosgene–pyridine conditions. For example, as shown in Table 4, α-phenyl and α-methyl phenethyl alcohols 10a,b readily underwent chlorination to provide the primary alkyl chlorides 11a,b in 81% and 86% yields, respectively. N-Boc prolinol 10c also produced the corresponding chloride 11c in 65% yield.
Table 4. Chlorination of α-Branched Primary Alcohols.
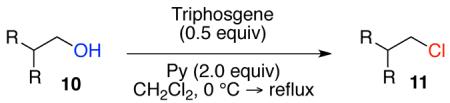
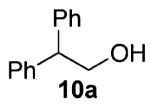
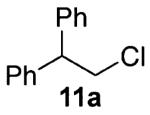
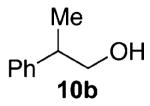
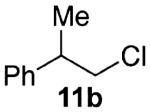
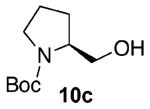
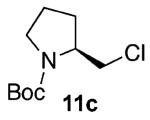
Alcohol 10b is racemic.
Yield based on product isolated by flash chromatography.
As an extension to the scope of substrate study, we continued our exploration to demonstrate the feasibility of our method to simultaneously introduce two carbon–chlorine bonds via global chlorination of multiple hydroxy centers. Scheme 2 details our initial attempts at this strategy using starting materials such as 1,3-diol 12 and 1,6-diol 14. Treatment of these compounds by doubling the amount of triphosgene and pyridine cleanly afforded alkyl dichlorides 13 and 15 in 55% and 73% yields, respectively. The lower isolated yields were most likely attributed to the high volatility of the resulting alkyl chlorides. These results suggested that, in the presence of excess triphosgene and pyridine, global chloroformylation of the two hydroxy groups preceded the potentially competitive intramolecular six-membered carbonate cyclization resulting from monochloroformylation.24,25 This hypothesis was clearly supported by the fact that exposure of 1,3-diol 12 to the typical 0.5 equiv of triphosgene and 2.0 equiv of pyridine resulted in a mixture of chlorination at the primary position, i.e. chloro alcohol 16, in 14% yield and cyclic carbonate 17 in 66% yield. These observations were consistent with our previous report.9
Scheme 2.

Reactivity of 1,3- and 1,6-Diols
The mechanism of this chlorination reaction is proposed as follows (Scheme 3). It is well precedented that activation of alcohol with a triphosgene–pyridine mixture leads to formation of the chloroformate functionality: viz., 18.16,19-22 It is reasonable to presume that excess pyridine then readily adds to the chloroformate to generate the putative N-acylpyridinium ion intermediate 19.26-28 This carbonyl activation increases the reactivity at the electrophilic secondary carbon center, which allows for SN 2 nucleophilic substitution by chloride ions. This process releases CO2 and pyridine, while generating the alkyl chloride with an inversion of stereochemistry.
Scheme 3.
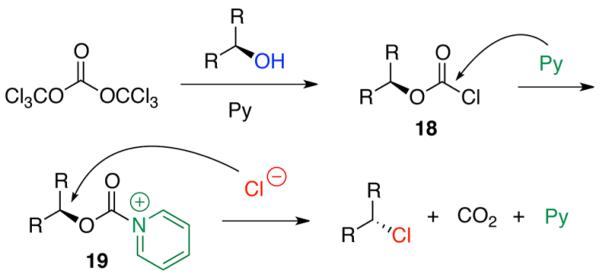
Proposed Reaction Mechanism
CONCLUSION
In summary, we have successfully addressed the previously unresolved issues concerning chlorination of aliphatic alcohols. We discovered that activation of unactivated aliphatic secondary alcohols with triphosgene and pyridine in dichloromethane at reflux cleanly produced the desired alkyl chlorides while eliminating the diethylcarbamoylation byproducts. These new reaction conditions are very useful and will strongly compliment the already existing chlorination methods. Our method is operationally simple, high yielding, stereospecific, mild, and well tolerated by a wide array of common functional and protecting groups. More importantly, our chlorination reaction does not produce any reactive or nuisance waste products, and the typical crude materials cleanly contain the target alkyl chloride. An extension of this work to other halogenation reactions and their applications toward stereoselective syntheses of chlorine-containing complex natural products are ongoing in our laboratories. Results from these efforts will be reported in due course.
EXPERIMENTAL SECTION
All materials, unless otherwise stated, were purchased from commercial sources and utilized without further purification. Anhydrous reactions were conducted in oven-dried glassware, which was then cooled under vacuum and purged with nitrogen gas. Anhydrous solvents (dichloromethane, toluene, acetonitrile, diethyl ether, and tetrahydrofuran) were filtered through activated 3 Å molecular sieves under nitrogen in a solvent purification system. Reactions were monitored either by analytical thin-layer chromatography (TLC silica gel 60 F254, glass plates) and analyzed using 254 nm UV light and anisaldehyde–sulfuric acid or potassium permanganate stains or via gas chromatography–mass spectrometry (GC-MS). The column for the GC-MS system was 5% phenyl methyl siloxane, measuring 30 m in length with an internal diameter of 250 μm and film thickness of 0.25 μm. Low and high mass readings were set to 40 to 800 m/z, respectively. Oven, inlet, and detector temperatures were set to 250 °C, and helium was used as the inert carrier gas. Column chromatography was completed using silica gel. Unless otherwise noted, all 1H and 13CNMR spectra were recorded in CDCl3 using a spectrometer operating at 400 MHz for 1H and 100 MHz for 13C or at 250 MHz for 1H and 62.5 MHz for 13C. Chemical shifts (δ) are reported in ppm relative to residual CHCl3 as an internal reference (1H, 7.26 ppm; 13C, 77.00 ppm). Coupling constants (J) are reported in hertz (Hz). Peak multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), x (septet), h (heptet), b (broad), and m (multiplet). FT-IR spectra were recorded using thin films, and absorption frequencies are reported in reciprocal centimeters (cm−1). High-resolution mass spectrometry (HRMS) analyses were performed using electron spray ionization–time of flight (ESI-TOF) methods.
General Chlorination Procedure
Unless otherwise noted, the alcohol (2.0 mmol) was placed in an oven-dried round-bottomed flask and dissolved in anhydrous dichloromethane (15 mL). The solution was then cooled to 0 °C. Pyridine (0.32 mL, 4.0 mmol) was then added via syringe, followed by triphosgene (297 mg, 1.0 mmol) in one portion. The solution was stirred for 5 min and then warmed to gentle reflux overnight. The reaction mixture was then poured into a separatory funnel containing 1 M HCl aqueous solution (30 mL), and the biphasic mixture was shaken vigorously. Upon separation of layers, the aqueous layer was re-extracted with dichloromethane (2 × 30 mL). Organic extracts were collected, dried over MgSO4, filtered, and concentrated under vacuum. The resulting crude material was purified using flash column chromatography with silica gel as the stationary phase and a hexanes/ethyl acetate, pentane/diethyl ether, or pentane/dichloromethane mixture as the mobile phase.
(+)-(S)-(2-Chloropropyl)benzene (9a)
Alcohol 8a (272 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 9a in 89% yield as a colorless oil (273 mg, 1.77 mmol). The purified product was eluted with 100% hexanes. 1H NMR (250 MHz, CDCl3): δ (ppm) 7.37–7.21 (5H, m), 4.24 (1H, x, J = 6.8 Hz), 3.11 (1H, dd, J = 13.8, 7.0 Hz), 2.98 (1H, dd, J = 13.9, 7.0 Hz), 1.53 (3H, d, J = 6.5 Hz). 13C NMR (62.5 MHz, CDCl3): δ (ppm) 137.9, 129.2, 128.3, 126.7, 58.4, 46.6, 24.6. GC-MS: M+ 154.6 calculated for C9H11Cl, experimental 154.0. [α]25D = +23.12° (c = 2.2 in CHCl ). Compound 9a is known:23 literature [α]25D = +23.19° (c = 5 in CHCl3) for enantiomerically pure (S)-(2-chloropropyl)benzene.
(2-Chloropropane-1,3-diyl)dibenzene (9b)
Alcohol 8b (420 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 9b in 87% yield as a colorless oil (397 mg, 1.73 mmol). The purified product was eluted with 100/0 → 90/10 hexanes/EtOAc. 1H NMR (250 MHz, CDCl3): δ (ppm) 7.38–7.23 (10H, m), 4.34 (1H, dddd, J = 8.0, 8.0, 5.6, 5.2 Hz), 3.16 (2H, dd, J = 14.1, 5.6 Hz), 3.04 (2H, dd, J = 14.2, 8.0 Hz). 13C NMR (62.5 MHz, CDCl3): δ (ppm) 137.8, 129.3, 128.3, 126.7, 63.9, 44.2. IR (cm−1): ν 3088, 3064, 3029, 1603, 1498, 1455, 911, 741, 700, 670. GC-MS: (M – H)+ 229.7 calculated for C15H14Cl, experimental 229.9. Compound 9b is known.29
N-Boc-4-chloropiperidine (9c)
Alcohol 8c (403 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 9c in 88% yield as a pale yellow oil (387 mg, 1.76 mmol). The purified product was eluted with 90/10 → 80/20 pentane/diethyl ether. 1H NMR (400 MHz, CDCl3): δ (ppm) 4.20 (1H, m), 3.73–3.68 (2H, m), 3.32–3.26 (2H, m) 2.06–1.99 (2H, m), 1.86–1.76 (2H, m), 1.46 (9H, s). 13C NMR (100 MHz, CDCl3): δ (ppm) 154.5, 79.6, 56.8, 41.3 (b), 34.9, 28.3. IR (cm−1): ν 2977, 2870, 2839, 1692, 1478, 1419, 1366, 1264, 1218, 1165, 1110, 1001, 895, 767, 719. HRMS-ESI: (M + Na)+ 242.0918 calculated for C10H18ClNNaO2, experimental 242.0918.
Ethyl (±)-3-Chloro-4-phenylbutyrate (9d)
Alcohol 8d 30 (417 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 9d in 82% yield as a pale yellow oil (372 mg, 1.64 mmol). The purified product was eluted with 100% pentane. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.34–7.22 (5H, m), 4.51 (1H, dt, J = 13.4, 7.3 Hz), 4.16 ppm (2H, q, J = 6.8 Hz), 3.09 (2H, dd, J = 6.7, 4.6 Hz), 2.75–2.71 (2H, m), 1.27 (3H, t, J = 7.1 Hz). 13C NMR (100 MHz, CDCl3): δ (ppm) 170.1, 137.0, 129.5, 128.6, 127.1, 60.9, 57.7, 44.3, 42.6, 14.2. IR (cm−1): ν 3065, 3030, 2983, 2905, 1737, 1654, 1304, 1150, 1096, 910, 747, 650. HRMS-ESI: (M + H)+ 227.0833 calculated for C12H16ClO2, experimental 227.0835.
(±)-4-Chloro-5-phenylpent-1-ene (9e)
Alcohol 8e 31 (324 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 9e in 94% yield as a colorless oil (340 mg, 1.88 mmol). The purified product was eluted with 100% pentane. 1H NMR (400, MHz, CDCl3): δ (ppm) 7.36–7.24 (5H, m), 5.93 (1H, m), 5.20–5.15 (2H, m), 4.17 (1H, m), 3.10 (1H, dd, J = 14.1, 6.2 Hz), 3.04 (1H, dd, J = 14.1, 6.4 Hz), 2.59 (1H, m), 2.48 (1H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 137.8, 134.0, 129.4, 128.5, 126.8, 118.3, 62.5, 44.2, 41.8. IR (cm−1): ν 3080, 3030, 2981, 2952, 1643, 1604, 1543, 1433, 1284, 1031, 993, 920, 700, 618. GC-MS: M+ 180.1 calculated for C11H13Cl, experimental 180.0. Compound 9e is known.6
(±)-(E)-4-Chloro-1-phenylhepta-1,6-diene (9f)
Alcohol 8f (377 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 9f in 82% yield as a yellow oil (339 mg, 1.64 mmol). The purified product was eluted with 100% pentane. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.38–7.17 (5H, m), 6.48 (1H, d, J = 15.8 Hz), 6.25 (1H, m), 5.94–5.83 (1H, m), 5.18–5.10 (2H, m), 4.03 (1H, m), 2.72–2.61 (2H, m), 2.59–2.50 (2H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 137.1, 134.0, 133.2, 128.6, 127.4, 126.2, 125.5, 118.2, 61.5, 42.0, 41.2. IR (cm−1): ν 3081, 3061, 3028, 2980, 2946, 1644, 1495, 1449, 1289, 967, 918, 744, 694. HRMS-ESI: (M + H)+ 207.0935 calculated for C13H16Cl, experimental 207.0936.
(±)-4-((tert-Butyldimethylsilyl)oxy)-2-chloro-1-phenylbutane (9g)
Alcohol 8g (560 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 9g in 90% yield as a colorless oil (536 mg, 1.80 mmol). The purified product was eluted with 100/0 → 90/10 hexanes/EtOAc. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36–7.22 (5H, m), 4.33 (1H, ddt, J = 10.1, 9.8, 3.4 Hz), 3.85–3.73 (2H, m), 3.08 (2H, d, J = 6.8 Hz), 2.03 (1H, m), 1.81 (1H, ddt, J = 14.3, 9.8, 4.4 Hz), 0.89 (9H, s), 0.05 (6H, d, J = 7.1 Hz). 13C NMR (100 MHz, CDCl3): δ (ppm) 137.9, 129.4, 128.4, 126.7, 60.3, 59.8, 45.1, 40.6, 25.9, 18.3, −5.4. IR (cm−1): ν 3030, 2954, 2930, 2857, 1472, 1256, 1110, 910, 837, 778. HRMS-ESI: (M + H)+ 299.1592 calculated for C16H28ClOSi, experimental 299.1599.
(±)-4-Benzyloxy-2-chloro-1-phenylbutane (9h)
Alcohol 8h (256 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 9h in 84% yield as a colorless oil (462 mg, 1.69 mmol). The purified product was eluted with 98/2 hexanes/EtOAc. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.34–7.21 (10H, m), 4.54–4.45 (2H, m), 3.88 (1H, m), 3.69–3.60 (2H, m), 2.97 (1H, dd, J = 13.5, 6.0 Hz), 2.80 (1H, dd, J = 13.6, 6.4 Hz), 1.96–1.90 (2H, m). 13C NMR (100, CDCl3): δ (ppm) 138.3, 138.1, 129.5, 128.4, 128.0, 127.8, 126.4, 77.1, 72.1, 41.7, 40.6, 37.4. IR (cm−1): ν 3063, 3023, 2925, 2866, 1496, 1456, 1350, 1289, 1073, 1029, 910, 737, 699, 651. HRMS-ESI: (M + Na)+ 297.1017 calculated for C17H19ClNaO, experimental 297.1019.
(±)-2-Chloro-4-((4-methoxybenzyl)oxy)-1-phenylbutane (9i)
Alcohol 8i (572 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol), and triphosgene (297 mg, 1.00 mmol) to produce 9i in 82% yield as a colorless oil (498 mg, 1.64 mmol). The purified product was eluted with 90/10 hexanes/EtOAc. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.33–7.19 (7H, m), 6.87 (2H, d, J = 8.6 Hz), 4.42 (2H, q, J = 10.9 Hz), 3.86 (1H, m), 3.81 (3H, s), 3.67–3.57 (2H, m), 2.95 (1H, dd, J = 13.6, 6.1 Hz), 2.78 (1H, dd, J = 13.7, 6.4 Hz), 1.96–1.86 (2H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 159.3, 138.2, 130.4, 129.5, 128.4, 126.4, 113.9, 76.8, 71.8, 55.3, 41.7, 40.7, 37.4. IR (cm−1): ν 3063, 3030, 2936, 2870, 1612, 1513, 1248, 1076, 1034, 822, 740, 701. HRMS-ESI: (M + Na)+ 327.1122 calculated for C18H21ClNaO2, experimental 327.1133.
(±)-3-Chloro-4-phenylbutyl 4-Methylbenzoate (9j)
Alcohol 8j (568 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 9j in 85% yield as a colorless oil (514 mg, 1.70 mmol). The purified product was eluted with 90/10 hexanes/EtOAc. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.91 (2H, d, J = 8.2 Hz), 7.35–7.24 (5H, m), 4.55 (1H, ddd, J = 11.2, 6.2, 4.7 Hz), 4.47 (1H, m), 4.32 (1H, m), 3.13 (2H, d, J = 6.8 Hz), 2.42 (3H, s), 2.31 (1H, m), 2.09 (1H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 166.5, 143.8, 137.4, 129.6, 129.4, 129.1, 128.5, 127.4, 127.0, 61.8, 59.9, 45.0, 36.7, 21.7. IR (cm−1): ν 3087, 3062, 2964, 2861, 1715, 1270, 1177, 1109, 1021, 752, 700. HRMS-ESI: (M + H)+ 303.1152 calculated for C18H20ClO2, experimental 303.1145.
2-Chloro-1,1-diphenylethane (11a)
Alcohol 10a (397 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 11a in 81% yield as a light yellow oil (350 mg, 1.62 mmol). The purified product was eluted with 100% hexanes. 1H NMR (250 MHz, CDCl3): δ (ppm) 7.35–7.21 (10H, m), 4.34 (1H, t, J = 7.8 Hz), 4.07 (2H, d, J = 7.8 Hz). 13C NMR (62.5 MHz, CDCl3): δ (ppm) 141.2, 128.5, 127.9, 126.9, 53.5, 47.1. IR (cm−1): ν 3062, 3029, 1494, 1452, 910, 737, 699. GC-MS: M+ 216.1 calculated for C14H13Cl, experimental 216.0. Compound 11a is known.32
(±)-1-Chloro-2-phenylpropane (11b)
Alcohol 10b (0.28 mL, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 11b in 86% yield as a colorless oil (266 mg, 1.73 mmol). The purified product was eluted with 100% hexanes. 1H NMR (250 MHz, CDCl3): δ (ppm) 7.39–7.19 (5H, m), 3.72 (1H, dd, J = 10.7, 6.2 Hz), 3.61 (1H, dd, J = 10.7, 7.8 Hz), 3.12 (1H, x, J = 7.0 Hz), 1.42 (3H, d, J = 7.0 Hz). 13C NMR (62.5 MHz, CDCl3): δ (ppm) 143.2, 128.5, 127.1, 126.8, 50.7, 42.2, 18.9. IR (cm−1): ν 3030, 2970, 2875, 1494, 1454, 1015, 910, 762, 720, 699. GC-MS: (M)+ 154.1 calculated for C9H11Cl, experimental 154.0. Compound 11b is available commercially.
(S)-N-Boc-2-(chloromethyl)pyrrolidine (11c)
Alcohol 10c (345 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 11c in 65% yield as a colorless oil (244 mg, 1.11 mmol). The purified product was eluted with 90/10 hexanes/EtOAc. 1H NMR (400 MHz, CDCl3): δ (ppm) 4.04 (0.5H, b), 3.95 (0.5H, b), 3.75 (0.5H, b, d, J = 9.9 Hz), 3.66 (0.5H, b, d, J = 9.9 Hz), 3.54 (0.5H, t, J = 9.0 Hz), 3.46–3.33 (2.5H, m), 2.00 (2H, t, J = 6.4 Hz), 1.88–1.78 (2H, m), 1.46 (9H, s). 13C NMR (100, MHz, CDCl3): δ (ppm) 154.5, 154.2, 79.8, 79.5, 58.1, 58.0, 47.3, 46.8, 45.5, 45.3, 29.2, 28.4, 23.6, 22.8. IR (cm−1): ν 2977, 2879, 1695, 1392, 1171, 1118, 910, 733. [α]25D = −7.29° (c = 1.6 in CHCl ). HRMS-ESI: (M + Na)+ 242.0924 calculated for C10H18ClNNaO2, experimental 242.0917.
(±)-(2,4-Dichlorobutyl)benzene (13)
Alcohol 12 33 (332 mg, 2.00 mmol) was utilized along with pyridine (0.65 mL, 8.00 mmol) and triphosgene (593 mg, 2.00 mmol) in 30 mL of CH2Cl2 to produce 13 in 55% yield as a colorless oil (223 mg, 1.10 mmol). The purified product was eluted with 100/0 → 90/10 hexanes/EtOAc. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36–7.22 (5H, m), 4.35 (1H, m), 3.78–3.69 (2H, m), 3.13 (1H, dd, J = 14.1, 7.3 Hz), 3.06 (1H, dd, J = 14.0, 6.6 Hz), 2.20 (1H, m), 2.08 (1H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 137.1, 129.4, 128.5, 127.0, 60.0, 44.8, 41.7, 40.1. IR (cm−1): ν 3031, 2965, 1496, 1454, 1318, 1285, 910, 735, 701. GC-MS: M+ 202.0 calculated for C10H12Cl2, experimental 202.0. HRMS-ESI: (M – H – 2Cl)+ 131.0855 calculated for C10H11, experimental 131.0857.
(±)-4,8-Dichlorooct-1-ene (15)
Alcohol 14 34 (288 mg, 2.00 mmol) was utilized along with pyridine (0.64 mL, 8.00 mmol) and triphosgene (593 mg, 2.00 mmol) in 30 mL of CH2Cl2 to produce 15 in 73% yield as a colorless oil (264 mg, 1.47 mmol). The purified product was eluted with 100/0 → 90/10 hexanes/EtOAc. 1H NMR (400 MHz, CDCl3): δ (ppm) 5.85 (1H, m), 5.16–5.12 (2H, m), 3.93 (1H, m), 3.55 (2H, t, J = 6.4 Hz), 2.53–2.49 (2H, m), 1.87–1.66 (6H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 134.0, 118.1, 62.2, 44.7, 42.7, 37.0, 32.1, 23.8. IR (cm−1): ν 3081, 2950, 2868, 1643, 995, 911, 735, 650. GC-MS: (M)+ 180.0 calculated for C8H14Cl2, experimental 179.9. HRMS-ESI: (M – 2H – 2Cl + Na)+ 131.0831 calculated for C8H12Na, experimental 131.0838.
(±)-4-Chloro-1-phenylbutan-2-ol (16) and (±)-4-Benzyl-1,3-dioxan-2-one (17)
Alcohol 12 33 (332 mg, 2.00 mmol) was utilized along with pyridine (0.32 mL, 4.00 mmol) and triphosgene (297 mg, 1.00 mmol) to produce 17 in 66% yield as a colorless oil (254 mg, 1.32 mmol) and 16 in 14% yield as a colorless oil (50 mg, 0.27 mmol). The purified products were eluted with 90/10 → 80/20 → 70/30 hexanes/EtOAc. Data for the less polar product (16) are as follows. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.38–7.22 (5H, m), 4.08 (1H, ddd, J = 15.8, 7.9, 4.7 Hz), 3.80–3.65 (2H, m), 2.86 (1H, dd, J = 13.5, 4.2 Hz), 2.71 (1H, dd, J = 13.5, 8.6 Hz), 2.01–1.93 (2H, m), 1.66 (1H, d, J = 3.0 Hz). 13C NMR (100 MHz, CDCl3): δ (ppm) 137.7, 129.3, 129.6, 126.6, 69.4, 43.9, 41.7, 39.1. IR (cm−1): ν 3425, 3378, 3063, 3029, 2943, 2920, 1946, 1454, 1081, 742, 701. HRMS-ESI: (M + Na)+ 207.0553 calculated for C10H13ClNaO, experimental 207.0543. Data for the more polar product (17) are as follows. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.37–7.22 (5H, m), 4.67 (1H, m), 4.45–4.27 (2H, m), 3.15 (1H, dd, J = 13.8, 5.7 Hz), 2.92 (1H, dd, J = 13.8, 7.3 Hz), 2.03–1.83 (2H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 148.6, 135.0, 129.4, 128.7, 127.1, 79.5, 66.8, 41.3, 26.2. IR (cm−1): ν 3352, 3028, 2973, 2932, 2875, 1753, 1409, 1250, 1188, 1124, 911, 738, 703. HRMS-ESI: (M − CO + 3H)+ 167.1072 calculated for C10H15O2, experimental 167.1055.
Preparation of Secondary Alcohols 8f–j
(±)-(E)-1-Phenylhepta-1,6-dien-4-ol (8f)
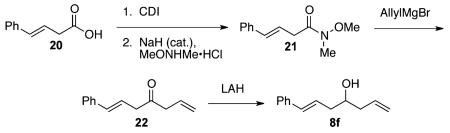
trans-Styrylacetic acid (20; 3.30 g, 20.35 mmol) was dissolved in THF (50 mL), and carbonyldiimidazole (4.30 g, 26.45 mmol) was then added in one portion. The reaction mixture was then stirred overnight. The crude reaction mixture was concentrated in vacuo, diluted with Et2O (50 mL), and then washed with saturated brine solution (3 × 50 mL). Collected aqueous layers were extracted with Et2O (3 × 50 mL) and combined with the organic layer. The organic fractions were then dried over MgSO4, concentrated under vacuum, and taken to the next step without further purification. This crude material was then dissolved in THF (50 mL), and MeONHMe·HCl (1.85 g, 18.99 mmol) was added. A catalytic amount of sodium hydride (μ5 mg) was then added to the solution, and the reaction mixture was stirred for 3 h. The reaction was quenched with a half-saturated NH4Cl solution (50 mL). Upon separation of layers, the organic layer was washed with a saturated NaHCO3 solution (50 mL), which was then back-extracted with EtOAc (3 × 50 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The resulting crude material was purified with 80/20 hexanes/EtOAc to yield the Weinreb amide 21 in 57% yield (2.39 g, 11.66 mmol) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.38–7.20 (5H, m), 6.51 (1H, d, J = 15.9 Hz), 6.37 (1H, ddd, J = 15.8, 6.9, 6.8 Hz), 3.72 (3H, s), 3.39 (2H, d, J = 6.4 Hz), 3.21 (3H, s). 13C NMR (100 MHz, CDCl3): δ (ppm) 137.1, 133.1, 128.5, 127.4, 126.3, 122.8, 61.2, 36.5, 32.3 (b), 29.7. FT-IR (cm−1): ν 2936, 1749, 1722, 1448, 1419, 1177, 999, 968, 910, 735. HRMS-ESI: (M + H)+ 206.1176 calculated for C12H16NO2, experimental 206.1170.
The Weinreb amide 21 (1.16g, 5.65 mmol) was dissolved in dry THF (50 mL), and the solution was cooled to 0 °C. Allylmagnesium bromide (11.3 mL, 11.3 mmol, 1.0 M in Et2O) was then added slowly over 20 min. The reaction mixture was stirred for 1 h and then quenched with a half-saturated NH4Cl (50 mL) solution. Upon separation of layers, the aqueous layer was extracted with EtOAc (3 × 50 mL). The organic layers were combined and dried over MgSO4. The crude material was concentrated in vacuo and purified with 90/10 hexanes/EtOAc to afford ketone 22 in 82% yield (855 mg, 4.59 mmol) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.38–7.22 (5H, m), 6.48 (1H, d, J = 15.9 Hz), 6.31 (1H, ddd, J = 16.0, 7.1, 6.9 Hz), 6.00–5.89 (1H, m), 5.23–5.15 (2H, m), 3.36 (2H, d, J = 7.0 Hz), 3.27 (2H, d, J = 6.9 Hz). 13C NMR (100 MHz, CDCl3): δ (ppm) 206.4, 136.8, 133.9, 130.3, 128.6, 127.6, 126.3, 121.7, 119.2, 47.3, 46.4. FT-IR (cm−1): ν 3082, 3061, 3027, 2981, 1717, 1639, 1578, 1449, 1424, 1323, 1071, 993, 967, 912, 741, 695, 650. HRMS-ESI: (M + H)+ 187.1117 calculated for C13H15O, experimental 187.1117.
Ketone 22 (855 mg, 4.60 mmol) as a solution in Et2O (20 mL) was added via cannula to a cooled (0 °C) suspension of lithium aluminum hydride (209 mg, 5.50 mmol). The reaction mixture was then warmed to room temperature and set to reflux for 30 min. After the reaction mixture was cooled to 0 °C, deionized water (0.21 mL) was slowly added, which was followed by 15% aqueous sodium hydroxide solution (0.21 mL) and then deionized water (0.63 mL). This workup sequence resulted in the formation of white precipitates. The solution was then stirred for 1 h. The filtrate was collected using vacuum filtration and concentrated in vacuo. The crude material was then purified with 90/10 → 80/20 hexanes/EtOAc to give 8f with a yield of 94% (811 mg, 4.31 mmol) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.37–7.20 (5H, m), 6.49 (1H, d, J = 16.1 Hz), 6.25 (1H, ddd, J = 15.9, 7.1, 6.9 Hz), 5.87 (1H, m), 5.19–5.14 (2H, m), 3.78 (1H, m), 2.48–2.33 (3H, m), 2.24 (1H, m), 1.76 (1H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 137.2, 134.6, 133.1, 128.5, 127.3, 126.1, 118.2, 70.2, 41.4, 40.5, 29.7. FT-IR (cm−1): ν 3416, 3079, 3027, 2930, 1641, 1599, 1495, 1449, 1073, 997, 967, 912, 742. HRMS-ESI: (M + H)+ 189.1274 calculated for C13H17O, experimental 189.1275.
(±)-4-((tert-Butyldimethylsilyl)oxy)-1-phenylbutan-2-ol (8g)
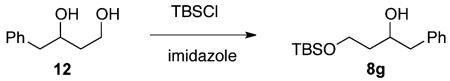
Diol 12 33 (479 mg, 2.88 mmol) was dissolved in CH2Cl2 (75 mL). Imidazole (432 mg, 6.34 mmol) was then added, and the reaction mixture was cooled to −42 °C. TBSCl (478 mg, 3.17 mmol) was then added in one portion. The reaction mixture was stirred overnight while being slowly warmed to room temperature and then quenched with a half-saturated NH4Cl solution (50 mL). Upon separation of layers, the aqueous layer was extracted with CH2Cl2 (3 × 30 mL), washed with brine, dried over MgSO4, filtered, and concentrated under vacuum. The crude material was purified with 90/10 hexanes/EtOAc to give 8g in 56% yield as a colorless oil (452 mg, 1.61 mmol). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.32–7.20 (5H, m), 4.08 (1H, m), 3.90 (1H, p, J = 4.8 Hz), 3.79 (1H, ddd, J = 10.4, 6.5, 6.3 Hz), 3.35 (1H, d, J = 2.0 Hz), 2.85 (1H, dd, J = 13.5, 7.0 Hz), 2.74 (1H, dd, J = 13.5, 6.2 Hz), 1.71–1.66 (2H, m), 0.91 (9H, s), 0.08 (6H, s). 13C NMR (100 MHz, CDCl3): δ (ppm) 138.8, 129.4, 128.4, 126.3, 73.0, 62.6, 44.0, 37.6, 25.9, 18.2, −5.5. IR (cm−1): ν 3455, 2954, 2930, 2857, 1472, 1255, 1085, 909, 836, 777, 740, 700. HRMS-ESI: (M + H)+ 281.1931 calculated for C16H29O2Si, experimental 281.1922.
(±)-4-Benzyloxy-1-phenylbutan-2-ol (8h)
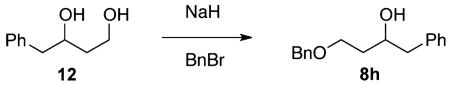
Diol 12 33 (2.78g, 16.8 mmol) in a solution of DMF (20 mL) was slowly added via cannula to a cooled suspension (0 °C) of NaH (0.426 g, 18.42 mmol) in DMF (20 mL). After the mixture was stirred for 30 min, benzyl bromide (1.99 mL, 16.8 mmol) was added. The reaction mixture was then warmed to room temperature and stirred overnight. After the reaction was quenched with a half-saturated NH4Cl solution, the mixture was then extracted with EtOAc (3 × 30 mL), and the collected organic layers were washed with water and dried over MgSO4. The crude material was purified with 90/10 → 80/20 → 70/30 hexanes/EtOAc, and alcohol 8h was isolated in 24% yield (1.08 g, 3.95 mmol) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36–7.19 (10H, m), 4.59 (1H, d, J = 11.4 Hz), 4.48 (1H, d, J = 11.4 Hz), 3.86 (1H, m), 3.81–3.70 (2H, m), 3.04 (1H, dd, J = 13.4, 5.8 Hz), 2.79 (1H, dd, J = 13.6, 7.0 Hz), 2.22 (1H, t, J = 5.0 Hz), 1.67–1.81 (2H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 138.4, 138.1, 129.5, 128.5, 128.4, 128.0, 127.8, 126.3, 79.6, 71.6, 60.7, 40.5, 30.1. IR (cm−1): ν 3410, 3086, 3063, 3029, 2493, 2875, 1604, 1496, 1454, 1056, 1029, 910, 738, 699. HRMS-ESI: (M + H)+ 257.1536 calculated for C17H21O2, experimental 257.1529.
(±)-4-((4-Methoxybenzyl)oxy)-1-phenylbutan-2-ol (8i)
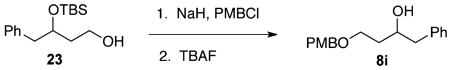
Alcohol 23 9 (2.88g, 10.3 mmol) was dissolved in THF (150 mL) and cooled to 0 °C. NaH (1.12 g, 46.8 mmol) was then added slowly, and the suspension was stirred for 30 min. PMBCl (2.31 g, 14.8 mmol) was then added, and the reaction mixture was brought to room temperature and stirred overnight. The reaction was then slowly quenched with methanol (60 mL), followed by addition of a half-saturated brine solution (50 mL). Upon separation of layers, the aqueous solution was extracted with EtOAc (3 × 50 mL). The organic layers were combined and washed with saturated NaCl, dried over MgSO4, filtered, and concentrated under vacuum. The resulting crude material was dissolved in THF (50 mL) and treated with 1.0 M TBAF solution in THF (20.5 mL, 20.56 mmol). After this solution was stirred overnight, EtOAc (100 mL) was added. This organic solution was washed sequentially with deionized H2O and brine, and it was then dried over MgSO4, filtered, and concentrated under vacuum. Purification of the resulting crude material with 80/20 → 70/30 → 60/40 hexanes/EtOAc gave alcohol 8i in 23% yield as a colorless oil (664 mg, 2.32 mmol). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.32–7.20 (7H, m), 6.91–6.83 (2H, m), 4.52 (1H, d, J = 11.1 Hz), 4.41 (1H, d, J = 11.0 Hz), 3.85 (1H, m), 3.81 (3H, t, J = 4.2 Hz), 3.78–3.66 (2H, m), 3.03 (1H, dd, J = 13.5, 5.8 Hz), 2.77 (1H, dd, J = 13.5, 5.8 Hz), 1.81–1.62 (2H, m). 13C NMR (100 MHz, CDCl3): δ (ppm) 159.3, 138.4, 130.2, 129.6, 128.7, 128.4, 126.3, 113.9, 79.4, 71.2, 60.7, 55.3, 40.5, 36.1. IR (cm−1): ν 3417, 2941, 1613, 1514, 1249, 1035, 822, 741, 703. HRMS-ESI: (M + Na)+ 309.1467 calculated for C18H22NaO3, experimental 309.1465.
(±)-3-Hydroxy-4-phenylbutyl 4-Methylbenzoate (8j)
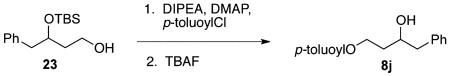
Alcohol 23 9 (1.12g, 4.00 mmol) was dissolved in CH2Cl2 (40 mL) and cooled to 0 °C. DIPEA (3.56 mL, 20.0 mmol), DMAP (0.24 g, 2.00 mmol), and p-toluoyl chloride (0.63 mL, 4.80 mmol) were sequentially added. After it was stirred for 3 h, the reaction mixture was quenched with 2 M HCl solution (40 mL). Upon separation of layers, the aqueous solution was extracted with CH2Cl2 (3 × 30 mL), dried over MgSO4, filtered, and concentrated under vacuum. The resulting crude material was then dissolved in THF (25 mL) and treated with 1.0 M TBAF solution in THF (8.0 mL, 8.00 mmol). After the mixture was stirred overnight, EtOAc (100 mL) was added. This organic solution was washed sequentially with deionized H2O and brine, and it was then dried over MgSO4, filtered, and concentrated under vacuum. Purification of the resulting crude material with 80/20 hexanes/EtOAc gave 8j in 65% yield as a colorless oil (744 mg, 2.62 mmol). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.01–7.89 (2H, m), 7.34–7.23 (7H, m), 4.59 (1H, ddd, J = 11.1, 8.6, 5.2 Hz), 4.43 (1H, p, J = 5.6 Hz), 4.01 (1H, ddt, J = 8.6, 8.6, 4.3 Hz), 2.88 (1H, dd, J = 13.6, 4.6 Hz), 2.78 (1H, dd, J = 13.6, 8.2 Hz), 2.44 (1H, s), 2.42 (3H, s), 2.03 (1H, m), 1.89 (1H, m). 13C NMR (400 MHz, CDCl3): δ (ppm) 167.0, 143.7, 138.1, 130.2, 129.7, 129.5, 129.1, 128.6, 127.4, 126.6, 69.6, 62.0, 44.0, 36.0, 21.7. IR (cm−1): ν 3482, 3029, 2957, 2919, 1712, 1612, 1455, 1274, 1178, 1111, 1021, 972, 842, 753, 701. HRMS-ESI: (M + H)+ 285.1491 calculated for C18H21O3, experimental 285.1494.
Supplementary Material
ACKNOWLEDGMENTS
Generous financial support from Louisiana State University is greatly appreciated. A.V. thanks the National Science Foundation for the Bridge to the Doctorate Project (BDP) Fellowship (NSF1141152). C.E.A. thanks the Louisiana Board of Regents for the BOR Fellowship (LEQSF(2011-16)-GF-03). We thank Professor George Stanley for kindly allowing us to use the GC-MS instrument in his laboratories.
ASSOCIATED CONTENT
Supporting Information
Figures giving GC-MS chromatograms for Tables 1 and 2 and NMR (1H and 13C) spectra for characterized compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Footnotes
Undergraduate research participant.
REFERENCES
- (1).Cahiez G, Lefevre N, Poizat M, Moyeux A. Synthesis. 2013;45:231. [Google Scholar]
- (2).Denton RM, An J, Adeniran B, Blake AJ, Lewis W, Poulton AM. J. Org. Chem. 2011;76:6749. doi: 10.1021/jo201085r. [DOI] [PubMed] [Google Scholar]
- (3).Braddock DC, Pouwer RH, Burton JW, Broadwith P. J. Org. Chem. 2009;74:6042. doi: 10.1021/jo900991z. [DOI] [PubMed] [Google Scholar]
- (4).Shibuya GM, Kanady JS, Vanderwal CD. J. Am. Chem. Soc. 2008;130:12514. doi: 10.1021/ja804167v. [DOI] [PubMed] [Google Scholar]
- (5).Yasuda M, Shimizu K, Yamasaki S, Baba A. Org. Biomol. Chem. 2008;6:2790. doi: 10.1039/b804589e. [DOI] [PubMed] [Google Scholar]
- (6).Dubey A, Upadhyay AK, Kumar P. Tetrahedron Lett. 2010;51:744. [Google Scholar]
- (7).Nilewski C, Carreira EM. Eur. J. Org. Chem. 2012:1685. [Google Scholar]
- (8).Bedke DK, Vanderwal CD. Nat. Prod. Rep. 2011;28:15. doi: 10.1039/c0np00044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ayala CE, Villalpando A, Nguyen AL, McCandless GT, Kartika R. Org. Lett. 2012;14:3676. doi: 10.1021/ol301520d. [DOI] [PubMed] [Google Scholar]
- (10).Eckert H, Forster B. Angew. Chem., Int. Ed. Engl. 1987;26:894. [Google Scholar]
- (11).Damle SB. Chem. Eng. News. 1993;71:4. [Google Scholar]
- (12).Cotarca L. Org. Process Res. Dev. 1999;3:377. [Google Scholar]
- (13).Lemoucheux L, Rouden J, Ibazizene M, Sobrio F, Lasne MC. J. Org. Chem. 2003;68:7289. doi: 10.1021/jo0346297. [DOI] [PubMed] [Google Scholar]
- (14).Banwell MG, Coster MJ, Harvey MJ, Moraes JJ. Org. Chem. 2003;68:613. doi: 10.1021/jo0263622. [DOI] [PubMed] [Google Scholar]
- (15).Igarashi J, Kobayashi Y. Tetrahedron Lett. 2005;46:6381. [Google Scholar]
- (16).Roestamadji J, Mobashery S, Banerjee A. Encyclopedia of Reagents for Organic Synthesis. Wiley; Hoboken, NJ: 2006. [Google Scholar]
- (17).Kuna S, Pawlak Z, Tusk M. J. Chem. Soc., Faraday Trans. 1. 1982;78:2685. [Google Scholar]
- (18).Goren Z, Heeg MJ, Mobashery S. J. Org. Chem. 1991;56:7186. [Google Scholar]
- (19).Fu LQ, Liu X, Ling CY, Cheng JJ, Guo XS, He HL, Ding S, Yang YS. Bioorg. Med. Chem. Lett. 2012;22:814. doi: 10.1016/j.bmcl.2011.12.063. [DOI] [PubMed] [Google Scholar]
- (20).Fang LJ, Yang JO, Yang F. Org. Lett. 2010;12:3124. doi: 10.1021/ol1011423. [DOI] [PubMed] [Google Scholar]
- (21).Hemantha HP, Sureshbabu VV. Synlett. 2008:496. [Google Scholar]
- (22).Hajra S, Bhowmick M, Maji B, Sinha D. J. Org. Chem. 2007;72:4872. doi: 10.1021/jo070614n. [DOI] [PubMed] [Google Scholar]
- (23).Masuda S, Nakajima T, Suga S. Bull. Chem. Soc. Jpn. 1983;56:1086. [Google Scholar]
- (24).Burk RM, Roof MB. Tetrahedron Lett. 1993;34:395. [Google Scholar]
- (25).Anderson EA, Davidson JEP, Harrison JR, O’Sullivan PT, Burton JW, Collins I, Holmes AB. Tetrahedron. 2002;58:1943. [Google Scholar]
- (26).Wolfe BH, Libby AH, Al-Awar RS, Foti CJ, Comins DL. J. Org. Chem. 2010;75:8564. doi: 10.1021/jo1019688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Comins DL, Joseph SP, Goehring RR. J. Am. Chem. Soc. 1994;116:4719. [Google Scholar]
- (28).Sahn JJ, Bharathi P, Comins DL. Tetrahedron Lett. 2012;53:1347. [Google Scholar]
- (29).Kalyani D, Sanford MS. J. Am. Chem. Soc. 2008;130:2150. doi: 10.1021/ja0782798. [DOI] [PubMed] [Google Scholar]
- (30).Kiyooka S, Shirouchi M. J. Org. Chem. 1992;57:1. [Google Scholar]
- (31).Canova S, Bellosta M, Mignani S, Bigot A, Cossy J. Org. Lett. 2006;8:2091. doi: 10.1021/ol060529m. [DOI] [PubMed] [Google Scholar]
- (32).Pollastri MP, Sagal JF, Chang G. Tetrahedron Lett. 2001;42:2459. [Google Scholar]
- (33).Medlik-Balan A, Klein J. Tetrahedron. 1980;36:299. [Google Scholar]
- (34).Lin MH, Hung SF, Lin LZ, Tsai WS, Chuang TH. Org. Lett. 2011;13:332. doi: 10.1021/ol102825t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.