

Abstract
A Robinson annulation, van Leusen homologation, and a desymmetrizing C–H oxidation enabled an enantiospecific synthesis of the neurotrophic natural product jiadifenolide. From a pulegone-derived building block, a key propellane intermediate was constructed through the use of simple reagents in a highly diastereoselective fashion. A short series of oxidations of this tricylic framework allowed progression to the natural product.
Keywords: total synthesis, natural products, C–H activation, terpenoids, chiral pool
The investigations of the chemical constituents of the flowering plant Illicium jiadifengpi by Fukuyama and coworkers led to the isolation of a new metabolite that was given the name jiadifenolide.[1] High-resolution mass spectrometric and infrared spectroscopic analyses revealed the molecular formula (C15H18O7) and functional group-content of jiadifenolide, respectively, while interpretations of spectroscopic data suggested a structural analogy to the known sesquiterpene jiadifenin.[2] A single-crystal X-ray crystallographic analysis confirmed this structural analogy and revealed that jiadifenolide is a seco-prezizaane-type sesquiterpene with the novel, cage-like architecture shown in Figure 1. Thirteen of the fifteen carbons and three oxygen atoms of jiadifenolide (1) define an intricate ring system comprising two γ-lactones, a 5-membered ring hemi-ketal, and seven contiguous stereocenters.
Figure 1.
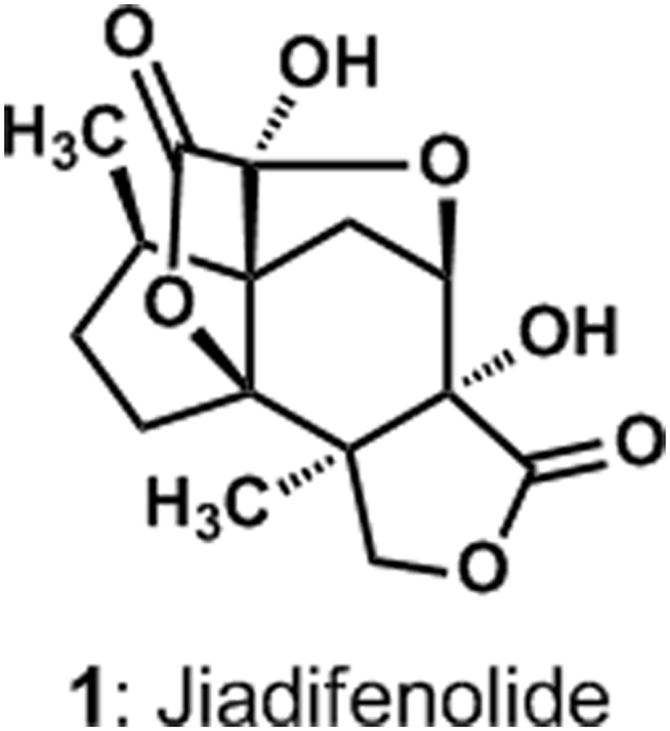
The molecular structure of jiadifenolide (1).
Jiadifenolide was also found to significantly potentiate neurite outgrowth in primary cultured rat cortical neurons at a concentration of 10 nM.[1] This compound belongs to a growing class of natural products that offers new entities with promising neurotrophic properties, as well as compelling objectives for undertakings in chemical synthesis.[3] In 2011, Theodorakis and coworkers described their impressive, pioneering synthesis of jiadifenolide.[4] Herein, we describe the ideas and reactions that permitted our laboratory to achieve a concise, enantiospecific synthesis of this pentacyclic natural product.
There is a correspondence between the highlighted domain of jiadifenolide (1) and the seven highlighted carbons of β-keto ester 2 (Figure 2). This insight, enabled by an exercise in pattern recognition,[5] identified the pulegone-derived, chiral building block 2[6] as the foundation for the synthesis. This functionalized compound, which possesses the needed cyclopentane frame and the C-1 secondary methyl group, would allow a rapid advance to late-stage intermediates in a “left-to-right” ring annulation strategy. The reliable chemistry associated with 1,3-dicarbonyl compounds would thus be leveraged in a synthesis of an intermediate of the type shown as 4, wherein the unspecified directing group (DG) would serve the synthesis by guiding a pivotal oxidation of one of the C–H bonds in the highlighted methyl group.[7] This concept was, in fact, the heart of our design for synthesis, and we were mindful that Sanford’s powerful, catalytic method for achieving directed, site-selective acetoxylations of unactivated C–H bonds[8] might be ideally suited for enabling the desired oxidation of 4 to a compound of the type 5. A successful Sanford oxidation in the structural context of 4 (in which the unspecified DG would be a trigonal oxime functional group) would simplify the strategy because it would allow us to carry simple geminal methyl groups through the early and middle phases of our synthesis; the key, directed oxidation would differentiate those methyl groups and introduce the oxygen atom that is attached to C-14 in jiadifenolide (1). The manner in which these ideas were brought to fruition is outlined in Scheme 1 and described below.
Figure 2.
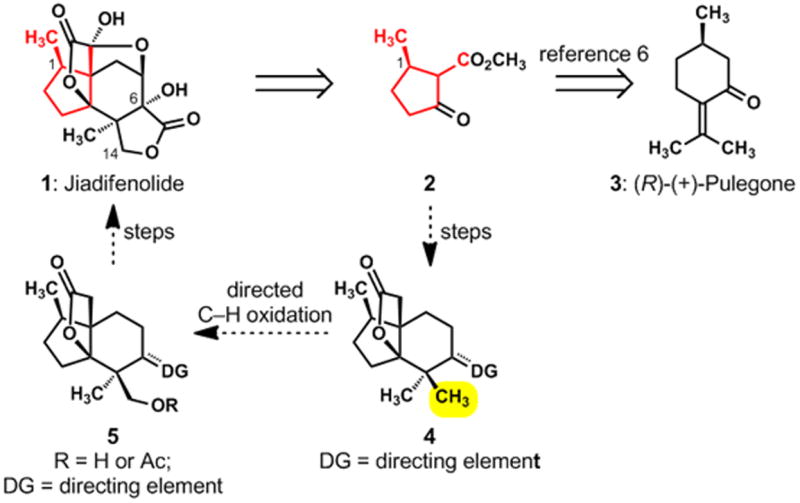
Toward a strategy for synthesizing jiadifenolide (1): the pulegone connection and a concept for differentiating geminal methyl groups via a directed, catalytic C–H oxidation.
Scheme 1.
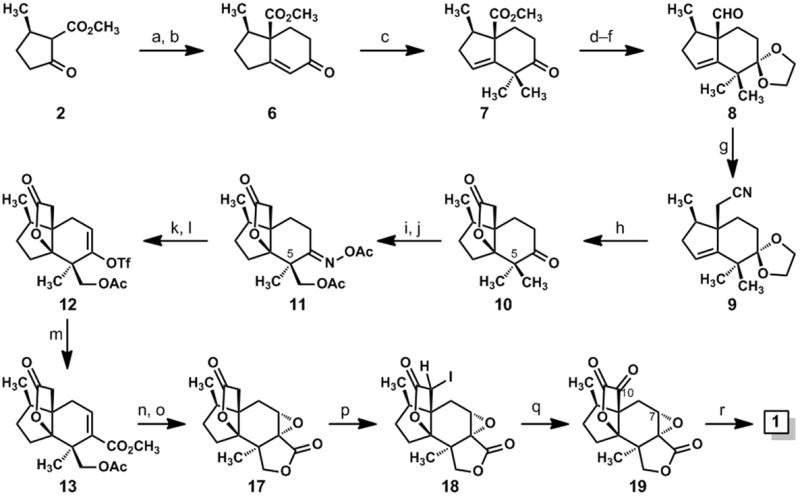
Synthesis of jiadifenolide: (a) methyl vinyl ketone (1.2 eq.), DBU (0.25 eq.), EtOH, rt, 97%; (b)p-TsOH (0.8 eq.), benzene, reflux, 85%; (c) CH3I (6 eq.), KOt-Bu (3.1 eq.), t-BuOH, rt, 91%; (d) ethylene glycol (6 eq.), p-TsOH (0.1 eq.), benzene, reflux; (e) DIBAL-H (3.1 eq.), CH2Cl2, –78 °C→ 0 °C, 77% (over two steps); (f) (COCl)2 (1.5 eq.), DMSO (3 eq.); then Et3N (5 eq.), CH2Cl2, –78 °C → rt, 90%; (g) TosMIC (1.7 eq.), KOt-Bu (2.6 eq.), THF, –50 °C, then CH3OH, 65 °C, 90%; (h) H2SO4, CH3OH, 100 °C, 73%; (i) NH2OH•HCl (1.5 eq.), pyridine, 80 °C, 91%; (j) Pd(OAc)2 (0.05 eq.), PhI(OAc)2 (1.5 eq.), 1:1 Ac2O/AcOH, 100 °C, 12 h, 22% of 11; (k) Fe (10 eq.), AcOH (cat.), Me3SiCl (cat.), THF, rt, 89%; (l) KN(SiMe3)2 (1.1 eq.), Comins reagent (1.2 eq.), THF, –78 °C; (m) Pd(OAc)2 (0.1 eq.), Ph3P (0.2 eq.), Et3N (2 eq.), CO (1 atm), CH3OH, DMF, 40 °C, 49% (over two steps); (n) K2CO3 (2 eq.), CH3OH, rt; (o) 3 M NaOH (3 eq.), H2O2 (10 eq.), CH3OH, 0 °C→ rt, 61% (over two steps); (p) Me3SiCl (2.5 eq.), LiN(SiMe3)2 (2.1 eq.), THF, –78 °C, then NIS (1.1 eq.); (q) DMDO (0.05 M in acetone, 6 eq.), CH2Cl2, rt; (r) LiOH (2 eq.), THF, rt, 40% (over three steps). DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; p-TsOH = para-toluene sulfonic acid; DIBAL-H = diisobutylaluminum hydride; DMSO = dimethyl sulfoxide; TosMIC = para-toluenesulfonylmethyl isocyanide; DMF = dimethylformamide; NIS = N-iodosuccinimide; DMDO = dimethyldioxirane.
To achieve the desired Robinson annulation[9,10] of the central six-membered ring of jiadifenolide, we modified a known, two-step method for merging the pulegone-derived β-keto ester 2 with methyl vinyl ketone;[11] this process, in which the Michael addition and ring-forming aldol condensation steps are performed in sequence, afforded bicyclic enone 6, which was subsequently advanced to compound 7 by a base-mediated, deconjugative dimethylation.[12] After a protection of the keto group in the form of a dioxolane ketal, the carbomethoxyl group was reduced with diisobutylaluminum hydride and the resulting primary alcohol was oxidized to aldehyde 8 via the Swern method.[13] This synthesis of 8 from β-keto ester 2 proceeded with an overall yield of 52% and could be conducted on large scales.
Despite its sterically crowded nature, the aldehyde function of 8 reacted at –50 °C with the conjugate base of tosylmethyl isocyanide (TosMIC) and gave rise to the homologated nitrile 9 in 90% yield after methanolysis of the putative N-formyl iminoketene.[14] This method, which was among the few that enabled the needed one-carbon homologation of 8,[15] afforded a nitrile that could be hydrolyzed in acid with a concomitant, desired lactone ring formation. In fact, a simple exposure of compound 9 to concentrated sulfuric acid in wet methanol brought about three desired changes en route to compound 10: the hydrolysis of the nitrile function, the desired heterocyclization of the γδ-unsaturated acid,[16] and the hydrolysis of the dioxolane ketal. The manner in which this simple procedure served the synthesis was pleasing.
With an effective synthesis of the [4.3.3] propellane 10, we approached the pivotal CH3 oxidation step with cautious optimism. Molecular models revealed the unique, “Y-shaped” architectures of 10[17] and its derived oxime and that it would be difficult to make a confident prediction about site-selectivity in a directed C–H oxidation event; we considered these compounds to be unique contexts for Sanford’s catalytic C–H oxidation method, and our plan for using this method was at least bolstered by prior examples of oxime-directed oxidations of unactivated methyl groups.[8] Thus, with the aim of differentiating the C-5 geminal methyl groups in the course of a directed, C–H oxidation reaction,[8] we heated a mixture of the oxime derived from ketone 10 (not shown), (diacetoxyiodo)benzene, and a catalytic amount of Pd(OAc)2 in Ac2O/AcOH to 100 °C for 12 hours. This reaction accomplished the desired methyl oxidation, although it afforded a 1:1 mixture of the desired oxime acetate 11 and a diastereoisomeric oxime acetate, epimeric at C-5, in addition to a small amount of a compound having two acetoxymethyl groups attached to C-5. In other structural contexts, this method actually yielded greater amounts of the undesired C-5 epimer of 11.[18]
We observed the desired reactivity, although the lack of diastereoselectivity was a concern. We hypothesized that the high temperature requirement for this reaction and the conformational flexibility of the six-membered ring in our [4.3.3] propellane system led to an equal sampling of the methyl groups by the palladium catalyst. This supposition was realized by performing the reaction under conditions developed previously by the Baldwin group,[19] which allow for a stoichiometric, palladium-mediated activation of the C–H bond at room temperature. In this experiment, a preference for one diastereoisomer was observed; however, it was the undesired C-5 epimer that was the major component.[20] While the isolated yield of the desired oxime acetate 11 was only 22%, Sanford’s catalytic oxidation method permitted the needed methyl group oxidation, and it was straightforward to resolve the mixture of C–H oxidation products by silica gel chromatography. In one experiment, we obtained 1.4 grams of compound 11 from 4.46 grams of the oxime derived from 10.
With access to significant quantities of compound 11, we could address the annulation of the second γ-lactone ring and the final three oxidations required for completion of the synthesis. After a reductive cleavage of the oxime acetate group in 11 by the method of Weinreb,[21] the resulting C-6 ketone was advanced to vinyl triflate 12 by a reaction of the derived enolate ion with Comins’s reagent.[22] A palladium-catalyzed carbomethoxylation[23] of 12 afforded 13 and was followed by a base-induced methanolysis of the acetate ester with concomitant lactone ring formation. Interestingly, this method for lactonizing 13 afforded a mixture of compounds 14, 15, and 16 (Scheme 2); however, this three-component mixture was transformed to a single epoxy bislactone, compound 17, on treatment with hydrogen peroxide and sodium hydroxide. We assume that compounds 15 and 16 are converted to 14 under the basic conditions of this reaction and that the nucleophilic epoxidation of 14 selectively forms the cis-fused epoxy lactone shown as 17 (Scheme 1).
Scheme 2.
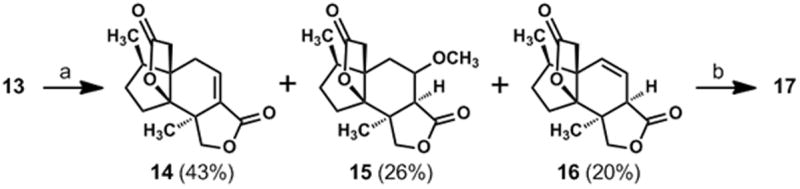
(a) K2CO3 (2 eq.), CH3OH, rt; (b) 3 M NaOH (3 eq.), H2O2 (10 eq.), CH3OH, 0 °C→ rt, 61% (over two steps).
From epoxy bislactone 17, the ascent to jiadifenolide was achieved in three transformations (Scheme 1). As we contemplated potential end-game sequences, we were drawn to the teachings of Danishefsky and Cox[24] who had demonstrated that the active methylene of a structurally complex γ-lactone could be oxidized to the corresponding ketone under mild conditions by way of an ostensible “iodoso-Pummerer” rearrangement.[25] In the case at hand, an iodination of the silyl ketene acetal derived from compound 17 furnished α-iodo lactone 18 in a diastereoselective fashion, although the configuration of the newly formed, iodine-bearing stereocenter was not assigned. This stereochemical matter was, however, inconsequential because a subsequent exposure of 18 to dimethyldioxirane in methylene chloride at room temperature resulted in the formation of the desired α-keto lactone 19. In the final transformation, the electrophilic carbons at positions 7 and 10 were bridged through an oxygen atom via a simple reaction of 19 with lithium hydroxide; a neutralization of this reaction with aqueous acid furnished jiadifenolide (1).[26] The spectroscopic data that we accumulated for this substance matched the data reported previously by Fukuyama and coworkers.[1]
From a (R)-(+)-pulegone-derived building block that incorporates the C-1 secondary methyl group, this synthesis of the highly oxygenated, cage-like structure of jiadifenolide (1) is reliant on the time-honored Robinson annulation and rather simple reagents for inducing three heterocyclizations. This synthesis also offers what we believe to be the first application of Sanford’s powerful, catalytic method for achieving directed C–H oxidations to a problem in natural product synthesis. While the yield for this application is only moderate, this method for oxidizing a methyl group afforded significant amounts of an advanced intermediate en route to jiadifenolide (1). This method also yielded a stereoisomeric intermediate that could serve syntheses of new, structurally unique relatives of the natural product. Our effort to achieve that aim and expand the class of jiadifenolide-related neurotrophic agents is underway.
Footnotes
This work was supported by the National Institute of General Medical Sciences (GM065483) and Princeton University. We also acknowledge helpful discussions with Prof. Huw Davies and the CCI Center for Selective C–H Functionalization supported by the National Science Foundation (CHE-1205646). We thank Dr. István Pelczer, Ken Conover, and Dr. John Eng of Princeton University for assistance with NMR spectroscopic analyses, the collection of NMR data, and the collection of HRMS data, respectively.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- 1.Kubo M, Okada C, Huang J-M, Harada K, Hioki H, Fukuyama Y. Org Lett. 2009;11:5190–5193. doi: 10.1021/ol9021029. [DOI] [PubMed] [Google Scholar]
- 2.Yokoyama R, Huang J-M, Yang C-S, Fukuyama Y. J Nat Prod. 2002;65:527–531. doi: 10.1021/np010571k. [DOI] [PubMed] [Google Scholar]
- 3.For a recent review, see: Xu J, Lacoske MH, Theodorakis EA. Angew Chem. 2014;126:972–1004. doi: 10.1002/anie.201302268.; Angew Chem Int Ed. 2014;53:956–987. doi: 10.1002/anie.201302268..
- 4.a) Xu J, Trzoss L, Chang WK, Theodorakis EA. Angew Chem. 2011;123:3756–3760. doi: 10.1002/anie.201100313. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:3672–3676. doi: 10.1002/anie.201100313. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Trzoss L, Xu J, Lacoske MH, Mobley WC, Theodorakis EA. Chem Eur J. 2013;19:6398–6408. doi: 10.1002/chem.201300198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson RM, Danishefsky SJ. J Org Chem. 2007;72:4293–4305. doi: 10.1021/jo070871s. [DOI] [PubMed] [Google Scholar]
- 6.a) Marx JN, Norman LR. J Org Chem. 1975;40:1602–1606. [Google Scholar]; b) Niwa H, Kunitani K, Nagoya T, Yamada K. Bull Chem Soc Jpn. 1994;67:3094–3099. [Google Scholar]
- 7.For discussions about strategies and methods for achieving site-selective C–H functionalization reactions, see: Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731.; Davies HML, Manning JR. Nature. 2008;451:417–424. doi: 10.1038/nature06485.; Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e..
- 8.a) Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; b) Neufeldt SR, Sanford MS. Org Lett. 2010;12:532–535. doi: 10.1021/ol902720d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Neufeldt SR, Sanford MS. Acc Chem Res. 2012;45:936–946. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rapson WS, Robinson R. J Chem Soc. 1935:1285–1288. [Google Scholar]
- 10.For reviews of the Robinson annulation and related reactions, see: Gawley RE. Synthesis. 1976:777–794.; Jung ME. Tetrahedron. 1976;32:3–31..
- 11.Vettel PR, Coates RM. J Org Chem. 1980;45:5430–5432. [Google Scholar]
- 12.a) Woodward RB, Patchett AA, Barton DHR, Ives DAJ, Kelly RB. J Am Chem Soc. 1954;76:2852–2853. [Google Scholar]; b) Ringold HJ, Rosenkranz G. J Org Chem. 1957;22:602–605. [Google Scholar]; c) Ringold HJ, Malhotra SK. J Am Chem Soc. 1962;84:3402–3403. [Google Scholar]
- 13.Mancuso A, Brownfain D, Swern D. J Org Chem. 1979;44:4148–4150. [Google Scholar]
- 14.a) Oldenziel OH, van Leusen D, van Leusen AM. J Org Chem. 1977;42:3114–3118. [Google Scholar]; b) van Leusen AM, Oomkes PG. Syn Commun. 1980;10:399–403. [Google Scholar]; c) van Leusen D, van Leusen AM. Org React. 2001;57:417–666. [Google Scholar]; d) Hampel T, Brückner R. Org Lett. 2009;11:4842–4845. doi: 10.1021/ol9018979. [DOI] [PubMed] [Google Scholar]
- 15.A Peterson olefination of aldehyde 8 with (methoxymethyl)trimethylsilane was also successful; however, hydrolysis of the resulting enol ether was problematic and resulted in a longer sequence leading to tricycle 10.
- 16.Multiple mechanistic possibilities exist for this transformation, including attack by the oxygen atom of a protonated imino carboxylic acid on a tertiary carbocation, followed by hydrolysis of the resulting cyclic imidate. An acid-induced, Markovnikov addition of water to the trisubstituted alkene, followed by a Pinner cyclization could also lead to the same cyclic imidate.
- 17.For a review of propellanes, see: Altman J, Babad E, Itzchaki J, Ginsburg D. Tetrahedron. 1966;Suppl. 8(part I):279–304..
- 18.The structure of the C-5 epimer of 11, including full characterization data, are provided in the supporting information.
- 19.Baldwin JE, Nájera C, Yus M. J Chem Soc Chem Commun. 1985:126–127. [Google Scholar]
- 20.See the supporting information.
- 21.Majireck MM, Witek JA, Weinreb SM. Tetrahedron Lett. 2010;51:3555–3557. doi: 10.1016/j.tetlet.2010.04.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Comins DL, Dehghani A. Tetrahedron Lett. 1992;33:6299–6302. [Google Scholar]
- 23.a) Schoenberg A, Bartoletti I, Heck RF. J Org Chem. 1974;39:3318–3326. [Google Scholar]; b) Hidai M, Hikita T, Wada Y, Fujikura Y, Uchida Y. Bull Chem Soc Jpn. 1975;48:2075–2077. [Google Scholar]; c) Stille JK, Wong PK. J Org Chem. 1975;40:532–534. [Google Scholar]
- 24.Cox C, Danishefsky SJ. Org Lett. 2000;2:3493–3496. doi: 10.1021/ol006531+. [DOI] [PubMed] [Google Scholar]
- 25.Reich HJ, Peake SL. J Am Chem Soc. 1978;100:4888–4889. [Google Scholar]
- 26.This enantiospecific synthesis required 18 steps from β-keto ester 2 and 21 steps from(R)-(+)-pulegone (3).