

Abstract
Apolipoprotein E plays a crucial role in inhibiting chronic neurodegenerative processes. However, its impact on neurological function following diffuse brain injury is still unclear. This study was designed to evaluate the therapeutic effects and mechanisms of action of apolipoprotein E mimetic peptide on diffuse brain injury. Apolipoprotein E mimetic peptide was administered into the caudal vein of rats with diffuse brain injury before and after injury. We found that apolipoprotein E mimetic peptide significantly decreased the number of apoptotic neurons, reduced extracellular signal-regulated kinase1/2 phosphorylation, down-regulated Bax and cytochrome c expression, decreased malondialdehyde content, and increased superoxide dismutase activity in a dose-dependent manner. These experimental findings demonstrate that apolipoprotein E mimetic peptide improves learning and memory function and protects against diffuse brain injury-induced apoptosis by inhibiting the extracellular signal-regulated kinase1/2-Bax mitochondrial apoptotic pathway.
Keywords: nerve regeneration, brain injury, apolipoprotein E, diffuse brain injury, learning and memory, extracellular signal-regulated kinase, Bax, mitochondria, reactive oxygen species, apoptosis, Scientific Research and Development Plan of Hebei Province in China, neural regeneration
Introduction
Because the majority of patients with diffuse brain injury are not suitable candidates for surgery, neuroprotective agents are of great importance[1]. The numerous functions of apolipoprotein E include regulation of neuronal tubulin metabolism, neurotrophic support, repairing and regenerating nerve tissue, neuroimmunomodulation, and protection against oxidative and inflammatory stress. Apolipoprotein E exerts a neuroprotective effect against brain injury, but synthetic apolipoprotein E cannot cross the blood-brain barrier, thus limiting its application. It has been reported that apolipoprotein E (138–149) mimetic peptide can cross the blood-brain barrier in both normal and injured brain, and has robust neuroprotective effects in models of closed head injury, perinatal hypoxia-ischemia and subarachnoid hemorrhage[2,3,4], and improves histological and functional outcomes. Apolipoprotein E mimetic peptides, derived from the receptor binding region (130–150) of apolipoprotein E, such as apolipoprotein E (138–149), retain the ability to bind apolipoprotein E cell surface receptors and have functional activity similar to the intact protein. In the current study, we used the small 12-residue apolipoprotein E mimetic peptide (apolipoprotein E-1410) in an effort to evaluate the protective effects and mechanisms of action of small apolipoprotein E mimetic peptides on diffuse brain injury.
Extracellular signal-regulated kinase 1/2 (ERK1/2), the key member of the mitogen-activated protein kinase family, is activated by a number of stimuli. In turn, activated ERK1/2 can phosphorylate downstream substrates, such as c-Raf-1, MEK, Elk-1 and c-Fos, which regulate a variety of genes and proteins involved in apoptosis, such as bcl-2, caspase-3 and FasL[5,6]. Increasing evidence suggests that ERK1/2 plays a harmful role in traumatic brain injury. For example, Clausen et al.[7] proposed that traumatic brain injury-induced apoptosis-like cell death was mediated by oxygen free radical-dependent activation of ERK1/2, and that U0126 preconditioning may protect the brain against focal traumatic brain injury by down-regulating ERK1/2 phosphorylation in the cerebral cortex. Furthermore, it has been reported that reactive oxygen species can activate growth factor receptors, leading to Ras-Raf-ERK1/2 signal pathway activation[8,9]. Thus, the ERK1/2 signaling pathway is a promising target for novel therapeutic approaches in the treatment of brain injury.
Given the important role of reactive oxygen species and the ERK1/2 pathway in apoptosis after brain injury, and the robust anti-oxidative effects of apolipoprotein E mimetic peptides, we explored whether a comparatively small apolipoprotein E mimetic peptide could improve learning and memory function and protect neurons against traumatic brain injury-induced neuronal apoptosis in the hippocampal CA1 region by inhibiting the ERK1/2/Bax mitochondrial apoptotic pathway. We assessed the effects of low and high doses of apolipoprotein E mimetic peptide on diffuse brain injury by examining changes in ERK1/2 phosphorylation, Bax and cytochrome c expression, and malondialdehyde content and superoxide dismutase activity.
Results
Quantitative analysis of animals
A total of 173 Sprague-Dawley rats were divided into the following groups: sham (n = 28), trauma (n = 45), low-dose apolipoprotein E mimetic peptide (low-dose apolipoprotein E: n = 43) and high-dose apolipoprotein E mimetic peptide (high-dose apolipoprotein E: n = 41) groups. Intravenous injections of 0.6 mg/kg (low dose group) and 1.2 mg/kg of apolipoprotein E peptide (high dose group) or PBS (trauma group) via the tail vein were given before and after diffuse brain injury. There were 17, 15 and 13 deaths, respectively, in the trauma, and low- and high-dose apolipoprotein E groups during model establishment. A total of 128 rats were involved in the final analysis, with 28 rats in each group. At 1, 6 and 24 hours after injury, two rats were decapitated for histological and ultrastructural examination. At 6, 24 and 48 hours after injury, two rats were decapitated for immunohistochemistry, and five rats were used for malondialdehyde and superoxide dismutase assays and western blot analysis. At 72 hours after injury, five rats were given the radial arm maze test and subjected to neurological deficit assessment, and then decapitated for the TUNEL assay. To clearly evaluate the protective effect of apolipoprotein E, the assays were performed when changes were apparent after brain injury.
Physiological parameters
There were no significant differences in arterial blood pressure, PaO2 or PaCO2 among the sham, trauma and low- and high-dose apolipoprotein E groups before and after brain injury (P > 0.05; Table 1).
Table 1.
Physiological parameters 30 minutes before and after diffuse brain injury (n = 5)
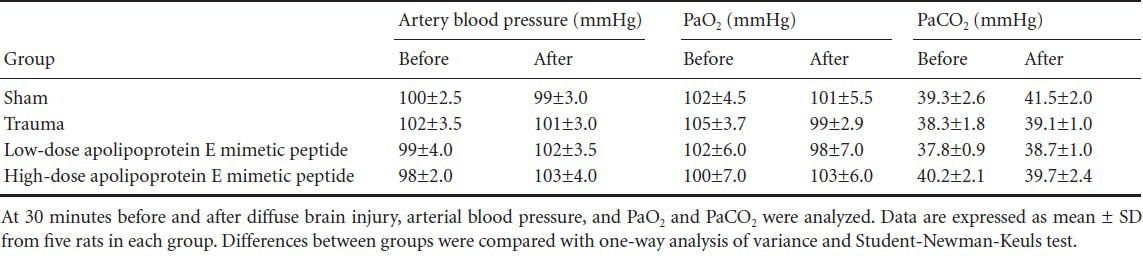
Effects of apolipoprotein E mimetic peptide on brain tissue histology in rats with diffuse brain injury
At 1 hour after injury, brain tissue showed spots of bleeding on the surface, with no obvious contusion. Under light microscopy, the parietal cortex exhibited extensive edema and vascular congestion. Subarachnoid cavity hemorrhage or yellowish staining was observed. In addition, shrunken cells with pyknotic nuclei, representing degenerating or necrotic neurons, were visible after trauma. Under the electron microscope, perturbed axonal arrangement, swollen and ruptured axons, degeneration of neurofilaments in axons, vacuolization, and infolding and stratification of the myelin sheath were clearly visible. In addition, edema of the capillaries and a substantial aggregation of organelles in swollen and degenerating neurons were observed. These observations are consistent with the pathological changes in Marmarou's traumatic brain injury model[10], indicating that our diffuse brain injury model was successful (Figure 1).
Figure 1.
Brain tissue histology in rats in the trauma group 1 hour after diffuse brain injury.
(A) Hemorrhage (arrow) was noted in the subarachnoid cavity (hematoxylin-eosin staining, × 200). (B) Shrunken cells with pyknotic nuclei (arrow) were noted (hematoxylin-eosin staining, × 200). (C) Infolding and stratification of myelin sheath (arrow) were seen under the electron microscope (× 20,000). (D) Capillary walls were rough and the basement membrane was loose (arrow), as seen under the electron microscope (× 20,000).
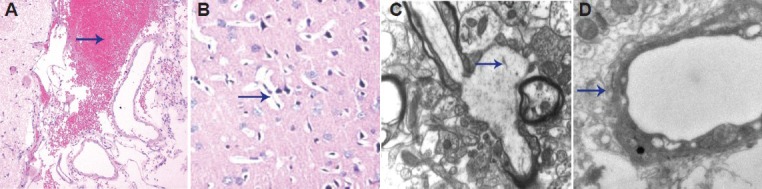
The period up to 6 hours after injury was considered the acute phase of brain injury. As the result of direct violent impact, there were numerous necrotic nerve cells and ruptured axons, and apolipoprotein E had no impact on the synapse (data not shown). At 24 hours after injury, in the sham group, synaptic structure was intact with clearly visible pre-synaptic membranes, synaptic cleft and post-synaptic membranes. Large numbers of closely spaced spherical vesicles and intact mitochondria were seen in the pre-synaptic membrane. In the trauma group, the mitochondria lost their typical structure and giant mitochondria were visible. There were only a few synaptic vesicles with an indistinct structure and reduced content. In the apolipoprotein E peptide groups, the synaptic structures were comparatively more numerous, and a few indistinct synaptic vesicles were visible. Mitochondria were round with an indistinct structure (Figure 2).
Figure 2.
Effects of apolipoprotein E mimetic peptide on synaptic structure in the cortex of rats with diffuse brain injury 24 hours after injury under the transmission electron microscope (uranyl acetate and lead citrate staining, × 20,000).
(A) The synaptic cleft and the post-synaptic membrane were clearly visible in the sham group. (B) Synaptic vesicles decreased in number and the synaptic cleft was unclear in the trauma group. (C) Synaptic structure showed comparative integrity in the low-dose apolipoprotein E group. (D) Synaptic vesicles were more numerous in the high-dose apolipoprotein E group. Arrows show synaptic vesicles.
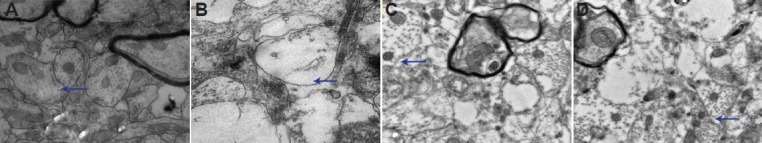
Apolipoprotein E mimetic peptide inhibited expression of phosphorylated ERK1/2 in the hippocampus of rats with diffuse brain injury
ERK1/2 phosphorylation, an indicator of ERK1/2 signal pathway activation, increased in the early phase after injury (within 24 hours)[7]. Consequently, we examined the effects of apolipoprotein E mimetic peptide on ERK1/2 phosphorylation 6 and 24 hours after injury. Immunohistochemistry showed that ERK1/2 phosphorylation localized in the nuclei and was mainly distributed in the hippocampal CA1 and CA2 regions, although some labeling was visible in the CA3 and dentate gyrus. Western blot analysis revealed that treatment with apolipoprotein E mimetic peptide significantly decreased ERK1/2 phosphorylation 6 and 24 hours after injury in a dose-dependent manner (Figure 3). ERK1/2 activation was not significant at 48 hours (data not shown).
Figure 3.
Effects of apolipoprotein E (ApoE) mimetic peptide on extracellular signal-regulated kinase 1/2 (ERK1/2) activation in the hippocampus of rats with diffuse brain injury.
Immunohistochemical staining showed ERK1/2 phosphorylation in neurons in the trauma group (A), low-dose ApoE mimetic peptide group (B) and high-dose ApoE mimetic peptide group (C) at 24 hours after injury (× 200). A few yellow-stained neurons positive for ERK1/2 phosphoryla-tion are visible in the hippocampus in the trauma group, and these neurons are fewer in the low- (low ApoE) and high-dose ApoE mimetic peptide groups (high ApoE). Arrows point to neurons positive for phosphorylated ERK1/2. (D) Phosphorylated ERK1/2 levels were examined by western blot analysis. (E) Bands corresponding to phosphorylated ERK1/2 were scanned and the intensities were normalized to β-actin. Data are expressed as mean ± SD from five rats in each group. Differences between groups were compared with one-way analysis of variance and Student-New-man-Keuls test. aP < 0.05, vs. sham group; bP < 0.05, vs. trauma group; cP < 0.05, vs. low ApoE group. h: Hours.
Apolipoprotein E mimetic peptide inhibited Bax expression in the hippocampus of rats with diffuse brain injury
Our previous studies indicate that the ERK1/2 signaling pathway plays an important role in neuronal apoptosis by regulating Bax expression[11]. Furthermore, ERK1/2 inhibition attenuates the apoptotic signaling pathway by decreasing Bax protein levels following brain injury[12]. Raghupathi et al.[13] showed that traumatic brain injury induces an increase in Bax expression at 6 hours. Our previous studies indicate that Bax expression increases after injury and peaks at 48 hours[10]. Thus, we examined whether apolipoprotein E peptide could attenuate the increase in Bax levels following injury.
Immunohistochemistry showed Bax immunoreactivity was localized in the cytosol, and was mainly distributed in the hippocampal CA1 and CA2 regions. Western blot analysis showed that apolipoprotein E peptide significantly decreased Bax expression 24 and 48 hours after injury in a dose-dependent manner (Figure 4). Bax expression showed no apparent change 6 hours after injury (data not shown).
Figure 4.
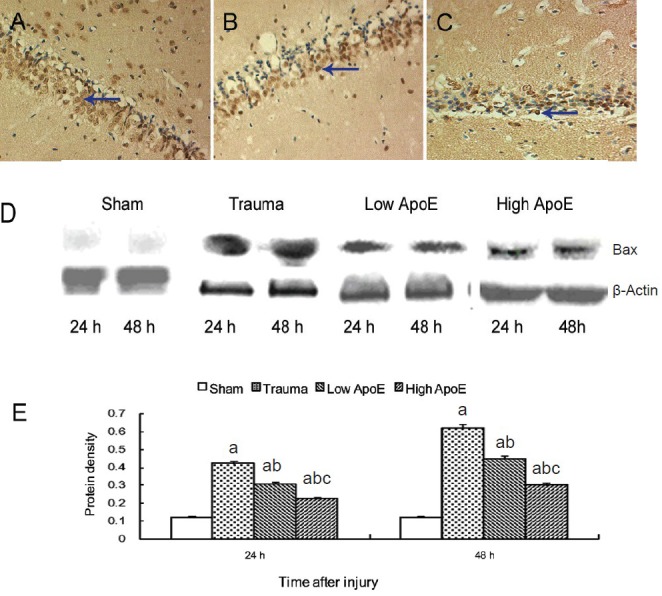
Effects of apolipoprotein E (ApoE) peptide on Bax expression in the hippocampus of rats with diffuse brain injury.
Immunohistochemical staining showing Bax-positive neurons in the trauma group (A), low-dose ApoE peptide group (low ApoE; B) and high-dose ApoE peptide group (high ApoE; C) 48 hours after injury (× 200). Yellow-stained Bax-positive neurons are visible in the hippocampus in the trauma group, and these neurons are fewer in the low and high ApoE groups. Arrows point to Bax-positive neurons. (D) Bax expression was exam-ined by western blot analysis. (E) Bands corresponding to Bax were scanned and the intensities were normalized to β-actin. Data are expressed as mean ± SD from five rats in each group. Differences between groups were compared with one-way analysis of variance and Student-Newman-Keuls test. aP < 0.05, vs. sham group; bP < 0.05, vs. trauma group; cP < 0.05, vs. low ApoE group. h: Hours.
Apolipoprotein E mimetic peptide inhibited cytochrome c expression in the hippocampus of rats with diffuse brain injury
The release of cytochrome c into the cytoplasm is a characteristic feature of mitochondrial apoptotic pathway activation. Previous studies indicate that cytochrome c is released from mitochondria into the cytosol following brain injury, with cytosolic levels increasing from 3 hours to 3 days after the trauma[14,15]. Thus, we examined the effects of apolipoprotein E peptide on cytochrome c levels 24 and 48 hours after injury.
Immunohistochemistry showed cytochrome c immunoreactivity was situated in the cytosol, and was mainly distributed in the hippocampal CA1 and CA2 regions. Western blot analysis showed that, compared with the sham group, cytochrome c levels increased in the trauma group. Treatment with apolipoprotein E peptide significantly decreased cytochrome c levels in a dose-dependent manner in the apolipoprotein E peptide groups (Figure 5). Cytochrome c levels were not significantly changed 6 hours after injury (data not shown).
Figure 5.
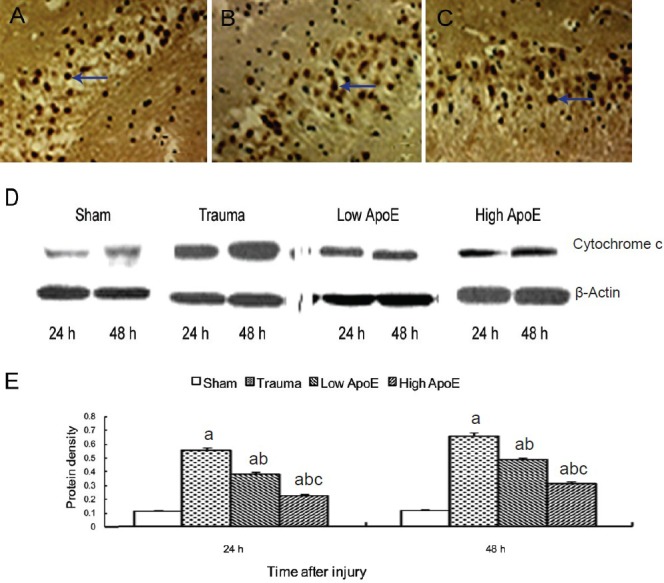
Effects of apolipoprotein E (ApoE) peptide on cytochrome c levels in the hippocampus of rats with diffuse brain injury.
Immunohistochemical staining showing cytochrome c-positive neurons in the trauma group (A), low-dose ApoE peptide group (low ApoE; B) and high-dose ApoE peptide group (high ApoE; C) 48 hours after injury (× 200). Yellow-stained cytochrome c-positive neurons are visible in the hip-pocampus in the trauma group, and these neurons are fewer in the low and high ApoE groups. Arrows point to cytochrome c-positive neurons. (D) Cytochrome c levels were examined by western blot analysis. (E) Bands corresponding to cytochrome c were scanned and the intensities were nor-malized to β-actin. Data are expressed as mean ± SD from five rats in each group. Differences between groups were compared with one-way analy-sis of variance and Student-Newman-Keuls test. aP < 0.05, vs. sham group; bP < 0.05, vs. trauma group; cP < 0.05, vs. low ApoE group. h: Hours.
Apolipoprotein E mimetic peptide inhibited apoptosis in the hippocampus of rats with diffuse brain injury
Some apoptotic neurons showed a characteristic appearance, such as shrunken, condensed nuclei and apoptotic bodies. However, others had lightly stained and swollen nuclei. We excluded these cells from the analysis of TUNEL-positive cells because they may be undergoing necrosis. We found that TUNEL-positive cells were mainly distributed in the hippocampal CA1 and CA2, with a few cells in the CA3 and dentate gyrus, similar to the distribution of ERK1/2 phosphorylation and Bax (Figure 6).
Figure 6.
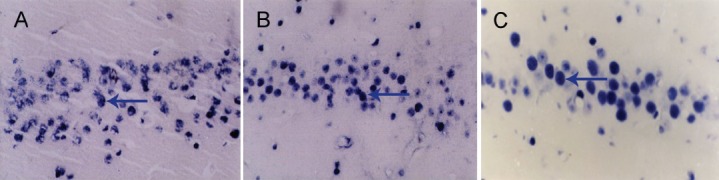
TUNEL staining of the hippocampus 3 days after diffuse brain injury.
TUNEL staining showing apoptotic neurons in the trauma group (A), and in the low- (B) and high-dose (C) apolipoprotein E groups 72 hours after injury (× 200). Apoptotic neurons are visible in the hippocampus in the trauma group, and are fewer in the low- and high-dose apolipoprotein E groups. Arrows point to apoptotic neurons.
To further investigate whether apolipoprotein E mimetic peptide provides neuroprotection against brain injury-induced apoptosis, we examined the effects of the peptide on the number of apoptotic cells 3 days after injury. Compared with the trauma group, apolipoprotein E mimetic peptide significantly decreased the number of TUNEL-positive cells in a dose-dependent manner.
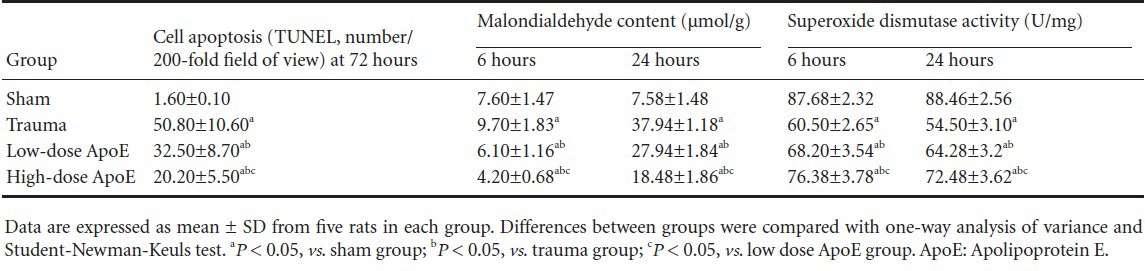
Effects of apolipoprotein E mimetic peptide on malondialdehyde content and superoxide dismutase activity in the hippocampus of rats with diffuse brain injury
Previous studies indicate that reactive oxygen species can activate growth factor receptors that induce Ras-Raf and ERK1/2 signal pathway activation after brain injury[16]. To further investigate how apolipoprotein E mimetic peptide inhibits brain injury-induced ERK1/2 activation, we examined the effects of the peptide on malondialdehyde content (an indicator of reactive oxygen species levels) and superoxide dismutase activity 6 and 24 hours following diffuse brain injury. Compared with the sham group, malondialdehyde content increased and superoxide dismutase activity decreased in the trauma group. Apolipoprotein E mimetic peptide significantly decreased malondialdehyde content and increased superoxide dismutase activity in a dose-dependent manner in the apolipoprotein E mimetic peptide groups (Table 2).
Table 2.
Quantitative analysis of the effects of apolipoprotein E peptide on diffuse brain injury-induced neuronal apoptosis, malondialdehyde content and superoxide dismutase activity
Neuroprotective effects of apolipoprotein E mimetic peptide on memory impairment and neurological deficit in rats with diffuse brain injury
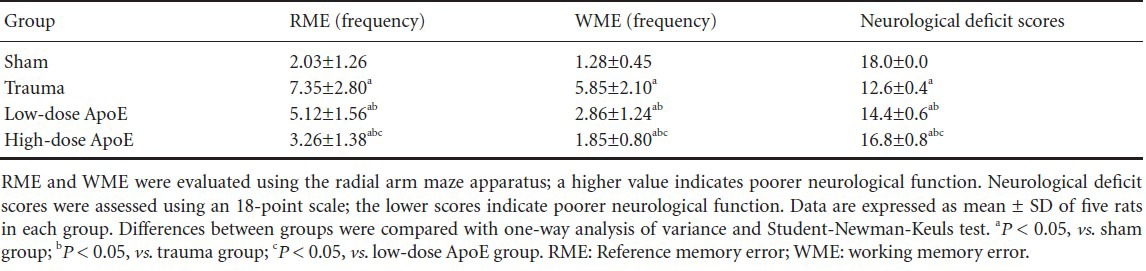
The reference memory and working memory errors 72 hours after injury are shown in Table 3. Compared with the sham group, reference memory errors and working memory errors were increased in the trauma group. Apolipoprotein E mimetic peptide significantly improved memory function and decreased the number of errors compared with the trauma group. Compared with the sham group, neurological deficit scores were increased in the trauma group. Neurological function was substantially improved and neurological deficit scores were reduced in the apolipoprotein E peptide mimetic group compared with the trauma group.
Table 3.
Quantitative analysis of the protective effects of apolipoprotein E (ApoE) peptide against memory impairment and neurological deficits in rats with diffuse brain injury
Discussion
Previous studies show that endogenous apolipoprotein E, acting as a neurotrophic factor, stabilizes the microenvironment of nerve cells and promotes nerve repair following injury[17,18]. The present study, indicates that apolipoprotein E mimetic peptide suppresses the ERK1/2 pathway and improves learning and memory function after diffuse brain injury.
Mitogen-activated protein kinase is a central signaling factor connecting extracellular signals, membrane receptors, transcription factors and gene expression. Family members include ERK1/2, C-Jun JNK and p38 MAPK. Previous studies indicate that ERK1/2 can be activated by cerebral injury and that regulation of ERK1/2 activation effects behavioral outcome in brain-injured animals[19,20]. In this study, apolipoprotein E mimetic peptide improved learning and memory abilities and reduced synaptic structural damage in a dose-dependent manner, and down-regulated ERK1/2 activation. This illustrates that apolipoprotein E mimetic peptide may improve learning and memory abilities by regulating ERK1/2 activity. It has been reported that ERK1/2 impacts long-term potentiation and learning and memory function by regulating the transcription of various factors and cytokines[21].
Growing evidence indicates that ERK1/2 is a key mediator of mitochondrial apoptosis. For example, Alessandrini et al.[22,23] demonstrated that ERK1/2 phosphorylation and cytochrome c levels increased after cerebral ischemia-reperfusion, and that ERK1/2 phosphorylation and cytochrome c colocalized in nerve cells. Clausen et al.[7] observed that inhibition of ERK1/2 activation, using the ERK1/2 inhibitor U0126, decreased caspase-3 expression and the apoptotic rate following closed brain injury. Moreover, some researchers have suggested that ERK1/2 mediates apoptosis through the mitochondrial apoptotic pathway by regulating Bcl-2 family members[24,25]. The Bcl-2 family comprises proapoptotic and anti-apoptotic proteins; some proteins within this family, such as Bcl-2 and Bcl-XL, enhance cell survival after ischemia by maintaining mitochondrial membrane integrity, whereas others, including Bad, Bax and Bid, promote ischemic cell death by releasing apoptogenic factors such as cytochrome c. It is known that the translocation of Bax plays an important role in mitochondrial apoptosis. In our previous study, inhibition of ERK1/2 activation using the ERK1/2 inhibitor U0126 decreased Bax and cytochrome c levels in a model of severe traumatic brain injury[11]. ERK1/2 activation increases the translocation of Bax to mitochondria, leading to cell death. ERK1/2 activation causes an increase in p53 activity, which in turn increases Bax levels. Moreover, ERK1/2 activation can activate calpain, a calcium-dependent cysteine protease which cleaves Bcl-XL. This leads to dissociation of Bcl-XL from Bax, resulting in an increase in free Bax[26,27]. Our immunohistochemistry data show that the distribution of phosphorylated ERK1/2 and cytochrome c were mostly within TUNEL-positive cells, most of which were distributed in the hippocampal CA1 and CA2 regions. Furthermore, we found that apolipoprotein E mimetic peptide reduced ERK1/2 activation and Bax levels, and lowered cytochrome c levels and the number of TUNEL-positive cells in a dose-dependent manner. Taken together, our findings suggest that apolipoprotein E mimetic peptide protects against diffuse brain injury-induced neuronal apoptosis by inhibiting the ERK1/2-Bax mitochondrial apoptotic pathway.
Apolipoprotein E (138–149) mimetic peptide protects neurons from excitotoxic injury, inflammation and oxidative stress in different types of brain injury models[2,3,28]. Our malondialdehyde and superoxide dismutase findings suggest that apolipoprotein E mimetic peptide reduces oxidative stress and increased antioxidant capacity. The antioxidative effects of apolipoprotein E holoprotein and apolipoprotein E peptide have been demonstrated in a focal cerebral ischemia/reperfusion injury model[29,30]. Recent studies show that reactive oxygen species can activate the Raf-ERK1/2 signaling pathway and suppress oxidative stress, and that regulating the activation of the MAP kinase signaling pathway can effectively protect neurons against ischemic damage. For example, Noshita et al.[31] observed that over-expressed superoxide dismutase decreased phosphorylated ERK1/2 levels and reduces death of nerve cells in a focal cerebral ischemia model. Clausen et al.[7] reported that reducing reactive oxygen species levels decreases phosphorylated ERK1/2 levels and the death of nerve cells in a closed traumatic brain injury model. In our previous study, edaravone suppressed ERK1/2 pathway activation and improved outcome after severe traumatic brain injury in rats, and the effects of edaravone on ERK1/2 activation depended on the dosage of the drug[32,33]. Taken together, our findings suggest that apolipoprotein E mimetic peptide suppresses ERK1/2 activation and reduces neuronal death in diffuse brain injury by reducing oxidative stress.
In summary, apolipoprotein E-derived therapeutic peptide improved synaptic stability and improved learning and memory performance after diffuse brain injury. Furthermore, apolipoprotein E mimetic peptide protected against brain injury by reducing oxidative stress, thereby suppressing the ERK1/2-Bax mitochondrial apoptotic pathway and decreasing neuronal death. These findings suggest that apolipoprotein E and its mimetic peptides have substantial therapeutic potential in the treatment of central nervous system diseases.
Materials and Methods
Design
A randomized controlled animal experiment.
Time and setting
This experiment was conducted at the Experimental Center of the Affiliated Hospital of Hebei United University, China from March 2011 to July 2012.
Materials
Animals
Sprague-Dawley male rats, weighing 270 ± 30 g, were provided by Beijing Experimental Animal Center, Chinese Academy of Science (Beijing, China; license No. SCXK (Beijing) 2002-003). Rats were given free access to food and water before surgery. Temperature of the laboratory was 22–26°C and the relative humidity was 40–70%. Protocols were approved by the Animal Use and Care Advisory Committee of Health Science Center of Hebei University, China.
Drugs
Apolipoprotein E mimetic peptide 1410 (Cetyl-AS-Aib-LRKL-Aib-KRLL-amide) is derived from apolipoprotein E residues 138–149 with Aib (aminoisobutyric acid) substitutions at positions 104 and 145[28]. It was synthesized by the University of North Carolina (Chapel Hill, NC, USA), with a purity of 95%.
Methods
Induction of diffuse brain injury
Closed diffuse brain injury was induced according to Marmarou's method as previously described[10]. In brief, the animal was anesthetized with diethyl ether for 70–150 seconds and positioned in a stereotactic device (Zhenghua Science and Technology Co., Ltd., Huaibei, Anhui Province, China), the scalp was incised and the skull exposed. A concave 3-mm metallic disc was glued to the skull immediately caudal to the bregma. A 2.0-mm diameter pneumatic impactor (Lu Da Technology Co., Ltd., Beijing, China) was used to deliver a single midline impact to the disc surface. The impactor was discharged at 6.8 ± 0.2 m/second with a head displacement of 3 mm. After impact, the animals were allowed to recover spontaneous ventilation, and the tracheas were extubated. Following recovery, rats were allowed free access to food and water. At 30 minutes before and 30 minutes after brain injury, arterial blood pressure was monitored in the awake state with the tail-measured volume method using an RBP-II-type blood pressure and heart rate measuring instrument (Huaibei Zhenghua Science and Technology Co., Ltd.). Before measuring, the rat was placed in a greenhouse at 37–38°C for 10 minutes, and after the tail arteries were dilated, the tail was set on the pressure-sensitive sensor. After repeatedly pinching the mercury manometer to inflate the balloon until the pulse disappeared, it was slowly deflated. When the normal sound was heard, the blood pressure scale indicated systolic blood pressure. Blood pressure was continuously measured three times, and the average value was noted. 30 minutes before and after the injury, 0.5 mL blood was extracted from the femoral artery to analyze blood gas using a GEM3000 automatic blood gas analyzer (America Experimental Instrument Co., Ltd, Boston, MA, USA).
Drug administration
Low-dose (0.6 mg/kg) or high-dose (1.2 mg/kg) apolipoprotein E mimetic peptide was dissolved in PBS. Drug infusion was performed by intravenous injections from the tail vein using a micro-injector (Xinhua Medical Instrument Co., Ltd., Shangdong Province, China) as previously described[28,34]. Intravenous injections of apolipoprotein E mimetic peptide or PBS were given 30 minutes before injury, and continued over 3 days at 12-hour intervals.
Tissue preparation
Rats were decapitated under anesthesia and brain tissues were separated and fixed with 4% paraformaldehyde solution and embedded in paraffin. Paraffin-embedded brain tissues were cut into 5-µm coronal sections using a microtome. These brain tissues were prepared for histology, immunohistochemistry and TUNEL detection. For ultrastructure detection, rats were decapitated under anesthesia and the frontal cortex was separated and fixed with a mordant solution of 0.1 mol/L phosphate buffer, 4% paraformaldehyde and 10% tannic acid.
For malondialdehyde and superoxide dismutase detection and western blot analysis, rats were decapitated under anesthesia and the hippocampal region was separated and rapidly frozen in liquid nitrogen. The frozen hippocampal tissue samples were homogenized in 1:10 (w/v) ice-cold homogenization buffer A containing 10 mmol/L HEPES, pH 7.9, 0.5 mmol/L MgCl2, 10 mmol/L KCl, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 50 mmol/L NaF, 5 mmol/L dithiothreitol, 10 mmol/L β-glycerophosphate, 1 mmol/L sodium orthovanadate, 1% NP-40 and enzyme inhibitors (1 mmol/L benzamidine, p-nitrodomains phenyl phosphate, phenylmethylsulfonyl fluoride, and 5 µg/mL each of aprotinin, leupeptin and pepstatin A) and then centrifuged at 1,000 × g for 15 minutes at 4°C. Supernatants containing the cytosolic fraction were collected and protein concentrations were determined.
Histological evaluation
The 5-µm coronal sections were deparaffinized with xylene and rehydrated using an ethanol gradient (100–70% v/v), followed by washing with water. Sections were stained with hematoxylin-eosin, and examined with light microscopy (Olympus, Tokyo, Japan).
Cortical tissues were cut into sections (1 mm × 1 mm × 1 mm), fixed with 4% glutaraldehyde, and then washed twice with 0.1 mol/L cacodylic acid buffer solution, fixed with 1% osmium tetroxide, and washed. Afterwards, these tissue sections were dehydrated using acetone, step by step, saturated with epoxy resin, embedded, cut into ultrathin slices, stained with uranyl acetate and lead citrate, and then observed under a transmission electron microscope (H-7650 Hitachi; Days-US Science and Technology Co., Ltd., Beijing, China).
Immunohistochemical analysis
After incubation in 5% normal goat serum, 5-µm coronal sections were incubated with rat-anti-rat phosphorylated ERK1/2 or rat-anti-rat Bax or rat-anti-rat cytochrome c antibody (1:200; Jinqiao Biological Technology Co., Ltd., Beijing, China) overnight at 4°C. An equivalent of PBS was used in place of primary antibody for the negative controls. Subsequently, the sections were incubated in biotinylated secondary antibody (1:500; Jinqiao Biological Technology Co. Ltd) for 90 minutes at room temperature, followed by incubation in avidin-biotin complex for 90 minutes. Finally, the sections were developed with 3,3′-diaminobenzidine, and nuclei were counterstained with hematoxylin. Brown-stained positive cells were observed with an optical microscope (Olympus).
Western blot analysis
Samples were separated by 10% or 7.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis and electrotransferred onto nitrocellulose membranes. After blocking with 3% bovine serum albumin for 3 hours, membranes were probed with primary antibodies at 4°C overnight. The primary antibodies were monoclonal mouse anti-rat phosphorylated ERK1/2 (1:1,000; Cell Signaling Biotechnology, Danvers, MA, USA), rabbit polyclonal anti-Bax (1:1,000; Cell Signaling Biotechnology), rabbit polyclonal anti-cytochrome c (1:1,000; Cell Signaling Biotechnology) and rabbit polyclonal anti-β-actin (1:1,000; Cell Signaling Biotechnology). The secondary antibodies were alkaline phosphatase-conjugated goat anti-rat IgG (1:10,000; Sigma, St. Louis, MO, USA) and goat anti-rabbit IgG (1:10,000; Sigma), which were incubated with the blots for 2 hours at room temperature. Immunoreactivity was detected with an NBT/BCIP assay kit (Sigma) according to the manufacturer's instructions. Membranes were scanned and analyzed with an image analyzer (LabWorks Software; UVP Inc, Upland, CA, USA).
TUNEL staining
TUNEL staining was performed using an ApopTag Peroxidase In Situ Apoptosis Detection Kit (Jinqiao Biological Technology Co. Ltd) according to the manufacturer's protocol with minor modifications. The 5-µm coronal sections were treated with protease K at 20 µg/mL for 15 minutes at room temperature and then incubated with reaction buffer containing TdT enzyme at 37°C for 1 hour. After washing with stop/wash buffer, sections were treated with anti-digoxigenin conjugate for 30 minutes at room temperature and subsequently incubated with peroxidase substrate for color development. The nuclei were lightly counterstained with BCIP/NBT. The number of TUNEL-positive pyramidal cells in the hippocampal CA1 region was counted[35]. Briefly, four sections from each specimen were harvested and each section was divided into three equal parts, and four fields were examined for each part. TUNEL-positive nerve cells were counted in each field (200 × magnification) by optical microscopy, and the average number of positive cells under each field was recorded.
Malondialdehyde and superoxide dismutase detection
The frozen hippocampal tissue samples were homogenized and centrifuged at 800 × g for 15 minutes at 4°C. Malondialdehyde content and superoxide dismutase activities were measured with the corresponding kit (Nanjing Jiancheng Biological Engineering Institute, Nanjing, Jiangsu Province, China). Malondialdehyde, a lipid peroxide degradation product, undergoes condensation with thiobarbituric acid to form a red product with a maximum absorbance peak at 532 nm, which can be quantified with a spectrophotometer.
Superoxide dismutase activity was detected using the xanthine oxidase method. The anion free radical produced by the xanthine oxidase-superoxide dismutase system reduced nitrites, leading to the formation of hydroxylamine. The absorbance value of the detected tube was lower than the absorbance value of the control tube, and the activity of superoxide dismutase was calculated. Definition of unit: when 1 mg tissue protein was incubated in 1 mL reaction solution, and the rate of superoxide dismutase inhibition was 50%, the amount of the corresponding superoxide dismutase is a superoxide dismutase unit.
Neurological function assessment
Spatial memory was measured with the radial arm maze apparatus. Briefly, the radial maze was made of gray polyvinylchloride and consisted of eight arms (length: 32 cm; width: 8 cm; height: 19 cm) extending radially from an octagonal central area (20 cm across). Rats were trained in the radial maze for 2 consecutive days with two trials per day. Each rat was placed in the centre of the maze with all arm entries closed. After 10 seconds, the doors were opened and the rat was permitted to enter any of the eight arms. Only three of the eight arms contained water. The rationale for using three instead of four baited arms was to increase the sensitivity of the task in measuring reference memory errors by decreasing the probability that a correct choice was made by chance. The three arms containing water were randomly determined for each rat and were unchanged over the 2-day learning period before injury. A trial was terminated after either all baits were consumed or after 10 minutes, whichever occurred first. A previous study show that basic mental state and neurological function in rats recovered to a greater extent 72 hours after injury. Consequently, in this study, memory function was assessed at 72 hours, as previously described[36]. An experimenter who was blinded to experimental groups scored each rat for (1) reference memory error: entering an arm which was never baited; and (2) working memory error: re-entering an arm already visited.
Neurological deficit scores were evaluated using an 18-point scale, as previously described[37]. The neurobehavioral scale consisted of the following six tests: (1) spontaneous activity (0 to 3 points); (2) symmetry in the movement of four limbs (0 to 3 points); (3) forepaw outstretching (0 to 3 points); (4) climbing (1 to 3 points); (5) body proprioception (1 to 3 points); and (6) response to vibrissae touch (1 to 3 points). The score for each rat at the completion of the evaluation was the summation of all six individual test scores. Three is the minimum neurological score and eighteen is the score for normal animals.
Statistical analysis
Measurement data were expressed as mean ± SD and were analyzed using SPSS 17.0 software (SPSS, Chicago, IL, USA). Differences between groups were compared with one-way analysis of variance and Student-Newman-Keuls test. P values less than 0.05 were considered significant.
Footnotes
Conflicts of interest: None declared.
Funding: This study was supported by Scientific Research and Development Plan of Hebei Province, No. 20276102D; Key Project of Scientific Research in Universities of Hebei Province in China, No. ZD2010106.
Peer review: The role of apolipoprotein E mimic peptide 1410 in diffuse brain injury is rarely reported. In this study, we detected the ERK1/2 signal pathway and learning-memory indicators, in an effort to explore the preventive and therapeutic effect of apolipoprotein E mimic peptide 1410 in diffuse brain injury, thus guiding the clinics.
Copyedited by Patel B, Norman C, Fei ZM, Huang HL, Mu WJ, Yang Y, Li CH, Song LP, Zhao M
References
- [1].Dijkhuizen RM. Advances in MRI-based detection of cerebrovascular changes after experimental traumatic brain injury. Transl Stroke Res. 2011;12(4):524–532. doi: 10.1007/s12975-011-0130-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lynch JR, Tang W, Wang H, et al. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. 2003;278(49):48529–48533. doi: 10.1074/jbc.M306923200. [DOI] [PubMed] [Google Scholar]
- [3].Lynch JR, Wang H, Mace B, et al. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp Neurol. 2005;192(1):109–116. doi: 10.1016/j.expneurol.2004.11.014. [DOI] [PubMed] [Google Scholar]
- [4].McAdoo JD, Warner DS, Goldberg RN, et al. Intrathecal administration of a novel apoE-derived therapeutic peptide improves outcome following perinatal hypoxic-ischemic injury. Neurosci Let. 2005;381(3):305–308. doi: 10.1016/j.neulet.2005.02.036. [DOI] [PubMed] [Google Scholar]
- [5].Satoh T, Nakatsuka D, Watanabe Y, et al. Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultures cortical neurons. Neurosci Lett. 2000;288(2):163–166. doi: 10.1016/s0304-3940(00)01229-5. [DOI] [PubMed] [Google Scholar]
- [6].Kulich SM, Chu CT. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: Implications for Parkinson's disease. J Neurochem. 2001;77(4):1058–1066. doi: 10.1046/j.1471-4159.2001.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Clausen F, Lundqvist H, Ekmark S, et al. Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. Neurotrauma. 2004;21(9):1168–1182. doi: 10.1089/neu.2004.21.1168. [DOI] [PubMed] [Google Scholar]
- [8].Dash P, Mach S, Moore A. The role of extracellular signal-regulated kinase in cognitive and motor deficits following experimental traumatic brain injury. Neuroscience. 2002;114(3):755. doi: 10.1016/s0306-4522(02)00277-4. [DOI] [PubMed] [Google Scholar]
- [9].Irving EA, Barone FC, Reith AD, et al. Differential activation of MAPK/ERK and p38/SAPK in neurons and glia following focal cerebral ischaemia in the rat. J Mol Brain Res. 2003;77(1):65–75. doi: 10.1016/s0169-328x(00)00043-7. [DOI] [PubMed] [Google Scholar]
- [10].Marmarou A, Foda MA, van den Brink W, et al. A new model of difuse brain injury in rats. Part I: Pathophysiology and biomechanics. J Neurosurg. 1994;80(2):291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- [11].Zhao YN, Gao JL, Rao YZ, et al. Changes of ERKl/2 signal pathway after severe diffuse brain injury in rats. Zhongguo Bingli Shengli Zazhi. 2010;26(3):487–491. [Google Scholar]
- [12].Otani N, Nawashiro H, Fukui S, et al. Temporal and spatial profile of phosphorylated mitogen-actived protein kinase pathways after lateral fluid percussion injury in the cortex of the rats brain. J Neurotrauma. 2002;12(19):1587–1596. doi: 10.1089/089771502762300247. [DOI] [PubMed] [Google Scholar]
- [13].Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17(10):927–938. doi: 10.1089/neu.2000.17.927. [DOI] [PubMed] [Google Scholar]
- [14].Balaratnasingam C, Pham D, Morgan WH. Mitochondrial cytochrome c oxidase expression in the central nervous system is elevated at sites of pressure gradient elevation but not absolute pressure increase. J Neurosci Res. 2009;87(13):2973–2982. doi: 10.1002/jnr.22120. [DOI] [PubMed] [Google Scholar]
- [15].Dai W, Cheng HL, Huang RQ, et al. Quantitative detection of the expression of mitochondrial cytochrome coxidase subunits mRNA in the cerebral cortex after ex-perimental traumatic brain injury. J Brain Res. 2009;28(1251):287–295. doi: 10.1016/j.brainres.2008.11.034. [DOI] [PubMed] [Google Scholar]
- [16].Lu KT, Sun CL, Wo PY, et al. Hippocampal neurogenesis after traumatic brain injury is mediated by vascular endothelial growth factor receptor-2 and the Raf/MEK/ERK cascade. J Neurotrauma. 2011;28(3):441–450. doi: 10.1089/neu.2010.1473. [DOI] [PubMed] [Google Scholar]
- [17].Holtzman DM, Pitas PE, Kilbridge J, et al. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc Natl Acad Sci U S A. 1995;92(21):9480–9484. doi: 10.1073/pnas.92.21.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mace BE, Wang H, Lynch JR, et al. Apolipoprotein E modifies the CNS response to injury via a histamine-mediated pathway. Neurol Res. 2007;29(3):243–250. doi: 10.1179/016164107X158974. [DOI] [PubMed] [Google Scholar]
- [19].Wang WY, Yang R, Hu SF, et al. N-stearoyl-L-tyrosine ameliorates sevoflurane induced neuroapoptosis via MEK/ERK1/2 MAPK signaling pathway in the developing brain. Neurosci Lett. 2013;29(541):167–172. doi: 10.1016/j.neulet.2013.02.041. [DOI] [PubMed] [Google Scholar]
- [20].Chen T, Cao L, Dong W, et al. Protective effects of mGluR5 positive modulators against traumatic neuronal injury through PKC-dependent activation of MEK/ERK pathway. J Neurochem Res. 2012;37(5):983–990. doi: 10.1007/s11064-011-0691-z. [DOI] [PubMed] [Google Scholar]
- [21].Berkeley JL, Comeza J, Wess J, et al. M1 muscarinic acctylcholine receptors activate extracellular signial regulated kinase in CA1 pyram idal neurons in mouse hippocampal slices. Mol Cell Neurosci. 2001;18(5):512–524. doi: 10.1006/mcne.2001.1042. [DOI] [PubMed] [Google Scholar]
- [22].Alessandrini A, Namura S, Bonventre JV, et al. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci U S A. 1999;96(22):12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mori T, Wang XY, Jung JC, et al. Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects. J Cereb Blood Flow Metab. 2002;22(4):444–452. doi: 10.1097/00004647-200204000-00008. [DOI] [PubMed] [Google Scholar]
- [24].Biswas SC, Greene LA. Nerve growth factor (NGF) down-regulates the Bcl-2 homology3 (BH3) domain-only protein Bim and suppresses its proapoptotic activity by phosphorylation. J Biol Chem. 2002;277(5):49511–49516. doi: 10.1074/jbc.M208086200. [DOI] [PubMed] [Google Scholar]
- [25].Kim YK, Kim HJ, Kwon CH, et al. Role of ERK activation in cisplatin-induced apoptosis in OK renal epithelial cells. J Appl Toxicol. 2005;25(9):374–382. doi: 10.1002/jat.1081. [DOI] [PubMed] [Google Scholar]
- [26].Brown L, Benchimol S, Klesse LJ, et al. The involvement of MAPK signaling pathways in determining the cellular response to p53 activation: cell cycle arrest or apoptosis. J Biol Chem. 2007;281(155):3832–3840. doi: 10.1074/jbc.M507951200. [DOI] [PubMed] [Google Scholar]
- [27].Kang JQ, Chong ZZ, Maiese K, et al. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003;64(12):557–569. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- [28].Gao J, Wang H, Sheng H, et al. A novel apoE-derived therapeutic reduces vasospasm and improves outcome in a murine model of subarachnoid hemorrhage. Neurocrit Care. 2006;4(1):25–31. doi: 10.1385/NCC:4:1:025. [DOI] [PubMed] [Google Scholar]
- [29].Colton CA, Brown CM, Cook D, et al. APOE and the regulation of microglial nitric oxide production: a link between genetic risk and oxidative stress. J Neurobiol Aging. 2002;23(5):777–785. doi: 10.1016/s0197-4580(02)00016-7. [DOI] [PubMed] [Google Scholar]
- [30].Wang D, El-Amouri SS, Dai M, et al. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc Natl Acad Sci U S A. 2013;110(8):2999–3004. doi: 10.1073/pnas.1222742110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Noshita N, Sugawara T, Hayashi T, et al. Copper/zinc superoxide dismutase attenuates neuronal cell death by preventing extracellular signal-regulated kinase activation after transient focal cerebral ischemia in mice. J Neurosci. 2002;22(18):7923–7930. doi: 10.1523/JNEUROSCI.22-18-07923.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li JM, Zhao YN, Chen CX, et al. Protective effects of Edaravone of the diffuse injury of brain in rat. Zhonghua Jizhen Yixue Zazhi. 2010;19(11):1171–1175. [Google Scholar]
- [33].Zhao YN, Guo X, Gao JL, et al. Effects of edaravone in extracellular signal-regulated kinase 1/2 pathway following severe traumatic brain injury in rats. Zhongguo Weizhong Bing Jijiu Yixue. 2010;22(4):230–233. [PubMed] [Google Scholar]
- [34].Monceau V, Meziani L, Strup-Perrot C, et al. Enhanced sensitivity to low dose irradiation of ApoE-/- mice mediated by early proinflammatory profile and delayed activation of the TGFβ1 cascade involved in fibrogenesis. PLoS One. 2013;8(2):e57052. doi: 10.1371/journal.pone.0057052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhu Y, Yang G, Ahlemeyer B, et al. Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22(10):3898–3909. doi: 10.1523/JNEUROSCI.22-10-03898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zlomuzica A, Ruocco LA, Sadile AG, et al. Histamine H1 receptor knockout mice exhibit impaired spatial memory in the eight-arm radial maze. Br J Pharmacol. 2009;157(1):86–91. doi: 10.1111/j.1476-5381.2009.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rancan S, Cavanaugh JM, Ozaktay AC, et al. The effect of varying impact enery on diffuse axonal injury in the rat brain: a preliminary study. Exp Brain Res. 2003;148(4):419–424. doi: 10.1007/s00221-002-1307-2. [DOI] [PubMed] [Google Scholar]