

Abstract
Besides functioning as the plasma transporter of retinol and thyroxine, TTR (transthyretin) is a protease, cleaving apoA-I (apolipoprotein A-I) after a phenylalanine residue. In the present study, we further investigated TTR substrate specificity. By using both P-diverse libraries and a library of phosphonate inhibitors, a TTR preference for a lysine residue in P1 was determined, suggesting that TTR might have a dual specificity and that, in addition to apoA-I, other TTR substrates might exist. Previous studies revealed that TTR is involved in the homoeostasis of the nervous system, as it participates in neuropeptide maturation and enhances nerve regeneration. We investigated whether TTR proteolytic activity is involved in these functions. Both wild-type TTR and TTRprot− (proteolytically inactive TTR) had a similar effect in the expression of peptidylglycine α-amidating mono-oxygenase, the rate-limiting enzyme in neuropeptide amidation, excluding the involvement of TTR proteolytic activity in neuropeptide maturation. However, TTR was able to cleave amidated NPY (neuropeptide Y), probably contributing to the increased NPY levels reported in TTR-knockout mice. To assess the involvement of TTR proteolytic activity in axonal regeneration, neurite outgrowth of cells cultivated with wild-type TTR or TTRprot−, was measured. Cells grown with TTRprot− displayed decreased neurite length, thereby suggesting that TTR proteolytic activity is important for its function as a regeneration enhancer. By showing that TTR is able to cleave NPY and that its proteolytic activity affects axonal growth, the present study shows that TTR has natural substrates in the nervous system, establishing further its relevance in neurobiology.
Keywords: neurite outgrowth, neuropeptide Y (NPY), phosphonate inhibitor, proteolysis, substrate specificity, transthyretin
INTRODUCTION
TTR (transthyretin) is the plasma homotetrameric carrier of T4 (thyroxine) and retinol, in the latter case through binding to RBP (retinol-binding protein) [1]. In the plasma, a small TTR fraction is carried in HDL (high-density lipoprotein), through binding to apoA-I (apolipoprotein A-I) [2]. Besides being a transporter, TTR is able to cleave the C-terminus of apoA-I, being a novel cryptic protease [3]. The relevance of apoA-I cleavage by TTR in lipoprotein metabolism was determined: upon TTR cleavage, HDL displays a reduced capacity to promote cholesterol efflux, and cleaved apoA-I displays increased amyloidogenicity [4]. Several features, including an optimum pH for activity of approx. 7, inhibition by serine protease inhibitors and cleavage in apoA-I after a phenylalanine residue, strongly indicate that TTR is a serine protease [3]. However, its catalytic mechanism remains to be solved. The cryptic nature of TTR proteolytic activity derives not only from the fact that it lacks canonical structural protease determinants, but also because its physiological function is apparently unrelated to proteolysis [5].
The two major sites of TTR expression are the liver and the epithelial cells of the choroid plexus of the brain, which are the sources of the protein in the plasma and in the CSF (cerebrospinal fluid) respectively [6]. The conservation of TTR expression in the choroid plexus from reptiles to mammals led to the hypothesis that the expression of this gene first arose in the brain of reptiles [7]. During human embryonic development, TTR is first expressed in the tela choroidea, the precursor of the choroid plexus, followed by expression in the liver [8,9]. This pattern of TTR expression in the choroid plexus, conserved throughout evolution and starting early in embryonic development, points to a pivotal role for TTR in the brain. It has been suggested that TTR is involved in thyroid hormone homoeostasis and hormone delivery to the brain [10]. However, this issue has been a subject of some controversy. Although studies with TTR-KO (knockout) mice revealed that TTR is not necessary for thyroid hormones to be normally distributed [11], an association between the lack of TTR and decreased apoptosis of neural stem cells of the subventricular zone (a brain region which is in close contact with the CSF) has been described recently [12]. Nevertheless, the pattern of TTR expression suggests that the relevance of TTR is more than that initially described in the literature, as a carrier of thyroid hormones.
A number of TTR mutations are related to a neurodegenerative disease, FAP (familial amyloid polyneuropathy) [13], characterized by the deposition of TTR fibrils particularly in the PNS (peripheral nervous system) [14]. Under physiological conditions, TTR has access to the nerve through the blood and the CSF. A function for TTR in nerve biology could explain its preferential deposition, when mutated, in the PNS. Several studies using TTR-KO mice [15] revealed new TTR functions specifically related to the nervous system. At the functional level, TTR-KO mice present a sensorimotor impairment [16], and the absence of TTR is associated with reduced signs of depressive-like behaviour [17]. At the molecular level, TTR was shown to regulate the overexpression of PAM (peptidylglycine α-amidating mono-oxygenase) mRNA. PAM, the rate-limiting enzyme in neuropeptide maturation, is overexpressed in the PNS and CNS of TTR-KO mice, as well as in cell cultures grown in the absence of TTR [18]. PAM is the only enzyme that C-terminally amidates peptides, and 50 %of all known neuropeptides require amidation to be active. NPY (neuropeptide Y) is the major neuropeptide present in the mammalian brain and its activation requires C-terminal α-amidation by PAM. As such, as a consequence of PAM mRNA overexpression, TTR-KO mice, as well as cell cultures in which TTR is absent from the culture medium, present increased levels of amidated NPY [18]. Finally, after injury, TTR enhances nerve regeneration: in vivo, following sciatic nerve crush, TTR-KO mice present a decreased regenerative capacity, and in vitro, cells grown in the presence of TTR display increased neurite outgrowth, namely an approx. 20 % increase in the length of the longest neurite [16]. As it is possible that apoA-I may not be the major TTR substrate and given the phenotypes of TTR-KO mice (which can be related to the absence of TTR proteolytic activity and not to the absence of the protein itself), it is important to analyse the physiological relevance of TTR proteolytic activity in the nervous system.
In the present study, we investigated the substrate specificity of TTR and assessed whether some of the phenotypes described in TTR-KO mice, namely TTR involvement in PAM expression and nerve regeneration, might be related to the lack of TTR proteolytic activity. If so, this would indicate that a TTR substrate should exist in the nervous system. The identification of novel TTR substrates would allow the further characterization of the physiological relevance of the proteolytic activity of TTR, and the identification of its mechanism of action in the nervous system.
EXPERIMENTAL
Proteins
Recombinant wt (wild-type) TTR and glutathionylated TTR were produced in BL-21 pLys Escherichia coli cells transformed with pET plasmids carrying TTR cDNA. Glutathionylated TTR was detected by MALDI (matrix-assisted laser-desorption ionization)-MS and was shown to be proteolytically inactive (designated TTRprot−) (M. A. Liz and M. M. Sousa, unpublished work) using a TTR fluorigenic peptide as described below. After bacterial lysis, proteins were isolated and purified as described previously [3].
PS-SCLs (positional scanning synthetic combinatorial libraries)
To profile TTR preferences at substrate positions P4–P1, we used PS-SCLs of fluorigenic peptides [19]. To determine the P1 preference of TTR, we used a P1-diverse tetrapeptide library of 160 000 substrates each containing the fluorigenic leaving group ACC (7-amino-4-carbamoylmethylcoumarin). The P1-diverse library consists of 20 wells in which only the P1 residue remains constant at each one of the 20 proteinogenic amino acids, excluding cysteine and including norleucine. The P2, P3 and P4 positions consist of an equimolar mixture of the 20 amino acids, for a total of 8000 substrate sequences per well. The extended P4–P2 specificity was profiled with tetrapeptide libraries in which the P1 position was held constant. Three P1-fixed sublibraries denoting the second fixed position (P4, P3 and P2) and consisting of 19 wells addressing a fixed amino acid (omitting cysteine and norleucine substituted for methionine) were screened (361 compounds per well and 6859 compounds per library). Assays were performed at 37 °C in 50 mM Tris/HCl (pH 7.0). The final concentration for each substrate compound was 10 nM. Substrate hydrolysis was initiated by addition of enzyme (5 μM) and monitored fluorimetrically with excitation at 380 nm and emission at 450 nm for 3 h.
TTR screening with a library of phosphonate inhibitors
A total number of 160 compounds from a library of peptide phosphonate inhibitors of serine proteases was used in the screening [20]. Library screening was carried out in a dose–response manner with inhibitor concentrations ranging from 1 to 50 μM. TTR (5 μM) was incubated with the inhibitors for 30 min at 37 °C, in 50 mM Tris/HCl (pH 7.0). After enzyme–inhibitor incubation, 5 μM Abz-ESFKVS-EDDnp (aminobenzoyl-Glu-Ser-Phe-Lys-Val-Ser-ethylenediamine-2,4-dinitrophenol) substrate was added, and the kinetics of the reactions were followed at 37 °C for 1 h as described previously [3]. The above substrate was designed in order to encompass the apoA-I sequence cleaved by TTR (cleavage occurs following the phenylalanine residue in the hexapeptide) as reported previously [3]. Relative fluorescence values were converted into percentages of residual activity relative to uninhibited controls.
Assessment of TTR proteolytic activity
TTR proteolytic activity was tested with the fluorigenic peptide Abz-ESFKVS-EDDnp as described above. Specificity rate constants (kcat/Km) were determined under pseudo-first-order conditions. Pseudo-first-order rate constants were obtained from the linear plots where the y-axis corresponds to Ln [(Fmax − Ftime)/Fmax], where Fmax is the fluorescence corresponding to total degradation of 5 μM substrate and Ftime is the fluorescence measured at each time point, and the x-axis corresponds to the time of reaction. The slope of the linear plots, corresponding to the first-order rate constants, was divided by the total enzyme concentration to provide kcat/Km.
T4-binding assays
Binding of wt TTR and TTRprot− to T4 was assayed quantitatively by a gel-filtration procedure as described previously [21]. Briefly, a 30 nM solution of TTR (either wt TTR or TTRprot−) in 0.1 M Tris/HCl (pH 8.0), 0.1 M NaCl and 0.001 M EDTA was incubated with a constant amount of [125I]T4 (~50 000 c.p.m.; PerkinElmer) and increasing concentrations of unlabelled T4 (0–1000 nM; Sigma) overnight at 4 °C. [125I]T4 bound to TTR was separated from unbound T4 by gel-filtration chromatography through a Bio-Gel P6-DG column (Bio-Rad Laboratories). The eluates containing bound T4 were collected and counted in a Wizard 1470 gamma counter (Wallac Oy). Binding was expressed as the ratio between bound T4 and total T4. All samples were run in triplicate and the data were analysed using GraphPad Prism software.
RBP-binding assays
wt TTR was iodinated using the iodogen method [2]. Briefly, 15 μg of wt TTR was added to reaction tubes coated with 10 μg of iodogen (Sigma) and containing 100 μl of 0.25 M phosphate buffer (pH 7.5) and 1 mCi of Na125I (PerkinElmer). The reaction was allowed to proceed in an ice bath for 20 min. Labelled TTR was separated from free iodide in a 5 ml Sephadex G50 column (GE Healthcare). RBP was isolated from human serum as described previously [3]. For binding assays, 96-well plates (Maxisorp, Nunc) were coated with rabbit polyclonal anti-RBP (The Binding Site; 1:500 dilution) in coating buffer (0.1 M bicarbonate/carbonate buffer, pH 9.6) and incubated overnight at 4 °C. 125I-labelled wt TTR (500 000 c.p.m./well) mixed previously with RBP (400 nM/well) was incubated with the plates alone or with the unlabelled competitors, either wt TTR or TTRprot− at concentrations ranging from 0 to 5 μM, in binding buffer [0.1 % dried skimmed milk powder, 10 mM Hepes in minimal essential medium (Invitrogen)] for 2 h at 37 °C with gentle shaking. Binding was determined after five washes in ice-cold PBS with 0.05 % Tween 20 (0.2 ml/wash). Then, 0.1 ml of elution buffer (0.1 M NaCl containing 1 %Nonidet P40) was added for 5 min at 37 °C, and the content of the wells was aspirated and counted in a Wizard 1470 gamma counter. All samples were run in quadriplicate, and results were analysed using GraphPad Prism software.
Uptake of TTR by a human hepatoma cell line
The SAHep cell line (human hepatoma without TTR expression) was kindly provided by Professor João Monjardino (School of Medicine, Imperial College London, London, U.K.). Cells were grown to confluence in 24-well plates in DMEM (Dulbecco’s modified Eagle’s medium) (Invitrogen) supplemented with 10 % FBS (fetal bovine serum) (Sigma), 50 units/ml penicillin and 50 μg/ml streptomycin (Invitrogen). For the competition experiments, cells were incubated with 125I-labelled wt TTR (100 000 c.p.m./well) in the presence of increasing concentrations of unlabelled TTR (0–50 μg/ml; either wt TTR or TTRprot−), in DMEM with 0.1 % ovalbumin (Sigma) for 4 h at 4 °C. Cell-associated radioactivity was determined by measuring radioactivity of the PBS-washed cell layer solubilized in 0.1 M NaOH. All samples were run in triplicate, and results were analysed using GraphPad Prism software.
DIGE (differential fluorescence gel electrophoresis) proteome profiling
In order to identify additional plasma substrates for TTR, two distinct DIGE experiments were performed. In the first experiment, 50 μl of plasma from TTR-KO mice was incubated with 75 μg of either wt TTR or TTRprot− in 50 mM Tris/HCl (pH 7.0) overnight at 37 °C. The sample incubated with wt TTR was labelled with Cy3 (indocarbocyanine) and the one incubated with TTRprot− was labelled with Cy5 (indodicarbocyanine). In a second experiment, 50 μl of plasma from TTR-KO mice were incubated with (labelled with Cy5) or without (labelled with Cy3) 75 μg of wt TTR in 50 mM Tris/HCl (pH 7) overnight at 37 °C. In both experiments, 50 μg of each sample was loaded in the two-dimensional gel. DIGE was performed as a service by the W.M. Keck Facility, Yale University, New Haven, CT, U.S.A., according to the Ettan™ DIGE manual (Amersham) available at http://keck.med.yale.edu/pdfs/Ettan%20DIGE%20User%20Manual.pdf. Briefly, one sample was labelled with Cy3 and the other with Cy5; an equal amount of each sample was labelled and run in the DIGE experiment. Then, 24 cm, pH 3–10 Immobiline (IPG) Drystrips (GE Healthcare) were rehydrated overnight with the two samples, and isoelectric focusing was carried out. SDS/PAGE (12.5 % gels) was run, and, immediately afterwards, gels were scanned using a GE Healthcare Typhoon 9410 Imager. Gels were then fixed and stained with Sypro Ruby. GE Healthcare DeCyder software was used to quantify and to identify the differentially expressed protein spots with a fold change higher than 1.5. These spots were excised, MALDI-MS/MS spectra were acquired on each target (using an Applied Biosystems 4800 Tof/Tof instrument), and the resulting peptide masses were subjected to database searching using Mascot algorithms (http://www.matrixscience.com).
Influence of TTR proteolytic activity in PAM expression
Neuropeptide processing was assessed by quantification of PAM expression in PC12 cells (European Collection of Cell Cultures), as reported previously [18]. Briefly, cells were grown in six-well plates in William’s medium E (Invitrogen) supplemented with 2 mM L-glutamine (Sigma) and 10 % FBS (Invitrogen). When cells reached 50 %confluence, FBS was withdrawn and William’s medium E was supplemented with 100 ng/ml 2.5S nerve growth factor (Sigma) and 10 % TTR-KO mouse serum or 10 % TTR-KO mouse serum containing 300 μg/ml recombinant TTR (either wt or TTRprot−). After 72 h, total RNA was extracted, and PAM levels were determined by RT (reverse transcription)–PCR.
NPY cleavage by TTR
Amidated NPY (2 μg; Bachem) was incubated either with 5 μg of wt TTR or TTRprot−. Analysis of the reaction samples was performed using MALDI–TOF (time-of-flight)-MS, as a service performed at the IPATIMUP (Institute of Molecular Pathology and Immunology of the University of Porto) Proteomics Unit, Porto, Portugal. Assessment of NPY cleavage by TTR was performed by comparing the molecular mass of NPY either incubated with active wt TTR or with TTRprot−. As a control, the molecular mass of NPY alone was also determined using MALDI–TOF-MS.
Influence of TTR proteolytic activity in nerve regeneration
The effect of TTR proteolytic activity in nerve regeneration was assessed by performing neurite outgrowth assays [16]. PC12 cells were grown in six-well plates in DMEM supplemented with 10 % FBS. When cells reached 50 % confluence, FBS was withdrawn and DMEM was supplemented with 10 % of either wt or TTR-KO mouse serum, or 10 % TTR-KO mouse serum containing 300 μg/ml of recombinant TTR (either wt or TTRprot−), i.e. the physiological concentration of TTR in the plasma. After 48 h, PC12 cells were fixed in 2 %neutral buffered formalin for 30 min, washed with PBS and kept at 4 °C until further analysis. Neurite size was determined from 20× magnified fields using ImageJ software (http://rsbweb.nih.gov/ij/). For each cell, the average neurite length was determined, and at least 100 cells were analysed for each condition.
RESULTS
Libraries of combinatorial substrates and phosphonate inhibitors revealed a preference for a lysine residue in P1 for TTR proteolysis
In order to determine the P1–P4 specificity for TTR cleavage, we used combinatorial peptide libraries [19]. The P1 site preference for TTR cleavage was determined as being Lys > Ala > Arg > Leu ≥ Met > Phe (Figure 1a). To define the extended substrate specificities of TTR, we performed a screening for P2–P4 positions with libraries in which the P1 was fixed to a lysine residue (Figures 1b–1d). The P2, P3 and P4 cleavage preferences were broad, which is common for some serine proteases, and, as a consequence, we were unable to identify TTR specificity for the P2–P4 sites. Moreover, by using a library of 160 peptide phosphonate inhibitors, which are irreversible inhibitors of serine proteases [20], TTR preference for a lysine residue in P1 was confirmed (Table 1). In a first screening, we tested the inhibitors at a concentration of 50 μM and selected the compounds presenting an inhibitory effect higher than 90 %(40 compounds were selected). In a second screening, we performed dose-dependent experiments using inhibitor concentrations ranging from 1 to 50 μM; the most potent inhibitors were selected as being the ones that did not show a dose–response effect as they presented a high inhibitory effect at all of the concentrations tested. At a concentration of 1 μM, the lowest concentration tested, those compounds were able to block more than 80 % of TTR activity (Table 1). Analysis of the structures of the most potent compounds showed that TTR was preferentially inhibited by compounds with a basic residue such as lysine in the P1 position and a proline residue in P2. Among the most potent inhibitors, only one presented a phenylalanine residue in P1; one of the compounds presented an amidine-modified phenylalanine which has a basic-like behaviour, similarly to lysine (Table 1).
Figure 1. Subsite preferences of TTR as determined by combinatorial P-diverse libraries.
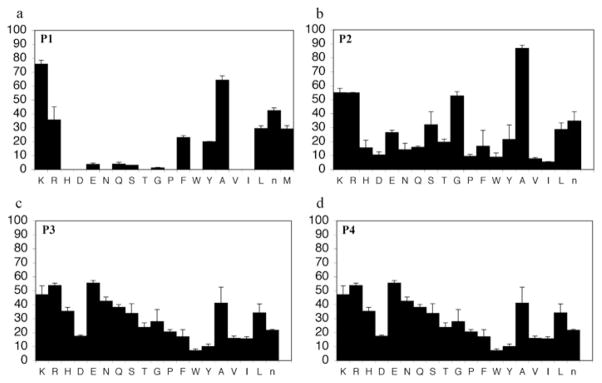
(a) P1 preference; (b) P2 preference; (c) P3 preference and (d) P4 preference. The y-axis represents the substrate-cleavage rates in picomolar concentrations of fluorophore released per second. The x-axis indicates the amino acid held constant at each position, designated by the one-letter code; n = norleucine. Results are means ± S.D.
Table 1. TTR screening with a library of phosphonate inhibitors.
Chemical structure of substrate-based phosphonate compounds that at 1 μM concentration yielded more than 80 % inhibition of TTR proteolytic activity.
| Structure | P1 | P2 |
|---|---|---|
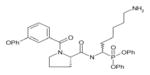
|
Homolysine | Proline |
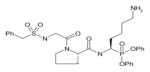
|
Lysine | Proline |
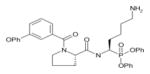
|
Lysine | Proline |
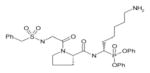
|
Homolysine | Proline |
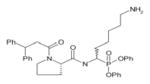
|
Homolysine | Proline |
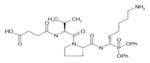
|
Homolysine | Proline |
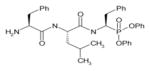
|
Phenylalanine | Leucine |
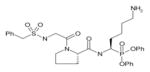
|
4-(Amidine)-phenylalanine | Proline |
Regarding the results for the P1 preference, it is noteworthy that TTR cleaves apoA-I after a phenylalanine residue and not after the preferred lysine residue. As such, it is possible that other unknown TTR substrates with P1 lysine, alanine, arginine, leucine and methionine exist.
ApoA-I is the sole plasma TTR substrate as confirmed by DIGE
In order to identify other putative natural TTR substrates in the plasma, we performed DIGE analysis of plasma from TTR-KO mice incubated with TTR. To determine the differences in proteome profiling derived from proteolytic cleavage of a possible TTR plasma substrate, we selected only the common spots between the experiment where TTR-KO plasma was incubated with either wt TTR or TTRprot− and the experiment where TTR-KO plasma was either run alone or following incubation with wt TTR. In both experiments, there was only one spot increased when TTR-KO plasma was incubated with wt TTR, corresponding to a fragment of a plasma protein originated by TTR cleavage (results not shown). This spot was analysed by MS and was identified as C-terminally cleaved mouse apoA-I. In agreement with this finding, we had shown previously that human TTR is able to cleave mouse apoA-I [4]. As such, despite the fact that we show that DIGE enables the identification of TTR substrates, we were unable to identify new putative TTR substrates in the plasma.
TTRprot− retains its ability to bind T4, RBP and its specific hepatic receptor
In DIGE, as well as in the following experiments, we used a preparation of glutathionylated TTR that was shown to be proteolytically inactive (TTRprot−). TTR proteolytic activity was assessed by determining the kinetic constants for the cleavage of the peptide Abz-ESFKVS-EDDnp. Whereas we determined a kcat/Km of 10.5 × 10−4 min−1 · μM−1 for wt TTR, in TTRprot− we observed complete loss of proteolytic activity. The modification by glutathione in this preparation was detected by MALDI–MS, by the presence of a mass peak corresponding to the mass of TTR with an increment of 305 Da, which corresponds to the molecular mass of glutathione. To confirm whether the biological functions of TTR, namely its ability to act as a plasma transporter, were maintained in the presence of the modification with glutathione in TTRprot−, we assessed the ability of this preparation to bind its major ligands T4 and RBP. No significant differences were observed between wt TTR and TTRprot− in binding to T4, as shown by the log EC50 of 1.6 ± 0.1 and 1.7 ± 0.1 nM respectively (Figure 2a); the same finding was observed for binding to RBP, as both TTR preparations competed similarly with 125I-labelled wt TTR (Figure 2b), with a log EC50 of 0.9 ± 0.1 nM for wt TTR and 0.9 ± 0.2 nM for TTRprot−. TTR uptake by its hepatic receptor was also assessed in experiments performed with human hepatomas, which were shown to internalize TTR by a specific yet unidentified receptor [22]. We determined that both wt TTR and TTRprot− bind similarly to the hepatic receptor, as no significant differences in 125I-labelled wt TTR cell association were observed by the presence of either of the proteins as shown by the log EC50 of 2.8 ± 0.2 ng/ml for wt TTR and 3.0 ± 0.3 ng/ml for TTRprot− (Figure 2c).
Figure 2. Comparison of the biological functions of wt TTR and TTRprot−.

(a) T4 binding to wt TTR and TTRprot−. Competition was performed with increasing concentrations of unlabelled T4 (0–1000 nM). (b) Binding of 125I-labelled wt TTR to RBP. Competition was performed with either unlabelled wt TTR or unlabelled TTRprot− at increasing concentrations ranging from 0 to 5 μM. (c) Cell association of 125I-labelled wt TTR in SAHep cells. Competition was performed with either unlabelled wt TTR or unlabelled TTRprot− at increasing concentrations ranging from 0 to 50 μg/ml. Results are means ± S.D.
TTR proteolytic activity is not relevant for its ability to regulate PAM mRNA expression
As the P1 preference of TTR suggests that it may have additional substrates besides apoA-I, we started to unravel the effect of TTR proteolytic activity in recently described TTR functions. To analyse the effect of TTR proteolytic activity in PAM expression, we assessed the expression levels of PAM in PC12 cells grown in the presence of TTR-KO serum or TTR-KO serum supplemented with either wt TTR or TTRprot−. Previous experiments performed with PC12 cells demonstrated that the addition of recombinant wt TTR to the cell growth medium leads to a decrease in PAM mRNA expression, when compared with cells grown in the absence of TTR [18]. If TTR proteolytic activity was involved in regulating PAM expression, it was expected that, similarly to cells grown in the absence of TTR, in the presence of TTRprot−, PAM expression would be increased. However, both wt TTR and TTRprot− led to a similar decrease of PAM mRNA levels in PC12 cultures (Figure 3), showing that TTR proteolytic activity does not influence PAM expression.
Figure 3. PAM expression in PC12 cells.
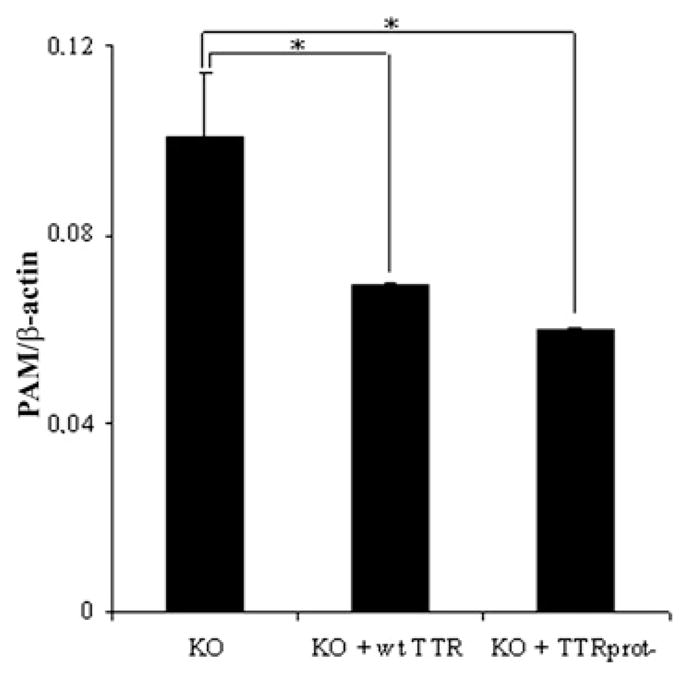
PAM semi-quantitative RT–PCR analysis of PC12 cells grown in the presence of TTR-KO mouse serum (KO), or TTR-KO mouse serum containing either wt TTR (KO + wt TTR) or TTRprot−(KO + TTRprot−). Results are means ±S.D. *P < 0.05.
In vitro, TTR is able to cleave NPY
Although we did not observe an effect of TTR proteolytic activity in PAM expression, we investigated whether in vitro TTR was able to cleave amidated NPY, as increased NPY levels are observed in the absence of TTR [18]. For this, we analysed the MS spectra of full-length NPY and NPY incubated with either wt TTR or TTRprot−. We observed that both full-length (Figure 4A) or TTRprot−-incubated NPY (Figure 4B) presented only one mass peak, corresponding approximately to the molecular mass of intact NPY (4272 Da). In the case of NPY incubated with wt TTR, we observed the fragmentation of the neuropeptide as shown by the appearance of two additional mass peaks, one with 3826 Da and a second with 4111 Da, besides the peak corresponding to intact NPY with an observed mass of 4273 Da (Figure 4C). By calculating the difference between the mass of intact NPY and the mass corresponding to each of the fragmentation products, we determined that the NPY fragments generated correspond to amino acid residues 1–33 and 1–35, showing that TTR cleaves NPY after Arg33 and Arg35 (Figure 4D). The TTR cleavage site on NPY is in agreement with a P1 arginine specificity determined with the P1-diverse libraries (P1 site preference for TTR cleavage Lys > Ala > Arg > Leu ≥ Met > Phe).
Figure 4. NPY cleavage by TTR.

MS spectra of (a) full-length NPY; (b) NPY incubated with TTRprot−; (c) NPY incubated with wt TTR; FL, full-length NPY; Fr, NPY fragments. (d) Full-length NPY sequence displaying TTR cleavage sites (arrows); calculated molecular mass of each NPY fragment is shown.
TTR proteolytic activity affects its ability to promote neurite outgrowth
Another TTR function in the nervous system is its participation in nerve regeneration [16]. In vitro, neurite outgrowth of PC12 cells is augmented in the presence of TTR, which probably underlies the increased regenerative capacity of wt mice when compared with TTR-KO mice [16]. To assess the putative involvement of TTR proteolytic activity in neurite outgrowth, we determined the average neurite length of PC12 cells cultivated in the presence of either wt or TTR-KO mouse serum, or TTR-KO mouse serum containing either wt TTR or TTRprot−. PC12 cells exposed to TTR-KO serum displayed approx. 20 % decreased average neurite length when compared with cells grown with wt serum, as reported previously [16]. Addition of wt TTR to TTR-KO serum was able to rescue the decrease in neurite length of PC12 cells. However, cells grown with TTRprot− displayed a similar neurite length to that of cells grown in the absence of TTR (Figure 5). These results suggest that lack of TTR proteolytic activity, and not only the absence of TTR itself, is responsible for a decreased neurite length in PC12 cells, thus demonstrating that the proteolytic activity of TTR is relevant for the ability of TTR to promote neurite outgrowth.
Figure 5. Neurite outgrowth in PC12 cells.

Average neurite length per cell of PC12 cells after exposure to either wt (WT) or TTR-KO serum (KO), or TTR-KO serum supplemented with either wt TTR (KO + wt TTR) or TTRprot− (KO + TTRprot−). Results are means ± S.E.M. *P < 0.05.
DISCUSSION
TTR was described as a protease able to cleave the C-terminus of apoA-I after the residue Phe225 [3]. In this work, we investigated the existence of other putative natural TTR substrates rather than apoA-I. We started by assessing the P1–P4 preference for TTR cleavage using P-diverse libraries [19], and observed the following P1 preference: Lys > Ala > Arg > Leu ≥ Met > Phe. This was confirmed further by performing a screening of TTR inhibition pattern using a library of peptide phosphonates [20], as TTR was inhibited by several phosphonate compounds, where the most potent inhibitors had a lysine residue in the P1 position and a proline residue in P2. TTR seems to follow a dual specificity preferring basic amino acids at P1, such as lysine and arginine (the latter residue was preferred over lysine in the case of NPY), but also being able to cleave after hydrophobic/aromatic amino acids (as determined for apoA-I). Similarly, cathepsin G also presents two opposite specificities [23]. Cathepsin G was initially thought to possess a strict chymotrypsin-like specificity, with preference for cleavage after phenylalanine residues. However, kinetic studies demonstrated that phenylalanine could be replaced by lysine, giving rise to similar kcat/Km values. Regarding TTR, cleavage in apoA-I occurs after a phenylalanine residue, and not after the preferred lysine determined in vitro. Although we cannot state that lysine would be the best P1 amino acid in a physiological context, our results strengthen the need to search for other natural TTR substrates. It is, however, noteworthy that results obtained using a substrate library composed of tetrapeptides must be interpreted with caution. The interaction of short peptides with a given enzyme will certainly differ widely from the one achieved in a particular physiological environment with its natural substrates, which may display a complex tridimensional structure and multiple binding and/or interaction sites with the protease.
New TTR functions related to nerve biology have been identified recently. In the present study, we assessed the involvement of TTR proteolytic activity in two of the described functions, namely TTR participation in regulating PAM mRNA expression [18] and the ability of TTR to promote nerve regeneration [16]. Although we demonstrated that TTR proteolytic activity is not necessary for TTR regulation of PAM expression, we determined that in vitro TTR is able to cleave NPY. As such, it is possible that in addition to the decreased levels of PAM expression, cleavage of amidated NPY by TTR might also contribute to the lower NPY levels observed in wt mice when compared with TTR-KO littermates [18]. Moreover, we determined that TTR cleavage of NPY occurs after an arginine residue; this P1 arginine preference is in accordance with the results obtained with the P-diverse libraries, which demonstrated a preference for basic residues at the P1 site, and suggests further a dual specificity of TTR observed both in vitro (using P-diverse libraries) and in vivo (cleavage of apoA-I and NPY occurs after a phenylalanine and lysine residue respectively).
To ascertain whether the proteolytic activity of TTR is related to its ability to enhance nerve regeneration, we measured neurite outgrowth in PC12 cells cultivated in the presence of wt TTR or TTRprot−. In these experiments TTR was used at 300 μg/ml, which is the physiological concentration of the protein in plasma. It is, however, noteworthy that in the settings of nerve injury, the blood–nerve barrier is disrupted enabling plasma to gain direct contact with the endonerium. Cells grown with TTRprot− displayed decreased neurite length compared with cells grown with proteolytically active wt TTR. This observation suggests that the proteolytic activity of TTR is relevant for its ability to promote neurite outgrowth. Although one cannot exclude the possibility that glutathionylation itself might affect neurite outgrowth, this hypothesis is hard to sustain as we have shown that glutathionylated TTR maintains its transport functions, being still able to bind T4 and RBP. Moreover, studies assessing TTR uptake by its hepatic receptor showed no major differences between the glutathionylated and the unmodified forms of the protein. As such, our results suggest that a TTR substrate probably exists in the nerve, and that its cleavage should enhance nerve regeneration.
Apart from the results of the present study, a putative effect of TTR proteolytic activity in the biology of the nervous system is supported further by the pattern of TTR expression throughout evolution and embryonic development. It is interesting to note that, in addition to the functions inferred from the TTR-KO phenotype, TTR has been suggested to protect against Aβ (amyloid β-peptide) deposition, the key pathological feature in Alzheimer’s disease [24]. When the nature of TTR–Aβ interaction was investigated further, TTR was found to be able to cleave Aβ in multiple positions with some of the generated Aβ peptides displaying lower amyloidogenic potential than the full-length counterpart [25]. The multiplicity of cleavage sites reported does not allow a direct comparison with the results of the present study. It is, however, interesting that cleavage in Aβ following phenylalanine, lysine and alanine residues is shown, in accordance with the TTR specificity described in the present paper. Additionally, the identification of Aβ as a TTR substrate stresses further the involvement of its proteolytic activity in neurobiology. Until recently, apoA-I was the only known TTR substrate [3,4]. Considering that TTR proteolytic activity might have been conserved during evolution, this functional property was highly unlikely to be maintained to increase the propensity for pathological conditions, as is the case of apoA-I cleavage which has an impact on the development of atherosclerosis and amyloidosis [4].
In summary, our results show that TTR proteolytic activity is related to the recently described TTR functions in the nerve: we identified NPY as a novel TTR substrate and determined that the proteolytic activity of TTR is important for the ability of this protein to enhance regeneration. As such, future work should address the identification of other TTR substrates in the nerve.
Acknowledgments
We thank Dr Hugo Osório (Proteomics Unit, IPATIMUP, Porto) and Dr Terence Wu (W. M. Keck Facility, Yale University, New Haven, U.S.A.) for performing the MALDI–MS and DIGE analyses respectively.
FUNDING
This project was supported by Fundação para a Ciênciae Tecnologia, Portugal [grant number PTDC/BIA-PRO/64437/2006], Association Française contre les Myopathies, France, National Institute of Health [grant number CA72006 (to Y. C. and C. S. C.)] and National Institutes of Health National Technology Center for Networks and Pathways [grant number U54 RR 020843 (to M. B.)]. M. A. L. is the recipient of a fellowship [grant number SFRH/BPD/34811/2007] from Fundação para a Ciência e Tecnologia, Portugal.
Abbreviations used
- Aβ
amyloid β-peptide
- Abz-ESFKVS-EDDnp
aminobenzoyl-Glu-Ser-Phe-Lys-Val-Ser-ethylenediamine-2,4-dinitrophenol
- apoA-I
apolipoprotein A-I
- CSF
cerebrospinal fluid
- Cy3
indocarbocyanine
- Cy5
indodicarbocyanine
- DIGE
differential fluorescence gel electrophoresis
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- HDL
high-density lipoprotein
- KO
knockout
- MALDI
matrix-assisted laser-desorption ionization
- NPY
neuropeptide Y
- PAM
peptidylglycine α-amidating mono-oxygenase
- PNS
peripheral nervous system
- PS-SCL
positional scanning synthetic combinatorial library
- RBP
retinol-binding protein
- RT
reverse transcription
- T4
thyroxine
- TOF
time-of-flight
- TTR
transthyretin
- TTRprot−
proteolytically inactive TTR
- wt
wild-type
References
- 1.Raz A, Goodman DS. The interaction of thyroxine with human plasma prealbumin and with the prealbumin–retinol-binding protein complex. J Biol Chem. 1969;244:3230–3237. [PubMed] [Google Scholar]
- 2.Sousa MM, Berglund L, Saraiva MJ. Transthyretin in high density lipoproteins: association with apolipoprotein A-I. J Lipid Res. 2000;41:58–65. [PubMed] [Google Scholar]
- 3.Liz MA, Faro CJ, Saraiva MJ, Sousa MM. Transthyretin, a new cryptic protease. J Biol Chem. 2004;279:21431–21438. doi: 10.1074/jbc.M402212200. [DOI] [PubMed] [Google Scholar]
- 4.Liz MA, Gomes CM, Saraiva MJ, Sousa MM. ApoA-I cleaved by transthyretin has reduced ability to promote cholesterol efflux and increased amyloidogenicity. J Lipid Res. 2007;48:2385–2395. doi: 10.1194/jlr.M700158-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Liz MA, Sousa MM. Deciphering cryptic proteases. Cell Mol Life Sci. 2005;62:989–1002. doi: 10.1007/s00018-005-4544-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickson PW, Howlett GJ, Schreiber G. Rat transthyretin (prealbumin): molecular cloning, nucleotide sequence, and gene expression in liver and brain. J Biol Chem. 1985;260:8214–8219. [PubMed] [Google Scholar]
- 7.Schreiber G. The evolution of transthyretin synthesis in the choroid plexus. Clin Chem Lab Med. 2002;40:1200–1210. doi: 10.1515/CCLM.2002.210. [DOI] [PubMed] [Google Scholar]
- 8.Harms PJ, Tu GF, Richardson SJ, Aldred AR, Jaworowski A, Schreiber G. Transthyretin (prealbumin) gene expression in choroid plexus is strongly conserved during evolution of vertebrates. Comp Biochem Physiol Part B Biochem Mol Biol. 1991;99:239–249. doi: 10.1016/0305-0491(91)90035-c. [DOI] [PubMed] [Google Scholar]
- 9.Richardson SJ, Bradley AJ, Duan W, Wettenhall RE, Harms PJ, Babon JJ, Southwell BR, Nicol S, Donnellan SC, Schreiber G. Evolution of marsupial and other vertebrate thyroxine-binding plasma proteins. Am J Physiol. 1994;266:R1359–R1370. doi: 10.1152/ajpregu.1994.266.4.R1359. [DOI] [PubMed] [Google Scholar]
- 10.Dickson PW, Aldred AR, Menting JG, Marley PD, Sawyer WH, Schreiber G. Thyroxine transport in choroid plexus. J Biol Chem. 1987;262:13907–13915. [PubMed] [Google Scholar]
- 11.Palha JA, Hays MT, Morreale de Escobar G, Episkopou V, Gottesman ME, Saraiva MJ. Transthyretin is not essential for thyroxine to reach the brain and other tissues in transthyretin-null mice. Am J Physiol. 1997;272:E485–E493. doi: 10.1152/ajpendo.1997.272.3.E485. [DOI] [PubMed] [Google Scholar]
- 12.Richardson SJ, Lemkine GF, Alfama G, Hassani Z, Demeneix BA. Cell division and apoptosis in the adult neural stem cell niche are differentially affected in transthyretin null mice. Neurosci Lett. 2007;421:234–238. doi: 10.1016/j.neulet.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 13.Saraiva MJ. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum Mutat. 2001;17:493–503. doi: 10.1002/humu.1132. [DOI] [PubMed] [Google Scholar]
- 14.Andrade C. A peculiar form of peripheral neuropathy: familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75:408–427. doi: 10.1093/brain/75.3.408. [DOI] [PubMed] [Google Scholar]
- 15.Episkopou V, Maeda S, Nishiguchi S, Shimada K, Gaitanaris GA, Gottesman ME, Robertson EJ. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc Natl Acad Sci USA. 1993;90:2375–2379. doi: 10.1073/pnas.90.6.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleming CE, Saraiva MJ, Sousa MM. Transthyretin enhances nerve regeneration. J Neurochem. 2007;103:831–839. doi: 10.1111/j.1471-4159.2007.04828.x. [DOI] [PubMed] [Google Scholar]
- 17.Sousa JC, Grandela C, Fernandez-Ruiz J, de Miguel R, de Sousa L, Magalhaes AI, Saraiva MJ, Sousa N, Palha JA. Transthyretin is involved in depression-like behaviour and exploratory activity. J Neurochem. 2004;88:1052–1058. doi: 10.1046/j.1471-4159.2003.02309.x. [DOI] [PubMed] [Google Scholar]
- 18.Nunes AF, Saraiva MJ, Sousa MM. Transthyretin knockouts are a new mouse model for increased neuropeptide Y. FASEB J. 2006;20:166–168. doi: 10.1096/fj.05-4106fje. [DOI] [PubMed] [Google Scholar]
- 19.Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA, Craik CS. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem. 2006;281:12824–12832. doi: 10.1074/jbc.M513331200. [DOI] [PubMed] [Google Scholar]
- 20.Arastu-Kapur S, Ponder EL, Fonovic UP, Yeoh S, Yuan F, Fonovic M, Grainger M, Phillips CI, Powers JC, Bogyo M. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat Chem Biol. 2008;4:203–213. doi: 10.1038/nchembio.70. [DOI] [PubMed] [Google Scholar]
- 21.Almeida MR, Macedo B, Cardoso I, Alves I, Valencia G, Arsequell G, Planas A, Saraiva MJ. Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem J. 2004;381:351–356. doi: 10.1042/BJ20040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sousa MM, Saraiva MJ. Internalization of transthyretin: evidence of a novel yet unidentified receptor-associated protein (RAP)-sensitive receptor. J Biol Chem. 2001;276:14420–14425. doi: 10.1074/jbc.M010869200. [DOI] [PubMed] [Google Scholar]
- 23.Hof P, Mayr I, Huber R, Korzus E, Potempa J, Travis J, Powers JC, Bode W. The 1.8 Å crystal structure of human cathepsin G in complex with Suc-Val-Pro-PheP-(OPh)2: a Janus-faced proteinase with two opposite specificities. EMBO J. 1996;15:5481–5491. [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarzman AL, Goldgaber D. Interaction of transthyretin with amyloid β-protein: binding and inhibition of amyloid formation. Ciba Found Symp. 1996;199:146–160. doi: 10.1002/9780470514924.ch10. [DOI] [PubMed] [Google Scholar]
- 25.Costa R, Ferreira-da-Silva F, Saraiva MJ, Cardoso I. Transthyretin protects against A-β peptide toxicity by proteolytic cleavage of the peptide: a mechanism sensitive to the Kunitz protease inhibitor. PLoS ONE. 2008;3:e2899. doi: 10.1371/journal.pone.0002899. [DOI] [PMC free article] [PubMed] [Google Scholar]