

Abstract
Pancreatic cancer is the most deadly neoplasm with a 5-year survival rate of less than 6%. Over 90% of cases harbor K-Ras mutations, which are the most challenging to treat due to lack of effective therapies. Here, we reveal that polyisoprenylated methylated protein methyl esterase (PMPMEase) is overexpressed in 93% of pancreatic ductal adenocarcinoma. We further present polyisoprenylated cysteinyl amide inhibitors (PCAIs) as novel compounds designed with structural elements for optimal in vivo activities and selective disruption of polyisoprenylation-mediated protein functions. The PCAIs inhibited PMPMEase with Ki values ranging from 3.7 to 20 µM. The 48 h EC50 values for pancreatic cancer Mia PaCa-2 and BxPC-3 cell lines were as low as 1.9 µM while salirasib and farnesylthiosalicylamide were ineffective at 20 µM. The PCAIs thus have the potential to serve as effective therapies for pancreatic and other cancers with hyperactive growth signaling pathways mediated by Ras and related G-proteins.
Keywords: PMPMEase, Pancreatic cancer, Polyisoprenylation, Prenylation, Polyisoprenylated cysteinyl amides, PCAIs
1 Introduction
Pancreatic ductal adenocarcinoma (PDA) accounts for approximately 95% of all pancreatic cancers (PC) and remains one of the deadliest neoplasms as the number of new diagnoses closely mirrors deaths. The poor prognosis is attributed to lack of early symptoms, with >50% of patients diagnosed at a late stage of the disease [1]. Epidermal growth factor receptor (EGFR) plays a vital role in the progression of PC [2] and is associated with poor prognosis [3]. Research to uncover the molecular aberrations in PC has revealed gene mutations in important oncogenic proteins such as K-Ras, which functions downstream of EGFR. Mutations in K-Ras are involved in >90% of PC cases [4]. Point mutations on this oncogene diminish or abolish its GTPase activity leaving it in the constitutively active state [5]. K-Ras is metabolized via the polyisoprenylation pathway (PP) and efforts to inhibit its maturation and localization have shown anticancer benefit in PC [6]. The PP encompasses a final reversible step that may possibly regulate the proteins’ function. Polyisoprenylated protein methyl transferase (PPMTase) esterifies the carboxyl terminus whereas polyisoprenylated methylated protein methyl esterase (PMPMEase) hydrolyzes the ester bond. Substrate analysis of PMPMEase [7, 8] led to the synthesis of high affinity, irreversible sulfonyl fluoride inhibitors (Figure 1) [9]. Meanwhile, polyunsaturated fatty acids (PUFAs), which are known to exhibit anticancer activity, have been shown to inhibit PMPMEase [10]. The decrease in cancer cell viability due to inhibition of PMPMEase with PUFAs, synthetic, and other chemopreventive agents highlights its potential as an anticancer target.
Figure 1.

Structure of PMPMEase (PMSF, L-28) and Ras (FTS and FTSA) inhibitors.
The study aims were three-fold. The first aim was to determine the expression of PMPMEase in PDA and in normal pancreatic tissue in view of the potential to utilize PMPMEase as a possible biomarker and therapeutic target. Second, to design and synthesize polyisoprenylated cysteinyl amide inhibitors (PCAIs), a first-in-class group of compounds to either inhibit PMPMEase and/or disrupt the polyisoprenylation-dependent interactions of the proteins it metabolizes. The design strategy incorporated three key elements to maximize selectivity, biochemical stability and bioavailability. These included, (1) the farnesyl group for high affinity/selective interactions, (2) a substituted amide bioisostere of the scissile ester bond found in the endogenous substrates, and (3) an ionizable appendage group designed to mitigate the excessive hydrophobicity of the farnesyl cysteinyl amide that constitutes the pharmacophore. Finally, to examine the effect of PMPMEase inhibition by PCAIs in PC cell lines.
The potential beneficial impact of these compounds was further investigated by examining the expression of PMPMEase in PDA relative to normal pancreatic tissues that might substantiate the use of the PCAIs as effective targeted therapies. PMPMEase overexpression in PDA would not only contribute to its validation as a putative drug target but also as a biomarker for potential early/companion diagnosis and prognosis. The results strongly suggest possible beneficial outcomes for the continuous development of the PCAIs as a novel class of targeted therapies for PC.
2 Results and Discussion
2.1 Chemistry
The design and synthesis of polyisoprenylated sulfonyl fluorides was the first successful attempt at developing inhibitors of PMPMEase [9]. Incorporation of a polyisoprenyl tail increased the inhibitory potency determined using pseudo-first order kinetics by 330-fold compared to the lead compound PMSF. Although these compounds are potent inhibitors, lack of polar groups means that they are very hydrophobic (ClogP ranged from 2.8 to 4.3). Additionally, the highly reactive sulfonyl fluoride functional group is moisture-sensitive. PCAIs address these former limitations by adding an ionizable functional group (pyrrolidine or N-methylpiperazine) to diminish the excessive hydrophobicity. The calculated LogD values ranged from 3.15 – 5.88 at the physiological pH of 7.4 whereas the calculated LogD value for the C15 polyisoprenylated sulfonyl fluoride was 5.04. In addition, the unstable, highly reactive sulfonyl fluoride group that irreversibly inhibits PMPMEase was substituted with an amide group that functions as a more stable bioisostere of the substrate ester bond. The resulting compounds, unlike the sulfonyl fluorides, are reversible PMPMEase inhibitors.
The PCAIs were prepared according to Scheme 1. Briefly, the starting alcohol is first converted to the bromide (2) using phosphorous tribromide. The resulting bromide (2) was used to alkylate L-cysteine methyl ester to form the alpha amino ester which underwent alkylation with bromoalkynoyl chloride to produce compound 4. Compound 4 then served as the alkylating agent for the selected amines (pyrrolidine and 1-methylpiperazine) designed to impart solubility, prior to the amidation of the ester function overnight as a neat reaction at 90°C. However, several low boiling amines and di-substituted amines showed no reaction and required the use of the corresponding acyl chlorides. The acyl chlorides were obtained by hydrolyzing the ester bonds and converting the products to the required acyl chlorides before reaction with the low boiling point amines. Reactions with the amines resulted in compounds 7a–g (pyrrolidines) and 8a–g (1-methylpiperazines).
Scheme 1.

Synthesis of polyisoprenylated cysteinyl amide inhibitors (PCAIs) of PMPMease. Reagents: (a) −10°C, PBr3, Et2O (b) TEA, L-cysteine methyl ester, Anh. MeOH (c) 6-bromohexanoyl chloride, TEA, DCM, (d) K2CO3, amine, toluene, (e) amine, 90°C, ON.
2.2 Docking Analysis
The in silico analysis of the PCAIs revealed compounds with AScore docking energies ranging from −17.21 to −13.35 kcal/mole (Table 1). A representative of the PCAIs, 8a is shown in the active site of PMPMEase in Figure 2. The pyrrolidine derivatives showed lower docking energies (−17.21 to −14.03 kcal/mole) compared to the N-methylpiperazine derivatives (−15.08 to −13.35 kcal/mole). Compound 7d had the lowest AScore docking energy of −17.21 kcal/mole versus −14.26 kcal/mole for its N-methylpiperazinyl derivative (8d).
Table 1.
Physicochemical, docking, and inhibition kinetics data for polyisoprenylated cysteinyl amide inhibitors of PMPMEase
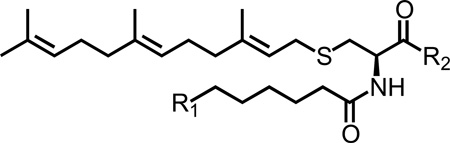 | |||||||
|---|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | LogD | AScore (kcal/ mole) |
Ki (µM) |
MIAPaCa-2 48h EC50 (µM) |
BxPC-3 48h EC50 (µM) |
| 7a | 3.15 | −14.03 | 17.5 | >20 | >20 | ||
| 7b | 4.04 | −16.93 | 3.7 | 3.2 | 6.8 | ||
| 7c | 4.48 | −15.78 | 11.2 | 5.6 | 12.7 | ||
| 7d | 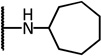 |
4.93 | −17.21 | 11.6 | 6.8 | 5.8 | |
| 7e | 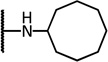 |
5.37 | −16.73 | 11.9 | 1.9 | 4.0 | |
| 7f | 3.31 | −16.10 | 9.7 | 10.8 | 6.5 | ||
| 7g | 3.76 | −14.70 | 11.0 | 12.4 | 8.7 | ||
| 8a | 3.66 | −14.24 | 13.8 | 10.4 | 6.3 | ||
| 8b | 4.55 | −13.66 | 10.1 | 10.9 | >20 | ||
| 8c | 4.99 | −13.35 | 16.9 | 5.9 | 12 | ||
| 8d | 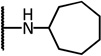 |
5.44 | −14.26 | 17.6 | 2.7 | 6.2 | |
| 8e | 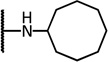 |
5.88 | −15.08 | 15.2 | 2.5 | 7.5 | |
| 8f | 3.82 | −14.53 | 20.0 | 6.0 | 3.2 | ||
| 8g | 4.27 | −14.32 | 13.0 | 14.3 | 8.8 | ||
| FTS | - | - | 3.43 | ND | ND | >20 | >20 |
| FTSA | - | - | 6.03 | ND | ND | >20 | >20 |
Figure 2.

Docking of PCAIs in PMPMEase. A) A representative PCAI, compound 8a, is shown in the active site of PMPMEase. The amide bond (X) is in close proximity to the catalytic serine (SER221, Y) and the methylpiperazine ring (Z) is located outside of the active site of the protein. B) Docking pose of compound 8a is shown surrounded by the active site hydrophobic residues.
2.3 Biological Evaluation
The clinicopathologic data of the PC cases are summarized in Table 2. The age range of the donors was 1 month to 78 years with a mean age of 53 years. Intense PMPMEase immunoreactivity was observed in pancreatic tumors. Figure 3 is a PC TMA showing varying degrees of inter- and intra-slice brown staining indicative of the varying tissue expression levels of PMPMEase in various PC cases. The intra-tissue slice distributions appear to be concentrated in tumors as the areas of intense brown staining, indicative of high levels of PMPMEase, are accompanied by larger numbers of blue-stained nuclei. The scoring of PMPMEase expression in various pancreatic tissues is summarized in Table 3. PC normal adjacent tissue (NAT) and normal pancreatic tissue (NT) showed trace to weak PMPMEase staining with mean scores of 145.0 ± 8.6 and 115 ± 7.6, respectively. With the exception of only three PDA cores, all the cores (93%) showed intermediate to very strong PMPMEase staining with a mean score of 393.6 ± 14.4. The positive expression of PMPMEase in the PDA cores was significantly higher than both NAT and NT (p<0.0001). PMPMEase expression compared to tumor grade, tumor status, and metastasis (Table 3) was also significantly higher compared to both NAT and NT (p<0.0001).
Table 2.
Clinicopathologic data for the 98 pancreatic tissue donors. Grade, tumor status, nodal status, and metastasis are taken from the 46 carcinoma samples.
| Characteristics | Donors |
Characteristics | Donors |
||||
|---|---|---|---|---|---|---|---|
| N | (%) | N | (%) | ||||
| Age | Grade | ||||||
| ≤ 53 | 41 | 41.8 | 1 | 8 | 17.4 | ||
| > 53 | 57 | 58.2 | 2 | 15 | 32.6 | ||
| Sex | 3 | 13 | 28.3 | ||||
| Male | 57 | 58.2 | Not determined | 10 | 21.7 | ||
| Female | 41 | 41.8 | Tumor status | ||||
| Histology | 1 | 2 | 4.3 | ||||
| Adenosquamous carcinoma | 3 | 3.1 | 2 | 27 | 58.7 | ||
| Chronic inflammation | 2 | 2.0 | 3 | 14 | 30.4 | ||
| Chronic pancreatitis | 2 | 2.0 | 4 | 2 | 4.3 | ||
| Duct adenocarcinoma | 42 | 42.9 | Not determined | 1 | 2.2 | ||
| Hyperplasia | 2 | 2.0 | Nodal status | ||||
| Islet cell carcinoma | 1 | 1.0 | 0 | 36 | 78.3 | ||
| Islet cell tumor | 10 | 10.2 | 1 | 8 | 17.4 | ||
| Mild chronic inflammation | 6 | 6.1 | Not determined | 2 | 4.3 | ||
| Type | Metastasis | ||||||
| Normal | 10 | 10.2 | 0 | 42 | 91.3 | ||
| NAT | 20 | 20.4 | 1 | 3 | 6.5 | ||
| Benign | 10 | 10.2 | Not determined | 1 | 2.2 | ||
| Hyperplasia | 2 | 2.0 | |||||
| Inflammation | 10 | 10.2 | |||||
| Malignant | 46 | 46.9 | |||||
Figure 3.

PMPMEase immunoreactivity in pancreatic tumor and normal tissue. I) Selected pancreatic cancer TMA cores of ductal adenocarcinoma (B–D), adenosquamous cell carcinoma (E and F) and benign islet cell carcinoma (G and H) showing varying degrees of brown staining indicating varying expression levels of PMPMEase. A represents an image of normal pancreatic tissue slice. II) Magnification of selected pancreatic cancer TMA cores of duct adenocarcinoma (A2 to B1), adenosquamous cell carcinoma (B2 and B3) and benign islet cell carcinoma (C1 to C3) showing varying degrees of brown staining indicating varying expression levels of PMPMEase. A1 represents an image of normal pancreatic tissue.
Table 3.
PMPMEase expression in various pancreatic tissue. Mean ± SEM.
| Characteristic | PMPMEase Staining Intensity, N (%) | Normal | NAT | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1–100 Trace |
101–200 Weak |
201–300 Intermediate |
301–400 Strong |
401–500 Very strong |
Total cases |
Mean Score | p-value t-test |
p-value t-test |
|
| Histology | |||||||||
| Normal pancreatic tissue | 7 (70.0) | 3 (30.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 10 | 115.0 ± 7.6 | ||
| Normal adjacent tissue | 6 (30.0) | 14 (70.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 20 | 145.0 ± 8.6 | ||
| Adenosquamous carcinoma | 0 (0.0) | 2 (66.7) | 1 (33.3) | 0 (0.0) | 0 (0.0) | 3 | 183.3 ± 46.4 | 0.0268 | ns |
| Chronic inflammation | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (50.0) | 1 (50.0) | 2 | 412.5 ± 12.5 | <0.0001 | <0.0001 |
| Chronic pancreatitis | 1 (50.0) | 1 (50.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 | 112.5 ± 12.5 | ns | ns |
| Duct adenocarcinoma | 0 (0.0) | 3 (7.1) | 3 (7.1) | 17 (40.5) | 19 (45.2) | 42 | 393.6 ± 14.4 | <0.0001 | <0.0001 |
| Hyperplasia | 0 (0.0) | 1 (50.0) | 1 (50.0) | 0 (0.0) | 0 (0.0) | 2 | 212.5 ± 62.5 | 0.0059 | 0.0446 |
| Islet cell carcinoma | 0 (0.0) | 0 (0.0) | 1 (100.0) | 0 (0.0) | 0 (0.0) | 1 | 250.0 ± 0.0 | - | - |
| Islet cell tumor | 4 (40.0) | 1 (10.0) | 3 (30.0) | 1 (10.0) | 1 (10.0) | 10 | 225.0 ± 48.3 | 0.0372 | 0.0333 |
| Mild chronic inflammation | 2 (33.3) | 1 (16.7) | 2 (33.3) | 1 (16.7) | 0 (0.0) | 6 | 204.2 ± 48.5 | 0.0342 | ns |
| Type | |||||||||
| Benign | 4 (40.0) | 1 (10.0) | 3 (30.0) | 1 (10.0) | 1 (10.0) | 10 | 225.0 ± 48.3 | 0.0372 | 0.0333 |
| Hyperplasia | 0 (0.0) | 1 (50.0) | 1 (50.0) | 0 (0.0) | 0 (0.0) | 2 | 212.5 ± 62.5 | 0.0059 | 0.0446 |
| Inflammation | 3 (30.0) | 2 (20.0) | 2 (20.0) | 2 (20.0) | 1 (10.0) | 10 | 227.5 ± 43.4 | 0.0199 | 0.0171 |
| Malignant | 0 (0.0) | 5 (10.9) | 5 (10.9) | 18 (39.1) | 18 (39.1) | 46 | 374.5 ± 55.2 | <0.0001 | <0.0001 |
| Grade | |||||||||
| 1 | 0 (0.0) | 2 (25.0) | 1 (12.5) | 3 (37.5) | 2 (25.0) | 8 | 328.1 ± 39.9 | <0.0001 | <0.0001 |
| 2 | 0 (0.0) | 0 (0.0) | 1 (6.7) | 6 (40.0) | 8 (53.3) | 15 | 411.7 ± 19.6 | <0.0001 | <0.0001 |
| 3 | 0 (0.0) | 0 (0.0) | 1 (7.1) | 7 (50.0) | 6 (42.9) | 14 | 408.9 ± 22.2 | <0.0001 | <0.0001 |
| Tumor status | |||||||||
| 1 | 0 (0.0) | 0 (0.0) | 0 * (0.0) | 0 (0.0) | 2 (100.0) | 2 | 425.0 ± 25.0 | <0.0001 | <0.0001 |
| 2 | 0 (0.0) | 2 (7.4) | 3 (11.1) | 11 (40.7) | 11 (40.7) | 27 | 384.3 ± 19.2 | <0.0001 | <0.0001 |
| 3 | 0 (0.0) | 3 (20.0) | 1 (6.7) | 5 (33.3) | 6 (40.0) | 15 | 356.7 ± 31.9 | <0.0001 | <0.0001 |
| 4 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (50.0) | 1 (50.0) | 2 | 450.0 ± 50.0 | <0.0001 | <0.0001 |
| Nodal status | |||||||||
| 0 | 0 (0.0) | 2 (5.4) | 3 (8.1) | 16 (43.2) | 16 (43.2) | 37 | 390.5 ± 15.6 | <0.0001 | <0.0001 |
| 1 | 0 (0.0) | 3 (37.5) | 1 (12.5) | 2 (25.0) | 2 (25.0) | 8 | 325.0 ± 51.1 | 0.0003 | <0.0001 |
| Metastasis | |||||||||
| 0 | 0 (0.0) | 5 (11.6) | 4 (9.3) | 18 (41.9) | 16 (37.2) | 43 | 374.4 ± 16.3 | <0.0001 | <0.0001 |
| 1 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (100.0) | 3 | 458.3 ± 22.0 | <0.0001 | <0.0001 |
PMPMEase expression was increased in donors with chronic inflammation. Mild chronic inflammation (6 cases) showed a mean score of 204.2 ± 48.5 which is statistically significant compared to NT (p=0.0342), but was not significant compared to NAT (Table 3). Chronic inflammation (2 cases) showed a statistically higher mean score of 412.5 ± 12.5 compared to both NAT and NT (p<0.0001). Malignant tumor tissues also showed high levels of PMPMEase immunoreactivity. Other tumor types (benign, hyperplasia, and inflammation) showed a 1.5- to 2-fold increase in staining while malignant tumor tissues (median score=374.5 ± 55.2) showed about a 3-fold increase in PMPMEase staining intensity as compared to both controls (Table 3). All of the PCAIs inhibited PMPMEase in a concentration-dependent manner with Ki values ranging from 3.7 to 20 µM (Table 3). Of the secondary amides, ring size affected inhibitor potency. In the 5-membered pyrrolidinyl series (7a–e), cyclopentylamide analog (7b, Ki = 3.7 µM) was observed to be the most potent inhibitor of PMPMEase. Substituting the cyclopentyl moiety with a cyclopropyl group decreased the potency by 4.7-fold (7a, Ki = 17.5 µM) while increasing the ring size to a 6 (7c), 7 (7d) or 8-membered ring (7e) yielded Ki values of 11–12 µM. A similar trend was also observed with the N-methylpiperazine derivatives (8a–e). The cyclopentylamide derivative, compound 8b, was the most potent (Ki = 10.1 µM). The Ki values for the 3-, 6-, 7-, and 8-membered rings (8a, 8c, 8d, and 8e, respectively) were 13.8, 16.9, 17.6, and 15.2, respectively. Tertiary amide derivatives (7f, 7g, 8f, and 8g) had Ki values ranging from 9.7 to 20 µM and here again the pyrrolidinyl analogs showing a higher potency (Table 1).
The PCAIs were screened against MIA PaCa-2 pancreatic cancer cells that harbor the mutant K-Ras oncogene and the pancreatic cancer BxPC-3 cells that lack the mutation [11]. The cancer cells were cultured and treated in 5% FBS and treated with PCAIs at concentrations ranging from 0 to 20 µM for 48 h. The results show that the PCAIs induce cell degeneration in a concentration-dependent manner (Figure 4). The EC50 values for the effect of the PCAIs against MIA PaCa-2 cells ranged from 1.9 to >20 µM and the EC50 values against BxPC-3 cells ranged from 3.2 to >20 µM for 48 h (Table 1). On the other hand, the Ras inhibitors, Salirasib (FTS) and farnesylthiosalicylamide (FTSA, Figure 1), showed no observable effects on the viability of the cells at 20 µM concentrations under the same treatment conditions (Figure 5).
Figure 4.

Concentration-response curves of pancreatic cancer BxPC-3 (A) and MIA PaCa-2 (B) cells treated with 0 – 20 µM of PCAIs for 48 h in media containing 5% FBS. Salirasib (FTS, ▲) and FTSA (□) were ineffective at 20 µM.
Figure 5.

PCAIs induce cell death by apoptosis. MIAPaCa-2 cells were treated with 8e (0.5 – 2 µM) for 48h and stained with AO/EB 10 µg/mL. Viable cells do no absorb EB while AO is absorbed turning their intact nuclei green. Apoptotic cells pick up EB and are orange to red with highly condensed nuclei. The results presented are representative of triplicate experiments (N=4). BF: Bright Field; AO: Acridine Orange; EB: Ethidium Bromide. OL: overlay of AO and EB.
In order to determine the mechanism of cell death, the PC cells were treated with 8e for 48 h in the presence of 5% FBS and stained with acridine orange/ethidium bromide (AO/EB) according to a previously described method [12, 13]. As shown in Figure 5, signs of apoptosis were observed in MIA PaCa-2 cells treated with 0.5, 1 and 2 µM concentrations of compound 8e. AO permeated the live control cells as well as cells exposed to the lower concentration of 8e (1 µM) turning their nuclei green. Cells treated with concentrations of 2 µM 8e fluoresced red due to EB uptake. Early apoptotic cells with bright green fragmented nuclei were observed after 48 h treatment of MIA PaCa-2 cells with 8e.
The design, synthesis, and in vitro analysis of the current batch of PCAIs demonstrate that polyisoprenylated small molecules that effectively disrupt polyisoprenylated protein metabolism and/or functional interactions to address cancers with hyperactive G-proteins is entirely possible. Although the exact mechanism(s) of action of the PCAIs against the cancer cell lines remains to be fully explored, their effectiveness against the MIA PaCa-2 cells indicates that they would be effective against cancers with the oncogenic K-Ras mutations. Over 90% of PC cases [4] have this oncogenic transformation, which is the highest proportion that has been observed in any other type of cancer. The strategy successfully accomplished three key objectives to obtain (1) aqueous-stable polyisoprenylated small molecules that are (2) soluble in aqueous buffers and can (3) be effective against tumors with not only the K-Ras oncogene but also those with hyperactive EGFR signaling of which Ras is a key mediator.
MIA PaCa-2 and BxPC-3 PC cells treated with PCAIs resulted in significant cell degeneration. However, there is a lack of correlation between the Ki values for PMPMEase inhibition and EC50 values against the cell lines. This suggests that the compounds’ effects on cell viability may include mechanisms that involve PMPMEase inhibition and/or other disruptions of polyisoprenylation-mediated protein-protein interactions. Like FTS, PCAIs share the farnesyl moiety essential for protein-protein interactions [14]. The PCAIs may function in a similar manner to FTS, which is known to disrupt Ras function by dislodging it from the membrane [15]. For example, galectin-3 is a selective binding partner of activated, GTP-bound K-Ras [16] that forms Ras nanoclusters essential for cell signaling. Signaling is largely dependent on these nanoclusters and can be blocked if Ras localization is altered [17]. Relative biochemical instabilities between the PCAIs in the cancer cell lines may also account for some of the differences between the inhibition kinetics against the enzyme activity and the cell viability.
K-Ras mutations are common in PC and affects cell signaling downstream of EGFR. Mutations lead to constitutively active Ras and subsequent activation of the Ras/MAPK signaling pathway, which has been associated with anti-EGFR resistance [18, 19]. It has been reported that K-Ras mutations are indicative of resistance to anti-EGFR monoclonal antibodies and TKIs in PC [20, 21] and that K-Ras may serve as a plausible target for cancer treatment. KRas requires polyisoprenylation for proper localization, but potential drugs that inhibit its polyisoprenylation produced significant adverse side effects [22]. PMPMEase, which plays a vital role in regulating the function of polyisoprenylated proteins such as K-Ras, may serve as a viable alternative drug target for inhibiting or reversing PC tumor growth. It has recently been shown that siRNA targeting hCE1/PMPMEase disrupt F-actin organization by inhibiting the demethylation of RhoA leading to altered cell morphology [23]. This is consistent with our own studies showing the effects of PMPMEase inhibition-induced changes on cell morphology [9, 10]. This is further indication that the PCAIs may be effective not only at abrogating cell proliferation and tumor growth but also cell migration and invasion that promote metastasis, the most devastating feature of malignant tumors.
3 Conclusions
Targeting PMPMEase offers a potentially effective strategy for treating cancers with activating mutations and/or overexpression of both EGFR and monomeric G-proteins that confer resistance to current drug therapies. The PCAIs are a new class of agents that not only target a preponderant genetic aberration in PC but combines physico-chemical elements for their effective delivery. These have the potential to address the oncogenic Ras therapeutic void.
4 Experimental Section
4.1 Synthetic procedure
Reagents were purchased from Sigma-Aldrich (Milwaukee, WI), Acros (Morris Plains, NJ), TCI America (Portland, OR), Alfa Aesar (Ward Hill, MA), ChemImpex International (Wood Dale, IL) and Santa Cruz Biotechnology (Santa Cruz, CA) and used without further purification. Flash column chromatography was carried on 40–63µ SiO2. NMR spectra were obtained from a Varian Mercury 300 (300 MHz) in CDCl3, unless otherwise stated. Chemical shifts (δ) are given in ppm relative to the signal for the deuterated solvent and are reported consecutively as position (dH), relative integral, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, m = multiplet and where br = broad), coupling constant (J/Hz) and assignment. Salirasib (farnesylthiosalicylic acid or FTS) and farnesylthiosalicylamide (FTSA) were synthesized as previously described [15].
4.2 Preparative purification and MS analysis
Reaction mixtures were purified via reversed-phase high performance liquid chromatography (RP-HPLC) using a Shimadzu Prominence HPLC system with UV detection at 214 nm. Samples were loaded onto a Hamilton PRP-1 (21.5×150 mm ID) or Hamilton PRP-1 (10×250 mm ID) column and eluted with a linear gradient of acetonitrile in 0.1% trifluoroacetic acid at 5 mL/min or 3 mL/min, respectively. MS analysis of the fractions was achieved using 4800 MALDI/TOF on linear low mass mode. Fractions containing only the target compound were combined and the solvents removed under vacuum and dried at 40°C and 3 mm Hg.
4.3 Synthesis of trans, trans-Farnesyl bromide (2)
A solution of PBr3 (0.75 mL, 8 mmol) in anhydrous ether (5 mL) was added drop-wise to trans, trans-farnesol (1, 5 g, 22.9 mmol) and anhydrous ether (10 mL) at −15 to −10°C. When the addition was complete, the mixture was kept at 0°C for a further 10 min and then at 20°C for 30 min. The organic phase was decanted, washed with cold 5% NaHCO3, brine, dried over MgSO4 and the solvent removed at 40°C/3 mm Hg, yielding 2 as a pale-yellow liquid (6.4 g, 98%). 1H NMR: δ 1.60 (s, 6H), 1.67 (s, 3H), 1.72 (s, 3H), 1.95–2.15 (m, 8H), 4.16 (d, J=8.4 Hz, 2H), 5.09 (m, 2H), 5.54 (m, 1H).
4.4 Synthesis of L-S-(trans, trans-farnesyl) cysteine methyl ester (3)
A mixture of trans, trans-farnesyl bromide (2, 4.56 g, 16 mmol), L-cysteine methyl ester hydrochloride (2.75 g, 16 mmol) in anhydrous methanol (28 mL) and 7N anhydrous NH3 solution in methanol (92 ml) at −5°C was stirred at this temperature for 2 h. The solvent was evaporated under vacuum and the residue was then purified by flash chromatography, eluting with hexane-EtOAc 1:1 (1.69 g, 31%). Rf 0.2 (hexane-EtOAc 1:1). 1H-NMR: δ 1.48 (s, 3H), 1.51 (s, 3H), 1.59 (s, 3H), 1.66 (s, 3H), 1.65 (s, 2H), 1.80–2.10 (m, 8H), 2.61–2.77 (dd, 1H, J=7.5Hz), 3.09 (d, J=7.2Hz, 2H), 3.55 (dd, J=3.0Hz, 1H), 3.65 (s, 3H), 5.04 (br, 2H), 5.14 (t, J=1.2 Hz, 1H).
4.5 Synthesis of L-((pyrrolidinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine methyl ester (5)
6-Bromohexanoyl chloride (1.4 mL, 9.2 mmol) was added drop-wise to a stirred solution of L-S-(trans, trans-farnesyl) cysteine methyl ester (3, 3.0 g, 8.8 mmol) and TEA (1.5 mL, 10.5 mmol) at 0°C and the mixture stirred for 3 h at RT until completion of the reaction as determined by TLC. The mixture was then washed with cold 5% NaHCO3, brine, dried over MgSO4, and the solvent removed under vacuum resulting in a pale yellow oil (4). L-(6-bromohexanoyl)-S-(trans, trans-farnesyl) cysteine methyl ester (4) was added to a solution of pyrrolidine (0.82 mL, 10 mmol) and K2CO3 (1.1 g, 10 mmol) in 25 mL toluene and refluxed for 3 h. The mixture was filtered, solvent was removed and the residue was purified by HPLC to afford the product (5, 3.5 g, 80%). 1H-NMR: δ 1.18 – 1.51 (m, 3H), 1.51 – 1.86 (m, 17H), 1.90 – 2.17 (m, 12H), 2.19 – 2.37 (m, 2H), 2.84 (dt, J = 9.9, 8.5 Hz, 3H), 3.02 – 3.26 (m, 5H), 3.73 – 3.85 (m, 3H), 4.72 (ddd, J = 29.2, 15.6, 5.1 Hz, 1H), 5.09 (t, J = 6.0 Hz, 2H), 5.20 (t, J = 7.6 Hz, 1H), 6.77 (t, J = 18.1 Hz, 1H). MALDI/TOF-MS m/z = 507.61 [M+1] (calcd. for C29H50N2O3S = 506.78)
4.6 Synthesis of L-((4-methylpiperazinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine methyl ester (6)
6-Bromohexanoyl chloride (1.4 mL, 9.2 mmol) was added drop-wise to a stirred solution of L-S-( trans, trans-farnesyl) cysteine methyl ester (3, 3.0 g, 8.8 mmol) and TEA (1.5 mL, 10.5 mmol) at 0°C and the mixture stirred for 3 h at RT until completion of the reaction (monitored by TLC). The mixture was then washed with cold 5% NaHCO3, brine, dried over MgSO4, and the solvent removed under vacuum resulting in a pale yellow oil (4). L-(6-bromohexanoyl)-S-(trans, trans-farnesyl) cysteine methyl ester (4) was added to a solution of 1-methylpiperazine (1.1 mL, 10 mmol) and K2CO3 (1.1 g, 10 mmol) in 25 mL toluene and refluxed for 3 h. The mixture was filtered, solvent was removed and the residue was purified by HPLC to afford the product (6, 4 g, 85%). 1H-NMR: δ 1.17 – 1.44 (m, 2H), 1.55 – 1.76 (m, 17H), 1.89 – 2.15 (m, 10H), 2.26 (t, J = 7.3 Hz, 2H), 2.43 – 2.57 (m, 3H), 2.58 – 2.70 (m, 2H), 2.70 – 3.04 (m, 7H), 3.04 – 3.28 (m, 2H), 3.66 – 3.80 (m, 3H), 4.78 (td, J = 6.3, 4.9 Hz, 1H), 5.09 (t, J = 6.0 Hz, 2H), 5.20 (t, J = 7.8 Hz, 1H), 6.44 (d, J = 7.6 Hz, 1H). MALDI/TOF-MS m/z 536.67 [M+1] (calcd. for C30H53N3O3S = 535.83).
4.7 General procedure for the synthesis of compounds 7a–g
Compound 5 (100 mg, 0.20 mmol) and the respective amine (0.4 mL, 3.14 – 5.80 mmol) were heated as a neat reaction at 90°C overnight. The mixture was then purified on a HPLC system to afford the respective products.
4.7.1 L-((pyrrolidinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cyclopropylamide (7a, NSL-BA-037)
Yielded 30 mg, 28%. 1H NMR δ 0.54 (s, 2H), 0.75 (s, 2H), 0.82 – 1.06 (m, 2H), 1.07 – 1.55 (m, 18H), 1.55 – 1.85 (m, 5H), 1.99 – 2.29 (m, 5H), 2.80 (s, 5H), 3.12 (d, J = 32.0 Hz, 4H), 3.80 (s, 4H), 4.45 (s, 2H), 5.09 (s, 2H), 5.22 (s, 1H), 6.78 (s, 1H). MALDI/TOF-MS m/z 532.58 [M+1] (calcd. for C31H53N3O2S = 531.84).
4.7.2 L-((pyrrolidinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cyclopentylamide (7b, NSL-BA-038)
Yielded 35 mg, 31%. 1H NMR δ 1.34 – 1.52 (m, 4H), 1.52 – 1.87 (m, 20H), 2.03 (ddd, J = 24.6, 13.7, 5.9 Hz, 14H), 2.26 (t, J = 7.2 Hz, 2H), 2.79 (d, J = 6.7 Hz, 4H), 3.10 (dd, J = 14.5, 9.8 Hz, 2H), 3.16 – 3.29 (m, 2H), 3.81 (d, J = 4.9 Hz, 2H), 4.09 – 4.22 (m, 1H), 4.45 (q, J = 7.0 Hz, 1H), 5.05 – 5.50 (m, 2H), 5.23 (t, J = 7.4 Hz, 1H), 6.67 (d, J = 7.1 Hz, 1H), 7.09 (d, J = 7.2 Hz, 1H). MALDI/TOF-MS m/z 560.53 [M+1] (calcd. for C33H57N3O2S = 559.89).
4.7.3 L-((pyrrolidinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cyclohexylamide (7c, NSL-BA-039)
Yielded 34 mg, 30%. 1H NMR δ 1.13 – 1.44 (m, 12H), 1.71 (t, J = 37.1 Hz, 17H), 2.16 (d, J = 58.3 Hz, 12H), 2.79 (s, 4H), 3.14 (d, J = 47.6 Hz, 4H), 3.80 (s, 4H), 4.44 (s, 1H), 5.09 (s, 2H), 5.24 (s, 1H), 6.39 (s, 1H), 6.92 (s, 1H). MALDI/TOF-MS m/z 574.66 [M+1] (calcd. for C34H59N3O3S = 573.92).
4.7.4 L-((pyrrolidinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cycloheptylamide (7d, NSL-BA-040)
Yielded 43 mg, 37%. 1H NMR δ 1.30 – 1.84 (m, 3H), 1.85 – 2.22 (m, 12H), 2.26 (dd, J = 13.3, 6.3 Hz, 2H), 2.78 (d, J = 6.7 Hz, 4H), 3.09 (s, 2H), 3.26 – 3.17 (m, 2H), 3.85 (d, J = 24.2 Hz, 3H), 4.44 (d, J = 6.7 Hz, 1H), 5.06 – 5.13 (m, 2H), 5.23 (t, J = 7.3 Hz, 1H), 6.64 (d, J = 7.7 Hz, 1H), 7.13 (d, J = 7.0 Hz, 1H). MALDI/TOF-MS m/z 588.75 [M+1] (calcd. for C35H61N3O2S for 587.94).
4.7.5 L-((pyrrolidinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cyclooctylamide (7e, NSL-BA-055)
Yielded 45 mg, 37%. 1H NMR δ 1.24 – 1.47 (m, 2H), 1.57 (d, J = 16.7 Hz, 17H), 1.66 (d, J = 13.4 Hz, 8H), 1.80 (t, J = 20.0 Hz, 3H), 1.89 – 2.20 (m, 12H), 2.24 (q, J = 6.9 Hz, 2H), 2.80 (s, 4H), 3.07 (s, 3H), 3.22 (s, 2H), 3.81 (s, 24H), 3.95 (s, 2H), 4.43 (d, J = 6.8 Hz, 2H), 5.09 (dd, J = 6.7, 5.5 Hz, 2H), 5.24 (t, J = 7.6 Hz, 1H), 6.54 (d, J = 8.0 Hz, 1H), 7.00 (d, J = 6.9 Hz, 1H). MALDI/TOF-MS m/z 602.51 [M+1] (calcd. for C36H63N3O2S = 601.97).
4.7.6 N-((R)-1-oxo-1-(pyrrolidin-1-yl)-3-(trans, trans farnesyl)thio)propan-2-yl)-6-(pyrrolidin-1-yl)hexanamide (7f, NSL-BA-041)
Yielded 27 mg, 25%. 1H NMR δ 0.83 – 1.48 (m, 6H), 1.70 (dd, J = 37.3, 18.3 Hz, 14H), 1.81 – 2.29 (m, 16H), 2.80 (s, 4H), 3.12 (d, J = 40.5 Hz, 4H), 3.41 (d, J = 35.0 Hz, 2H), 3.54 – 3.93 (m, 4H), 4.88 (s, 1H), 5.09 (s, 2H), 5.22 (s, 1H), 6.82 (s, 1H). MALDI/TOF-MS m/z 546.34 [M+1] (calcd. for C32H55N3O2S = 545.86).
4.7.7 N-((R)-1-oxo-1-(piperidin-1-yl)-3-((trans, trans farnesyl)thio)propan-2-yl)-6-(pyrrolidin-1-yl)hexanamide (7g, NSL-BA-048)
Yielded 29 mg, 26%. 1H NMR δ 0.75 – 1.49 (m, 4H), 1.64 (d, J = 22.9 Hz, 19H), 1.88 – 2.36 (m, 14H), 2.79 (s, 3H), 3.06 (s, 7H), 3.54 (d, J = 18.5 Hz, 4H), 3.82 (s, 2H), 5.09 (s, 2H), 5.21 (s, 1H), 6.73 (s, 1H). MALDI/TOF-MS m/z 560.361 [M+1] (calcd. for C33H57N3O2S = 559.89).
4.8 General procedure for the synthesis of compounds 8a–g
Compound 6 (100 mg, 0.20 mmol) and the respective amine (0.4 mL, 3.14 – 5.80 mmol) were heated as a neat reaction at 90°C overnight. The mixture was then purified on a HPLC system to afford the respective products.
4.8.1 L-((4-methylpiperazinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cyclopropylamide (8a, NSL-BA-045)
Yielded 30 mg, 28%. 1H NMR δ 7.02 – 6.79 (m, 1H), 5.22 (s, 1H), 5.09 (s, 2H), 4.56 – 4.35 (m, 1H), 3.64 (s, 8H), 3.13 (d, J = 22.9 Hz, 4H), 2.88 (s, 3H), 2.82 – 2.64 (m, 3H), 2.25 (s, 2H), 2.17 – 1.85 (m, 7H), 1.81 – 1.12 (m, 19H), 0.76 (d, J = 6.9 Hz, 2H), 0.52 (s, 2H). MALDI/TOF-MS m/z 561.29 [M+1] (calcd. for C32H56N4O2S = 560.88).
4.8.2 L-((4-methylpiperazinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cyclopentylamide (8b, NSL-BA-049)
Yielded 32 mg, 29%. 1H NMR δ 0.76 – 0.96 (m, 2H), 1.17 – 1.35 (m, 5H), 1.43 (s, 3H), 1.67 (dd, J = 23.9, 18.5 Hz, 20H), 1.86 – 2.17 (m, 7H), 2.28 (s, 2H), 2.75 (s, 1H), 2.94 (s, 3H), 3.19 (s, 4H), 3.71 (s, 6H), 4.13 (s, 1H), 4.45 (m, 1H), 5.08 (d, J = 6.8 Hz, 2H), 5.21 (d, J = 7.1 Hz, 1H), 6.70 (d, J = 7.4 Hz, 1H), 7.20 (s, 1H). MALDI/TOF-MS m/z 589.94 [M+1] (calcd. for C32H56N4O2S = 588.93).
4.8.3 L-((4-methylpiperazinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cyclohexylamide (8c, NSL-BA-058/NSL-RD-035)
Yielded 25 mg, 21%. 1H NMR δ 1.09 – 1.52 (m, 12H), 1.67 (dd, J = 23.1, 19.4 Hz, 19H), 1.91 – 2.13 (m, 8H), 2.26 (s, 2H), 2.76 (d, J = 5.1 Hz, 1H), 2.92 (s, 2H), 3.08 – 3.26 (m, 4H), 3.68 (s, 8H), 4.44 (d, J = 6.7 Hz, 1H), 5.09 (t, J = 6.5 Hz, 2H), 5.23 (t, J = 7.5 Hz, 1H), 6.55 (d, J = 7.9 Hz, 1H), 7.05 (d, J = 6.9 Hz, 1H). MALDI/TOF-MS m/z 603.58 [M+1] (calcd. for C32H56N4O2S = 602.96).
4.8.4 L-((4-methylpiperazinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cycloheptylamide (8d, NSL-BA-4–57/NSL-RD-036)
Yielded 45 mg, 36%. 1H NMR δ 1.11 – 1.30 (m, 2H), 1.34 – 1794 (m, 29H), 1.86 – 2.15 (m, 8H), 2.28 (s, 2H), 2.75 (s, 2H), 2.94 (s, 3H), 3.18 (d, J = 10.8 Hz, 4H), 3.71 (s, 7H), 3.88 (s, 1H), 4.45 (d, J = 6.4 Hz, H), 5.09 (t, J = 6.2 Hz, 2H), 5.18 – 5.27 (m, 1H), 6.70 (d, J = 7.6 Hz, 1H), 7.20 (s, 1H). MALDI/TOF-MS m/z 617.64 [M+1] (calcd. for C32H56N4O2S = 616.98).
4.8.5 L-((4-methylpiperazinyl) hexanoyl)-S-(trans, trans-farnesyl) cysteine cyclooctylamide (8e, NSL-BA-056)
Yielded 35 mg, 29%. 1H NMR δ 0.76 – 1.02 (m, 2H), 1.12 – 1.31 (m, 2H), 1.37 – 1.83 (m, 32H), 2.02 (dd, J = 25.3, 6.5 Hz, 6H), 2.21 (d, J = 23.8 Hz, 2H), 2.83 (d, J = 39.2 Hz, 5H), 3.04 – 3.27 (m, 4H), 3.66 (s, 6H), 3.92 (s, 1H), 4.42 (s, 1H), 5.08 (d, J = 6.6 Hz, 2H), 5.24 (s, 2H), 6.55 (d, J = 7.4 Hz, 1H), 6.99 (s, 1H). MALDI/TOF-MS m/z 631.62 [M+1] (calcd. for C32H56N4O2S = 631.01).
4.8.6 N-((R)-1-oxo-1-(piperidin-1-yl)-3-((trans, trans farnesyl)thio)propan-2-yl)-6-(4-methylpiperazin-1-yl)hexanamide (8f, NSL-BA-007)
Yielded 27 mg, 25%. 1H NMR δ 1.26 (dd, J = 46.4, 31.1 Hz, 3H), 1.57 – 1.83 (m, 15H), 1.98 (ddd, J = 21.9, 13.6, 6.6 Hz, 12H), 2.25 (t, J = 6.9 Hz, 2H), 2.65 – 2.97 (m, 5H), 3.03 – 3.25 (m, 4H), 3.45 (t, J = 6.5 Hz, 2H), 3.53 – 3.81 (m, 10H), 4.87 (q, J = 7.4 Hz, 1H), 5.08 (dd, J = 9.5, 3.8 Hz, 2H), 5.21 (t, J = 7.2 Hz, 1H), 7.05 (d, J = 7.2 Hz, 1H). MALDI/TOF-MS m/z 575.43 [M+1] (calcd. for C33H58N4O2S = 574.90).
4.8.7 N-((R)-1-oxo-1-(piperidin-1-yl)-3-((trans, trans farnesyl)thio)propan-2-yl)-6-(4-methylpiperazin-1-yl)hexanamide (8g, NSL-BA-048)
Yielded 28 mg, 26%. 1H NMR δ 1.09 (dd, J = 83.9, 27.4 Hz, 4H), 1.43 (s, 5H), 1.67 (dd, J = 22.9, 18.3 Hz, 22H), 1.90 – 2.18 (m, 5H), 2.27 (s, 2H), 2.67 (s, 1H), 2.92 (s, 5H), 3.13 (s, 4H), 3.53 (s, 3H), 3.68 (s, 6H), 5.08 (s, 2H), 5.21 (s, 1H), 6.97 (s, 2H). MALDI/TOF-MS m/z 590.31 [M+1] (calcd. for C34H60N4O2S = 588.93).
4.9 Enzyme assays
PMPMEase enzyme inhibition analysis was conducted as previously described [9]. PCAIs were dissolved in water and RD-PNB substrate was dissolved in DMSO. After pre-incubation of the enzyme (1µg) and varying concentration of inhibitor (1–1000 µM) for 15 min, RD-PNB (1 mM) was added and incubated for 2 h at 37°C in 100 mM Tris-HCl, pH 7.4 in a total volume of 100 µL. The incubation was stopped by adding 200 µL methanol and stored at −20°C for 5 min followed by centrifugation at 5000xg for 5 min. The supernatants were analyzed by RP-HPLC on a Hamilton PRP-1 column (5 µm particles, 4.1 mm ID×50 mm) with UV detection at 260 nm. The mobile phase consisted of a linear gradient of 0.1% ethanolamine and increasing acetonitrile. Acetonitrile increased to 35% in 1.5 min and maintained for 1 min before 95% acetonitrile for 1 min and re-equilibrated with 0% acetonitrile for 1 min.
4.10 Statistical analysis
The data were expressed as mean ± S.E.M. Nonlinear regression analysis of the inhibitor-response curves were calculated using GraphPad Prism version 5.0 for Mac (San Diego, CA) and the concentrations that inhibit 50% of enzymatic activity (IC50) was determined. The inhibition constant (Ki) was then calculated from the previously reported kinetics analysis of RD-PNB substrate [7, 8].
4.11 Effect of PCAIs on pancreatic cancer cells
The pancreatic adenocarcinoma cell lines BxPC-3 and MIA PaCa-2 were obtained from the American Type Culture Collection (Manassas, VA). BxPC-3 and MIA PaCa-2 were routinely grown in RPMI 1640 and DMEM 1x growth medium (Invitrogen, Carlsbad, CA) respectively. These growth media were each supplemented with 10% fetal bovine serum, 100 units/mL penicillin and 100 µg/mL streptomycin (Atlanta Biological, Atlanta, GA). Cells were constantly maintained in a humidified incubator containing 5% CO2 at 37°C until about 80% confluent. Cells were trypsinized, counted and plated in 96-well plate at 1 × 104 cells/well in 100 µL experimental media containing 5% FBS. The cells were then treated with PCAIs (0 – 20 µM) for 24, 48 and 72 h. To determine cell viability, 20 µL of 0.02% resazurin reagent prepared in Dulbecco PBS was added into each well. This method is based on the fact that resazurin, a non-toxic, water soluble, redox-sensitive dye, changes from its blue/non-fluorescent state to a pink/highly-fluorescent product upon reduction by viable cells. Fluorescence was measured at an excitation wavelength of 560 nm and an emission wavelength of 590 nm using a Microplate Flourescence Reader FLx 800 after 2 h. The concentration of the inhibitor that inhibited 50% of the viability of each cell line (EC50) was determined for each inhibitor using the non-linear regression curve of concentration against response using the GraphPad Prism 5 software (San Diego, CA).
4.12 Apoptosis assay
The mode of cell death induced by the PCAIs was determined by the modified EB/AO[13] staining method [12, 24]. 5 × 104 MIA PaCa-2 cells were cultured in 8 well Lab-Tek II chamber slide and treated with 8e, (0.5, 1, and 2 µM) for 48 hours and mechanisms of cell death determined by staining cells with AO/EB (10 µg/mL) and cell images were visualized and captured by an Olympus fluorescent microscope (x40 objective) using 480/30 nm excitation filter fitted with an Olympus DP70 camera.
4.13 Docking Analysis of PCAIs
Docking analysis of the PCAIs was conducted as previously described [8, 9] with an increased potential binding area of 20×20×24 points. Poses were selected based on the interaction energy and the plausibility of the interactions between the PCAIs and the active site residues. For example, poses with the pyrrolidine and N-methylpiperazine ring inside the active site were deemed implausible because these moieties would be ionized at physiological pH and therefore be unlikely to be interacting with the hydrophobic amino acids lining the active site. On the other hand, poses with the polyisoprenyl tail outside of the active site would be thermodynamically unlikely in the aqueous environment, especially given that the ionizable polar groups would then be in the hydrophobic active site. Reported poses had (1) the farnesyl tail located in the hydrophobic pocket of the active site, (2) the charged ring structure located either outside or at the entrance of the binding site and (3) the amide linkage preferably in close proximity to the catalytic SER 221.
4.14 Pancreatic cancer tissue microarrays
The PC tissue microarray (TMA) consisted of 196 duplicate cores from 57 males and 41 females (Table 2). The 196 duplicate cores included 12 mild chronic inflammation (6 cases), 4 chronic inflammation (2 cases), 4 chronic pancreatitis (2 cases), 4 hyperplasia (2 cases), 84 duct adenocarcinoma (42 cases), 6 adenosquamous carcinoma (3 cases), 2 islet cell carcinoma (1 case), 20 islet cell tumor (10 cases), 40 cancer normal adjacent time (20 cases), and 20 normal tissue (10 cases).
4.15 Immunohistochemistry
The TMA and immunohistochemical staining were provided by US Biomax (Rockville, MD). These were processed and analyzed as previously described [25].
4.16 Immunohistochemistry scoring
The method used to score the PMPMEase immunoreactivity was adapted from that of [26]. The intensity of the staining was given scores of 0 (no staining), 1 (trace), 2 (weak), 3 (intermediate), 4 (strong) and 5 (very strong). The score of the staining intensity was then multiplied by the percentage of the immunoreactive tumor cells. The overall scores ranged between 0 and 500 with those between 1 and 100 described as trace, 101 to 200 as weak, 201 to 300 as intermediate, 301 to 400 as strong and 401 to 500 as very strong. The scoring was conducted by RD, FA, BJA and RP without prior knowledge of the diagnosis of the individual samples on the TMAs.
Supplementary Material
-
■
Design, syntheses and biological evaluation of PCAIs as potential anticancer drugs
-
■
PCAIs were more effective against pancreatic Mia PaCa-2 than BxPC-3 cells
-
■
PCAIs were over 10-fold more potent than salirasib and erlotinib against the cells
-
■
PMPMEase is overexpressed in 93% of pancreatic ductal carcinoma
-
■
PCAIs may thus be disrupting PMPMEase and/or polyisoprenylated protein function
ACKNOWLEDGMENT
The research reported in this publication was supported by the National Institute On Minority Health and Health Disparities (NIMHD) of the National Institutes of Health (NIH) under Award Number G12 MD007582 (previous award NIH/NCRR/RCMI G12 RR03020). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- EGFR
epidermal growth factor receptor
- FTS
S-t, t farnesylthiosalicylic acid (Salirasib)
- FTSA
S-t, t farnesylthiosalicylic amide
- NT
normal tissue
- NAT
normal adjacent tissue
- PC
pancreatic cancer
- PCAIs
polyisoprenylated cysteinyl amide inhibitors
- PDA
pancreatic ductal adenocarcinoma
- PMPMEase
polyisoprenylated methylated protein methyl esterase
- PMSF
phenylmethylsulfonyl fluoride
- PP
polyisoprenylation pathway
- PPMTase
polyisoprenylated protein methyl transferase
- PUFAs
polyunsaturated fatty acids
- RD-PNB
L-N-(4-nitrobenzoyl)-S-t, t-farnesyl-cysteine methyl ester
- TMA
tissue microarray
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics 2012, CA: a cancer journal for clinicians. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65:1566–1584. doi: 10.1007/s00018-008-7440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ueda S, Ogata S, Tsuda H, Kawarabayashi N, Kimura M, Sugiura Y, Tamai S, Matsubara O, Hatsuse K, Mochizuki H. The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas. 2004;29:e1–e8. doi: 10.1097/00006676-200407000-00061. [DOI] [PubMed] [Google Scholar]
- 4.Macaluso M, Russo G, Cinti C, Bazan V, Gebbia N, Russo A. Ras family genes: an interesting link between cell cycle and cancer. J Cell Physiol. 2002;192:125–130. doi: 10.1002/jcp.10109. [DOI] [PubMed] [Google Scholar]
- 5.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 6.Friday BB, Adjei AA. K-ras as a target for cancer therapy. Biochim Biophys Acta. 2005;1756:127–144. doi: 10.1016/j.bbcan.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Lamango NS, Duverna R, Zhang W, Ablordeppey SY. Porcine Liver Carboxylesterase Requires Polyisoprenylation for High Affinity Binding to Cysteinyl Substrates. Open Enzym Inhib J. 2009;2:12–27. doi: 10.2174/1874940200902010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duverna R, Ablordeppey SY, Lamango NS. Biochemical and docking analysis of substrate interactions with polyisoprenylated methylated protein methyl esterase. Curr Cancer Drug Targets. 2010;10:634–648. doi: 10.2174/156800910791859443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aguilar B, Amissah F, Duverna R, Lamango NS. Polyisoprenylation potentiates the inhibition of polyisoprenylated methylated protein methyl esterase and the cell degenerative effects of sulfonyl fluorides. Curr Cancer Drug Targets. 2011;11:752–762. doi: 10.2174/156800911796191015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amissah F, Taylor S, Duverna R, Ayuk-Takem L, Lamango NS. Regulation of polyisoprenylated methylated protein methyl esterase by polyunsaturated fatty acids and prostaglandins. Eur J Lipid Sci Technol. 2011;113:1321–1331. doi: 10.1002/ejlt.201100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayuk-Takem L, Amissah F, Aguilar BJ, Lamango NS. Inhibition of polyisoprenylated methylated protein methyl esterase by synthetic musks induces cell degeneration. Environ Toxicol. 2012 doi: 10.1002/tox.21773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribble D, Goldstein NB, Norris DA, Shellman YG. A simple technique for quantifying apoptosis in 96-well plates. BMC Biotechnol. 2005;5:12. doi: 10.1186/1472-6750-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: posttranslational modification of RAS. Nat Rev Mol Cell Biol. 2012;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldberg L, Haklai R, Bauer V, Heiss A, Kloog Y. New derivatives of farnesylthiosalicylic acid (salirasib) for cancer treatment: farnesylthiosalicylamide inhibits tumor growth in nude mice models. J Med Chem. 2009;52:197–205. doi: 10.1021/jm801165r. [DOI] [PubMed] [Google Scholar]
- 16.Elad-Sfadia G, Haklai R, Balan E, Kloog Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J Biol Chem. 2004;279:34922–34930. doi: 10.1074/jbc.M312697200. [DOI] [PubMed] [Google Scholar]
- 17.Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol. 2007;9:905–914. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- 18.Janmaat ML, Rodriguez JA, Gallegos-Ruiz M, Kruyt FA, Giaccone G. Enhanced cytotoxicity induced by gefitinib and specific inhibitors of the Ras or phosphatidyl inositol-3 kinase pathways in non-small cell lung cancer cells. Int J Cancer. 2006;118:209–214. doi: 10.1002/ijc.21290. [DOI] [PubMed] [Google Scholar]
- 19.Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, Zhang T, Marrano P, Whitehead M, Squire JA, Kamel-Reid S, Seymour L, Shepherd FA, Tsao MS. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008;26:4268–4275. doi: 10.1200/JCO.2007.14.8924. [DOI] [PubMed] [Google Scholar]
- 20.Laurent-Puig P, Taieb J. Lessons from Tarceva in pancreatic cancer: where are we now, and how should future trials be designed in pancreatic cancer? Curr Opin Oncol. 2008;20:454–458. doi: 10.1097/CCO.0b013e32830218d6. [DOI] [PubMed] [Google Scholar]
- 21.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 22.Yeh JJ, Der CJ. Targeting signal transduction in pancreatic cancer treatment. Expert Opin Ther Targets. 2007;11:673–694. doi: 10.1517/14728222.11.5.673. [DOI] [PubMed] [Google Scholar]
- 23.Cushman I, Cushman SM, Potter PM, Casey PJ. Control of RhoA methylation by carboxylesterase I. J Biol Chem. 2013;288:19177–19183. doi: 10.1074/jbc.M113.467407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amissah F, Poku RA, Aguilar BJ, Duverna R, Abonyo BO, Lamango NS. Polyisoprenylated Methylated Protein Methylesterase: The Fulcrum of Chemopreventive Effects of NSAIDs and PUFAs. In: Berhardt LV, editor. Advances in Medicine and Biology. Nova Biomedical. New York: 2013. pp. 119–144. [Google Scholar]
- 25.Amissah F, Duverna R, Aguilar BJ, Poku RA, Lamango NS. Polyisoprenylated methylated protein methyl esterase is both sensitive to curcumin and overexpressed in colorectal cancer: implications for chemoprevention and treatment. Biomed Res Int. 2013;2013:416534. doi: 10.1155/2013/416534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bremnes RM, Veve R, Gabrielson E, Hirsch FR, Baron A, Bemis L, Gemmill RM, Drabkin HA, Franklin WA. High-throughput tissue microarray analysis used to evaluate biology and prognostic significance of the E-cadherin pathway in non-small-cell lung cancer. J Clin Oncol. 2002;20:2417–2428. doi: 10.1200/JCO.2002.08.159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.