

Abstract
Neuroadaptations that participate in the ontogeny of alcohol dependence are likely a result of altered gene expression in various brain regions. The present study investigated brain region-specific changes in the pattern and magnitude of gene expression immediately following chronic intermittent ethanol (CIE) exposure and 8 hours following final ethanol exposure [i.e. early withdrawal (EWD)]. High-density oligonucleotide microarrays (Affymetrix 430A 2.0, Affymetrix, Santa Clara, CA, USA) and bioinformatics analysis were used to characterize gene expression and function in the prefrontal cortex (PFC), hippocampus (HPC) and nucleus accumbens (NAc) of C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME, USA). Gene expression levels were determined using gene chip robust multi-array average followed by statistical analysis of microarrays and validated by quantitative real-time reverse transcription polymerase chain reaction and Western blot analysis. Results indicated that immediately following CIE exposure, changes in gene expression were strikingly greater in the PFC (284 genes) compared with the HPC (16 genes) and NAc (32 genes). Bioinformatics analysis revealed that most of the transcriptionally responsive genes in the PFC were involved in Ras/MAPK signaling, notch signaling or ubiquitination. In contrast, during EWD, changes in gene expression were greatest in the HPC (139 genes) compared with the PFC (four genes) and NAc (eight genes). The most transcriptionally responsive genes in the HPC were involved in mRNA processing or actin dynamics. Of the few genes detected in the NAc, the most representatives were involved in circadian rhythms. Overall, these findings indicate that brain region-specific and time-dependent neuroadaptive alterations in gene expression play an integral role in the development of alcohol dependence and withdrawal.
Keywords: Alcohol dependence, alcohol withdrawal, brain regional adaptations, chronic intermittent alcohol exposure, gene expression profiling, microarray analysis
INTRODUCTION
Alcoholism is a chronic relapsing disease that continues to be a serious public health problem. Prolonged heavy drinking can lead to the development of dependence, which is characterized by complex and dynamic changes in brain function (for reviews, see Heilig & Koob 2007; Hansson et al. 2008; Koob and Le Moal, 2008; Vengeliene et al. 2008). The development of alcohol dependence is thought to reflect an allostatic state that is fueled by progressive dysregulation of the brain’s reward and stress systems beyond their normal homeostatic limits (Koob 2003). These neuroadaptive changes associated with dependence and withdrawal are postulated to significantly contribute to the transition from controlled alcohol use to more excessive, uncontrollable drinking.
Animal models have played a key role in advancing our knowledge of underlying neurobiological mechanisms and environmental factors associated with dependence. Nonetheless, a continued major challenge in studying ethanol dependence is to understand the numerous complex and interconnected molecular, cellular and neural circuitry adaptations that are fundamental to the addiction process. While genetic factors have long been known to influence ethanol sensitivity and response, genomic alterations in brain that are critical in driving cellular and neural network adaptations relevant to dependence remain to be elucidated (Lovinger & Crabbe 2005; Mayfield, Harris & Schuckit 2008). Microarray technologies have made it possible to monitor chronic ethanol-induced changes in the simultaneous response of thousands of genes in human alcoholics (Lewohl et al. 2000; Sokolov et al. 2003; Flatscher-Bader et al. 2005; Liu et al. 2006) and rodents (Daniels & Buck 2002; Rimondini et al. 2002; Saito et al. 2002; Repunte-Canonigo et al. 2007; Hashimoto & Wiren 2008). This approach has allowed for a relatively unbiased survey of transcriptional changes in brain that may be important in ethanol dependence.
Several studies have used gene arrays to profile differentially expressed genes in alcoholic and non-alcoholic human post-mortem brain (Mayfield et al. 2008). For example, unique alterations in gene expression were reported in prefrontal cortex (PFC) of alcoholics compared with matched controls (CTL) (Liu et al. 2006). Some of these changes (e.g. genes involved in myelination, ubiquitination, apoptosis and cell adhesion) suggest compromised neuroadaptations in the alcoholic brain. Gene expression profiling in post-mortem human alcoholic brain also has revealed transcriptional changes in other cortical regions (e.g. motor cortex) as well as subcortical regions [e.g. nucleus accumbens (NAc)]. Interestingly, relatively little overlap in alcohol-related differentially expressed genes has been observed in comparative profiling of PFC with motor cortex (Liu et al. 2004) or NAc (Flatscher-Bader et al. 2005). Collectively, these findings provide evidence for unique brain region-specific patterns of altered gene expression in human brain following a history of long-term alcohol consumption. A major issue for studies involving human postmortem analyses is the relatively small sample sizes and the accuracy of relevant clinical data, including demographic variables, drinking history, other dug use and other co-morbid conditions (Mayfield et al. 2008). Indeed, smoking and concomitant liver cirrhosis have been shown to impact gene expression profiles in alcoholics (Flatscher-Bader & Wilce 2006; Liu et al. 2007).
Studies also have employed microarray procedures to examine differential gene expression in various animal models (Mulligan et al. 2006; Hu et al. 2008; Mayfield et al. 2008; Tabakoff et al. 2008). This includes analysis of alcohol-naïve rodents with known genetically defined differences in the propensity to drink (Arlinde et al. 2004; Kerns et al. 2005; Worst et al. 2005; Kimpel et al. 2007) as well as differences in sensitivity to ethanol (Xu et al. 2002; Tabakoff, Bhave & Hoffman 2003). Alterations in gene expression have been demonstrated in several brain regions following chronic ethanol exposure in rats (Rimondini et al. 2002; Saito et al. 2002; Repunte-Canonigo et al. 2007). Likewise, significant transcriptional changes in different brain regions have been reported during acute withdrawal from chronic ethanol exposure in different inbred mouse strains (Daniels & Buck 2002) and mice selectively bred for differential sensitivity to withdrawal seizures (Hashimoto & Wiren 2008). However, no studies have directly compared gene expression profiles associated with chronic exposure to ethanol with those related to withdrawal from chronic ethanol across different brain regions in mice. Chronic intermittent ethanol (CIE) exposure mimics human patterns of ethanol consumption and it has been shown to induce long-lasting physiological and biochemical changes (Becker & Hale 1993; Rimondini et al. 2002; Veatch & Becker 2002; Hansson et al. 2008). Moreover, linking this model of dependence with self-administration procedures, we have shown significant escalation of voluntary ethanol drinking in C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME, USA) (Becker & Lopez 2004; Lopez & Becker 2005; Griffin, Lopez & Becker 2009a), suggesting that CIE may trigger neuroadaptations that contribute to the development of excessive ethanol consumption associated with dependence. Here we used oligonucleotide microarrays and bio-informatics tools to examine transcriptional events following CIE and 8 hours following final ethanol exposure [i.e. early withdrawal (EWD)] in PFC, hippocampus (HPC) and NAc, three brain regions implicated in neuroplastic events associated with various aspects of alcohol (and other drug) addiction (Koob & Volkow 2010). The overall objective was to profile time-related and brain region-specific changes in gene expression that may play a significant role in alcohol dependence.
MATERIALS AND METHODS
Subjects
Experimentally naïve adult male C57BL/6J mice (Jackson Laboratories) were use din all experiments. After a 2-week acclimation period, mice were individually housed under a 12-hour light/dark cycle in a temperature-controlled and humidity-controlled AAALAC-accredited animal facility. The animals had free access to food (Teklad rodent diet) and water throughout the experiments. All studies were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina (MUSC) and conducted in accordance with the guidelines of the NIH Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80–23, revised, 1996).
CIE exposure and EWD
Mice were separated into three groups. Two groups received CIE treatment involving four bouts of 16-hour exposure to ethanol vapor in inhalation chambers, with each bout separated by 8 hours of withdrawal. Mice were then returned to the colony room and left undisturbed for 1 week. Thereafter, mice were re-exposed to a second cycle of CIE vapor exposure (16 hours/day for 4 days). One group (CIE; n = 8) was sacrificed immediately following final ethanol exposure while the second group (EWD; n = 8) was sacrificed at peak withdrawal (8 hours following the final removal from the inhalation chambers). The third group served as CTL (n = 8/ethanol treatment) and these mice were treated identically, except that these animals were placed in control (air vapor) chambers at times corresponding to ethanol exposure for the CIE and EWD groups. This dependence model (involving repeated cycles of CIE) was employed to study dependence and withdrawal-related gene expression changes because the model has been shown to produce significant escalation of voluntary ethanol consumption that yields more than a twofold increase in resultant blood ethanol concentrations (BECs) (Becker & Lopez 2004; Lopez & Becker 2005) as well as significant elevation in brain ethanol levels (Griffin et al. 2009a).
Ethanol (or air) vapor exposure was delivered in Plexiglass inhalation chambers (60×36×60 cm) as previously described (Becker & Lopez 2004). Briefly, ethanol (95%) vapor was mixed with fresh air and delivered to the chambers at a rate of 10 l/minute, which maintained the ethanol concentration in the chamber in the range of 15–20 mg/l of air. We have demonstrated that these conditions yield stable blood ethanol levels during each bout of intoxication in C57BL/6J mice (Lopez & Becker 2005). Before entry into the ethanol inhalation chambers for each 16-hour exposure period, intoxication was initiated in CIE and EWD groups by administration of ethanol (1.6 g/kg; 8% w/v) and blood ethanol levels were stabilized by administration of the alcohol dehydrogenase inhibitor pyrazole (1 mmol/kg). The two drugs were injected intraperitoneally in a volume of 20 ml/kg body weight. Control mice received injections of saline and pyrazole. The housing conditions in the inhalation chambers were identical to that in the colony room (food and water freely available).
Ethanol samples and measurement
Chamber ethanol concentrations were monitored daily. Air samples from the ethanol and control (air) inhalation chambers (2 ml) were collected with a 5-ml syringe through a port in the chamber wall. The air samples were then transferred to Venoject tubes for later analysis using an enzymatic spectrophotometric assay procedure previously described (Becker & Hale 1993). The average chamber ethanol concentration for the present study was (mean ± SEM: 19.93 ± 0.66 mg/l air; range: 16.31– 22.81 mg/l air). Immediately following the final ethanol vapor exposure session, 40-µl blood samples were obtained from trunk for determination of BEC using a heparinized microcapillary tube. The blood samples were transferred to microcentrifuge tubes and centrifuged at 10 000 g for 10 minutes for phase separation. Five microliter aliquots of plasma were injected into an Analox Instruments alcohol analyzer (Lunenburg, MA, USA) for determination of BEC (detected by measuring oxygen uptake generated by the oxidation of ethanol into acetaldehyde and hydrogen peroxide by alcohol oxidase). In the present study, ethanol concentrations in the inhalation chambers were set to maintain blood ethanol levels in the range of 275–325 mg/dl for CIE and EWD groups during chronic ethanol exposure.
Tissue dissection and sample preparation
CIE (n = 8) and EWD (n = 8) mice along with respective CTL (n = 8/ethanol treatment) mice were removed from the inhalation chambers at the appropriate time and killed by decapitation. Mouse brains were immediately extracted, chilled on ice and sectioned (freehand) using a mouse brain mold. Coronal sections of 1-mm thickness were plated over dry ice and dissected bilaterally using a 1.25-mm micropuncher (Stoelting Co., Wooddale, IL, USA) with guidance based on established anatomical coordinates from the Franklin & Paxinos (2008) mouse brain atlas. Tissue punches of medial PFC (including pre-limbic and infralimbic subregions), HPC (CA1 and dentate gyrus regions) and NAc (shell and core) were placed into individual tubes using a spring-loaded nylon expeller with an electropolished sharp end for smooth cuts and minimal tissue adhesion. All samples were frozen immediately in dry ice and stored at 80°C until isolation of total RNA.
Total RNA was isolated according to the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). In order to isolate sufficient amounts of RNA for tissue hybridization, it was necessary to pool two subjects from each experimental condition. Pooled brain tissue samples (i.e. n = 4/brain region/group) were initially homogenized in Trizole Reagent (Invitrogen, Carlsbad, CA, USA) using a Tekmar homogenizer (Teledyne Tekmar, Mason, OH, USA). RNA quality was assured by microfluidics analysis using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). Aliquots of total RNA (5 µg) derived from each pooled sample were converted into double-stranded complimentary DNA (cDNA) using a T7-(dT) 24 primer (Genset, San Diego, CA, USA) and the Custom SuperScriptTM cDNA synthesis kit (Invitrogen). Biotin-labeled cRNA was synthesized from cDNA by in vitro transcription (Enzo BioArrayTM HighYield RNA Transcript Labeling Kit, Enzo Life Sciences, Farmingdale, NY, USA). After purification with RNeasy columns (Qiagen, Valencia, CA, USA), labeled cRNA was fragmented and then evaluated by agarose gel electrophoresis to ensure appropriate size distribution.
GeneChip hybridization
Duplicate pooled samples (n = 4/group) were hybridized to an individual microarray chip for each of the three brain regions studied. Labeled cRNA samples were analyzed on oligonucleotide microarray (GeneChip Mouse Genome 430A 2.0, Affymetrix, Santa Clara, CA, USA) that contained ~14 000 well-characterized genes and expressed sequence tags. Posthybridization washing, fluorescence staining and scanning were performed according to the standard protocols supplied by the manufacturer (Affymetrix) in the MUSC DNA Microarray and Bioinformatics Core Facility. Array quality control was assessed by accepting only arrays with a scaling factor < 2.0 and a 3′-5′ actin ratio < 4, and by examining chip validity and linearity of intensity values, according to robust multi-chip average guidelines. For the PFC, two arrays from CTL and one array from CIE mice were not included in the analysis (i.e. actin ratio > 4); whereas for the HPC, one array from CIE mice was excluded because of an actin ratio > 4. All arrays from the NAC met the recommended quality control criterion for analysis.
Microarray data analysis
Microarray data were analyzed using gene chip robust multi-array average (GCRMA), which normalizes and summarizes probe-level intensity measurements after background correction (Wu & Irizarry 2004). Expression intensity summaries produced by GCRMA were compared across treatment groups using significance analysis of microarrays (SAM) of the ‘Siggenes’ package of Bioconductor to determine significant changes in gene expression for CIE (n = 3–4/region) and EWD (n = 3–4/ region) groups compared with the CTL condition (n = 6–8/region). All SAM analyses were filtered using a false discovery rate and q-value of < 10%. This relatively stringent statistical criterion was adopted to enhance confidence in the veracity of identified changes in gene expression (i.e. reduce likelihood of false positives). For bioinformatics analyses, the expression patterns for genes that showed significant changes were further analyzed using GeneMesh, a web-based analysis tool for relating differentially expressed genes to medical subject headings (MeSH) hierarchical index (Jani et al. 2010). The expression intensity values of groups of genes that cluster in relation to a given MeSH category (i.e. biological processes) are displayed as heat maps of Z score-normalized hybridization intensity values (Jani et al. 2010). GeneMesh was developed at the MUSC and is freely available online at http://proteogenomics.musc.edu/genemesh/.
Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) assays
The remaining RNA derived from each sample isolated for microarray hybridizations was used for qRT-PCR analysis. cDNA synthesis and PCR amplification were carried out using 1 µg of total RNA by reverse transcription using an iScript One-Step RT-PCR kit (Bio-Rad, Hercules, CA, USA) according to the instructions of the manufacturer. Individual RNA samples were used to make the cDNA preparation for each treatment group (n = 4–6/group). qRT-PCR was performed using the iCycle iQ system (Bio-Rad) according to the instructions from the manufacturer for SYBR Green I-based detection (Molecular Probes, Eugene, OR, USA). The occurrence of non-specific amplification products, such as primer–dimer formation, was checked by melt-curve analysis for each pair of primers. Prior to statistical analysis, the qRT-PCR data were normalized to total mRNA levels by generating standard curves prepared from a dilution series of control-RNA templates of known concentrations. As detailed in Supporting Information Table S1, forward (F) and reverse (R) primers were designed according to UniSTS, a comprehensive database of sequenced tagged sites corresponding to GenBank accession numbers for the following genes: early growth response 1 (Egr1); brain-derived neurotrophic factor (Bdnf); opioid receptor-like 1 (Oprl1); vasoactive intestinal peptide (Vip); neuropeptide Y receptor Y1 (Npy1r); calcium channel, voltage-dependent, alpha2/delta subunit 1 (Cacna2d1); alpha thalassemia/mental retardation syndrome X-linked homolog (Atrx); and Staufen homolog 2 (Stau2). Data from qRT-PCR were analyzed using a one-way analysis of variance comparing all three treatment groups, followed by a Tukey’s B post hoc comparison.
RESULTS
Differential gene expression induced by CIE and EWD conditions
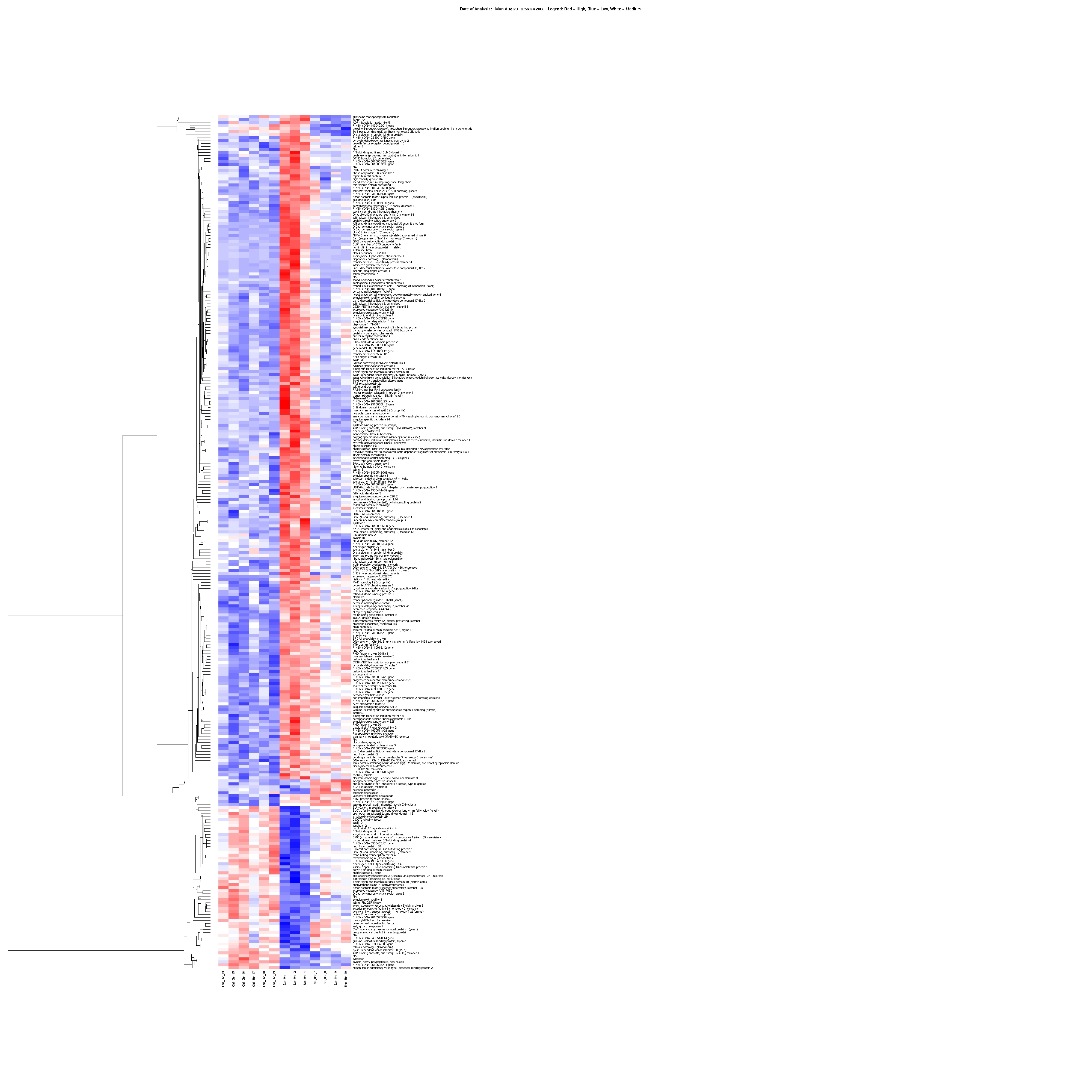
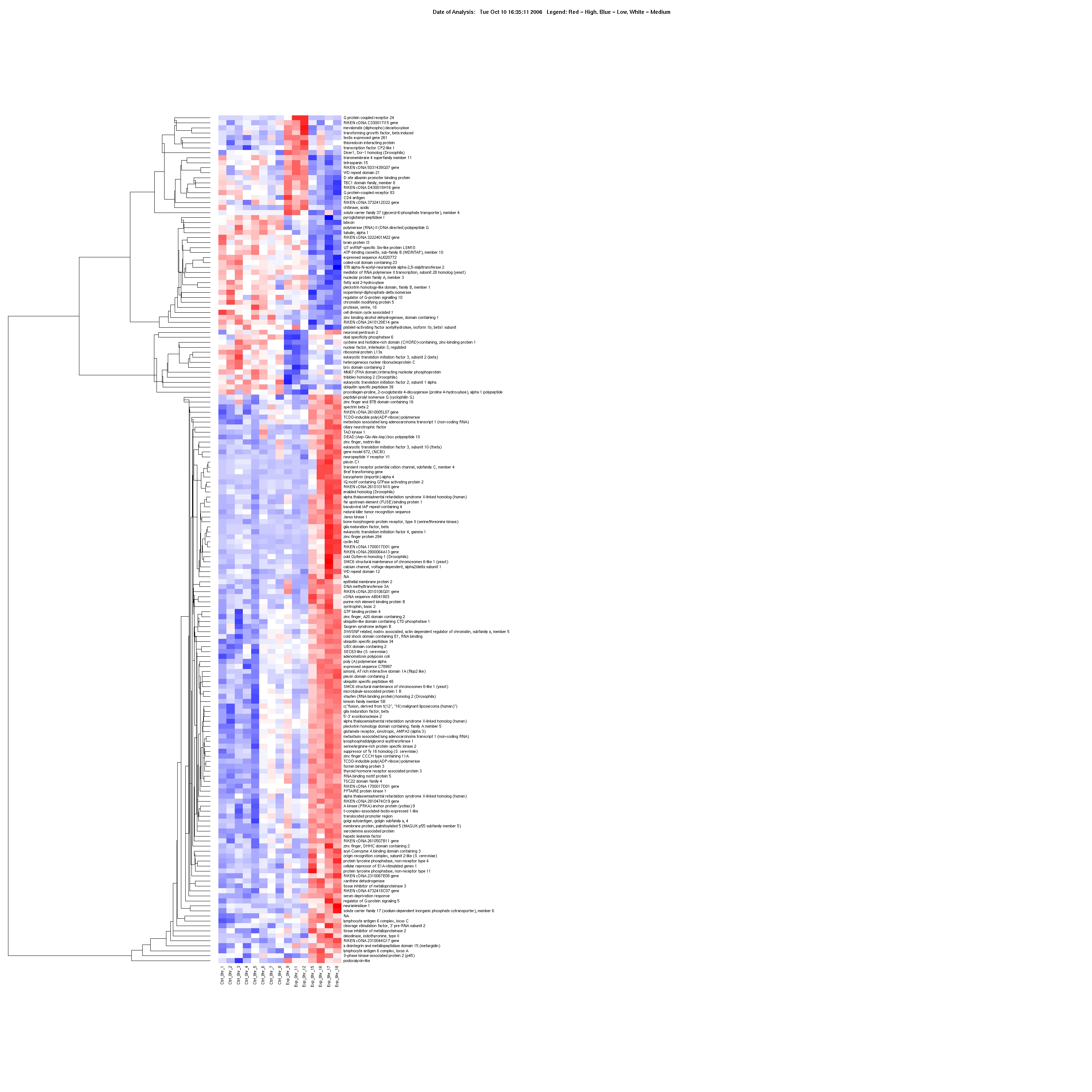
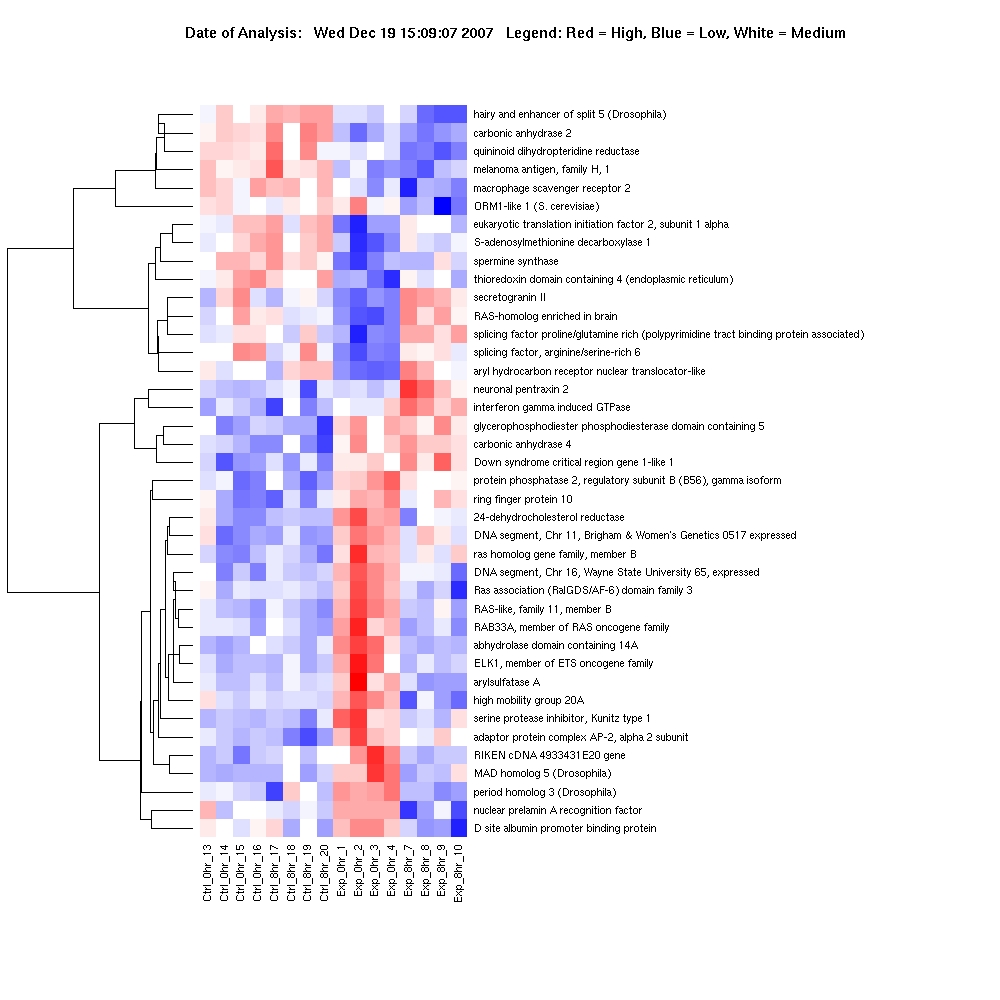
Multivariate analysis detected significant changes in expression (up-regulation or down-regulation) for a total of 288 PFC, 170 HPC and 40 NAc genes in CIE and EWD groups compared with CTL (see Supporting Information Tables S2–S4 for a list of all genes and expression values). For the CIE condition, the mean average fold change was 1.94 ± 0.06, 1.31 ± 0.03 and 1.45 ± 0.05 in the PFC, HPC and NAc, respectively. For the EWD condition, the mean average fold change was 1.24 ± 0.01, 2.80 ± 0.17 and 1.27 ± 0.05 in the PFC, HPC and NAc, respectively. Hierarchical clustering of genes specifically associated with CIE and EWD conditions versus CTL was generated in each of the brain regions examined. As shown in Fig. 1, several unique gene expression profiles were revealed as a function of brain region:
The number of significant changes in gene expression for the CIE condition (i.e. ‘CIE responsive’ denoted as gene cluster 1) was markedly greater in the PFC compared with the other brain regions, whereas the number of significant changes in gene expression for the EWD condition (i.e. ‘EWD responsive’ denoted as gene cluster 2) was greater in the HPC relative to the other brain regions examined.
Relatively few transcriptional changes related to the EWD condition were observed in the PFC, whereas relatively few transcriptional changes related to the CIE condition were observed in the HPC.
There were overall relatively few significant changes in gene expression detected in the NAc following CIE or EWD.
Of the relatively small number of genes altered in the NAc, the majority was CIE responsive.
Figure 1.

Expression profile (heat map) of the total number of genes associated with chronic intermittent ethanol (CIE) exposure and ethanol early withdrawal (EWD) in the prefrontal cortex (287 genes), hippocampus (177 genes) and nucleus accumbens (38 genes) of C57BL/6J mice. Duplicate pooled samples were hybridized to an individual microarray chip for each of the three brain regions studied. Control samples were collapsed and compared across conditions [i.e. controls (n = 6–8) versus CIE (n = 3–4) versus EWD (n = 4)] using multivariate significance analysis of microarrays with a false discovery rate of < 10%. Genes are hierarchically clustered and colored according to their expression values on a continuous scale from blue (under-expression) to red (over-expression). Red-blue midtones (e.g. white) indicate basal expression of genes compared with controls. Gene cluster 1:genes associated with CIE (i.e. EtOH responsive). Gene cluster 2:genes associated with EWD (i.e. withdrawal responsive). Over-responsive genes of functional relevance are listed on the side of each heat map and described further in Tables 1–3. The complete list of all genes and expression values for each of the brain regions is listed in Supporting Information Tables S2–S4
Brain regional differences in gene expression profiles for CIE and EWD conditions are also illustrated in Fig. 2. About 98% of genes altered in PFC were related to CIE treatment whereas over 90% of genes altered in HPC were associated with the EWD treatment condition. In NAc, a larger proportion (~80%) of the genes altered was related to the CIE condition. Thus, the preponderance of changes in gene expression associated with CIE occurred in PFC with the order of magnitude: PFC >> NAc > HPC.
Figure 2.

Number of genes identified by significance analysis of microarrays that were significantly altered by chronic intermittent ethanol (CIE) exposure and ethanol early withdrawal (EWD) in the prefrontal cortex (PFC), hippocampus (HPC) and nucleus accumbens (NAc) of C57BL/6J mice. The number of transcriptional alterations during CIE and EWD treatment was markedly greater in the PFC (i.e. 98% CIE responsive) and HPC (90% EWD responsive), respectively. There were relatively few significant changes in gene expression in the NAc following CIE or EWD
In contrast, a substantially larger proportion of changes in gene expression related to peak withdrawal (i.e. EWD responsive) occurred in HPC with the order of magnitude: HPC >> NAc > PFC. Additionally, more genes were up-regulated than down-regulated by CIE and EWD in the PFC and HPC, respectively (Fig. 2).
Validation of select CIE- and EWD-responsive genes
We employed qRT-PCR with RNA samples remaining from microarray hybridizations to confirm expression levels of select genes of interest in the PFC and HPC of CIE and EWD treatment groups, respectively. The NAc was excluded from qRT-PCR analysis because of the limited RNA samples available. A total of four genes in PFC (Egr1, Bdnf, Oprl1 and Vip) and four genes in HPC (Npy1r, Cacna2d1, Atrx and Stau2) were selected for primer design (see Supporting Information Table S1) and analysis. These genes were selected primarily because they represent a group of genes that was significantly altered by the CIE and EWD treatment in the PFC and HPC, respectively. Using normalized percent of control values, qRT-PCR analyses confirmed changes in gene expression identified by microarray analyses for most of the genes tested. Both the level and the direction of change in gene regulation were qualitatively similar in microarray and qRT-PCR analyses. In PFC (Fig. 3a), significant down-regulation of Egr1 and Bdnf and up-regulation of Oprl1 transcriptional activity were confirmed for the CIE condition. Furthermore, mRNA levels for Egr1 and Oprl1 in the EWD condition did not significantly differ from control levels as revealed by the microarray analysis. Although an increase in Vip expression was indicated by microarray analysis for both CIE and EWD conditions, qRT-PCR analysis indicated an increase in Vip mRNA only for the EWD condition. In HPC (Fig. 3b), up-regulation of ‘withdrawal-responsive’ genes revealed by microarray analysis was supported by qRT-PCR analysis for all genes examined, except Stau2 (mRNA levels of Npy1r, Cacna2ad1 and Atrx were significantly elevated compared with CTL for the EWD condition). Moreover, and consistent with the microarray results, no significant alterations were revealed for Npy1r, Cacna2d1, Atrx and Stau2 during the CIE condition.
Figure 3.

Confirmation of select genes of interest by quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) in the (a) prefrontal cortex and (b) hippocampus of chronic intermittent ethanol exposure (CIE) and ethanol early withdrawal (EWD)-treated mice. The complete primer sequences for the gene selected are listed in Supporting Information Table S1. Total RNA isolated for microarray hybridizations was used for mRNA expression by qRT-PCR. Gene and mRNA expression values were normalized as percent of controls. For most of the genes tested, changes in gene expression identified with microarray analyses for CIE and EWD conditions were confirmed by qRT-PCR analyses. Significance was determined by one-way analysis of variance (*P < 0.05; n = 4/group)
Bioinformatics analysis of CIE- and EWD-responsive genes
To identify select functional categories associated with the brain region-specific gene expression patterns presented in Fig. 1, we used a multi-layered strategy of bio-informatics analysis (Jani et al. 2010). In addition to time-dependent and brain region-specific gene expression patterns, the bioinformatics analysis revealed brain region-specific functional properties. Hence, a preferential number of CIE-responsive genes in the PFC was associated with Ras/MAPK signaling, notch signaling and ubiquitination (Table 1), whereas EWD-responsive genes in the HPC were primarily involved with mRNA processing and actin dynamics (Table 2). Interestingly, the majority of genes (albeit limited) in the NAc of CIE- and EWD-treated mice were primarily associated with circadian rhythms (Table 3).
Table 1.
Bioinformatics of ethanol-responsive genes in the prefrontal cortex of C57BL/6J mice.
| Change | Gene symbol (gene name) | Proposed function | P value |
|---|---|---|---|
| Ras/MAPK signaling pathway | |||
| ↑ | Elk1 (ELK1, member of ETS oncogene family) | Up-regulation of IEG | 1.3E-06 |
| ↑ | Ulk1 (Unc-51-like kinase 1) | Axon formation | 1.1E-04 |
| ↑ | Rhob (ras homolog gene family, member B) | Endosome transport | 1.3E-04 |
| ↑ | Hrasls (HRAS-like suppressor) | Regulation of cell growth | 2.1E-04 |
| ↑ | Erk1 (extracellular signal related protein 1) | Synaptic plasticity | 2.3E-04 |
| ↓ | Egr1 (early growth response 1) | Inhibition of gene transcription | 5.5E-03 |
| ↑ | Nras (neuroblastoma ras oncogene) | Regulation of cell cycle | 6.3E-03 |
| ↓ | Prkca (protein kinase C, alpha) | Inhibition of ERK1 | 6.3E-03 |
| ↓ | Bdnf (brain derived neurotrophic factor) | Neuron development | 6.3E-03 |
| ↑ | Rab8a (RAB8A, member of RAS oncogene family) | Protein transport | 6.4E-03 |
| ↑ | Oprl1 (Opioid receptor-like 1) | Ras/MAPK activator | 7.0E-03 |
| Notch signaling pathway | |||
| ↑ | Sel1h (suppressor of lin-12 homolog 1) | Transcription repressor | 5.5E-03 |
| ↓ | Aph1a (anterior pharynx defective 1a homolog) | Gamma-secratase complex | 5.5E-03 |
| ↑ | Adam 10 (a disitegrin and metallopeptidase 10) | Proteolytic cleavage | 5.5E-03 |
| ↑ | Adam 19 (a disitegrin and metallopeptidase 19) | Proteolytic cleavage | 6.3E-03 |
| ↓ | Dtx2 (deltex 2 homolog) | Transcriptional regulator | 7.7E-03 |
| Ubiquitylation | |||
| ↑ | Ube2l3 (ubiquitin-conjugating enzyme E2L 3) | Protein modification | 5.5E-03 |
| ↑ | Ufd1l (ubiquitin fusion degradation 1-like) | Protein catabolic process | 5.5E-03 |
| ↑ | Anapc7 (anaphase promoting complex 7) | Protein catabolic process | 6.1E-03 |
| ↑ | Ufc1 (ubiquitin-fold conjugating enzyme 1) | Protein catabolic process | 7.0E-03 |
| ↑ | Ube2i (ubiquitin-conjugating enzyme E2I) | Protein modification | 7.0E-03 |
| ↑ | Nedd4 (neuronal developmental down-regulated 4) | Protein modification | 7.4E-03 |
| ↑ | Ube2g2 (ubiquitin-conjugating enzyme E2G 2) | Protein modification | 7.4E-03 |
| ↑ | Rbx1 (ring-box 1) | DNA repair | 7.5E-03 |
P values indicate the level of significance in gene expression as determined by significance analysis of microarrays and the proposed function as determined by GeneMesh. Arrows indicate the direction (up- or down-regulation) of changes in gene expression.
Table 2.
Bioinformatics of withdrawal-responsive genes in the hippocampus of C57BL/6J mice.
| Change | Gene symbol (gene name) | Proposed function | P values |
|---|---|---|---|
| RNA processing | |||
| ↑ | Jak1 (Janus kinase 1) | RNA binding | 1.2E-05 |
| ↑ | Ptpn4 (protein tyrosine phosphatase, nonreceptor 4) | Hydrolase activity | 6.3E-05 |
| ↑ | Eif4g1 (eukaryotic translation factor 4, gamma 1) | Translation initiation factor | 1.8E-04 |
| ↑ | Papola (poly A polymerase alpha) | Transferase activity | 2.1E-04 |
| ↑ | Gm672 (gene model 672) | RNA binding | 2.3E-04 |
| ↑ | Zfml (zinc finger, matrin-like) | Nuclear mRNA splicing | 3.3E-04 |
| ↓ | Srpk2 (serine/arginine protein kinase 2) | Nuclear mRNA splicing | 6.1E-04 |
| ↑ | Cstf2 (cleavage stimulation factor, 3′ pre-RNA 2) | mRNA polyadenylation | 7.0E-04 |
| ↑ | Ssb (Sjogren syndrome antigen B) | RNA-nucleus export | 7.6E-04 |
| ↑ | Stau2 (Staufen homolog 2) | RNA binding | 7.6E-04 |
| ↑ | Rbm5 (RNA binding motif protein 5) | mRNA binding | 1.0E-03 |
| ↑ | Ddx10 (DEAD box polypeptide 10) | Helicase activity | 1.2E-03 |
| ↑ | Csde1 (cold shock domain containing E1) | RNA binding | 1.2E-03 |
| ↓ | Nola3 (nuclear protein family A, member 3) | rRNA processing | 2.8E-03 |
| Actin dynamics | |||
| ↑ | Gmfb (glia maturation factor, beta) | Actin binding | 1.8E-04 |
| ↑ | Sntb2 (syntrophin, basic 2) | Actin binding | 3.5E-04 |
| ↓ | Med28 (mediator of RNA polymerase II, subunit 28) | Actin binding | 4.7E-04 |
| ↑ | Braf (Braf transforming gene) | Actin nucleation | 4.9E-04 |
| ↑ | Kif5b (kinesin family member 5B) | Axonal transport | 8.1E-04 |
| ↑ | Enah (enabled homolog) | Actin polymerization | 1.0E-03 |
| ↑ | Spnb2 (spectrin beta 2) | Actin filament capping | 1.0E-03 |
P values indicate the level of significance in gene expression as determined by significance of analysis microarrays and the proposed function as determined by GeneMesh. Arrows indicate the direction (up- or down-regulation) of changes in gene expression.
Table 3.
Bioinformatics of ethanol and withdrawal-responsive genes in the nucleus accumbens.
| Change | Gene symbol (gene name) | Proposed function | P values |
|---|---|---|---|
| Circadian rhythms | |||
| ↓ | Car2 (Carbonic anhydrase 2) | Circadian oscillations | 2.3E-05 |
| ↓ | Arntl (Aryl receptor nuclear translocator-like) | Transcription factor | 1.7E-04 |
| ↑ | Car4 (Carbonic anhydrase 4) | Circadian oscillations | 3.5E-04 |
| ↑ | Dbp (D site albumin promoter binding protein) | Circadian oscillations | 3.7E-04 |
| ↑ | Per3 (period homolog 3) | Rhythmic behavior | 3.7E-04 |
P values indicate the level of significance in gene expression as determined by significance of analysis microarrays and the proposed function as determined by GeneMesh. Arrows indicate the direction (up- or down-regulation) of changes in gene expression.
PFC
As shown in Table 1, a number of key genes that encode proteins of the Ras/MAPK signaling pathway were altered in the PFC of CIE-treated mice. These included up-regulation of Oprl1 and down-regulation of Bdnf, which encode ligands that activate Ras/MAPK signaling. A number of members of the Ras oncogene family, which trigger the activity of Raf and in turn phosphorylate and activate MAPKs, were also shown to be CIE responsive. These included up-regulation of Rab8A, Nras, Hrasls, Rhob and Ulk1. Notably, Ulk1 (also known as Unc51.1) has been shown to govern axon formation via Ras-like GTPase signaling (Tomoda et al. 2004). A major downstream target of Ras signaling is Erk1, which is a key modulator of MAPKs and a central element for transcription and translation (Samuels, Burn & Chinnery 2009).
Erk1 was up-regulated, whereas Prkca, a negative modulator of Erk1 expression (Rucci et al. 2005), was robustly reduced in the PFC of CIE mice. A further downstream target of Erk1 is the immediate early gene Elk1, which was up-regulated in the PFC of CIE-treated mice. Elk1 forms a serum response element/binding complex with the promoter of its target genes, which include c-fos and c-jun. In addition, Egr1, a well-established immediate early gene modulated by Ras/MAPK signaling, was down-regulated in CIE mice. Egr1 is known to play a role in various forms of neuroplasticity and pathology in response to numerous environmental stimuli (Hughes & Dragunow 1995; Davis, Bozon & Laroche 2003).
The notch signaling pathway is involved in various neuronal functions including the control of neurogenesis, cell differentiation and proliferation (for review, see Bray 2006). Briefly, ligand binding promotes two proteolytic cleavage events in the notch receptor. The first cleavage is catalyzed by the ADAM-family metalloproteases, including Adam 10 and Adam 19, which were both up-regulated in the PFC of CIE-treated mice (Table 1). The second cleavage is mediated by gamma secretase, an enzyme complex that contains presenilin, nicastrin and anterior pharynx defective (Aph1), which releases the notch intracellular domain to promote transcription. The stability of the gamma-secretase enzyme complex involves Aph1a (Lee et al. 2004), which was down-regulated in the PFC of CIE mice. Additional CIE-responsive genes included the up-regulation of Sel1h, a corepressor of notch signaling, and down-regulation of Dtx2, a transcriptional regulator downstream of the notch receptor. Future studies will need to examine whether changes in adult neurogenesis following chronic ethanol exposure (Nixon, 2006; Crews & Nixon 2009) are mediated by alterations in notch signaling cascades (Imayoshi et al. 2010).
The ubiquitin system is a network of highly conserved regulatory proteins dedicated to the ubiquitination (e.g. E1-E3 enzyme cascade) of cellular targets and the subsequent control of numerous cellular functions (for review, see Hoeller & Dikic 2009). Although ubiquitin is the most well understood post-translational modifying enzyme, there is a growing family of ubiquitin-like proteins that modify cellular targets. Bioinformatics analysis revealed that various E2-like ubiquitin conjugating enzymes were up-regulated in the PFC of CIE-treated mice, including Ube2i, Ube2g2, Ube2l3 and Ufc1 (Table 1). The E3-like ubiquitin ligases Nedd4 and Ufd1l, which transfer the ubiquitinated proteins to the proteasome for degradation (Cao et al. 2007), were also up-regulated in the PFC. Intriguingly,Nedd4 belongs to the Nedd family of proteins that control the assembly of cullin-based ubiquitin E3 ligases (a process known as ‘neddylation’). Rbx1, a potent activator of the neddylation process (Kamura et al. 1999), was also up-regulated in the PFC of CIE-treated mice.
HPC
Protein synthesis involves a complex series of steps, including mechanisms involving the recruitment of ribosomal subunits to mRNA (i.e. mRNA binding) and the transport of mRNA from the nucleus to the cytoplasm (i.e. mRNA transport). In neurons, specialized mRNAs are transported from soma to dendrites where local translation occurs in response to external stimuli (for review, see Job & Eberwine 2001). As shown in Table 2, the analysis of the HPC revealed a number of changes in the expression of genes involved in the regulation of transcription, including mRNA binding (Rbm5, Ssb, Eif4g1, Zfml, Csde1, Cstf2 and Gm672) and mRNA transport (Papola and Stau2). Notably, Eif4g is crucial for the recruitment of mRNA binding to ribosomes and stabilizing the cap-binding process in preparation for translation (for review, see Prevot, Darlix & Ohlmann 2003). Our analysis revealed an up-regulation of Eif4g expression in the HPC that is indicative of enhanced mRNA processing during ethanol withdrawal. Furthermore, Zfml and Csde1, which were also up-regulated in EWD mice, have been shown to play an important role in regulating the stability of mRNA by protecting cap-binding processes (Dormoy-Raclet et al. 2005). With regard to mRNA transport, a robust up-regulation of Papola and Stau2 was observed in the HPC of EWD mice. These transporters have been shown to bind mRNA in the soma and shuttle it to dendritic spines where certain forms of synaptic plasticity may occur (for review, see Miki, Takano & Yoneda 2005).
An over-representation of genes associated with actin dynamics was also revealed in the HPC of EWD mice (Table 2). Regulation of the actin cytoskeleton is crucial for synaptic plasticity, especially structural remodeling of dendritic spines after chronic drug self-administration (Russo et al. 2009). A robust up-regulation of Apc was observed in EWD mice. Apc is a key target of Wnt signaling and essential for axon elongation, pausing and remodeling of actin cytoskeleton (Purro et al. 2008). Notably, many of the signals that control dendritic morphology also do so via the Rho GTPase family of proteins that link extracellular signals to control actin cytoskeleton (Nakayama & Luo 2000). Braf, a potent regulator of Rho GTPases and neurite outgrowth, was up-regulated in EWD mice. Furthermore, an increase in gene expression was also observed in Kifb5, a motor adaptor protein essential for axon delivery and presynaptic assembly during hippocampal synaptic plasticity (Cai, Pan & Sheng 2007). The up-regulation of the spectrin Spnb2 was also revealed in the HPC of EWD mice. Spectrins play a pivotal role in axonal growth, neurite extension and the organization of synaptic vesicles (Tang et al. 2003). The up-regulation of Sntb2, also revealed in EWD mice, has been shown to link voltage-gated sodium channels to the actin cytoskeleton and the extracellular matrix (Gee et al. 1998). Finally, the actin regulatory protein Enah, which was up-regulated in EWD mice, has been shown to regulate the activity of F-actin and is required for filopodia formation in various cell types including neuronal growth cones (Krause et al. 2003).
NAc
Genetic disruptions in normal circadian gene functions have recently been linked to a variety of psychiatric conditions including alcohol addiction (Falcon & McClung 2009). As shown in Table 3, bioinformatics analysis revealed an over-representation of NAc genes that are crucial to most, if not all, circadian clocks, including Arntl (also known as BMAL1 and MOP3), Dbp, Per3 and Car2. Per3, which was up-regulated in CIE mice, belongs to the family of Period (Per) circadian genes and is involved in rhythmically counterbalancing the activity of Arntl (Ripperger & Schibler 2006). Arntl, which was down-regulated in CIE mice, is a transcription factor necessary to govern time of day-dependent gene expression (Ripperger & Schibler 2006). The transcription factor Dbp, which was up-regulated in EWD mice, shows high-amplitude circadian oscillations and mediates binding of the Arntl-Clock protein complex (Ripperger & Schibler 2006). Interestingly, down-regulation of Car2 was also observed in the NAc of EWD mice. Studies show that Car2-deficient mice have long circadian periods, which are restored to normal circadian function with a single copy of Car2 (Kernek et al. 2006).
DISCUSSION
Through the use of high-density oligonucleotide microarrays, the present study has identified brain region-specific patterns of gene expression associated with CIE and EWD conditions in the PFC, HPC and NAc of C57BL/6J mice. The preponderance of changes in gene expression associated with CIE occurred in PFC whereas the majority of changes in gene expression related to peak withdrawal (EWD condition) occurred in HPC. Compared with the PFC and HPC, relatively few genes were significantly altered in the NAc. These expression patterns were then linked to bioinformatics analysis, which further revealed discrete region-specific functions associated with CIE and EWD treatment. An over-representation of CIE-responsive genes in the PFC primarily belonged to the Ras/MAPK signaling pathway, whereas an over-representation of EWD-responsive genes in the HPC was involved in mRNA processing and actin dynamics. Interestingly, of the limited genes altered in NAc, a majority of them were involved in circadian function. Although additional work is needed to definitively link these genes with behavioral responses to ethanol, the identified expression patterns clearly contribute novel insight into region-specific and time-dependent brain mechanisms underlying the development of alcohol dependence.
Recruitment of the Ras/MAPK signaling cascade has long been implicated as a convergence point for numerous signaling inputs that control various forms of synaptic plasticity (for review, see Thomas & Huganir 2004). Additionally, a meta-analysis of data generated from several microarray studies indicated activation of numerous H-Ras transcripts in association with genetic predisposition for high ethanol consumption (Mulligan et al. 2006). In this study, we found that many of the CIE-responsive genes in the PFC belong to the Ras/MAPK signaling pathway including Egr1. Although a number of studies have implicated Egr1 as an important contributor to ethanol-induced liver injury (Pritchard & Nagy 2005), its precise role in the central nervous system during the development of ethanol dependence remains unknown. One study showed that Egr1-DNA binding activity was markedly reduced in cortical-nuclear and hippocampal-nuclear extracts from chronic ethanol-treated rats (Depaz, Goodenough & Wilce 2000). More recently and relevant to the present study, using microarrays, Repunte-Canonigo et al. (2007) reported a significant decrease in Egr1 gene expression in the PFC of CIE-treated rats. Further, recent evidence indicates that many Egr1-target genes are down-regulated following Egr1 induction, suggesting that Egr1 suppresses gene expression (James, Conway & Morris 2005). Consistent with this possibility, our microarray analysis revealed that the majority of Egr1-target genes (e.g. Prka, Ccdc5 and Prepl; see Supporting Information Table S2) were up-regulated in the PFC of CIE-treated mice, which further suggests that reduced expression of Egr1 may lead to the up-regulation of its target genes. Thus, the possibility exists that the overwhelming increase in the number of CIE-responsive genes in the PFC may be due, in part, to down-regulated Egr1.
Other Ras/MAPK-related genes altered in the PFC by CIE included Bdnf, ErK1 and Oprl1. Although a growing number of studies show that acute ethanol exposure elevates the expression of Bdnf in various brain regions (McGough et al. 2004; Kerns et al. 2005; Pandey et al. 2006), our study is the first to reveal that CIE exposure decreases the expression of Bdnf in PFC. Consistent with our findings, Logrip, Janak & Ron (2009) recently reported that the expression of Bdnf is significantly reduced in the PFC following continuous chronic ethanol exposure, but not following acute exposure to ethanol. Together, these findings are in keeping with the hypothesis that there may be a shift in Bdnf homeostasis following acute versus chronic ethanol exposure. Additional CIE-responsive genes in the PFC included up-regulation of Oprl1 and Erk1. Interestingly, the endogenous ligand of OPRL1, nociceptin, plays a role in the rewarding properties of various drugs of abuse, including ethanol (Economidou et al. 2008). Also, Oprl1 activates Erk1 via G-coupled opioid transmission, which may be a novel mechanism by which CIE conveys its synaptic signal to alter transcription of specific genes in the PFC.
A distinct gene expression profile emerged in the analysis of the HPC. The preponderance of transcriptional changes was identified at the 8-hour time point, which corresponds to peak withdrawal. The HPC has long been recognized as being highly sensitive to ethanol, and compromised structural and functional integrity of the HPC following chronic ethanol exposure has been extensively documented (Sullivan & Pfefferbaum 2005; Oscar-Berman & Marinkovic 2007). Nonetheless, there are limited studies defining gene expression profiles associated with ethanol withdrawal in the HPC of C57BL/6J mice.To date, the one study available shows that a significant number of withdrawal-responsive genes in the HPC are found to be associated with Ras/MAPK signaling (Daniels & Buck 2002). In the present study, only a small proportion of genes in the HPC of EWD mice belonged to Ras/MAPK signaling. The lack of congruity between these findings is probably the result of differences in the experimental paradigm and methodology (i.e. choice of microarray platform and continuous versus intermittent ethanol exposure). Nonetheless, the studies show that EWD (i.e. 7–8 hours post-ethanol exposure) significantly alters gene expression in the HPC. In our hands, bioinformatics analysis revealed that a significant number of EWD-responsive genes in the HPC were associated with mRNA processing and actin dynamics. Up-regulation of a number of mRNA-binding (e.g. Eif4g, Zfml and Csde1) and mRNA-transport (e.g. Papola and Stau2) genes detected in the HPC of EWD mice is of interest because of their noted roles in regulating mRNA processing and transporting that are integral to various forms of synaptic plasticity (Job & Eberwine 2001). An over-representation of genes associated with actin dynamics was also revealed in the HPC of EWD mice. Active mobilization and regulation of the actin cytoskeleton are crucial for synaptic plasticity, especially structural remodeling of dendritic spines after chronic ethanol and other drug exposure (e.g. Carpenter-Hyland & Chandler 2006; Zhou et al. 2007; Pandey et al. 2008; Russo et al. 2009).
While a relatively limited number of genes were found to be significantly altered in the NAc during CIE or EWD, the majority of those altered were associated with circadian rhythms. Genetic disruptions in normal circadian gene functions have recently been linked to a variety of psychiatric conditions including depression, bipolar disorders and alcohol use disorders (Falcon & McClung 2009). Studies indicate that the rhythm-generating molecular circuitry involved in circadian cycles is thought to rely on transcriptional activators and repressors that generate a negative feedback loop (Ripperger & Schibler 2006). These oscillating-type genes include Arntl (also known as Bmal1) and the Period (Per) genes, which are a family of genes thought to regulate several brain functions. In the NAc of CIE mice, we show an increase in Per3 and a decrease in Arntl. Interestingly, Spanagel et al. (2005) reported that Per2 mutant mice show reduced glutamate uptake and increased extracellular glutamate levels in the NAc, glutamatergic adaptations that we and others have shown to be associated with increased ethanol intake (e.g. Kapasova & Szumlinski 2008; Griffin et al. 2009b). Thus, a potential link may exist between glutamate homeostasis, excessive ethanol consumption and dysfunction of the circadian clock Per genes.
Considering the involvement of the NAc in various aspects of drug and alcohol addiction (Koob 2003), it was surprising to find relatively few gene alterations in the NAc of CIE or EWD mice. Similarly, Repunte-Canonigo et al. (2007) found the NAc to be markedly less responsive to gene alterations induced by chronic ethanol exposure compared with the PFC. However, gene expression changes were shown to be greater in the NAc compared with the PFC following acute ethanol challenge (Kerns et al. 2005). Interestingly, regions strongly reactive to acute ethanol exposure, including the NAc and lateral septum, showed no cFos induction following chronic ethanol exposure (Ryabinin et al. 1997). These observations have led to the notion that chronic ethanol exposure results in ‘desensitization’ or general tolerance to gene expression alterations (Vilpoux et al. 2009). It is also worth noting that the development of ethanol dependence most likely involves a dynamic shift in ethanol-responsive neuronal networks and brain regions over the course of chronic ethanol exposure (Kalivas & O’Brien 2008). Furthermore, the fact that CIE-responsive genes were predominantly evident in the PFC, whereas EWD-responsive genes were primarily in the HPC, makes it tempting to suggest that the development of ethanol dependence results in unique time-dependent sensitization of PFC and HPC gene expression and tolerance of NAc gene expression alterations. It should be noted that this dynamic profile of gene expression changes was produced following passive exposure to ethanol (via vapor inhalation). It is unclear at present whether self-administered ethanol over a prolonged period of time would produce alterations in gene expression distinct from those reported in this study. Overall, a better understanding of the neuronal circuits that are affected by chronic ethanol exposure and their adaptations during the development of ethanol dependence will provide new opportunities for developing appropriate therapies.
In conclusion, this study provides a number of candidate genes for future hypothesis-driven studies that can be characterized for their role within a single pathway or within a functional network as it pertains to the development of ethanol dependence. The study also shows that the neuronal adaptations associated with CIE and EWD engage different brain regions and functions in a time-dependent manner. These regional and functional molecular changes may play a critical role in mediating the transition from moderate controlled drinking to excessive drinking associated with ethanol dependence.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This study was supported by grants R01 AA013885 (H.C.B.), P50 AA010761 (H.C.B.) and P50 DA015369 (P.W.K.). The authors thank Rolf Fritz and Kay Fernandez for excellent technical assistance and Saurin Jani for assistance with bioinformatics analysis.
Footnotes
Conflict of Interest
The authors declare no conflict of interest in connection with this report.
Authors Contribution
HCB provided the overall coordination of the project. HCB, JFM and PWK were responsible for the study concept and design. RIM and HCB contributed to the acquisition of animal data. RIM, JFM, PWK and HCB assisted with data analyses and interpretation of findings. RIM and HCB drafted the manuscript and JFM and PWK provided critical intellectual input and content in preparation of the manuscript. All authors critically reviewed content and approved final version for publication.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
Table S1 Oligonucleotide primers used for qRT-PCR assays
Table S2 Gene expression alterations in the prefrontal cortex of C57BL/6J mice
Table S3 Gene expression alterations in the hippocampus of C57BL/6J mice
Table S4 Gene expression alterations in the nucleus accumbens of C57BL/6J mice
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Arlinde C, Sommer W, Bjork K, Reimers M, Hyytia P, Kiianmaa K, Heilig M. A cluster of differentially expressed signal transduction genes identified by microarray analysis in a rat genetic model of alcoholism. Pharmacogenomics J. 2004;4:208–218. doi: 10.1038/sj.tpj.6500243. [DOI] [PubMed] [Google Scholar]
- Becker HC, Hale RL. Repeated episodes of ethanol withdrawal potentiate the severity of subsequent withdrawal seizures: an animal model of alcohol withdrawal ‘kindling’. Alcohol Clin Exp Res. 1993;17:94–98. doi: 10.1111/j.1530-0277.1993.tb00731.x. [DOI] [PubMed] [Google Scholar]
- Becker HC, Lopez MF. Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res. 2004;28:1829–1838. doi: 10.1097/01.alc.0000149977.95306.3a. [DOI] [PubMed] [Google Scholar]
- Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- Cai Q, Pan PY, Sheng ZH. Syntabulin-kinesin-1 family member 5B–mediated axonal transport contributes to activity-dependent presynaptic assembly. J Neurosci. 2007;27:7284–7296. doi: 10.1523/JNEUROSCI.0731-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Wang J, Qi W, Miao HH, Wang J, Ge L, DeBose-Boyd RA, Tang JJ, Li BL, Song BL. Ufd1 is a cofactor of gp78 and plays a key role in cholesterol metabolism by regulating the stability of HMG-CoA reductase. Cell Metab. 2007;6:115–128. doi: 10.1016/j.cmet.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Carpenter-Hyland EP, Chandler LJ. Homeostatic plasticity during alcohol exposure promotes enlargement of dendritic spines. Eur J Neurosci. 2006;24:3496–3506. doi: 10.1111/j.1460-9568.2006.05247.x. [DOI] [PubMed] [Google Scholar]
- Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 2009;44:115–127. doi: 10.1093/alcalc/agn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GM, Buck KJ. Expression profiling identifies strain-specific changes associated with ethanol withdrawal in mice. Genes Brain Behav. 2002;1:35–45. doi: 10.1046/j.1601-1848.2001.00008.x. [DOI] [PubMed] [Google Scholar]
- Davis S, Bozon B, Laroche S. How necessary is the activation of the immediate early gene zif268 in synaptic plasticity and learning? Behav Brain Res. 2003;142:17–30. doi: 10.1016/s0166-4328(02)00421-7. [DOI] [PubMed] [Google Scholar]
- Depaz IM, Goodenough S, Wilce PA. Chronic ethanol has region-selective effects on Egr-1 and Egr-3 DNA-binding activity and protein expression in the rat brain. Neurochem Int. 2000;37:473–482. doi: 10.1016/s0197-0186(00)00060-7. [DOI] [PubMed] [Google Scholar]
- Dormoy-Raclet V, Markovits J, Jacquemin-Sablon A, Jacquemin-Sablon H. Regulation of Unr expression by 5′- and 3′-untranslated regions of its mRNA through modulation of stability and IRES mediated translation. RNA Biol. 2005;2:e27–e35. [PubMed] [Google Scholar]
- Economidou D, Hansson AC, Weiss F, Terasmaa A, Sommer WH, Cippitelli A, Fedeli A, Martin-Fardon R, Massi M, Ciccocioppo R, Heilig M. Dysregulation of nociceptin/orphanin FQ activity in the amygdala is linked to excessive alcohol drinking in the rat. Biol Psychiatry. 2008;64:211–218. doi: 10.1016/j.biopsych.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon E, McClung CA. A role for the circadian genes in drug addiction. Neuropharmacology. 2009;56(Suppl. 1):91–96. doi: 10.1016/j.neuropharm.2008.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatscher-Bader T, van der Brug M, Hwang JW, Gochee PA, Matsumoto I, Niwa S, Wilce PA. Alcohol-responsive genes in the frontal cortex and nucleus accumbens of human alcoholics. J Neurochem. 2005;93:359–370. doi: 10.1111/j.1471-4159.2004.03021.x. [DOI] [PubMed] [Google Scholar]
- Flatscher-Bader T, Wilce PA. Chronic smoking and alcoholism change expression of selective genes in the human prefrontal cortex. Alcohol Clin Exp Res. 2006;30:908–915. doi: 10.1111/j.1530-0277.2006.00106.x. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Amsterdam: Elsevier; 2008. [Google Scholar]
- Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998;18:128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Lopez MF, Becker HC. Intensity and duration of chronic ethanol exposure is critical for subsequent escalation of voluntary ethanol drinking in mice. Alcohol Clin Exp Res. 2009a;33:1893–1900. doi: 10.1111/j.1530-0277.2009.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Mulholland PJ, Deegan DL, Sanchez JT, Randall JS, Chandler LJ, Becker HC. Effects of chronic intermittent ethanol exposure on extracellular glutamate levels in the accumbens of C57BL/6J mice. Alcohol Clin Exp Res. 2009b;33:82. [Google Scholar]
- Hansson AC, Rimondini R, Neznanova O, Sommer WH, Heilig M. Neuroplasticity in brain reward circuitry following a history of ethanol dependence. Eur J Neurosci. 2008;27:1912–1922. doi: 10.1111/j.1460-9568.2008.06159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto JG, Wiren KM. Neurotoxic consequences of chronic alcohol withdrawal: expression profiling reveals importance of gender over withdrawal severity. Neuropsychopharmacology. 2008;33:1084–1096. doi: 10.1038/sj.npp.1301494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- Hu W, Saba L, Kechris K, Bhave SV, Hoffman PL, Tabakoff B. Genomic insights into acute alcohol tolerance. J Pharmacol Exp Ther. 2008;326:792–800. doi: 10.1124/jpet.108.137521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes P, Dragunow M. Induction of immediate-early genes and control of neurotransmitter-regulated gene expression within the nervous system. Pharmacol Rev. 1995;47:133–178. [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Yamaguchi M, Mori K, Kageyama R. Essential roles of notch signaling in maintenance of neural stem cells in developing and adult brains. J Neurosci. 2010;30:3489–3498. doi: 10.1523/JNEUROSCI.4987-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AB, Conway AM, Morris BJ. Genomic profiling of the neuronal target genes of the plasticity-related transcription factor—Zif268. J Neurochem. 2005;95:796–810. doi: 10.1111/j.1471-4159.2005.03400.x. [DOI] [PubMed] [Google Scholar]
- Jani SD, Argraves GL, Barth JL, Argraves WS. GeneMesh: a web-based microarray analysis tool for relating differentially expressed genes to MeSH terms. BMC Bioinformatics. 2010;11:166. doi: 10.1186/1471-2105-11-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Job C, Eberwine J. Localization and translation of mRNA in dendrites and axons. Nat Rev Neurosci. 2001;2:889–898. doi: 10.1038/35104069. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, O’Brien C. Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology. 2008;33:166–180. doi: 10.1038/sj.npp.1301564. [DOI] [PubMed] [Google Scholar]
- Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Jr, Elledge SJ, Conaway RC, Harper JW, Conaway JW. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- Kapasova Z, Szumlinski KK. Strain differences in alcohol-induced neurochemical plasticity: a role for accumbens glutamate in alcohol intake. Alcohol Clin Exp Res. 2008;32:617–631. doi: 10.1111/j.1530-0277.2008.00620.x. [DOI] [PubMed] [Google Scholar]
- Kernek KL, Trofatter JA, Mayeda AR, Lahiri DK, Hofstetter JR. A single copy of carbonic anhydrase 2 restores wild-type circadian period to carbonic anhydrase II-deficient mice. Behav Genet. 2006;36:301–308. doi: 10.1007/s10519-005-9032-9. [DOI] [PubMed] [Google Scholar]
- Kerns RT, Ravindranathan A, Hassan S, Cage MP, York T, Sikela JM, Williams RW, Miles MF. Ethanol-responsive brain region expression networks: implications for behavioral responses to acute ethanol in DBA/2J versus C57BL/6J mice. J Neurosci. 2005;25:2255–2266. doi: 10.1523/JNEUROSCI.4372-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpel MW, Strother WN, McClintick JN, Carr LG, Liang T, Edenberg HJ, McBride WJ. Functional gene expression differences between inbred alcohol-preferring and -non-preferring rats in five brain regions. Alcohol. 2007;41:95–132. doi: 10.1016/j.alcohol.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Alcoholism: allostasis and beyond. Alcohol Clin Exp Res. 2003;27:232–243. doi: 10.1097/01.ALC.0000057122.36127.C2. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- Lee SF, Shah S, Yu C, Wigley WC, Li H, Lim M, Pedersen K, Han W, Thomas P, Lundkvist J, Hao YH, Yu G. A conserved GXXXG motif in APH-1 is critical for assembly and activity of the gamma-secretase complex. J Biol Chem. 2004;279:4144–4152. doi: 10.1074/jbc.M309745200. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, Wang L, Miles MF, Zhang L, Dodd PR, Harris RA. Gene expression in human alcoholism: microarray analysis of frontal cortex. Alcohol Clin Exp Res. 2000;24:1873–1882. [PubMed] [Google Scholar]
- Liu J, Lewohl JM, Dodd PR, Randall PK, Harris RA, Mayfield RD. Gene expression profiling of individual cases reveals consistent transcriptional changes in alcoholic human brain. J Neurochem. 2004;90:1050–1058. doi: 10.1111/j.1471-4159.2004.02570.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Lewohl JM, Harris RA, Dodd PR, Mayfield RD. Altered gene expression profiles in the frontal cortex of cirrhotic alcoholics. Alcohol Clin Exp Res. 2007;31:1460–1466. doi: 10.1111/j.1530-0277.2007.00444.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Lewohl JM, Harris RA, Iyer VR, Dodd PR, Randall PK, Mayfield RD. Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology. 2006;31:1574–1582. doi: 10.1038/sj.npp.1300947. [DOI] [PubMed] [Google Scholar]
- Logrip ML, Janak PH, Ron D. Escalating ethanol intake is associated with altered corticostriatal BDNF expression. J Neurochem. 2009;109:1459–1468. doi: 10.1111/j.1471-4159.2009.06073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez MF, Becker HC. Effect of pattern and number of chronic ethanol exposures on subsequent voluntary ethanol intake in C57BL/6J mice. Psychopharmacology (Berl) 2005;181:688–696. doi: 10.1007/s00213-005-0026-3. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, Crabbe JC. Laboratory models of alcoholism: treatment target identification and insight into mechanisms. Nat Neurosci. 2005;8:1471–1480. doi: 10.1038/nn1581. [DOI] [PubMed] [Google Scholar]
- McGough NN, He DY, Logrip ML, Jeanblanc J, Phamluong K, Luong K, Kharazia V, Janak PH, Ron D. RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci. 2004;24:10542–10552. doi: 10.1523/JNEUROSCI.3714-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield RD, Harris RA, Schuckit MA. Genetic factors influencing alcohol dependence. Br J Pharmacol. 2008;154:275–287. doi: 10.1038/bjp.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Takano K, Yoneda Y. The role of mammalian Staufen on mRNA traffic: a view from its nucleocytoplasmic shuttling function. Cell Struct Funct. 2005;30:51–56. doi: 10.1247/csf.30.51. [DOI] [PubMed] [Google Scholar]
- Mulligan MK, Ponomarev I, Hitzemann RJ, Belknap JK, Tabakoff B, Harris RA, Crabbe JC, Blednov YA, Grahame NJ, Phillips TJ, Finn DA, Hoffman PL, Iyer VR, Koob GF, Bergeson SE. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci USA. 2006;103:6368–6373. doi: 10.1073/pnas.0510188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama AY, Luo L. Intracellular signaling pathways that regulate dendritic spine morphogenesis. Hippocampus. 2000;10:582–586. doi: 10.1002/1098-1063(2000)10:5<582::AID-HIPO8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Nixon K. Alcohol and adult neurogenesis: roles in neurodegeneration and recovery in chronic alcoholism. Hippocampus. 2006;16:287–295. doi: 10.1002/hipo.20162. [DOI] [PubMed] [Google Scholar]
- Oscar-Berman M, Marinkovic K. Alcohol: effects on neurobehavioral functions and the brain. Neuropsychol Rev. 2007;17:239–257. doi: 10.1007/s11065-007-9038-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Misra K. Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J Neurosci. 2006;26:8320–8331. doi: 10.1523/JNEUROSCI.4988-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Ugale R, Prakash A, Xu T, Misra K. Effector immediate-early gene arc in the amygdala plays a critical role in alcoholism. J Neurosci. 2008;28:2589–2600. doi: 10.1523/JNEUROSCI.4752-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevot D, Darlix JL, Ohlmann T. Conducting the initiation of protein synthesis: the role of eIF4G. Biol Cell. 2003;95:141–156. doi: 10.1016/s0248-4900(03)00031-5. [DOI] [PubMed] [Google Scholar]
- Pritchard MT, Nagy LE. Ethanol-induced liver injury: potential roles for egr-1. Alcohol Clin Exp Res. 2005;29:146S–150S. doi: 10.1097/01.alc.0000189286.81943.51. [DOI] [PubMed] [Google Scholar]
- Purro SA, Ciani L, Hoyos-Flight M, Stamatakou E, Siomou E, Salinas PC. Wnt regulates axon behavior through changes in microtubule growth directionality: a new role for adenomatous polyposis coli. J Neurosci. 2008;28:8644–8654. doi: 10.1523/JNEUROSCI.2320-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repunte-Canonigo V, Lutjens R, van der Stap LD, Sanna PP. Increased expression of protein kinase A inhibitor alpha (PKI-alpha) and decreased PKA-regulated genes in chronic intermittent alcohol exposure. Brain Res. 2007;1138:48–56. doi: 10.1016/j.brainres.2006.09.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimondini R, Arlinde C, Sommer W, Heilig M. Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB J. 2002;16:27–35. doi: 10.1096/fj.01-0593com. [DOI] [PubMed] [Google Scholar]
- Ripperger JA, Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat Genet. 2006;38:369–374. doi: 10.1038/ng1738. [DOI] [PubMed] [Google Scholar]
- Rucci N, DiGiacinto C, Orru L, Millimaggi D, Baron R, Teti A. A novel protein kinase C alpha-dependent signal to ERK1/2 activated by alphaVbeta3 integrin in osteoclasts and in Chinese hamster ovary (CHO) cells. J Cell Sci. 2005;118:3263–3275. doi: 10.1242/jcs.02436. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology. 2009;56(Suppl. 1):73–82. doi: 10.1016/j.neuropharm.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryabinin AE, Criado JR, Henriksen SJ, Bloom FE, Wilson MC. Differential sensitivity of c-Fos expression in hippocampus and other brain regions to moderate and low doses of alcohol. Mol Psychiatry. 1997;2:32–43. doi: 10.1038/sj.mp.4000206. [DOI] [PubMed] [Google Scholar]
- Saito M, Smiley J, Toth R, Vadasz C. Microarray analysis of gene expression in rat hippocampus after chronic ethanol treatment. Neurochem Res. 2002;27:1221–1229. doi: 10.1023/a:1020937728506. [DOI] [PubMed] [Google Scholar]
- Samuels DC, Burn DJ, Chinnery PF. Detecting new neurodegenerative disease genes: does phenotype accuracy limit the horizon? Trends Genet. 2009;25:486–488. doi: 10.1016/j.tig.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov BP, Jiang L, Trivedi NS, Aston C. Transcription profiling reveals mitochondrial, ubiquitin and signaling systems abnormalities in postmortem brains from subjects with a history of alcohol abuse or dependence. J Neurosci Res. 2003;72:756–767. doi: 10.1002/jnr.10631. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Pendyala G, Abarca C, Zghoul T, Sanchis-Segura C, Magnone MC, Lascorz J, Depner M, Holzberg D, Soyka M, Schreiber S, Matsuda F, Lathrop M, Schumann G, Albrecht U. The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nat Med. 2005;11:35–42. doi: 10.1038/nm1163. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Neurocircuitry in alcoholism: a substrate of disruption and repair. Psychopharmacology (Berl) 2005;180:583–594. doi: 10.1007/s00213-005-2267-6. [DOI] [PubMed] [Google Scholar]
- Tabakoff B, Bhave SV, Hoffman PL. Selective breeding, quantitative trait locus analysis, and gene arrays identify candidate genes for complex drug-related behaviors. J Neurosci. 2003;23:4491–4498. doi: 10.1523/JNEUROSCI.23-11-04491.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabakoff B, Saba L, Kechris K, Hu W, Bhave SV, Finn DA, Grahame NJ, Hoffman PL. The genomic determinants of alcohol preference in mice. Mamm Genome. 2008;19:352–365. doi: 10.1007/s00335-008-9115-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–558. doi: 10.1101/gad.1151204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch LM, Becker HC. Electrographic and behavioral indices of ethanol withdrawal sensitization. Brain Res. 2002;946:272–282. doi: 10.1016/s0006-8993(02)02895-0. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Bilbao A, Molander A, Spanagel R. Neuropharmacology of alcohol addiction. Br J Pharmacol. 2008;154:299–315. doi: 10.1038/bjp.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilpoux C, Warnault V, Pierrefiche O, Daoust M, Naassila M. Ethanol-sensitive brain regions in rat and mouse: a cartographic review, using immediate early gene expression. Alcohol Clin Exp Res. 2009;33:945–969. doi: 10.1111/j.1530-0277.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- Worst TJ, Tan JC, Robertson DJ, Freeman WM, Hyytia P, Kiianmaa K, Vrana KE. Transcriptome analysis of frontal cortex in alcohol-preferring and nonpreferring rats. J Neurosci Res. 2005;80:529–538. doi: 10.1002/jnr.20496. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry RA. Preprocessing of oligonucleotide array data. Nat Biotechnol. 2004;22:656–658. doi: 10.1038/nbt0604-656b. [DOI] [PubMed] [Google Scholar]
- Xu Y, Demarest K, Hitzemann R, Sikela JM. Gene coding variant in Cas1 between the C57BL/6J and DBA/2J inbred mouse strains: linkage to a QTL for ethanol-induced locomotor activation. Alcohol Clin Exp Res. 2002;26:1–7. [PubMed] [Google Scholar]
- Zhou FC, Anthony B, Dunn KW, Lindquist WB, Xu ZC, Deng P. Chronic alcohol drinking alters neuronal dendritic spines in the brain reward center nucleus accumbens. Brain Res. 2007;1134:148–161. doi: 10.1016/j.brainres.2006.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.