

Abstract
T-2 toxin, one of the type A trichothecenes, presents a potential hazard to human and animal health. Our previous work demonstrated that porcine cytochrome P450 3A29 (CYP3A29) played an important role in the hydroxylation of T-2 toxin. To identify amino acids involved in this metabolic process, T-2 toxin was docked into a homology model of CYP3A29 based on a crystal structure of CYP3A4 using AutoDock 4.0. Nine residues of CYP3A29, Arg105, Arg106, Phe108, Ser119, Lys212, Phe213, Phe215, Arg372 and Glu374, which were found within 5 Å around T-2 toxin were subjected to site-directed mutagenesis. In the oxidation of nifedipine, the CL int value of R106A was increased by nearly two-folds compared with the wild-type CYP3A29, while the substrate affinities and CL int values of S119A and K212A were significantly reduced. In the hydroxylation of T-2 toxin, the generation of 3′-OH-T-2 by R105A, S119A and K212A was significantly less than that by the wild-type, whereas R106A slightly increased the generation of 3′-OH-T-2. These results were further confirmed by isothermal titration calorimetry analysis, suggesting that these four residues are important in the hydroxylation of T-2 toxin and Arg105 may be a specific recognition site for the toxin. Our study suggests a possible structure-function relationship of CYP3A29 in the hydroxylation of T-2 toxin, providing with new insights into the mechanism of CYP3A enzymes in the biotransformation of T-2 toxin.
Introduction
T-2 toxin is a fungal sesquiterpenoid metabolite belonging to the type A trichothecenes, which are widely distributed in corns, oats and mixed feeds. Feeding with feeds contaminated by these toxins may lead to growth retard and production decline of livestock. Trichothecenes can also make animal sick and even cause death when the situation is serious, posing a potential threat to human health. The toxicity of trichothecenes is most likely a result of their ability to inhibit protein synthesis and partially inhibit RNA and DNA synthesis in cells [1].
T-2 toxin is metabolized in organisms to form various metabolites, resulting in detoxification of this toxin. HT-2 toxin, neosolaniol (NEO), 3′-OH-T-2, 3′-OH-HT-2, T-2 triol, T-2 tetraol, and some de-epoxy products are the common phase I metabolites of T-2 toxin [2], [3]. Biotransformation of T-2 toxin to HT-2 toxin in Vero cells and rat spleen lymphocytes resulted in a reduced inhibition of protein synthesis [4]. The two hydroxylated metabolites, 3′-OH-T-2 and 3′-OH-HT-2, were far less cytotoxic to Reuber hepatoma cells than the parent compound [5]. The decreasing order of the apoptosis-inducing activity of T-2 toxin and its metabolites was found to be T-2 toxin>3′-OH T-2 toxin> HT-2 toxin>3′-OH HT-2 toxin [6]. When T-2 toxin was biotransformed into NEO, there was a substantial decrease in the level of toxic activity [6], [7]. De-epoxy T-2 toxin was one-400th as toxic as T-2 toxin in a rat skin irritation bioassay [8]. Therefore, it is of vital significance to study the metabolism of T-2 toxin for the prevention of its potential hazard.
It is presently known that carboxylesterases are involved in the biotransformation of T-2 toxin into HT-2 toxin, cytochrome P450s take part in the 3′-hydroxylation of isovaleryl group of T-2 toxin, and epoxide hydrolases contribute most in the de-epoxy metabolism [9]. Previous studies demonstrated that cytochrome P450 3As (CYP3As) were involved in the hydroxylation of T-2 toxin in pigs. CYP3A22 and CYP3A46 mainly catalyzed T-2 toxin to generate 3′-OH T-2 [10], [11]. Recombinant porcine CYP3A29 was able to convert T-2 and HT-2 toxins into 3′-OH-T-2 and 3′-OH-HT-2, respectively [12]. Since CYP3A29 is highly expressed in the livers and small intestines of pigs and serves as the major CYP3A contributor [13], it can easily draw the conclusion that CYP3A29 contributes most in the hydroxylation of T-2 toxin among CYP3As in pigs. However, the catalytic mechanism of CYP3A29 metabolizing T-2 toxin is still unknown.
Cytochrome P450s are a superfamily of hemoproteins that play key roles in the metabolism of a wide variety of xenobiotic and endogenous compounds [14]. Although there is a wealth of information from X-ray crystal structures of mammalian P450s, the deep understanding about how these enzymes recognize a variety of structurally diverse chemicals is still limited. Among CYP3A subfamily, only crystal structures of human CYP3A4 have been resolved. The overall structure of CYP3A4 conforms to the fold that is characteristic of the P450 superfamily, with a C-terminal domain that contains the heme and the active site [15]. The crystal structures of CYP3A4 in complex with or without ligands demonstrate that the heme iron is ligated by a conserved cysteine (Cys442) and the propionates of the heme interact with the side chains of Arg105, Trp126, Arg130, Arg375, and Arg440 [16], [17]. Phe213 and Phe215 point toward the active site and form a “Phe-cluster” together with other five phenylalanines (Phe108, Phe219, Phe220, Phe241, and Phe304), which are involved in the initial recognition of the substrates [16].
Since P450 enzymes are highly dynamic and flexible, undergoing large conformational changes to allow substrate access and product release [18], it may not be necessary to study each enzyme-ligand interaction by crystallization due to the tedious procedure. The method of homology modeling and molecular docking in conjunction with site-directed mutagenesis has been extensively employed to study the substrate orientation within the active site of P450 enzyme and to identify potential residues involved in the positioning and catalytic mechanisms. Some results between crystallization or mutagenesis studies and modeling and docking studies were proven in satisfactory agreement [19]–[21].
In this study, in order to investigate the mechanism of porcine CYP3A29 in the hydroxylation of T-2 toxin, the homology model of CYP3A29 was constructed based on the crystal structure of ligand-free CYP3A4 (PDB ID: 1tqn) [17], which exhibits 76% amino acid sequence identity with CYP3A29 [22]. Molecular docking between CYP3A29 model and T-2 toxin molecule was performed to screen the possible amino acids responsible for the binding or catalyzing of T-2 toxin, followed by site-directed mutagenesis and enzymatic activity assay to investigate the functions of these residues in the hydroxylation of the toxin. It was demonstrated that Arg105 might be a specific recognition site of CYP3A29 for T-2 toxin. Besides, Arg106, Ser119 and Lys212 played important roles in the hydroxylation of T-2 toxin and the oxidation of nifedipine. This study sheds light on a possible structure-function relationship of porcine CYP3A29 in the hydroxylation of T-2 toxin, which may provide a deeper understanding for the metabolism of T-2 toxin in pigs.
Materials and Methods
Chemicals
Nifedipine (NIF), oxidized nifedipine (ONIF), glucose 6-phosphate, and anti-human CYP3A4 monoclonal antibody were purchased from Sigma (St. Louis, MO, USA). The goat anti-mouse IgG labeled with horseradish peroxidase (HRP) was from TIANGEN (Beijing, China). Immobilon Western HRP substrate was obtained from Millipore (Billerica, MA, USA). Bicinchoninic acid protein kit was purchased from Pierce (Rockford, IL, USA). The β-nicotinamide adenine dinucleotide phosphate sodium salt (β-NADP+) was from Roche (Beijing, China). T-2 toxin, Grace’s insect medium and fetal bovine serum were obtained from Invitrogen (Carlsbad, CA, USA). Insect GeneJuice Transfection Reagent was purchased from Merck (KgaA, Darmstadt, Germany). HPLC-grade methanol was purchased from Fisher Scientific (Waltham, MA, USA). All other chemicals and reagents commercially available were of the highest analytical grade.
Cells and plasmids
Spodoptera frugiperda (Sf9) cells and baculovirus transfer vector pFastBac HTb were obtained from BD Gentest (Franklin Lakes, New Jersey, USA). Esherichia coli DH 5α, E. coli DH10Bac and pMD 18-T vector were purchased from Takara (Dalian, China). The plasmids pFastBac1-CYP3A29, pFastBac1-pNPR and pFastBac1-pb5 were previously constructed and stored in our lab [22].
Homology modeling and molecular docking
Amino acid sequence alignment was performed with Clustal X [23]. The amino acid sequence of CYP3A29 was screened against the PDB structure database (http://www.rcsb.org/pdb/home/home.do). From a series of templates, the crystal structure of ligand-free CYP3A4 (PDB ID: 1tqn) [17], exhibiting the highest sequence identity of 76% with CYP3A29, was selected as the modeling template. A three dimensional model of CYP3A29 was constructed by homology modeling tool Modeller [24]. The model with the highest score was validated by Procheck [25], which evaluated the stereochemical quality of a protein structure by analyzing residue-by-residue geometry and overall structural geometry.
The structure model of T-2 toxin, whose energy was minimized using the molecular mechanics method (MM2), was generated by ChemDraw (CambridgeSoft, Waltham, MA, USA). The docking on the monomer model of CYP3A29 with T-2 toxin was performed using Autodock 4.0 [26], which is based on Lamarckian Genetic Algorithm (LGA), a hybrid genetic algorithm with local optimization that uses a parameterized free-energy scoring to estimate the binding energy. In this docking simulation, semi-flexible docking protocols were used, in which the target protein CYP3A29 was kept as rigid. The Kollman charges, solvation parameters and polar hydrogen were added into the water free CYP3A29 model for the preparation of protein in docking simulation. The substrate T-2 toxin was treated as flexible ligand by modifying their rotatable torsions. The rigid root of the T-2 toxin ligand was defined automatically. All rotatable dihedrals in the ligand were assigned by Auto-Tors program and were allowed to rotate freely. AutoGrid 4.0 program was employed for generating grid maps for the ligands. The grid box was fixed in the catalytic active region between heme moiety and Ser 119 of CYP3A29. The box size was set as 60 Å×60 Å×60 Å to cover the whole active site and let the ligand rotate freely. The spacing between grid points was 0.375 Å. The LGA was utilized to search for the best conformer of the ligand. The individual LGA executions were clustered and ranked to generate the final docking model, and the potential binding sites were defined as amino acids in distances of less than 5 Å to T-2 toxin.
Site-directed mutagenesis
Using plasmid pFastBac1-CYP3A29 [22] as template, the mutants were constructed by one step site-directed mutagenesis [27] by using the primers shown in Table 1. The constructed plasmids were sequenced by GenScript (Nanjing, China) and confirmed to be correct.
Table 1. Primers for site-directed mutagenesis.
| Primer | Sequence (from 5′ to 3′ end) |
| R105A | Fwd: CTGTCTTCACAAACGCGAGGTCTTTTGGTCC |
| Rev: GGACCAAAAGACCTCGCGTTTGTGAAGACAG | |
| R106A | Fwd: TATTCTGTCTTCACAAACCGGGCGTCTTTTGGTCCATTG |
| Rev: CAATGGACCAAAAGACGCCCGGTTTGTGAAGACAGAATA | |
| F108P | Fwd: CTTCACAAACCGGAGGTCTGCTGGTCCATTGGGCGCTA |
| Rev: TAGCGCCCAATGGACCAGCAGACCTCCGGTTTGTGAAG | |
| S119A | Fwd: TATGAGAAACGCTCTCGCTCTGGCTGAGGATGAAG |
| Rev: CTTCATCCTCAGCCAGAGCGAGAGCGTTTCTCATA | |
| K212A | Fwd: CCCCTTTGTGGAAAACAGCAAGAAGCTCTTAGCCTTTAGTTTCTTTG |
| Rev: CAAAGAAACTAAAGGCTAAGAGCTTCTTGCTGTTTTCCACAAAGGGG | |
| F213A | Fwd: CCCCTTTGTGGAAAACAGCAAGAAGCTCTTAAAAGCGAGTTTCTTTG |
| Rev: CAAAGAAACTCGCTTTTAAGAGCTTCTTGCTGTTTTCCACAAAGGGG | |
| F215A | Fwd: GCAAGAAGCTCTTAAAATTTAGTGCCTTTGATCCATTCCTTCTCTC |
| Rev: GAGAGAAGGAATGGATCAAAGGCACTAAATTTTAAGAGCTTCTTGC | |
| R372A | Fwd: CCCAATTGCTGCTGCACTTGAGAGGGCCTGTAAG |
| Rev: CTTACAGGCCCTCTCAAGTGCAGCAGCAATTGGG | |
| E374A | Fwd: GCTAGACTTGCGAGGGCCTGTAAGAAGGATG |
| Rev: CATCCTTCTTACAGGCCCTCGCAAGTCTAGC |
Note: The mutated sites are underlined in bold and italic.
Expression of CYP3A29 and its mutants
The empty vector, pFastBac HTb, and the recombinant vectors, pFastBac1-CYP3A29 [22] (wild-type) and its variants, were transformed into E. coli DH10Bac cells, respectively. The positive clones were selected as white color, using 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) as the substrate. The recombinant baculovirus DNA (Bacmid-CYP3A29 or its mutants) was isolated from E. coli DH10Bac positive clones and then transfected into Sf9 cells using Insect GeneJuice Transfection Reagent. The medium containing viruses was collected three to four days post transfection and stored as virus stock.
For each batch of enzyme preparation, Sf9 cells were cultured to 2×107 cells/liter in Grace’s insect medium, and the cells were infected with recombinant viruses at six times higher concentration than that under expression conditions. The cells were co-infected with viruses encoding pNPR (porcine NADPH-P450 reductase) and pb5 (porcine cytochrome b5) as described previously [22]. The Sf9 cell lines were maintained in flasks (75 cm2) at 27°C in Grace’s medium containing 10% fetal bovine serum and 6 µM heme complexes of hemoglobin. The Sf9 cells were harvest after culturing for 72 h, and the microsomes were prepared by ultrasonication and differential speed centrifugation (10,000 and 105,000 g).
The microsomal protein content was determined according to Lowry et al. [28] using the bicinchoninic acid protein assay (Pierce). The content of P450 was measured using the carbon monoxide-difference spectrum [29], the co-expressed pNPR activity was measured using the cytochrome c reduction assay [30], and the concentration of co-expressed pb5 was estimated according to the method described previously [31]. After quantification, the microsomal proteins were frozen and stored at −70°C until use.
SDS-PAGE and immunoblot analysis
The SDS-PAGE was carried out with glycine-Tris buffer system [32]. The gel was stained with Coomassie brilliant blue G-250. For immunoblot analysis, microsomal proteins separated by SDS-PAGE were electro-transferred to a polyvinylidene difluoride membrane. The blots were developed with the primary antibody raised against human CYP3A4 (MAb HL3, dilution 1∶1000) followed by the goat anti-mouse IgG labeled with HRP (dilution 1∶20000). The individual blots were visualized on X-film using luminol chemiluminescent reagent of Immobilon Western HRP substrate.
Assay for enzymatic activity of CYP3A29 and its mutants
NIF oxidation activity was measured as previously described [22]. The reaction mixture (200 µL) for the assay consisted of NIF (0∼80 µM) as substrate, recombinant CYP3A29 or its mutants (5 pmol of P450), and the NADPH-generating system (2 mM NADP, 20 mM glucose 6-phosphate, 2 U/mL glucose 6-phosphate dehydrogenase and 5 mM MgCl2) in 50 mM potassium phosphate buffer (pH 7.4). All incubations were conducted in triplicate. The reactions were initiated by addition of the NADPH-generating system after preincubation at 37°C for 5 min. After incubation at 37°C for 10 min, the reaction was terminated by addition of 50 µL of ice-cold 15% (w/v) trichloroacetic acid, followed by centrifugation for 20 min at 10,000 g to precipitate the proteins. The supernatants were subjected to HPLC analysis.
The HPLC condition was set as previously described [22]. The formation of ONIF in the reaction mixture was determined based on calibration curves constructed from a series of standards of 0.01∼1 µM ONIF. Intraday (n = 5) and interday (n = 5) precisions did not exceed 10% in any of the assays. Kinetic parameters for the oxidation of NIF were calculated by fitting the data to the Hill equations, v = V max [S]n/(K m+[S]n), using nonlinear regression analysis. Equations were selected by goodness of fit based on R2 values and least residual sum of squares. The intrinsic clearance (CL int) was considered as the ratio of V max to K m.
Identification and semi-quantitation of T-2 toxin metabolites
The reaction mixture (200 µL) for the assay consisted of 40 µM of T-2 toxin as substrate, recombinant CYP3A29 or its mutants (10 pmol of P450), and the NADPH-generating system cited above in 50 mM Tris-HCl buffer (pH 7.4). All incubations were conducted in triplicate. The reaction mixture was initiated by the addition of the NADPH-generating system. The reaction mixture was maintained at 37°C for 90 min, and the reaction was terminated by addition of 50 µL of ice-cold 15% (w/v) trichloroacetic acid. Equal volume of ice-cold acetonitrile (100 µL) was added to terminate the reaction. The mixture was vortexed and centrifuged at 15,000 g for 15 min. The supernatant was filtered through a 0.22 µm microbore cellulose membrane, and 10 µL of aliquot was analyzed by ESI-IT-TOF mass spectrometry coupled with a high-performance liquid chromatography system (LC/MS-IT-TOF, Shimadzu, Kyoto, Japan) according to Wu et al [12] to identify the metabolites. The peak area of hydroxylation products was used for semi-quantitative analysis.
Isothermal titration calorimetry (ITC) measurement
Calorimetry pool was filled with CYP3A29 or its mutants (10 pmol of P450) and 1 mM NADPH, and the pool was thermostated to 37°C and stirred at 502 rpm. After the calorimeter was balanced for 60 s, 100 µM T-2 toxin was titrated by single drop. Then the toxin was continuously titrated by 20 drops, each drop for 10 µL with an interval of 300 s, and the heat and the time were recorded.
The ITC experiment directly measured the thermal power (P). Each peak represented the thermal effect associated with each injection. The heat evolved (Q) from each injection was calculated according to the following formula: P = dQ/dt. The Q generated by the enzymatic conversion of substrate to product was calculated as the difference between Q of the enzyme sample and Q of the dilution buffer.
Statistical analyses
Descriptive statistical parameters such as mean and standard deviation were calculated using Microsoft Excel 2003. Statistical analyses were performed with Student’s t-test and Bonferroni revision, and differences were considered to be statistically significant when the p-value was <0.05 or <0.01.
Results
Homology model of porcine CYP3A29 and docking of T-2 toxin
Amino acid sequence alignment demonstrated that the second structures and the substrate recognition sites of porcine CYP3A29 (GenBank ID: Z93099), CYP3A22 (GenBank ID: AB006010), CYP3A46 (GenBank ID: EF625347) and human CYP3A4 (GenBank ID: AF182273) were substantially identical (Fig. S1). The X-ray crystal structure of CYP3A4 (PDB ID: 1tqn) was chosen as template since it exhibited highest protein sequence identity (76%, 382 aa/503 aa) to that of CYP3A29. Among the five models generated by the MODELER program, the model with the lowest probability density function (PDF) total energy and discrete optimized potential energy (DOPE) score was selected and further refined by energy minimization. The quality of the refined model was assessed by PROCHECK Ramachandran plot, demonstrating that 91.5% of the residues were in the most favored regions, 6.9% in additional allowed regions, and 0.5% in generously allowed regions. Overall, the refined model of CYP3A29 was all well within the acceptable range with high quality.
When T-2 toxin was docked into CYP3A29, nine residues, Arg105, Arg106, Phe108, Ser119, Lys212, Phe213, Phe215, Arg372 and Glu374, were found to be located in a distance within 5Å to T-2 toxin, among which, Arg105 and Lys212 formed hydrogen bonds with the toxin molecule (Fig. 1).
Figure 1. The binding pocket of porcine CYP3A29 docked with T-2 toxin.
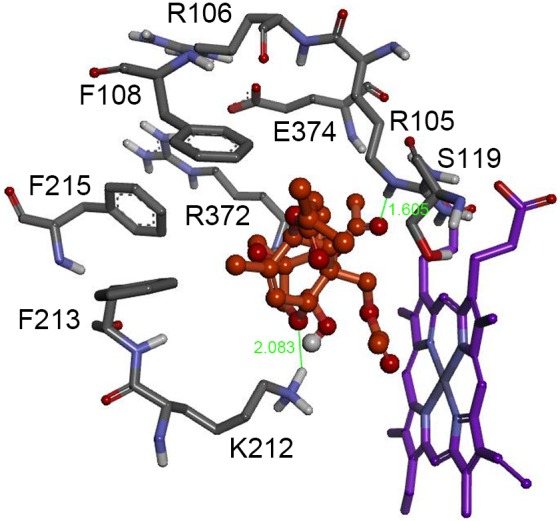
CYP3A29 interaction residues, Arg105, Arg106, Phe108, Ser119, Lys212, Phe213, Phe215, Arg372 and Glu374, are in distances within 5Å to T-2 toxin molecule. The T-2 toxin is colored in orange. The heme is represented in violet color. The green lines and numbers denote hydrogen bonds and bond lengths.
Heterologous expression of CYP3A29 and its variants in Sf9 insect cells
The screened nine amino acids, including 4 basic residues (Arg105, Arg106, Lys212 and Arg372), 1 acidic residue (Glu374), 1 polar residue (Ser119), and 3 phenylalanines (Phe108, Phe213 and Phe215) were respectively replaced with the small non-polar residue, alanine. Porcine CYP3A29 or its mutants were co-expressed with pNPR and pb5 in Sf9 insect cells, and about 50 pmol of P450, 200 units of pNPR and 110 pmol of pb5 were detected per 106 cells. The expression of each CYP3A29 variant, as presented in the microsomal proteins, was detected by immunoblot analysis using anti-CYP3A4 antibodies (Fig. S2), and the molecular mass of each mutant was about 55 kDa, in consistence with the theoretical molecular mass of CYP3A29 of 57188.7 Da.
Kinetic analysis of CYP3A29 and its mutants in the oxidation of nifedipine
As shown in Fig. 2, when NIF was incubated with Sf9 microsomes containing the recombinant CYP3A29, the oxidized product, ONIF, was generated (Fig. 2C), while no ONIF was generated when NIF was incubated with the microsomes of Sf9 cells transfected with the empty vector (Fig. 2D) or with microsomes containing the recombinant CYP3A29 but in the absence of NADPH-generating system (Fig. 2E), demonstrating that the heterogeneously expressed CYP3A29 was of biological activity. Equal amount (5 pmol of P450 in 200 µL reaction mixture) of each CYP3A29 variant was used for the subsequent enzymatic activity assays.
Figure 2. HPLC chromatograms of the metabolite of nifedipine (NIF) catalyzed by recombinant CYP3A29.

(A) NIF standard. (B) Oxidized nifedipine (ONIF) standard. (C) NIF incubated with Sf9 microsomes containing the recombinant CYP3A29 at 37°C for 30 min in the presence of NADPH-generating system. (D) NIF incubated with Sf9 microsomes free of the recombinant CYP3A29 at 37°C for 30 min in the presence of NADPH-generating system. (E) NIF incubated with Sf9 microsomes containing the recombinant CYP3A29 at 37°C for 30 min in the absence of NADPH-generating system.
The kinetic data for the generation of ONIF by wild-type and each variant CYP3A29 were examined. The relationship between substrate concentration and reaction velocity is shown in Fig. S3, and the V max and K m values are listed in Table 2. The V max/K m values, representing intrinsic clearance (CL int), were calculated and compared between wild-type CYP3A29 (V max, 8.53±0.69 nmol/min/nmol P450; K m, 15.68±2.96 µmol/L; CL int, 0.54±0.03 mL/nmol P450/min) and each mutant. The V max of S119A was slightly increased (11.03±0.85 nmol/min/nmol P450) compared with the wild-type, but the K m value (24.15±3.68 µmol/L) was predominantly increased by nearly 1.6 folds, resulting in a weak decrease of CL int value (0.45±0.01 mL/nmol P450/min). Similarly, K212A also exhibited a higher K m (21.71±1.83 µmol/L) and a lower CL int (0.46±0.03 mL/nmol P450/min). In contrast, the NIF oxidation activity of CYP3A29 was significantly increased by replacing Arg106 with Ala. R106A exhibited a slight increase in V max (9.86±0.37 nmol/min/nmol P450), a notable decrease in K m (10.63±0.93 µmol/L), and accordingly the CL int (0.93±0.02 mL/nmol P450/min) was nearly twice as much as that of the wild-type. No statistically significant differences were identified in other mutants.
Table 2. Enzyme kinetic parameters of recombinant CYP3A29 and its mutants oxidizing nifedipine.
| Enzymes | SRS | V max (nmol/min/nmol P450) | K m (µmol/L) | CL int (mL/nmol P450/min) | n |
| Wild-type | 8.53±0.69 | 15.68±2.96 | 0.54±0.03 | 1.10 | |
| R105A | SRS1 | 8.42±0.71 | 14.60±3.07 | 0.58±0.07 | 1.09 |
| R106A | SRS1 | 9.86±0.37* | 10.63±0.93* | 0.93±0.02** | 1.34 |
| F108A | SRS1 | 7.54±0.57 | 13.43±2.37 | 0.56±0.03 | 1.26 |
| S119A | SRS1 | 11.03±0.85* | 24.15±3.68* | 0.45±0.01** | 1.41 |
| K212A | 10.02±0.89 | 21.71±1.83* | 0.46±0.03* | 1.48 | |
| F213A | 8.45±0.61 | 14.50±2.45 | 0.58±0.04 | 1.25 | |
| F215A | 9.46±1.03 | 17.67±4.45 | 0.53±0.08 | 1.18 | |
| R372A | SRS5 | 8.58±0.78 | 15.88±3.60 | 0.54±0.05 | 1.07 |
| E374A | SRS5 | 8.48±0.72 | 17.05±3.08 | 0.50±0.05 | 1.41 |
Note: The values are expressed as means ± standard deviations of the results of three independent experiments. n indicates the Hill coefficients.
**(p<0.01) and *(p<0.05) indicate statistically significant difference between the wild-type and the mutant.
Metabolizing of T-2 toxin by CYP3A29 and its mutants
Previous study demonstrated that 3′-OH-T-2 and NEO were detected as the metabolites of T-2 toxin when incubating with recombinant pig CYP3A29 [12]. Similarly, T-2 toxin was shown in this study to be metabolized to 3′-OH-T-2 and NEO by the Sf9 microsomes containing recombinant CYP3A29 (Fig. 3) or its mutants (Fig. S4), while nearly no 3′-OH-T-2 or NEO was found by the microsomes containing no expressed recombinant CYP3A29 (vector control). The relative amount of 3′-OH-T-2 was much higher than that of NEO (Fig. 3 and Fig. S4), suggesting that the hydroxylated metabolite was the major metabolite.
Figure 3. Identification of the metabolites of T-2 toxin after incubation with recombinant CYP3A29.

(A) Accurate extracted ion chromatograms of the metabolites of T-2 toxin after incubation with CYP3A29. (B) Accurate MS spectrum of 3′-OH-T-2. (C) Accurate MS spectrum of NEO. (D) Structural illustration of the metabolites of T-2 toxin metabolized by CYP3A29.
Unfortunately, there was no 3′-OH-T-2 standard compound and the yield of 3′-OH-T-2 could not be calculated by the substrate disappearance due to that the biotransformation of T-2 toxin by CYP3A29 did not result in only one metabolite (i.e. 3′-OH-T-2). Therefore, the production of 3′-OH-T-2 by CYP3A29 or its mutants was semi-quantitated and compared according to the peak areas of the LC/MS data. As shown in Fig. 4, the yields of 3′-OH-T-2 by R105A (1.48-fold change), S119A (1.37-fold change) and K212A (1.28-fold change) were decreased significantly, while was slightly increased by R106A (1.11-fold change), compared with the wild-type. The hydroxylation of T-2 toxin by other mutants had no significant changes.
Figure 4. The peak area of the mass spectra of 3′-OH-T-2 generated by CYP3A29 metabolizing T-2 toxin.

The data were expressed as means ± standard deviations (error bars) (n = 3). **(p<0.01) and *(p<0.05) indicate statistically significant difference between the wild-type CYP3A29 and each mutant.
The above four mutants affecting the metabolism of T-2 toxin were further confirmed by ITC measurement. As shown in Fig. 5, the thermal powers of R105A, S119A and K212A in the metabolism of T-2 toxin were significantly reduced compared with the wild-type, while the thermal power of R106A was increased slightly. Heat evolved (Qenzyme-Qdilution) from the metabolism of T-2 toxin by R105A, S119A and K212A also changed significantly. The heats evolved by R105A (9.35 mJ, 3.59-fold change), S119A (15.18 mJ, 2.21-fold change) and K212A (19.31 mJ, 1.74-fold change) were decreased significantly compared with the wild-type (33.54 mJ). The heat evolved by R106A (36.16 mJ, 1.08-fold change) was slightly increased compared with that of the wild-type. Since the ITC measured the total heat produced by the enzymatic reaction, the ITC data manifested the biotransformation of T-2 toxin to both 3′-OH-T-2 and NEO. The NEO formation (hydrolysis of the C8-isovaleryloxy ester of T-2 toxin) was a heat absorbed reaction but not a major reaction catalyzed by CYP3A29, thus the fold changes of these four residues were different between the ITC data and the LC/MS data but with a similar tendency.
Figure 5. The thermogram of CYP3A29 and its mutants metabolizing T-2 toxin.

Discussion
This study demonstrates that porcine CYP3A29 is able to metabolize T-2 toxin to form 3′-OH-T-2 and NEO, implying that CYP3A29 is involved in the hydroxylation of the isovaleryl group and the hydrolysis of the C8-isovaleryloxy ester of T-2 toxin. These two biotransformation processes make T-2 toxin more polar and water-soluble to exclude from the body. Hydroxylation and hydrolysis metabolism of T-2 toxin are thought to reduce the toxic effects of the toxin [5], [33], therefore, CYP3A29 is very important in the detoxification of T-2 toxin. Similarly, previous studies demonstrated that T-2 toxin could be metabolized to 3′-OH-T-2 by porcine CYP3A22 and CYP3A46 [10], [11], but those studies did not test the carboxylesterase activity (hydrolysis of the ester bond) of porcine CYP3As for T-2 toxin. It is reported that human CYP3A4 can mediate the hydrolysis of the ester bond [34], [35], inferring that human CYP3A4 and porcine CYP3A29 are orthologous proteins. Based on the metabolic study of T-2 toxin in pigs, the metabolic process of T-2 toxin in the human body can be speculated. Nevertheless, the hydroxylation of T-2 toxin was the main reaction catalyzed by CYP3A29, and the hydrolysis reaction was quite weak. Conclusively, porcine CYP3As play a vital role in the hydroxylation of T-2 toxin in pigs.
Although the homology model of porcine CYP3A29 is very similar to the crystal structure of human CYP3A4, CYP3A29 has a relatively lower NIF oxidase activity than CYP3A4 [22]. The most distinct differences in the amino acid sequences of these two enzymes were found to be located in the helices F–G region (Fig. S1), and this region was considered important in determination of the substrate specificity of CYP3As [36]. In CYP3A4, Leu210 is implicated in the effector binding as well as the stereo- and regio-selectivity profile and both Leu211 and Asp214 are implicated in the cooperativity of the enzyme [37], [38]. Furthermore, a variety of studies have concentrated upon the ability of CYP3A4 to accommodate more than one substrate molecule simultaneously and the resultant activation/enhancer effects [38]–[41]. It is also reported that CYP3A29 displays ligand binding that does not follow the Michaelis-Menten kinetics, but in the sigmoidal or autoactivation kinetic behaviors with Hill coefficients (n) >1 [22]. Sigmoidal kinetics can be interpreted by the allosteric effect hypothesis. CYP3As usually undergo dramatic conformational changes upon ligand binding with an increasing volume in the active site, which could enhance the adaptability of enzyme to the ligands [42]. In this study, the mutants of CYP3A29 displayed different Hill coefficients in the oxidation of NIF, but all with n>1 (Table 2 and Fig. S3). The Hill coefficients of R106A (n = 1.34), S119A (n = 1.41), K212A (n = 1.48) and E374A (n = 1.41) were increased significantly compared with the wild-type (n = 1.10), indicating that these mutations enlarged the volume of the active site.
In the metabolism of NIF, among the amino acids determined by docking T-2 toxin into CYP3A29, R106A exhibited an increased Vmax and a reduced Km, resulting in that the metabolic capacity of R106A improved significantly by nearly two-fold compared with that of the wild-type. Arg106 is located in SRS1 (SRS, substrate recognition site), when it was mutated into the small residue, alanine, the steric hinderance was reduced as well as the charge was eliminated, thus NIF might be more easily to get into the active center. In contrast, S119A substitution weakened the ability of CYP3A29 metabolizing NIF, with higher K m thus lower CL int. Ser119 is conservative among CYP3A subfamily members. Studies show that Ser119 of CYP3A4 has a major impact on the substrate stereoselectivity [43], [44], therefore, S119A substitution may affect the orientation of NIF in the active center of the enzyme, thus affect its oxidation. Similarly, K212A also exhibited a higher Km and a lower CL int. Helices F-G are important for the substrate specificity of CYP3As [36]. This region is recognized to adopt different conformations dependent on the presence of different ligands. It was shown that broad substrate specificity of CYP3A4 stemed from the malleability of the F–F’ loop (residues 211–218, between helix F and F’) that resided in the vicinity of the channel connecting the active site and bulk solvent [45]. Arg212, located in this loop, was found in the active site in a ligand-free structure of CYP3A4, but moved towards the surface of the enzyme in the ketoconazole-CYP3A4 complex [42]. Therefore, K212A substitution might affect the transportation of NIF to the active center of CYP3A29.
In the hydroxylation of T-2 toxin by CYP3A29, since there was no standard compound of 3′-OH-T-2, only semi-quantitative assay was done by using LC/MS. R105A, S119A and K212A substitutions significantly reduced the generation of the hydroxylation product, 3′-OH-T-2, while R106A substitution slightly increased the hydroxylation rate (Fig. 4). This result was further confirmed by the isothermal titration calorimeter analysis based on the heat evolved produced by CYP3A29 and its mutants (Fig. 5). According to the results of molecular docking, T-2 toxin form hydrogen bonds with Arg105 and Lys212 in CYP3A29. Hydrogen bonding, electrostatic interaction, and π-π-conjugated contribute to the orientation of the substrate in the active center [46]–[48]. Mutation at these two sites might destroy the formation of hydrogen bonds between T-2 toxin and CYP3A29, influencing the orientation of the toxin molecule in the active-site cavity of the enzyme, thus the yields of 3′-OH-T-2 by R105A or K212A were much lower than that by the wild-type. In addition, S119A also exhibited a weaker metabolic capacity in the hydroxylation of T-2 toxin, and Ser119 might affect the orientation of T-2 toxin in the active center as in the case of NIF oxidation. Compared with the wild-type, the catalytic capability of R106A in the metabolism of NIF was increased more obviously than in the metabolism of T-2 toxin (Table 2 and Fig. 4). This may be due to the different kinetic characteristics of the enzyme for these two different substrates. The oxidation of NIF by CYP3A29 followed an auto-activation kinetic behavior as discussed above and R106A substitution might make the substrate access the active center more easily, while the metabolism of T-2 toxin by CYP3A29 was likely to be a hyperbolic saturation kinetics process. Combining the results of CYP3A29 metabolizing NIF and T-2 toxin, it can be speculated that Arg105, Arg106, Ser119 and Lys212 of CYP3A29 are crucial to the substrate positioning and binding. Furthermore, since R105A substitution did not influence the oxidation of NIF, Arg105 might be a unique recognition site of CYP3A29 for T-2 toxin.
Other sites, including Phe108, Phe213, Phe215, Arg372 and Glu374 did not show significant impact on the metabolism of NIF or T-2 toxin. Previous studies showed that Phe108, Phe213 and Phe215 formed the “Phe-cluster” on the ceiling of the active-site cavity of CYP3A4 [15]. When binding large substrates, phenylalanine residues in the “Phe-cluster” were repositioned, resulting in an extension of helix F and a larger active site [16], [42]. However, F108A, F213A and F215A substitutions did not change the metabolic capacity for either NIF or T-2 toxin in our study. Correspondingly, the Hill coefficients of these mutants didn’t alter much when compared with that of the wild-type (Table 2). This may be due to the small molecular sizes of the NIF (M r 346.34) and T-2 toxin (M r 466.53), which might not cause great conformation changes in the active center of CYP3A29, thus mutation of “Phe-cluster” did not influence the metabolism of these two substrates. This is in consistence with the case of progesterone (M r 314.47), which induced very little conformational change in CYP3A4 [16]. R372A and E374A, although located in SRS5, did not have significant impact on the metabolic capability either, suggesting they are not the functional sites of CYP3A29 for metabolizing NIF or T-2 toxin.
In conclusion, our study revealed that Arg105, Arg106, Ser119, Lys212 might be important to the function of CYP3A29 in the metabolism of NIF or T-2 toxin, and Arg105 might be a unique binding site of CYP3A29 for T-2 toxin. These findings provide a possible interpretation for the structure-function relationship of CYP3A29 in the hydroxylation of T-2 toxin, giving a deeper understanding of the metabolic processes of T-2 toxin by cytochrome P450s. Since the hydroxylation and hydrolysis of T-2 toxin by CYP3A29 are detoxification reactions, CYP3A29 could be used as a detoxification enzyme. Moreover, our study indicates a protein engineering direction for this detoxification enzyme in the future, which may improve the efficiency of the prevention of T-2 toxin mediated hazard.
Supporting Information
Amino acid sequence alignment of porcine CYP3A29, CYP3A22, CYP3A46 and human CYP3A4. Helices (red) are indicated by letters, and β-sheets (blue) are indicated by numbers above the sequences. The substrate recognition sites (SRSs) are indicated by pane (pink). Mutated sites are marked with fresh green.
(TIF)
Immunoblot analysis of recombinant CYP3A29 and its mutants. Microsomal proteins from Sf9 cells expressing R105A, R106A, F108A, S119A, K212A, F213A, F215A, R372A and E374A (from lanes 1 to 9) were subjected to SDS-PAGE. After electrophoresis, the proteins were transferred to a polyvinylidene fluoride membrane and probed with anti-human CYP3A4 immunoglobulin as described in Materials and Methods. Arrow indicates CYP3A29 and its mutants. M, protein molecular mass standard.
(TIF)
Nifedipine oxidation kinetics of recombinant CYP3A29 and its mutants. Nifedipine at concentration of 0, 2, 4, 8, 1, 20, 40, 60 and 80 µM were respectively incubated with 25 pmol CYP3A29 or its mutants at 37°C for 10 min as described in Materials and Methods. The Hill equation (v = Vmax [S]n/(Km+[S]n)) was fitted by the data points. The solid red lines through the experimental data showed the best fits for the non-linear regression analysis using the Hill equation for sigmoidal kinetics. The standard deviations of three replicates did not exceed 10% of the mean values.
(TIF)
Accurate extracted ion chromatograms of the metabolites of T-2 toxin after incubation with recombinant CYP3A29 or its mutants. The CYP3A29 mutants include R105A, R106A, F108A, S119A, K212A, F213A, F215A, R372A and E374A.
(DOC)
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the National Natural Science Foundation of China (grant no. 31272614) and the Fundamental Research Funds for the Central Universities (program no. 2013QC002). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No additional external funding was received for this study.
References
- 1. Wu Q, Dohnal V, Kuca K, Yuan Z (2013) Trichothecenes: structure-toxic activity relationships. Curr Drug Metab 14: 641–660. [DOI] [PubMed] [Google Scholar]
- 2. Wu Q, Dohnal V, Huang L, Kuca K, Yuan Z (2010) Metabolic pathways of trichothecenes. Drug Metab Rev 42: 250–267. [DOI] [PubMed] [Google Scholar]
- 3. Dohnal V, Jezkova A, Jun D, Kuca K (2008) Metabolic pathways of T-2 toxin. Curr Drug Metab 9: 77–82. [DOI] [PubMed] [Google Scholar]
- 4. Thompson WL, Wannemacher RW Jr (1986) Structure-function relationships of 12,13-epoxytrichothecene mycotoxins in cell culture: comparison to whole animal lethality. Toxicon 24: 985–994. [DOI] [PubMed] [Google Scholar]
- 5. Kobayashi J, Horikoshi T, Ryu JC, Tashiro F, Ishii K, et al. (1987) The cytochrome P-450-dependent hydroxylation of T-2 toxin in various animal species. Food Chem Toxicol 25: 539–544. [DOI] [PubMed] [Google Scholar]
- 6. Islam Z, Nagase M, Ota A, Ueda S, Yoshizawa T, et al. (1998) Structure-function relationship of T-2 toxin and its metabolites in inducing thymic apoptosis in vivo in mice. Biosci Biotechnol Biochem 62: 1492–1497. [DOI] [PubMed] [Google Scholar]
- 7. Anderson DW, Black RM, Lee CG, Pottage C, Rickard RL, et al. (1989) Structure-activity studies of trichothecenes: cytotoxicity of analogues and reaction products derived from T-2 toxin and neosolaniol. J Med Chem 32: 555–562. [DOI] [PubMed] [Google Scholar]
- 8. Swanson SP, Helaszek C, Buck WB, Rood HD Jr, Haschek WM (1988) The role of intestinal microflora in the metabolism of trichothecene mycotoxins. Food Chem Toxicol 26: 823–829. [DOI] [PubMed] [Google Scholar]
- 9. He J, Zhou T, Young J, Boland G, Scott P (2010) Chemical and biological transformations for detoxification of trichothecene mycotoxins in human and animal food chains: A review. Trends in Food Sci Technol 21: 67–76. [Google Scholar]
- 10.Wang J, Jiang J, Zhang H, Wang J, Cai H, et al.. (2011) Integrated transcriptional and proteomic analysis with in vitro biochemical assay reveal the important role of CYP3A46 in T-2 toxin hydroxylation in porcine primary hepatocytes. Mol Cell Proteomics 10: M111 008748. [DOI] [PMC free article] [PubMed]
- 11. Ge X, Wang J, Liu J, Jiang J, Lin H, et al. (2010) The catalytic activity of cytochrome P450 3A22 is critical for the metabolism of T-2 toxin in porcine reservoirs. Catalysis Communications 12: 71–75. [Google Scholar]
- 12. Wu Q, Huang L, Liu Z, Yao M, Wang Y, et al. (2011) A comparison of hepatic in vitro metabolism of T-2 toxin in rats, pigs, chickens, and carp. Xenobiotica 41: 863–873. [DOI] [PubMed] [Google Scholar]
- 13. Yao M, Dai M, Liu Z, Cui W, Li D, et al. (2012) mRNA expression profiles of P450 3A enzymes in the liver and small intestine of the domestic pig. Res Vet Sci 93: 360–365. [DOI] [PubMed] [Google Scholar]
- 14. Guengerich FP, Wu ZL, Bartleson CJ (2005) Function of human cytochrome P450s: characterization of the orphans. Biochem Biophys Res Commun 338: 465–469. [DOI] [PubMed] [Google Scholar]
- 15. Scott EE, Halpert JR (2005) Structures of cytochrome P450 3A4. Trends Biochem Sci 30: 5–7. [DOI] [PubMed] [Google Scholar]
- 16. Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, et al. (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305: 683–686. [DOI] [PubMed] [Google Scholar]
- 17. Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, et al. (2004) The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J Biol Chem 279: 38091–38094. [DOI] [PubMed] [Google Scholar]
- 18. Li H, Poulos TL (2004) Crystallization of cytochromes P450 and substrate-enzyme interactions. Curr Top Med Chem 4: 1789–1802. [DOI] [PubMed] [Google Scholar]
- 19. Lewis DFV, Ito Y, Goldfarb PS (2005) Cytochrome P450 structures and their substrate interactions. Drug Develop Res 66: 19–24. [Google Scholar]
- 20. Zhao C, Gao Q, Roberts AG, Shaffer SA, Doneanu CE, et al. (2012) Cross-linking mass spectrometry and mutagenesis confirm the functional importance of surface interactions between CYP3A4 and holo/apo cytochrome b(5). Biochemistry 51: 9488–9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moore CD, Shahrokh K, Sontum SF, Cheatham TE 3rd, Yost GS (2010) Improved cytochrome P450 3A4 molecular models accurately predict the Phe215 requirement for raloxifene dehydrogenation selectivity. Biochemistry 49: 9011–9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yao M, Dai M, Liu Z, Huang L, Chen D, et al. (2011) Comparison of the substrate kinetics of pig CYP3A29 with pig liver microsomes and human CYP3A4. Biosci Rep 31: 211–220. [DOI] [PubMed] [Google Scholar]
- 23. Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, et al. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31: 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815. [DOI] [PubMed] [Google Scholar]
- 25. Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8: 477–486. [DOI] [PubMed] [Google Scholar]
- 26. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, et al. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30: 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zheng L, Baumann U, Reymond JL (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275. [PubMed] [Google Scholar]
- 29. Omura T, Sato R (1964) The Carbon Monoxide-Binding Pigment of Liver Microsomes. I. Evidence for Its Hemoprotein Nature. J Biol Chem 239: 2370–2378. [PubMed] [Google Scholar]
- 30. Strobel HW, Dignam JD (1978) Purification and properties of NADPH-cytochrome P-450 reductase. Methods Enzymol 52: 89–96. [DOI] [PubMed] [Google Scholar]
- 31. Holmans PL, Shet MS, Martin-Wixtrom CA, Fisher CW, Estabrook RW (1994) The high-level expression in Escherichia coli of the membrane-bound form of human and rat cytochrome b5 and studies on their mechanism of function. Arch Biochem Biophys 312: 554–565. [DOI] [PubMed] [Google Scholar]
- 32. King J, Laemmli UK (1971) Polypeptides of the tail fibres of bacteriophage T4. J Mol Biol 62: 465–477. [DOI] [PubMed] [Google Scholar]
- 33. Konigs M, Mulac D, Schwerdt G, Gekle M, Humpf HU (2009) Metabolism and cytotoxic effects of T-2 toxin and its metabolites on human cells in primary culture. Toxicology 258: 106–115. [DOI] [PubMed] [Google Scholar]
- 34. Zhang D, Ly VT, Lago M, Tian Y, Gan J, et al. (2009) CYP3A4-mediated ester cleavage as the major metabolic pathway of the oral taxane 3′-tert-butyl-3′-N-tert-butyloxycarbonyl-4-deacetyl-3′-dephenyl-3′-N-debenzoyl-4-O-methoxycarbonyl-paclitaxel (BMS-275183). Drug Metab Dispos 37: 710–718. [DOI] [PubMed] [Google Scholar]
- 35. Yang D, Wang X, Chen YT, Deng R, Yan B (2009) Pyrethroid insecticides: isoform-dependent hydrolysis, induction of cytochrome P450 3A4 and evidence on the involvement of the pregnane X receptor. Toxicol Appl Pharmacol 237: 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Torimoto N, Ishii I, Toyama K, Hata M, Tanaka K, et al. (2007) Helices F-G are important for the substrate specificities of CYP3A7. Drug Metab Dispos 35: 484–492. [DOI] [PubMed] [Google Scholar]
- 37. Harlow GR, Halpert JR (1997) Alanine-scanning mutagenesis of a putative substrate recognition site in human cytochrome P450 3A4. Role of residues 210 and 211 in flavonoid activation and substrate specificity. J Biol Chem 272: 5396–5402. [DOI] [PubMed] [Google Scholar]
- 38. Harlow GR, Halpert JR (1998) Analysis of human cytochrome P450 3A4 cooperativity: construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc Natl Acad Sci U S A 95: 6636–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Domanski TL, He YA, Harlow GR, Halpert JR (2000) Dual role of human cytochrome P450 3A4 residue Phe-304 in substrate specificity and cooperativity. J Pharmacol Exp Ther 293: 585–591. [PubMed] [Google Scholar]
- 40. Fowler SM, Riley RJ, Pritchard MP, Sutcliffe MJ, Friedberg T, et al. (2000) Amino acid 305 determines catalytic center accessibility in CYP3A4. Biochemistry 39: 4406–4414. [DOI] [PubMed] [Google Scholar]
- 41. Wang H, Dick R, Yin H, Licad-Coles E, Kroetz DL, et al. (1998) Structure-function relationships of human liver cytochromes P450 3A: aflatoxin B1 metabolism as a probe. Biochemistry 37: 12536–12545. [DOI] [PubMed] [Google Scholar]
- 42. Ekroos M, Sjogren T (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci U S A 103: 13682–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. He YA, Roussel F, Halpert JR (2003) Analysis of homotropic and heterotropic cooperativity of diazepam oxidation by CYP3A4 using site-directed mutagenesis and kinetic modeling. Arch Biochem Biophys 409: 92–101. [DOI] [PubMed] [Google Scholar]
- 44. Khan KK, Halpert JR (2000) Structure-function analysis of human cytochrome P450 3A4 using 7-alkoxycoumarins as active-site probes. Arch Biochem Biophys 373: 335–345. [DOI] [PubMed] [Google Scholar]
- 45. Park H, Lee S, Suh J (2005) Structural and dynamical basis of broad substrate specificity, catalytic mechanism, and inhibition of cytochrome P450 3A4. J Am Chem Soc 127: 13634–13642. [DOI] [PubMed] [Google Scholar]
- 46. Kirton SB, Murray CW, Verdonk ML, Taylor RD (2005) Prediction of binding modes for ligands in the cytochromes P450 and other heme-containing proteins. Proteins 58: 836–844. [DOI] [PubMed] [Google Scholar]
- 47. Lewis DF, Jacobs MN, Dickins M (2004) Compound lipophilicity for substrate binding to human P450s in drug metabolism. Drug Discov Today 9: 530–537. [DOI] [PubMed] [Google Scholar]
- 48. Wang JF, Wei DQ, Li L, Zheng SY, Li YX, et al. (2007) 3D structure modeling of cytochrome P450 2C19 and its implication for personalized drug design. Biochem Biophys Res Commun 355: 513–519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amino acid sequence alignment of porcine CYP3A29, CYP3A22, CYP3A46 and human CYP3A4. Helices (red) are indicated by letters, and β-sheets (blue) are indicated by numbers above the sequences. The substrate recognition sites (SRSs) are indicated by pane (pink). Mutated sites are marked with fresh green.
(TIF)
Immunoblot analysis of recombinant CYP3A29 and its mutants. Microsomal proteins from Sf9 cells expressing R105A, R106A, F108A, S119A, K212A, F213A, F215A, R372A and E374A (from lanes 1 to 9) were subjected to SDS-PAGE. After electrophoresis, the proteins were transferred to a polyvinylidene fluoride membrane and probed with anti-human CYP3A4 immunoglobulin as described in Materials and Methods. Arrow indicates CYP3A29 and its mutants. M, protein molecular mass standard.
(TIF)
Nifedipine oxidation kinetics of recombinant CYP3A29 and its mutants. Nifedipine at concentration of 0, 2, 4, 8, 1, 20, 40, 60 and 80 µM were respectively incubated with 25 pmol CYP3A29 or its mutants at 37°C for 10 min as described in Materials and Methods. The Hill equation (v = Vmax [S]n/(Km+[S]n)) was fitted by the data points. The solid red lines through the experimental data showed the best fits for the non-linear regression analysis using the Hill equation for sigmoidal kinetics. The standard deviations of three replicates did not exceed 10% of the mean values.
(TIF)
Accurate extracted ion chromatograms of the metabolites of T-2 toxin after incubation with recombinant CYP3A29 or its mutants. The CYP3A29 mutants include R105A, R106A, F108A, S119A, K212A, F213A, F215A, R372A and E374A.
(DOC)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.