

Abstract
Cystic fibrosis (CF) is a common and deadly inherited disease, caused by mutations in the CFTR gene that encodes a cAMP-activated chloride channel. One outstanding manifestation of the disease is the persistent bacterial infection and inflammation in the lung, which claims over 90% of CF mortality. It has been debated whether neutrophil-mediated phagocytic innate immunity has any intrinsic defect that contributes to the host lung defense failure. Here we compared phagosomal CFTR targeting, hypochlorous acid (HOCl) production, and microbial killing of the neutrophils from myeloid Cftr-inactivated (Myeloid-Cftr−/−) mice and the non-inactivated control (Cftrfl10) mice. We found that the mutant CFTR that lacked Exon-10 failed to target to the neutrophil phagosomes. This dysfunction resulted in impaired intraphagosomal HOCl production and neutrophil microbial killing. In vivo lung infection with a lethal dose of Pseudomonas aeruginosa caused significantly higher mortality in the myeloid CF mice than in the controls. The myeloid-Cftr−/− lungs were deficient in bacterial clearance, and had sustained neutrophilic inflammation and stalled transition from early to late immunity. These manifestations recapitulated the symptoms of human CF lungs. The data altogether suggest that myeloid CFTR expression is critical to normal host lung defense. CFTR dysfunction in neutrophils compromises the phagocytic innate immunity, which may predispose CF lungs to infection.
Introduction
Cystic Fibrosis (CF) is the most common genetic disease in Caucasians with an occurrence of 1/∼3000 live births [1], [2]. It is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene which encodes for a cAMP-activated chloride channel. Even though CF affects multiple organs and systems, the most severe and life-threatening pathology occurs in the lung, which claims over 90% of CF mortality. Clinical manifestations include persistent bacterial infection and inflammation, prominent neutrophil infiltration, and purulent small airway obstruction. These symptoms imply that CF lungs have an impaired host defense. However, the true link between the chloride channel defect and the host defense failure in CF lungs has not been fully established.
Host lung defense reflects the combined activities of lung resident cells, such as pulmonary epithelial cells and tissue macrophages, and lung-recruited immune cells, most notably neutrophils and monocytes. The lungs of CF patients are remarkably neutrophilic and inflamed [3], indicating that a successful inflammatory response can be mounted by the host. In spite of the robust host response, CF lungs cannot resolve infections. Thus, it is the quality, not the quantity, of the host defense that falls short in CF. Many aspects of functional disturbance or behavior aberrance in CF neutrophils have been previously recognized [4], [5], [6], [7], [8], including suboptimal activation [9], cleavage of CXCR1 [10], hyper-sensitivity to LPS stimulation [11], deviant production of reactive oxygen species [12], genome-wide gene expression perturbation [13], alteration in inflammatory signaling [14], hyper-production of IL-8 [15], [16], delayed apoptosis [17], abnormal extracellular trap formation [18], hyper-oxidation of glutathione [19], and lately abnormal granule release [20]. Neutrophils are professional phagocytes constituting ∼60–70% of the circulating leukocytes in humans. Their major function is to control and eradicate infections, especially extracellular bacterial infection. One of the pivotal microbial killing mechanisms in neutrophils is to produce microbicidal oxidants [21], [22], [23], such as O2 −, H2O2 and hypochlorous acid (HOCl). Among them, HOCl, the chlorine bleach, has the greatest potency due to its reactivity with almost all macromolecules from lipids to proteins to nucleic acids [24], [25]. Notably, neutrophils use chloride to synthesize HOCl in their phagosomes [22], [26], [27]. This biosynthesis is catalyzed by myeloperoxidase (MPO), an enzyme exclusively expressed in neutrophils [28]. Because chloride is a charged ion, it cannot permeate lipid membranes unless transported through channels or transporters. An early study by Yoshimura and colleagues indicates that CFTR mRNA is transcribed in mature human neutrophils [29]. We have demonstrated that the CFTR channel protein is expressed in human neutrophils [30] and specifically targets to the phagosomes [31]. Further studies have proved that CFTR defect in the neutrophils from the patients with CF impairs the intraphagosomal HOCl production and microbial killing by the phagocyte [30], [32], [33]. However, these findings from CF patients have not been validated in any CF animal models. In the present study, we have used the myeloid tissue-specific Cftr−/− mice to interrogate CFTR expression and function in phagocytic host defense. In vitro and in vivo experiments have corroborated the critical role of myeloid CFTR in phagocyte-mediated lung host defense. Impairment of such a mechanism alone is sufficient to damage the lung’s ability to defend against Pseudomonas infection.
Materials and Methods
Ethics statement
This research involves use of animals. The study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All procedures involving animal use were reviewed and approved by the Louisiana State University Health Sciences Center Animal Care and Use Committee (IACUC #2836). For anesthesia, isoflurane inhalation was used prior to lung bacterial intubation. Carbon dioxide suffocation was used to euthanize animals.
Chemicals and solutions
All common chemicals were obtained from Sigma-Aldrich. The physiological chloride Ringer’s buffer was composed of 122 mM NaCl, 1.2 mM MgCl2, 1.2 mM CaCl2, 2.4 mM K2HPO4, 0.6 mM KH2PO4, 20 mM HEPES (pH 7.3), and 10 mM dextrose. The gluconate chloride-free Ringer’s buffer was made by substitution of the chloride salts with equal molar concentration of gluconate salts except 4 mM of calcium gluconate was used to compensate for the mild calcium chelating effect of gluconate. The KCl relaxation buffer contained 50 mM KCl, 3 mM NaCl, 3.5 mM MgCl2, 0.5 mM EGTA, 1 mM ATP, 20 mM PIPES (pH 7.0), 1 mM PMSF and Sigma protease inhibitor cocktail.
Mice
Creation of the Cftr Exon 10-floxed mice, referred to here as Cftrfl10, was published [34]. As documented previously [35], the myeloid CFTR-Exon-10-deleted (Myeloid-Cftr−/−) mice were produced by breeding the Cftrfl10 mice with the LysMCre transgenic mice [36]. After repeated backcross and screen, the Cftrfl10 homozygous mice carrying the LysMCre transgene were selected by PCR genotyping for experiments.
Lung bacterial infection and animal survival assay
Pseudomonas aeruginosa bacteria (PsA, clinical isolate) were embedded in agarose beads for chronic lung infection. Preparation of the PsA-laden beads was modified from the previous description [37], [38]. Briefly, overnight culture of PsA in tryptic soy broth (TSB) was washed and resuspended in fresh TSB to give a final concentration of ∼1×108 cfu/ml. Low-melting temperature agarose was melted in the gluconate chloride-free Ringer’s buffer to make a final concentration of 2%, which was then equilibrated to 45°C and mixed well with PsA. Then, the mixture was poured into 45°C pre-warmed heavy mineral oil (Sigma-Aldrich). The mixture was cooled on ice gradually with agitation. The agarose slush was washed with the gluconate chloride-free Ringer’s buffer, and homogenized (3 strokes, 10 seconds per stroke at the full speed) with a polytron homogenizer (Omni International Inc., Warrenton, VA, USA). Serial dilutions were made and plated in duplicates on LB plates for colony-forming unit determination. After obtaining the bacterial titer, the PsA-laden beads were non-invasively instilled into mouse lungs under anesthesia [39]. For lethal challenge, a total of 5×106 cfu of the agarose-embedded PsA in 50 µl of suspension was intubated. The bacterium-challenged mice were monitored twice a day for 7 days. Behavior change and mortality were recorded. If animals showed severe pain or stress symptoms such as weight loss, dehydration, incontinence, eyes sunken, lids closed, wasting of muscles on back, sunken or distended abdomen, decreased vibrissae movement, unresponsive, ataxia, circling, hypothermia, analgesics (Buprenorphine; 0.1 mg/kg, SQ) was administered. Dying animals with weight loss up to 20% were considered mortality and euthanized by CO2 asphyxiation. All survived animals were euthanized a week after bacterial challenge.
Bronchoalveolar lavage (BAL) and cytodifferential determination
The PsA-infected mice, euthanized by CO2 inhalation, were dissected to expose their tracheas. Blunt needles (23G), were inserted into the tracheas to cannulate the lungs, and secured with a silk suture. Ice-cold PBS lavage solution (1 ml) containing the protease inhibitor cocktail (Roche) was used to flush each lung and held for 1 minute before aspirating out. Then, additional 0.5 ml lavage solution was used for a second lung wash. Typically, a total of ∼1.2 ml of BAL fluid per mouse was recoverable by the procedure. BAL cell differentials were obtained by cytospin, Giemsa staining and microscopic assessment.
Peripheral blood neutrophil purification
Peripheral blood neutrophils were isolated following the method modified from the previous description [40]. Briefly, mouse blood (350±50 µl per animal) was collected by retro-orbital bleeding into PBS-EDTA solution (PBS without Ca2+ and Mg2+, pH 7.2, 15 mM EDTA, and 1% bovine serum albumin). After centrifugation (400×g) for 10 min at 4°C, cells were resuspended in 0.2 ml PBS-EDTA solution. Six to ten mouse blood samples were pooled and laid onto a three-layer Percoll gradient (78%, 69%, and 52% Percoll PLUS) (GE Healthcare Biosciences, PA, USA). After centrifugation for 30 min at 1500×g at room temperature without braking, the neutrophil layer at the 69% and 78% interface was collected into a 1% BSA-coated tube. After one wash with 2 ml PBS-EDTA, the red cells were lysed by hypotonic shock. After a final wash with 2 ml PBS-EDTA, the neutrophils were resuspended in the chloride-free gluconate Ringer’s buffer to deplete chloride for 1 hour on ice for use.
Peritoneal cavity neutrophil and monocyte isolation
In the case of isolating peritoneal neutrophils and monocytes, we followed the published protocol [41]. Briefly, 9% casein solution was intraperitoneally injected 12 hours and 3 hours prior to peritoneal exudate harvest. Then, the collected peritoneal neutrophils and monocytes were further purified by Percoll-gradient centrifugation as described above. Giemsa staining and microscopic examination confirmed that this protocol resulted in over 90% purity.
Neutrophil CFTR immunostaining
Percoll-gradient purified neutrophils from peripheral blood were fixed with 4% paraformaldehyde for 30 minutes at room temperature, and permeabilized with 0.5% Triton X-100/PBS for 30 minutes. The cells were then blocked with Blocking buffer (5% normal goat serum in 0.1% Triton X-100/PBS) for 1 hour, and incubated with the CFTR-specific antibody [42] (mAb #660, CFTR Antibody Distribution Program, UNC/CFF Therapeutics, Inc.) or IgG2b isotype control antibody in Blocking buffer for 2 hours at room temperature. Next, the cells were washed with PBS and subsequently incubated with the PE-conjugated goat–anti-mouse IgG secondary antibody (Invitrogen, Camarillo, CA, USA) for 45 minutes. Expression levels were analyzed by flow cytometry.
Phagosomal CFTR immunostaining
Latex beads (3 µm, Sigma-Aldrich) were coated with BSA and anti-BSA antibody and serum-opsonized similarly as previously described [31]. Purified mouse peripheral blood neutrophils were mixed with the opsonized beads and incubated for 30 min at 37°C with shaking, followed by washes with PBS by low speed centrifugation at 100×g to remove the non-phagocytosed beads. Then the cells were incubated in protease inhibitor cocktail on ice for 10 min to inactivate serine proteases, then rinsed with 1 ml of the KCl relaxation buffer with 1 mM ATP. Finally, 0.2 ml of the same relaxation buffer containing protease inhibitor cocktail (Roche) was added. The cells were immediately subjected to homogenization by going through 27G needle for 5 times. The lysate was collected and centrifuged (400×g) at 4°C for 5 min to remove intact cells and nuclei. The phagosome-containing supernatant was recovered for immunostaining with the primary CFTR antibody or the isotype control, in the presence of 5% normal goat serum, followed by staining with the PE-conjugated goat anti-mouse IgG. The samples were post-fixed with the same volume of 2% paraformaldehyde/PBS and analyzed immediately by flow cytometry. The instrument was electronically gated on the clearly distinguishable 3-µm bead/phagosome population. Our preliminary study proved that ∼70–80% of the gated population was positive for Lysosomal-associated membrane protein-1, indicating that the majority of the gated events were mature phagosomes.
Intraphagosomal hypochlorous acid measurement
HOCl production in neutrophils was examined using the HOCl-specific fluorescent probe R19-S [43], [44]. R19-S in its natural state does not fluoresce. However, HOCl oxidation renders it fluorescent [43]. Neutrophils were fed serum-opsonized PsA at a ratio of 1∶20 in chloride-free Ringer’s buffer for 15 min. After low speed centrifugation, the cells with phagocytosed bacteria were resuspended in the Ringer’s buffer with or without chloride for 20 min. Then, R19-S (10 µM) was added to the cell suspension for additional 15 minutes. Fluorescence intensity of R19-S was analyzed by flow cytometry.
Neutrophil-mediated bacterial killing
Bacterial killing assay was performed using our published protocol with modifications [33]. Neutrophils purified from mouse peripheral blood were resuspended in the chloride-free Ringer’s buffer on ice for 1 hour to deplete the intracellular free chloride. The cells (5×105) were plated in a well of a 48-well plate for 20 min at 37°C. After cell adhesion, the supernatant was aspirated and the serum-opsonized PsA bacteria in the phagocytosis buffer (chloride-free Ringer’s buffer supplemented with 10% chloride-depleted mouse normal serum) were added for phagocytosis for 15 min at 37°C. Next, the non-phagocytosed bacteria were washed away with 0.5 ml ice-cold chloride-free Ringer’s buffer three times. Then the cells were incubated in either no-chloride (0 mM) or NaCl (122 mM) Ringer’s buffer for additional 40 minutes at 37°C. The number of viable bacteria after killing was determined by LB agar plating as described before [33].
Determination of lung bacterial clearance
Mice were intubated intratracheally with agarose-embedded PsA at the sub-lethal dose (1×106 cfu). The animals were then sacrificed at Day 0 and Day 3. The lungs were lavaged with ice-chilled PBS (1 ml) and the bacterial number in each BAL fluid was determined by LB agar plating. Then, the total bacterial number in each BAL fluid was back calculated.
Statistics
Data were statistically analyzed by Student’s t-test for differences between two comparing groups. The animal survival data were compared by Log-Rank test. Results were expressed as mean ± SD. Differences with P-values smaller than 0.05 were considered statistically significant.
Results
Specific deletion of CFTR Exon-10 in myeloid lineages in mice
Myeloid tissue-specific Cftr−/− mice have been established and characterized in the previous report [35]. These mice express functional CFTR in all tissues except myeloid lineages in which CFTR Exon-10 is specifically deleted. Notably, their intestinal and pulmonary epithelia have normal CFTR function [35]. Thus, this model would allow us to define the potential role of myeloid CFTR in phagocytic host defense in the lungs without interference of other cell types. To ensure that all animals used for the current study had the designed tissue-specific CFTR Exon-10 deletion, we PCR-genotyped all the pups from the myeloid Cftr−/− breeding colony. Shown in Figure 1A is a representative DNA gel of the PCR products from the tail segments of different animals, displaying that 10 of the 13 tested mice had the Exon-10 deletion, which gave rise to the small PCR-amplicon (148 bp). The large PCR-amplicon (408 bp) indicates an intact and floxed Exon-10. Because the tail segments contained epithelial, fibroblast and blood cells, a mixture of Exon-10 deletion and non-deletion was detected in each PCR reaction from the Cre-positive mice. To confirm the deletion was myeloid lineage-specific, we isolated and purified the peritoneal neutrophils and monocytes from both non-deletion Cftrfl10 control and Myeloid-Cftr−/− mice. PCR-genotyping of the phagocytes and the corresponding tail segments was performed and compared. As displayed in Figure 1B , all the Cftrfl10 samples exhibited an intact floxed CFTR Exon-10 with the large amplicon (408 bp). In contrast, the myeloid Cftr−/− samples demonstrated the desired Exon-10 deletion with the small PCR-product (148 bp) in neutrophils and monocytes.
Figure 1. Myeloid CFTR-inactivation in mice.
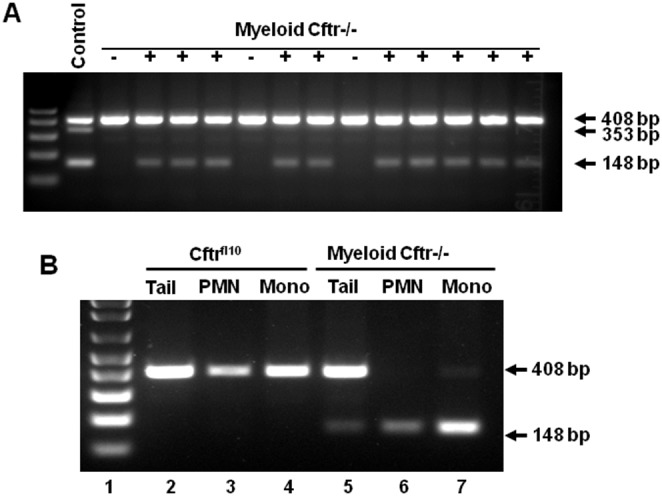
Myeloid tissue-specific CFTR-Exon-10 deletion mice (Myeloid-Cftr−/−) were produced by breeding the Cftrfl10 mice with the LysMCre transgenic mice. The Cftrfl10 mice had a double-allele modification of CFTR Exon-10 which was flanked by loxP sites, while the LysMCre mice had myeloid Cre-recombinase expression under the control of the LysM gene promoter. Myeloid-Cftr−/− mice were screened by PCR-genotyping using the specific primers to amply CFTR Exon-10. A) DNA gel of PCR products from the tail segments of two litters of pups. The 408-bp PCR amplicon indicates an intact and floxed CFTR Exon-10. In contrast, the 148-bp amplicon shows a deleted CFTR-Exon-10. The control lane shows the PCR products of a mixture of DNAs from the tails of wild-type, Cftrfl10 and Myeloid-Cftr−/− mice. The 353-bp amplicon represents the non-floxed, wild-type CFTR Exon-10. B) Confirmation of myeloid CFTR Exon-10 deletion. Purified neutrophils (PMN) and monocytes (Mono), and the corresponding tail segment (Tail) were used for PCR-genotyping. Both neutrophils and monocytes from the Myeloid-Cftr−/− mouse have the 148-bp product, indicating Exon-10 deletion, whereas the phagocytes from the control Cftrfl10 mouse show the 408-bp product. Contrarily, tails from both types of mice display the double bands due to the mixture of multiple cell types.
Myeloid CFTR Exon-10 deletion impairs CFTR presentation to neutrophil phagosomes
To determine whether the mutant CFTR without Exon-10 had any protein expression in myeloid cell lineages, we immunostained the peripheral blood neutrophils from Cftrfl10 control and Myeloid-Cftr−/− mice with the CFTR-specific antibody described in Materials and Methods. Flow cytometric analysis ( Fig. 2A ) demonstrated that there was no significant difference in the levels of overall CFTR expression in the phagocytes from the two types of mice. Next, we decided to compare CFTR targeting to the phagosomes of neutrophils. Peripheral blood neutrophils were fed serum-opsonized latex beads, homogenized to release their phagosomes which were then immunostained with the CFTR-specific antibody. As displayed ( Fig. 2B ), flow cytometry analysis revealed that the phagosomes from Myeloid-Cftr−/− neutrophils had little CFTR staining, while about half of the Cftrfl10 phagosomes were stained positive. These data suggest that the deletion of CFTR Exon-10 in myeloid lineages impairs CFTR presentation to the phagosomes, but not its overall CFTR protein expression in the cells.
Figure 2. Flow cytometric analysis of CFTR expression in neutrophils and phagosomes.

A) CFTR staining in neutrophils. Purified peripheral-blood neutrophils were immunostained for intracellular CFTR using the monoclonal CFTR antibody (mAb #660). Representative dot plots show that the neutrophils from Myeloid-Cftr−/− mice express CFTR (93%), and the cells from the control Cftrfl10 mice are 89% positive for CFTR. In contrast, the isotype control antibody shows little background staining. B) CFTR staining in phagosomes. Latex beads (3 µm) were phagocytosed by neutrophils from the control Cftrfl10 mice or the Myeloid-Cftr−/− mice. Phagosomes released by cell homogenization were immunostained with the same anti-CFTR antibody. The phagosomal population was gated by flow cytometry. Only the phagosomes from the control Cftrfl10 neutrophils show significant staining for CFTR.
Myeloid CFTR-Exon-10 deletion impairs intraphagosomal HOCl production in neutrophils
Neutrophils need chloride to synthesize HOCl in their phagosomes. Since CFTR is a chloride channel located to the phagosomal membrane, we predicted that its dysfunction might affect phagosomal chloride attainment and HOCl production, as found in human CF neutrophils [30]. To prove this prediction, we measured phagosomal oxidation of R19-S, a fluorescent probe specific for HOCl. Percoll-gradient purified peripheral blood neutrophils from both Cftrfl10 control and Myeloid Cftr−/− mice were kept on ice in the chloride-free Ringer’s buffer for 1 hour in order to deplete the intracellular free chloride. After phagocytosis of serum-opsonized PsA, the cells were resuspended in either chloride-free (0 mM) or physiological chloride Ringer’s buffer (122 mM) for 20 minutes, followed by R19-S application for additional 15 minutes. This protocol was designed to detect HOCl generated from the chloride acquired from the extracellular buffer after phagocytosis. R19-S fluorescence from HOCl oxidation was quantitatively analyzed by flow cytometry. Figure 3 shows that the mean fluorescence intensity (MFI) of the R19-S-positive neutrophils was significantly increased only in the case of control Cftrfl10 cells in 122 mM chloride Ringer’s buffer. In contrast, the neutrophils with the CFTR-Exon-10 deletion did not show any surge of HOCl production even with ample chloride supply in the 122 mM chloride medium. These data suggest a deficit in HOCl production by Myeloid-Cftr−/− neutrophils.
Figure 3. Impairment of phagosomal HOCl production in neutrophils from Myeloid Cftr−/− mice.

Neutrophil production of HOCl was assessed using the fluorescent HOCl probe R19-S. Phagocytosis was performed by mixing neutrophils with opsonized PsA in the chloride-free buffer (0 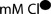 ) for 15 min, and the cells were further incubated in the buffer containing either 0 mM or 122
) for 15 min, and the cells were further incubated in the buffer containing either 0 mM or 122 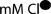 for 20 min. Then, R19-S was added to incubate for an additional 15 min. Fluorescence of R19-S produced by HOCl oxidation was analyzed by flow cytometry. Mean fluorescence intensity (MFI) of each sample was obtained and compared to their 0-min time point. Statistical difference was judged by Student’s t-test. Double asterisks indicate significant differences (p<0.01, n = 4).
for 20 min. Then, R19-S was added to incubate for an additional 15 min. Fluorescence of R19-S produced by HOCl oxidation was analyzed by flow cytometry. Mean fluorescence intensity (MFI) of each sample was obtained and compared to their 0-min time point. Statistical difference was judged by Student’s t-test. Double asterisks indicate significant differences (p<0.01, n = 4).
Myeloid CFTR Exon-10 deletion impairs neutrophil microbial killing
Phagosomal HOCl production in neutrophils plays a key role in neutrophil-mediated microbial killing [22], [26], [27], [45], [46], [47], [48]. Since the Myeloid-Cftr−/− neutrophils had an impaired HOCl production, we then examined the possible outcome of such a defect with regard to bacterial killing. Serum-opsonized PsA bacteria were phagocytosed in chloride-free Ringer’s buffer by neutrophils isolated from either Cftrfl10 control or Myeloid-Cftr−/− mice at a ratio of 1∶20 (PMN:PsA). After removal of the non-phagocytosed bacteria by repeated washing with the chloride-free Ringer’s buffer, further incubation was carried out in the physiological chloride Ringer’s buffer. After 40-min killing, viable PsA bacteria were recovered and cultured overnight on LB plates for colony counting. Figure 4 shows the bacterial survival rates relative to their initial time points (0 min after killing). Myeloid-Cftr−/− neutrophils had approximately twice as much PsA survival as that of the Cftrfl10 cells. Such a killing difference was not due to variation of bacterial uptake, because the numbers of bacteria phagocytosed by the two types of neutrophils, measured at the initial time point of killing, were comparable by Student’s t-test (p = 0.41, N = 3). Thus, myeloid CFTR inactivation undermines the neutrophil microbicidal capacity.
Figure 4. Bacterial killing deficiency of neutrophils from Myeloid-Cftr−/− mice.

Neutrophils, isolated from either Cftrfl10 mice or Myeloid-Cftr−/− mice, were incubated with serum-opsonized PsA at a ratio of 1∶20 in 0-mM chloride buffer. After washing away the non-phagocytosed bacteria, the cells were further incubated in the buffer containing 122 mM chloride for 40 minutes. After killing, viable PsA was released by lysing the neutrophils with 0.05% Saponion solution and cultured on LB plates for colony counting. Bacterial survival rate was obtained by comparing the colony forming units before and after neutrophil killing. Statistical differences were judged by Student’s t-test. Double asterisks indicate significant differences (p<0.01, N = 3).
Myeloid CF Mice have impaired lung host defense
To determine if the myeloid CFTR dysfunction alone significantly affects the lung host defense, we intratracheally inoculated Myeloid-Cftr−/− mice and Cftrfl10 control mice with the lethal dose of the agarose-embedded PsA (5×106 cfu). As shown in the Kaplan-Meier survival curve ( Fig. 5 ), casualties largely occurred over the period of Days 2 to 4 post inoculation. Under the applied level of challenge, Myeloid-Cftr−/− mice had a survival rate of ∼40%, whereas the non-deletion control mice had a survival rate of ∼70%, which was significantly higher. This result concurs with the published data [35], confirming the critical role of CFTR in myeloid cell-mediated lung host defense.
Figure 5. Lung host defense defect in Myeloid-Cftr−/− mice.

Myeloid-Cftr−/− mice and Cftrfl10 mice were infected with the agarose-embedded PsA at the lethal dose of (5×106 cfu) by lung intubation. Casualties were recorded and survival curves traced. The log-rank test was performed to check statistical difference in survival between the Cftrfl10 mice (n = 34) and the myeloid-Cftr−/− mice (n = 35). Double asterisks mean significant difference (p<0.01).
Myeloid CF mice are deficient in lung bacterial clearance
It is known that the myeloid CF mice have normal CFTR function in airway epithelia [35]. We asked whether the myeloid CFTR inactivation alone cripples the capacity of lung bacterial clearance. Myeloid Cftr−/− mice and Cftrfl10 control mice were intubated intratracheally with agarose-embedded PsA at the sub-lethal dose (1×106 cfu). Animals were sacrificed at Day 0 and Day 3. The lungs were lavaged and the bacterial load in each BAL fluid was determined by LB agar plating. As shown in Fig. 6 , bacterial numbers in the BAL fluids from both Cftrfl10 control and Myeloid-Cftr−/− mice were comparable at Day 0 post-inoculation. However, at Day 3 post-inoculation the bacterial numbers of the BAL fluids from the myeloid CF mice were ∼10-fold higher than those from the control mice, suggesting that the myeloid CF mice are deficient in lung bacterial clearance.
Figure 6. Deficiency in lung bacterial clearance in Myeloid-Cftr−/− mice.

Myeloid-Cftr−/− mice and Cftrfl10 mice were infected with the agarose-embedded PsA at the sub-lethal dose of (1×106 cfu) by lung intubation. BAL fluids from the animals were collected at Days 0 and 3 after inoculation, and cultured for bacterial quantitation. There was no statistical difference in the total bacterial numbers of the BAL fluids from both types of mice at Day 0 (p = 0.13, n = 4). However, the total BAL bacterial numbers from Myeloid-Cftr−/− mice were significantly higher than those from Cftrfl10 mice at day 3 post-infection by Student’s t-test (p<0.01, n = 5).
Sustained neutrophilic inflammation and stalled transition from early to late immunity in the lungs of myeloid CF mice
We next asked whether the infected Myeloid Cftr−/− lungs recapitulate the characteristic neutrophilic inflammation in CF patients’ lungs. The control Cftrfl10 and Myeloid-Cftr−/− mice were intratracheally instilled with the sub-lethal dose of the agarose-embedded PsA (1×106 cfu). At Days 2, 3 and 4, lung immune cells were harvested and differentials of these cells determined. Days 2–4 were chosen due to the high mortality rate during this time frame after lethal-dose PsA challenge, as shown in Fig. 5 . Dynamics of neutrophils, macrophages and lymphocytes in the control and myeloid Cftr−/− lungs are displayed ( Fig. 7 ). At Day 2, neutrophils were the predominant cells in all the lungs, occupying more than 80% of the total lung-recovered immune cells. There was no significant difference between the control and myeloid CF mice with regard to the percentage of each examined cell type. However, at Days 3 and 4 the control Cftrfl10 lungs exhibited receding of neutrophils and rising of macrophages and lymphocytes which became the major cell types. This represents a normal transition from early to late immunity, which is a typical process of resolution of infection and inflammation. Strikingly, the Myeloid-Cftr−/− lungs had a persistent neutrophilic inflammation and remained at the acute stage of immunity. This observation is consistent with the symptom of human CF lung disease. The data suggest that the myeloid CF mice have an intrinsic defect in neutrophil-mediated innate immunity, which contributes to the incomplete resolution of lung infection and infection.
Figure 7. Abnormal profile of lung immune cells in Myeloid-Cftr−/− mice in response to lung PsA infection.

Myeloid-Cftr−/− mice and Cftrfl10 control mice were intubated intratracheally with the agarose-embedded PsA at the sub-lethal dose (1×106 cfu). BAL cells were collected and evaluated for differentials by cytospin and Giemsa staining. At Day 2 after PsA infection, neutrophils were the main constituent of the BAL cells (∼85%) in the lungs of both control mice and myeloid CF mice. In contrast, at Days 3 and 4 neutrophil percent in the Cftrfl10 control lungs reduced to ∼15% and ∼7%, respectively. However, the percentage of neutrophils in the Myeloid-Cftr−/− lungs dropped only to ∼52% at Day 3 and ∼39% at Day 4, which still remained dominant. Asterisks indicate statistically significant differences by Student’s t-test (n = 4, p<0.01).
Discussion
CF lung disease is currently recognized as an epithelial and mucosal disease [49], in which immune cells are not considered as a primary player. Decades of research, centered on airway epithelia, has identified that airway CFTR dysfunction impairs transepithelial chloride and bicarbonate transport, which affects airway host defense. However, the epithelial defect alone cannot completely explain why CF lungs are predominantly infected by the opportunistic PsA, Staphylococcus aureus and Haemophilus influenzae, as opposed to the more virulent Streptococcus pneumoniae and Klebsiella pneumoniae, the common pathogens causing community-acquired pneumonias. We propose that the pathogen selection process involves host immune mechanisms.
CF lung disease must be triggered by lung infection, as fetal and newborn lungs of CF are functionally normal [50]. Thus, it is possible that CF lungs with defective host defense fail to eradicate the early invading microbes, leading to the development of chronic lung infection. Infected lungs are populated by not only the resident epithelial cells and tissue macrophages but also the mobilized immune cells. It is well established that the lung-recruited immune cells, largely neutrophils and monocytes, are an essential component in defending against extracellular bacterial infection [51]. The importance of this host defense mechanism is clearly exhibited by frequent or severe infection in patients with poor quantity or quality of circulating neutrophils such as in bone marrow failure [52], radio- or chemo-therapy [53], or inherited disorders of PMNs [54], [55]. Using the myeloid Cftr-inactivated mice, we have identified that the CFTR with Exon-10 deletion is expressed in neutrophils, but fails to target to the phagosomes. This engineered mutation closely mimics the common genetic ΔF508 mutation by affecting CFTR phagosomal targeting [31]. Myeloid-Cftr−/− mice have impaired neutrophil intraphagosomal HOCl production and compromised neutrophil microbial killing. These data thus validate our previous finding from CF patient neutrophils, demonstrating that CFTR chloride channel defect leads to microbicidal defect in neutrophils [30], [31], [32], [33].
The bacterial challenge and animal survival data ( Fig. 5 ) have revealed that the lung-infected Myeloid-Cftr−/− mice have ∼30% higher mortality than control Cftrfl10 mice, suggesting that myeloid CFTR dysfunction alone has significant impact on lung host defense. More importantly, this study has identified a striking neutrophil aberrance in the Myeloid-Cftr−/− lungs. As shown in Fig. 7 , the early stage of anti-infection immunity (Day 2) appears normal in Myeloid-Cftr−/− mice, as they can recruit a comparable number of neutrophils to the lungs as the control mice. However, at Day 3 and Day 4, neutrophil-mediated innate immunity fades away in the control lungs and is replaced by late immunity exemplified by predominant macrophages and lymphocytes. However, the Myeloid-Cftr−/− lungs fail to achieve such a transition. This is perhaps due to the incompetency of the neutrophil-mediated innate immunity which holds the lungs to a prolonged early immunity state. This postulation is supported by the long-observed pathology in CF lungs characterized by persistent neutrophilic inflammation.
In Myeloid-Cftr−/− mice, monocytes/macrophages also lack functional CFTR ( Fig. 1B , and [35]). Due to the focus of this study, we did not assess the function of the CF macrophages. It is possible that the macrophage defect may also contribute to the overall lung host defense defect. Previous studies have reported that CF lung macrophages have impairment in bacterial killing [56], [57], [58] and exaggerated inflammatory response [4], [59]. However, macrophages are more specialized in antigen presentation and immune modulation than in direct bacterial killing. The reason is that this type of phagocyte lacks the complete assortment of antimicrobial agents as in neutrophils and has very limited expression of MPO. Consequently, macrophages produce significantly less HOCl [60]. The exact role of macrophages in direct lung clearance of Pseudomonas in the myeloid CF mice awaits further characterization.
Neutrophils heavily invest in the MPO-halide-H2O2 host defense system, discovered by Klebanoff and colleagues about half a century ago [22], [26], by producing MPO to the magnitude of ∼5% of the total cellular proteins. Because of redundant and overlapping activities of multiple arms of host defense mechanisms, the MPO-halide-H2O2 system is not always pronouncedly manifested unless the pathogens overwhelms the capacity of the other host defense mechanisms [28]. According to the HOCl biosynthesis (H2O2+H++Cl−MPO→HOCl+H2O), there are four factors potentially limiting this reaction: H2O2, H+, Cl− and MPO. MPO-deficient neutrophils exhibit a profound defect in Candida clearance and bacterial killing [61], [62], [63], [64], [65], [66]. Our data here provide the first evidence indicating that limited attainability of chloride, one of the substrates, diminishes the HOCl production and neutrophil microbial killing, which consequently spoils the phagocytic host defense. This finding establishes a novel link between a chloride channelopathy and a host defense failure, reiterating the vital role of the MPO-halide-H2O2 system.
In summary, our current study using the myeloid tissue-specific Cftr-inactivation mice clearly demonstrates the significant role of CFTR in neutrophil-mediated innate immunity. CFTR dysfunction in myeloid cell lineages alone significantly affects the lung host defense. Thus, CF lung disease should be defined by the defects of multiple types of cells, including lung-recruited immune cells.
Acknowledgments
We thank Alyssa Fournett for her critical reading of the manuscript and acknowledge the Cystic Fibrosis Foundation reagent distribution program and Dr. John Riordan for the CFTR antibody used in this research.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are included within the paper.
Funding Statement
This work was supported by a LSUHSC research grant and an NIH grant (R01AI72327) to GW. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Collins FS (1992) Cystic fibrosis: molecular biology and therapeutic implications. Science 256: 774–779. [DOI] [PubMed] [Google Scholar]
- 2.Welsh MJ, Ramsey BW, Accurso F, Cutting G (2001) Cystic Fibrosis. In: Scriver CR, editor. Metabolic and Molecular Basis of Interited Disease. 8th ed. New York: McGraw-Hill. pp. 5121–5188.
- 3. Davis PB, Drumm M, Konstan MW (1996) Cystic fibrosis. Am J Respir Crit Care Med 154: 1229–1256. [DOI] [PubMed] [Google Scholar]
- 4. Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, et al. (2012) Innate immunity in cystic fibrosis lung disease. J Cyst Fibros 11: 363–382. [DOI] [PubMed] [Google Scholar]
- 5. Downey DG, Bell SC, Elborn JS (2009) Neutrophils in cystic fibrosis. Thorax 64: 81–88. [DOI] [PubMed] [Google Scholar]
- 6.Rieber N, Hector A, Carevic M, Hartl D (2014) Current concepts of immune dysregulation in cystic fibrosis. Int J Biochem Cell Biol. [DOI] [PubMed]
- 7. Hayes E, Pohl K, McElvaney NG, Reeves EP (2011) The cystic fibrosis neutrophil: a specialized yet potentially defective cell. Arch Immunol Ther Exp (Warsz) 59: 97–112. [DOI] [PubMed] [Google Scholar]
- 8. Witko-Sarsat V, Sermet-Gaudelus I, Lenoir G, Descamps-Latscha B (1999) Inflammation and CFTR: might neutrophils be the key in cystic fibrosis? Mediators Inflamm 8: 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sajjan U, Corey M, Humar A, Tullis E, Cutz E, et al. (2001) Immunolocalisation of Burkholderia cepacia in the lungs of cystic fibrosis patients. J Med Microbiol 50: 535–546. [DOI] [PubMed] [Google Scholar]
- 10. Hartl D, Latzin P, Hordijk P, Marcos V, Rudolph C, et al. (2007) Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med 13: 1423–1430. [DOI] [PubMed] [Google Scholar]
- 11.Su X, Looney MR, Su HE, Lee JW, Song Y, et al. (2011) Role of CFTR expressed by neutrophils in modulating acute lung inflammation and injury in mice. Inflamm Res. [DOI] [PMC free article] [PubMed]
- 12. Witko-Sarsat V, Allen RC, Paulais M, Nguyen AT, Bessou G, et al. (1996) Disturbed myeloperoxidase-dependent activity of neutrophils in cystic fibrosis homozygotes and heterozygotes, and its correction by amiloride. J Immunol 157: 2728–2735. [PubMed] [Google Scholar]
- 13. Adib-Conquy M, Pedron T, Petit-Bertron AF, Tabary O, Corvol H, et al. (2008) Neutrophils in cystic fibrosis display a distinct gene expression pattern. Mol Med 14: 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tirouvanziam R, Gernez Y, Conrad CK, Moss RB, Schrijver I, et al. (2008) Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc Natl Acad Sci USA 105: 4335–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reeves EP, Williamson M, O'Neill SJ, Greally P, McElvaney NG (2011) Nebulized hypertonic saline decreases IL-8 in sputum of patients with cystic fibrosis. Am J Respir Crit Care Med 183: 1517–1523. [DOI] [PubMed] [Google Scholar]
- 16. Corvol H, Fitting C, Chadelat K, Jacquot J, Tabary O, et al. (2003) Distinct cytokine production by lung and blood neutrophils from children with cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 284: L997–L1003. [DOI] [PubMed] [Google Scholar]
- 17. Moriceau S, Lenoir G, Witko-Sarsat V (2010) In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: evidence for an innate neutrophil disturbance. J Innate Immun 2: 260–266. [DOI] [PubMed] [Google Scholar]
- 18. Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, et al. (2010) CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med 16: 1018–1023. [DOI] [PubMed] [Google Scholar]
- 19.Kettle AJ, Turner R, Gangell CL, Harwood DT, Khalilova IS, et al. (2014) Oxidation contributes to low glutathione in the airways of children with cystic fibrosis. Eur Respir J. [DOI] [PubMed]
- 20.Pohl K, Hayes E, Keenan J, Henry M, Meleady P, et al. (2014) A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood. [DOI] [PMC free article] [PubMed]
- 21. Babior BM (1978) Oxygen-dependent microbial killing by phagocytes (first of two parts). N Engl J Med 298: 659–668. [DOI] [PubMed] [Google Scholar]
- 22. Klebanoff SJ (1968) Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J Bacteriol 95: 2131–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nauseef WM (2007) How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev 219: 88–102. [DOI] [PubMed] [Google Scholar]
- 24. Albrich JM, McCarthy CA, Hurst JK (1981) Biological reactivity of hypochlorous acid: implications for microbicidal mechanisms of leukocyte myeloperoxidase. Proc Natl Acad Sci USA 78: 210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang Q, Griffin DA, Barofsky DF, Hurst JK (1997) Intraphagosomal chlorination dynamics and yields determined using unique fluorescent bacterial mimics. Chem Res Toxicol 10: 1080–1089. [DOI] [PubMed] [Google Scholar]
- 26. Klebanoff SJ (1970) Myeloperoxidase: contribution to the microbicidal activity of intact leukocytes. Science 169: 1095–1097. [DOI] [PubMed] [Google Scholar]
- 27. Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ (2006) Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem 281: 39860–39869. [DOI] [PubMed] [Google Scholar]
- 28. Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM (2013) Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol 93: 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshimura K, Nakamura H, Trapnell BC, Chu CS, Dalemans W, et al. (1991) Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res 19: 5417–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Painter RG, Valentine VG, Lanson NA Jr, Leidal K, Zhang Q, et al. (2006) CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 45: 10260–10269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou Y, Song K, Painter RG, Aiken M, Reiser J, et al. (2013) Cystic fibrosis transmembrane conductance regulator recruitment to phagosomes in neutrophils. J Innate Immun 5: 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Painter RG, Marrero L, Lombard GA, Valentine VG, Nauseef WM, et al. (2010) CFTR-mediated halide transport in phagosomes of human neutrophils. J Leukoc Biol 87: 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, et al. (2008) The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol 83: 1345–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hodges CA, Cotton CU, Palmert MR, Drumm ML (2008) Generation of a conditional null allele for Cftr in mice. Genesis 46: 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonfield TL, Hodges CA, Cotton CU, Drumm ML (2012) Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J Leukoc Biol 92: 1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, et al. (2005) Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 25: 1647–1653. [DOI] [PubMed] [Google Scholar]
- 37. van Heeckeren AM, Schluchter MD (2002) Murine models of chronic Pseudomonas aeruginosa lung infection. Lab Anim 36: 291–312. [DOI] [PubMed] [Google Scholar]
- 38. Kukavica-Ibrulj I, Bragonzi A, Paroni M, Winstanley C, Sanschagrin F, et al. (2008) In vivo growth of Pseudomonas aeruginosa strains PAO1 and PA14 and the hypervirulent strain LESB58 in a rat model of chronic lung infection. J Bacteriol 190: 2804–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J, Merry AC, Nemzek JA, Bolgos GL, Siddiqui J, et al. (2001) Eotaxin represents the principal eosinophil chemoattractant in a novel murine asthma model induced by house dust containing cockroach allergens. J Immunol 167: 2808–2815. [DOI] [PubMed] [Google Scholar]
- 40. Boxio R, Bossenmeyer-Pourie C, Steinckwich N, Dournon C, Nusse O (2004) Mouse bone marrow contains large numbers of functionally competent neutrophils. J Leukoc Biol 75: 604–611. [DOI] [PubMed] [Google Scholar]
- 41.Luo Y, Dorf ME (2001) Isolation of mouse neutrophils. Curr Protoc Immunol Chapter 3: Unit 3 20. [DOI] [PubMed]
- 42. Cui L, Aleksandrov L, Chang XB, Hou YX, He L, et al. (2007) Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol 365: 981–994. [DOI] [PubMed] [Google Scholar]
- 43. Chen X, Lee KA, Ha EM, Lee KM, Seo YY, et al. (2011) A specific and sensitive method for detection of hypochlorous acid for the imaging of microbe-induced HOCl production. Chem Commun (Camb) 47: 4373–4375. [DOI] [PubMed] [Google Scholar]
- 44. Aiken ML, Painter RG, Zhou Y, Wang G (2012) Chloride transport in functionally active phagosomes isolated from Human neutrophils. Free Radic Biol Med 53: 2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Foote CS, Goyne TE, Lehrer RI (1983) Assessment of chlorination by human neutrophils. Nature 301: 715–716. [DOI] [PubMed] [Google Scholar]
- 46. Harrison JE, Schultz J (1976) Studies on the chlorinating activity of myeloperoxidase. J Biol Chem 251: 1371–1374. [PubMed] [Google Scholar]
- 47. Weiss SJ, Klein R, Slivka A, Wei M (1982) Chlorination of taurine by human neutrophils. Evidence for hypochlorous acid generation. J Clin Invest 70: 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chapman AL, Hampton MB, Senthilmohan R, Winterbourn CC, Kettle AJ (2002) Chlorination of bacterial and neutrophil proteins during phagocytosis and killing of Staphylococcus aureus. J Biol Chem 277: 9757–9762. [DOI] [PubMed] [Google Scholar]
- 49. Cohen TS, Prince A (2012) Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med 18: 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, et al. (2010) Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2: 29ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kantari C, Pederzoli-Ribeil M, Witko-Sarsat V (2008) The role of neutrophils and monocytes in innate immunity. Contrib Microbiol 15: 118–146. [DOI] [PubMed] [Google Scholar]
- 52. Leguit RJ, van den Tweel JG (2010) The pathology of bone marrow failure. Histopathology 57: 655–670. [DOI] [PubMed] [Google Scholar]
- 53. Bodey GP, Buckley M, Sathe YS, Freireich EJ (1966) Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med 64: 328–340. [DOI] [PubMed] [Google Scholar]
- 54. Malech HL, Nauseef WM (1997) Primary inherited defects in neutrophil function: etiology and treatment. Semin Hematol 34: 279–290. [PubMed] [Google Scholar]
- 55. Klein C (2011) Genetic defects in severe congenital neutropenia: emerging insights into life and death of human neutrophil granulocytes. Annu Rev Immunol 29: 399–413. [DOI] [PubMed] [Google Scholar]
- 56. Di A, Brown ME, Deriy LV, Li C, Szeto FL, et al. (2006) CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 8: 933–944. [DOI] [PubMed] [Google Scholar]
- 57. Deriy LV, Gomez EA, Zhang G, Beacham DW, Hopson JA, et al. (2009) Disease-causing mutations in the cystic fibrosis transmembrane conductance regulator determine the functional responses of alveolar macrophages. J Biol Chem 284: 35926–35938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Del Porto P, Cifani N, Guarnieri S, Di Domenico EG, Mariggio MA, et al. (2011) Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS One 6: e19970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bruscia EM, Zhang PX, Ferreira E, Caputo C, Emerson JW, et al. (2009) Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator−/− mice. Am J Respir Cell Mol Biol 40: 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deimann W (1984) Endogenous peroxidase activity in mononuclear phagocytes. Prog Histochem Cytochem 15: 1–58. [DOI] [PubMed] [Google Scholar]
- 61. Lehrer RI, Cline MJ (1969) Leukocyte myeloperoxidase deficiency and disseminated candidiasis: the role of myeloperoxidase in resistance to Candida infection. J Clin Invest 48: 1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lehrer RI, Hanifin J, Cline MJ (1969) Defective bactericidal activity in myeloperoxidase-deficient human neutrophils. Nature 223: 78–79. [DOI] [PubMed] [Google Scholar]
- 63. Hampton MB, Kettle AJ, Winterbourn CC (1996) Involvement of superoxide and myeloperoxidase in oxygen-dependent killing of Staphylococcus aureus by neutrophils. Infect Immun 64: 3512–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Aratani Y, Koyama H, Nyui S, Suzuki K, Kura F, et al. (1999) Severe impairment in early host defense against Candida albicans in mice deficient in myeloperoxidase. Infect Immun 67: 1828–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aratani Y, Kura F, Watanabe H, Akagawa H, Takano Y, et al. (2006) Contribution of the myeloperoxidase-dependent oxidative system to host defence against Cryptococcus neoformans. J Med Microbiol 55: 1291–1299. [DOI] [PubMed] [Google Scholar]
- 66. Aratani Y, Kura F, Watanabe H, Akagawa H, Takano Y, et al. (2000) Differential host susceptibility to pulmonary infections with bacteria and fungi in mice deficient in myeloperoxidase. J Infect Dis 182: 1276–1279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are included within the paper.