

Abstract
Background
Most strokes are due to blockage of an artery in the brain by a blood clot. Prompt treatment with thrombolytic drugs can restore blood flow before major brain damage has occurred and improve recovery after stroke in some people. Thrombolytic drugs, however, can also cause serious bleeding in the brain, which can be fatal. One drug, recombinant tissue plasminogen activator (rt‐PA), is licensed for use in selected patients within 4.5 hours of stroke in Europe and within three hours in the USA. There is an upper age limit of 80 years in some countries, and a limitation to mainly non‐severe stroke in others. Forty per cent more data are available since this review was last updated in 2009.
Objectives
To determine whether, and in what circumstances, thrombolytic therapy might be an effective and safe treatment for acute ischaemic stroke.
Search methods
We searched the Cochrane Stroke Group Trials Register (last searched November 2013), MEDLINE (1966 to November 2013) and EMBASE (1980 to November 2013). We also handsearched conference proceedings and journals, searched reference lists and contacted pharmaceutical companies and trialists.
Selection criteria
Randomised trials of any thrombolytic agent compared with control in people with definite ischaemic stroke.
Data collection and analysis
Two review authors applied the inclusion criteria, extracted data and assessed trial quality. We verified the extracted data with investigators of all major trials, obtaining additional unpublished data if available.
Main results
We included 27 trials, involving 10,187 participants, testing urokinase, streptokinase, rt‐PA, recombinant pro‐urokinase or desmoteplase. Four trials used intra‐arterial administration, while the rest used the intravenous route. Most data come from trials that started treatment up to six hours after stroke. About 44% of the trials (about 70% of the participants) were testing intravenous rt‐PA. In earlier studies very few of the participants (0.5%) were aged over 80 years; in this update, 16% of participants are over 80 years of age due to the inclusion of IST‐3 (53% of participants in this trial were aged over 80 years). Trials published more recently utilised computerised randomisation, so there are less likely to be baseline imbalances than in previous versions of the review. More than 50% of trials fulfilled criteria for high‐grade concealment; there were few losses to follow‐up for the main outcomes.
Thrombolytic therapy, mostly administered up to six hours after ischaemic stroke, significantly reduced the proportion of participants who were dead or dependent (modified Rankin 3 to 6) at three to six months after stroke (odds ratio (OR) 0.85, 95% confidence interval (CI) 0.78 to 0.93). Thrombolytic therapy increased the risk of symptomatic intracranial haemorrhage (OR 3.75, 95% CI 3.11 to 4.51), early death (OR 1.69, 95% CI 1.44 to 1.98; 13 trials, 7458 participants) and death by three to six months after stroke (OR 1.18, 95% CI 1.06 to 1.30). Early death after thrombolysis was mostly attributable to intracranial haemorrhage. Treatment within three hours of stroke was more effective in reducing death or dependency (OR 0.66, 95% CI 0.56 to 0.79) without any increase in death (OR 0.99, 95% CI 0.82 to 1.21; 11 trials, 2187 participants). There was heterogeneity between the trials. Contemporaneous antithrombotic drugs increased the risk of death. Trials testing rt‐PA showed a significant reduction in death or dependency with treatment up to six hours (OR 0.84, 95% CI 0.77 to 0.93, P = 0.0006; 8 trials, 6729 participants) with significant heterogeneity; treatment within three hours was more beneficial (OR 0.65, 95% CI 0.54 to 0.80, P < 0.0001; 6 trials, 1779 participants) without heterogeneity. Participants aged over 80 years benefited equally to those aged under 80 years, particularly if treated within three hours of stroke.
Authors' conclusions
Thrombolytic therapy given up to six hours after stroke reduces the proportion of dead or dependent people. Those treated within the first three hours derive substantially more benefit than with later treatment. This overall benefit was apparent despite an increase in symptomatic intracranial haemorrhage, deaths at seven to 10 days, and deaths at final follow‐up (except for trials testing rt‐PA, which had no effect on death at final follow‐up). Further trials are needed to identify the latest time window, whether people with mild stroke benefit from thrombolysis, to find ways of reducing symptomatic intracranial haemorrhage and deaths, and to identify the environment in which thrombolysis may best be given in routine practice.
Plain language summary
Clot‐dissolving drugs for treating ischaemic stroke in the early stages
Question
We wanted to compare the safety and efficacy of clot‐dissolving (thrombolytic) drugs versus placebo or no treatment in the early stages of ischaemic stroke to see if clot‐dissolving drugs improve outcome after stroke.
Background
Most strokes are due to blockage of an artery in the brain by a blood clot. Prompt treatment with clot‐dissolving (thrombolytic) drugs can restore blood flow before major brain damage has occurred and could mean that people are more likely to make a good recovery from their stroke. Thrombolytic drugs can also, however, cause serious bleeding in the brain, which can be fatal. Thrombolytic therapy has now been evaluated in many randomised trials in acute ischaemic stroke. The thrombolytic drug alteplase has been licensed for use within three hours of stroke in the USA and Canada, and within 4.5 hours in most European countries. The numbers of people receiving this treatment successively are increasing.
Study characteristics
We identified 27 trials with a total of 10,187 participants in searches conducted up to November 2013. Most data come from trials testing one drug (recombinant tissue Plasminogen Activator, rt‐PA) given into a vein up to six hours after acute ischaemic stroke, but several other drugs were also tested and at different times to treatment after stroke and given into an artery in the brain rather than into a vein in the arm. All trials compared a clot‐dissolving drug with a placebo (control) group. Most trials included participants with moderate to severe stroke. All trials took place in hospitals that were used to treating people with stroke. Differences between trials mean that not all trials contribute information to all outcomes, but we have used all available data. Most trials included participants after a computed tomography (CT) brain scan had excluded a brain haemorrhage as the cause of symptoms (a few trials used magnetic resonance brain scanning instead).
Key results
There is general agreement between the earlier trials and the one recent trial added in this update (IST‐3) for all main outcomes, and between the 12 trials that tested rt‐PA and the 15 trials that tested other clot‐dissolving drugs. The main difference between IST‐3 and earlier trials was that IST‐3 had many participants above 80 years. Clot‐dissolving treatment can reduce the risk of long‐term dependency on others for daily activities, in spite of there being an increased risk of bleeding in the brain which also increased the risk of early death. Once the early bleeding risk had passed, at three or six months after stroke, people given clot‐dissolving drugs were more likely to have recovered from their stroke and to be independent, especially if they had been treated within the first three hours after stroke. Older people benefited as much as younger people. Giving aspirin at the same time as clot‐busting drugs increased the risk of bleeding and should be avoided. Further analyses of individual patient data factors such as findings on brain scanning before treatment, and of different ways of giving the treatment, may give more information than the summary data that we used here. Meantime, people who think that they are experiencing a stroke should get to hospital quickly, be assessed by a stroke doctor, have a brain scan and receive clot‐dissolving treatment as fast as possible. They should not hesitate by thinking that they will be 'too old' for treatment. The treatment is very effective if started within three hours of stroke and definitely improves outcome if given up to 4.5 hours after stroke, but later than that the effects are less clear and are still being tested in trials. More information is needed from trials in people with mild stroke to see if the benefit of clot‐dissolving drugs outweighs the risk of haemorrhage.
Quality of the evidence
The evidence comes mostly from well‐conducted randomised trials run by stroke experts. Some trials (8/27) were run by companies that make the clot‐dissolving drugs, but most trials (19/27, including most participants) were funded by Government or charity sources independently of drug companies. These results apply to a wide range of people with a wide range of severities of stroke and other medical conditions.
Background
Description of the condition
Acute ischaemic stroke is a major cause of death and disability worldwide. Most strokes are due to blockage of an artery in the brain by a blood clot (ischaemic stroke) e.g. from the heart or neck arteries.
Description of the intervention
Thrombolytic drugs derive from naturally‐occurring enzymes that dissolve thrombus as part of the natural clotting cascade. Some are extracted from biological samples (e.g. urokinase, desmoteplase) and others are manufactured (e.g. recombinant tissue plasminogen activator (rt‐PA), or recombinant pro‐urokinase).
How the intervention might work
Clot‐dissolving (thrombolytic) drugs may reduce brain damage from a stroke by restoring the blood flow if given rapidly enough after stroke, but may also cause serious bleeding in the brain.
Why it is important to do this review
An overview of the literature on thrombolysis in acute ischaemic stroke in 1992 (Wardlaw 1992) identified six randomised trials of various thrombolytic drugs including a total of just 700 participants. A Cochrane review published in 1995 (Wardlaw 1995) updated the original 1992 review. It was updated again in 1999 (3478 participants in total) (Wardlaw 1999), in 2003 (5727 participants) (Wardlaw 2003b), in 2009 (7152 participants) (Wardlaw 2009) but even so, many essential questions remained unanswered: How big is the overall benefit? What is the latest time window in which the treatment is still beneficial? Which grades of stroke severity and which types of stroke, as judged clinically and on brain imaging, are more likely to respond favourably to treatment? Should people aged over 80 years receive thrombolysis? Which types of patients are most likely to be harmed by treatment, and which to benefit from it (e.g. with or without other major medical conditions like cardiac arrhythmias, diabetes, hypertension, or other disorders and concomitant medication) (Wardlaw 2002)? To answer these questions reliably, and in particular to be able to tailor treatment to the individual, more data are needed from new randomised controlled trials (RCTs).
Meanwhile, the thrombolytic drug alteplase (rt‐PA) was licensed for use within three hours of stroke in the USA and Canada, and up to 4.5 hours in most European countries, and an increasing number of people now receive the treatment. Guidelines recommended that thrombolysis should be delivered by a clinical team with suitable training and experience and in a setting with appropriate facilities (ESO Stroke Guidelines 2008; NICE Stroke Guideline 2008). A general review of the use of thrombolytic therapy in clinical practice and the clinical service required to deliver it was provided in a book on the management of stroke (Warlow 2008).
This updated review includes all trials completed and made public since 2009, as well as additional data published since 2009 from trials included in earlier versions of the review. The total number of participants is now 10,187, more than a 10‐fold increase since the review was initiated in 1990 and an increase of more than 40% since 2009. Although many of the data now come from trials testing intravenous rt‐PA within the first six hours after stroke onset, the more recent trials are exploring alternative methods for selecting participants and extending time windows, e.g. through use of advanced brain imaging. The upper age limit of 80 years, stroke severity and new imaging data are also analysed. This systematic review includes these data and provides a convenient and up‐to‐date summary of the evidence.
Objectives
To determine whether, and in what circumstances, thrombolytic therapy might be an effective and safe treatment for acute ischaemic stroke. We wished to determine whether:
-
thrombolytic therapy increases the risk of death:
within the first two weeks of stroke; or
at long‐term follow‐up;
thrombolytic therapy increases the risk of symptomatic or fatal intracranial haemorrhage, or symptomatic infarct swelling;
thrombolysis reduces the proportion of people dead or dependent at long‐term follow‐up, in spite of any early hazard, so that there is an overall net benefit.
We wished to undertake exploratory analyses to examine whether:
thrombolytic therapy interacts with antithrombotic therapy to increase the hazard;
the balance of risk and benefit with thrombolytic therapy may vary with the severity of the stroke;
the latest therapeutic time window for effective treatment can yet be determined;
whether the effect of thrombolysis is different in people aged over 80 compared with under 80 years of age;
whether people selected for treatment using MR diffusion/perfusion imaging had better effect of thrombolytic treatment than those selected using computed tomography (CT) brain imaging;
whether individual findings on CT brain imaging identified people in whom the effect of thrombolysis was different;
whether the effect of intra‐arterial therapy differed from intravenous therapy and explained any of the heterogeneity.
Methods
Criteria for considering studies for this review
Types of studies
We sought to identify all truly randomised unconfounded trials of thrombolytic therapy compared with placebo or open control in people with acute ischaemic stroke. We excluded trials that were not truly randomised, such as dose‐range‐finding studies, and trials that included thrombolytic treatment in the control group. We included trials in which the exact method of randomisation was unknown, even after correspondence with the authors, if the available information suggested that the randomisation was not likely to be biased. We also included trials that were not originally analysed on an intention‐to‐treat basis if information on outcome could be obtained for all randomised participants, thus allowing us to perform an intention‐to‐treat analysis.
Types of participants
We included trials of participants with a definite acute ischaemic stroke (CT or magnetic resonance (MR) scanning having excluded intracranial haemorrhage prior to randomisation).
Types of interventions
We included all types of thrombolytic drug, given in any dose, by the intravenous or intra‐arterial route: urokinase (UK, also known as u‐PA), recombinant pro‐urokinase (rpro‐UK), streptokinase (SK), recombinant tissue plasminogen activator (rt‐PA) including duteplase, lumbrokinase (LK), and desmoteplase.
We excluded trials that were confounded by the treatment or control group receiving another active therapy which had not been factored in to the randomisation (for example, thrombolytic drug plus another agent versus placebo, or thrombolytic drug versus another agent).
Types of outcome measures
The primary outcome measures were death or dependency, as defined by modified Rankin score of 3 to 6, and death at the end of follow‐up. We considered all other outcomes as secondary.
We assessed the following.
Deaths from all causes within the first seven to 10 days after treatment.
Symptomatic intracranial haemorrhage (SICH): either symptomatic (that is, temporally associated with a deterioration in the person's neurological state), or fatal (that is, leading directly to death), and occurring within the first seven to 10 days. Note that symptomatic intracranial haemorrhage includes haemorrhagic transformation of the infarct, haemorrhage elsewhere in the brain remote from the infarct, and haemorrhage into the spaces surrounding the brain. Definitions of SICH vary between trials and therefore we have used the SICH data as defined by each trial's primary definition rather than attempting to standardise the definition.
Fatal intracranial haemorrhage.
Deaths within the first seven to 10 days not due to intracranial haemorrhage.
Symptomatic infarct swelling (oedema).
Deaths occurring between the end of the first seven to 10 days and three to six months.
Deaths from all causes during the whole trial follow‐up period.
Poor functional outcome at the end of follow‐up. This was the primary outcome measure for the review and was defined as death or dependency, measured by the modified Rankin or Barthel scales, at the end of the trial follow‐up period. Poor functional outcome (the converse of good functional outcome) is the most clinically relevant and important measure of outcome, since the aim of treatment should be not merely to avoid death but also to decrease dependency among the survivors; that is, to increase the proportion of independent survivors and conversely to reduce the risk of survival with serious disability. Dependency in the present analysis was defined as a score of between 3 and 5 inclusive on the modified Rankin Scale (mRS). Some would prefer a definition of 'good outcome' (independence) including Rankin 0 and 1 only; therefore, wherever possible we sought data on the number of participants in each individual Rankin category so as to compare poor functional outcome defined as mRS 2 to 6 with the definition of 3 to 6. Where data were not available for mRS 3 to 6, we used mRS 2 to 6 instead, rather than excluding the trial from analysis.
Search methods for identification of studies
See the 'Specialized register' section in the Cochrane Stroke Group module. We searched for all trials in all languages using the following overlapping methods, and arranged translation of relevant papers published in languages other than English.
Electronic searches
We searched the Cochrane Stroke Group's Trials Register, which was last searched by the Managing Editor on 18 November 2013. In addition, we carried out comprehensive searches of MEDLINE (Ovid) (1966 to November 2013) (Appendix 1) and EMBASE (Ovid) (1980 to November 2013) (Appendix 2). We developed the search strategies for MEDLINE and EMBASE with the help of the Cochrane Stroke Group Trials Search Co‐ordinator.
Searching other resources
We handsearched the following conference proceedings and stroke and neurological journals: Stroke, Cerebrovascular Diseases, International Journal of Stroke, Journal of Stroke and Cerebrovascular Diseases, Neurology and Journal of Neurology, Neurosurgery and Psychiatry published to March 2014.
We checked multiple international conference proceedings on stroke and specifically on thrombolysis since 1991. These include all European Stroke Conferences (since 1992, annual since 1994), all International Stroke Conferences hosted by the American Heart Association (annual), all World Stroke Conferences (biannual), all Thrombolysis in Acute Ischaemic Stroke symposia (biannual).
We examined reference lists quoted in thrombolytic therapy papers.
We made direct contact with principal investigators of trials in Europe, North America, Japan, China, and Australasia.
We have been in regular contact with the manufacturer of rt‐PA, and other companies involved in ongoing studies of thrombolysis identified from the Washington Internet Stroke Center Register of ongoing trials (www.strokecenter.org).
For previous versions of this review:
We handsearched the following journals from 1979 to April 1994: Japanese Journal of Stroke, Clinical Evaluation, Japanese Journal of Pharmacology & Therapeutics, and Rinsho Ketsueki (we obtained translations of the non‐English language publications from people in whose native language the paper was published);
We contacted 321 pharmaceutical companies for more information about trials known to exist from the above efforts, and for information on any trials which were so far unknown to us (the last systematic contact was made in December 1997); all companies except one (which was known to be doing a trial in any case) responded, and no trials were identified that we did not already know about.
Data collection and analysis
This review builds on a continuous data collection process that started in 1989.
Selection of studies
Two review authors (JW, VM) screened the records obtained from the electronic searches and excluded obviously irrelevant studies. We obtained the full paper copy of the remaining studies and the same two review authors selected truly randomised trials comparing a thrombolytic drug with placebo or open control in people with acute ischaemic stroke, brain imaging having excluded cerebral haemorrhage and other structural causes of stroke‐like symptoms. We sought additional unpublished information from the principal investigators of all the trials that appeared to meet our inclusion criteria. We resolved any disagreements by discussion. The selection for 2003 update was verified by EB.
Data extraction and management
Two review authors (JM and VM) checked the data extraction and resolved any discrepancies or uncertainties by discussion or clarification with the principal investigator. We aimed to extract the number of participants originally allocated to each treatment group in each trial to allow an intention‐to‐treat analysis if the trial had not already been presented in this way.
Assessment of risk of bias in included studies
We assessed risk of bias as specified in the Cochrane Handbook for Systematic Reviews of Interventions, Version 5.1.0 (March 2011), Chapter 8 (Higgins 2011). We assessed whether the method of randomisation would allow allocation concealment, the adequacy of efforts to blind treatment administration and outcome assessment. For each included trial we collected information about:
the method of randomisation (including information on allocation concealment);
blinding of treatment administration;
blinding of outcome assessment; and
whether an intention‐to‐treat analysis was done, or could possibly be done.
We provide detailed 'Risk of bias' tables for the trial included since the last update.
Measures of treatment effect
We extracted the number of participants in the treated and control groups who had:
died within the first seven to 10 days;
developed any intracranial haemorrhages, symptomatic or fatal intracranial haemorrhage early after the stroke (within the first seven to 10 days);
developed symptomatic (including fatal) infarct swelling;
died by the end of the trial follow‐up; and
were dependent on others in activities of daily living (mRS 3 to 5) by the end of the trial follow‐up period (the converse is the number who were alive and independent at the end of follow‐up).
We also extracted data to perform subgroup analyses on time to treatment, age, stroke severity, prior or concomitant antithrombotic drug use, and attempted to find information on pretreatment brain imaging findings, blood pressure, and diabetes (details below).
Unit of analysis issues
Our definition of SICH included people who died or deteriorated clinically as a result of intracranial haemorrhage. This could be either secondary bleeding into the infarct or new bleeding at an anatomically separate site elsewhere in the brain or its surrounding spaces after randomisation, confirmed by CT or MR scanning or post‐mortem examination. We have defined 'early after the stroke' as within the first seven to 10 days, as the trials each tended to use a slightly different time point, but all had collected information on intracranial haemorrhage certainly within the first 10 days. Many symptomatic haemorrhages actually occurred within the first few days of the stroke. It is difficult to estimate the exact number of SICHs because some people died without a CT scan or post‐mortem examination. Thus the true number with SICH may be higher than that suggested by these data. On the other hand, heightened awareness of an association between haemorrhagic transformation and thrombolysis may mean that the investigators too readily attributed any neurological deterioration following treatment to intracranial haemorrhage, even if the amount of blood was small. A review of published CT findings suggests that, at least for some trials, SICH included people with very large swollen and oedematous infarcts with trivial amounts of haemorrhage within them (ECASS 1995; NINDS 1995). Therefore, it is also possible that the risk of intracranial haemorrhage has been overestimated (Von Kummer 2002). The ECASS trial (ECASS 1995) did not report the number with SICH, but whether the radiological appearance of the haemorrhage suggested haemorrhagic transformation of an infarct or parenchymatous haematoma (and its size). Most parenchymatous haemorrhages were associated with symptoms, so we used the number of participants with parenchymatous haematoma as the number with symptomatic haemorrhages.
Dealing with missing data
We contacted trial investigators to obtain all unpublished missing data where possible. Where data were still missing or had not been collected in the original trial, then that trial did not contribute to the relevant outcome. We clarified missing or unclear data with the principal investigator. The outcomes in two studies were very clearly described in the original texts and verification with the principal investigators was not necessary (Haley 1993; Morris 1995).
Assessment of heterogeneity
We tested for heterogeneity between trial results using the I² statistic. Heterogeneity might arise from a wide variety of factors, such as the design of the trials, the type of participants included, the use of concomitant treatments like aspirin or heparin, ancillary care during the acute treatment period or rehabilitation, lack of availability of certain data for a particular trial so that a trial appears as missing for a particular outcome, or simply by the play of chance, particularly in small trials (Deeks 2001; Higgins 2003).
Assessment of reporting biases
We have endeavoured to include data from all trials on all prespecified outcomes, obtained from secondary publications or the trial investigators if unpublished. We assessed the likelihood of missing trials using a funnel plot.
Data synthesis
We calculated odds ratios (ORs) for each outcome (that is, the ratio of the odds of an unfavourable outcome among treatment‐allocated participants to the corresponding odds amongst controls), which we calculated using the Peto fixed‐effect method (APT 1994), and the random‐effects method for outcomes if there was significant heterogeneity between trials. We calculated absolute numbers of events avoided (or caused) per 1000 patients treated using the risk differences method provided in the Review Manager 5 software (RevMan 2012) and also as the straight percentages calculated from the number of events per number randomised in the treated and control groups. However, please note that these events per 1000 treated data should be regarded with caution as they may produce misleading results, since the absolute risk amongst controls varies between trials.
Subgroup analysis and investigation of heterogeneity
We examined the effect of stroke severity, age under or over 80 years, time from stroke to treatment and the effect of having a large infarct on plain CT (ASPECT score 7 or less) on outcome after thrombolysis. We assessed the effect of time by several approaches: we examined the effect of time in all trials regardless of what time windows they contributed to, then in only those trials that contributed to all time windows, and then by latest time to randomisation. These approaches were used to maximise use of available data and minimise bias by excluding some trials from some analyses (e.g. the NINDS 1995 trial only recruited participants up to three hours and therefore would not contribute to an analysis comparing treatment administered within three hours with that administered between three and six hours, where inclusion was restricted to trials which included participants in both time windows). We used the proportion who died in the control group to estimate the severity of stroke. We assessed:
effect of time to treatment; the number of participants who had symptomatic intracranial haemorrhage, died or were dependent at the end of follow‐up according to whether they had been treated within three hours of the stroke or later than three hours (in trials which randomised participants beyond three hours after the stroke);
the number of participants aged over or under 80 years who had symptomatic intracranial haemorrhage, died or were dependent at the end of follow‐up;
the number of participants who were dead or dependent at the end of follow‐up according to whether they had been assessed for inclusion in a trial using CT scanning or MR diffusion‐ and perfusion‐weighted imaging (DWI/PWI);
the number of participants alive and independent (mRS 0 to 1) at end of follow‐up according to whether they had visible or not‐visible or small or large infarction on plain CT, measured by the ASPECT score.
Sensitivity analysis
We examined primarily the effect of the thrombolytic drug in all studies for all drugs combined. However, we also examined the effect of different thrombolytic drugs (streptokinase, urokinase, rt‐PA). We assessed whether the effect of thrombolysis on functional outcome varied with the definition of dependency (mRS 2 to 5, instead of 3 to 5). Additionally, we compared trials which included participants on the basis of plain CT scanning versus those which used diffusion/perfusion MR imaging or perfusion/angiography CT imaging. We stratified trials by the proportion of participants given aspirin or heparin within the treatment period by time after stroke;
Results
Description of studies
Results of the search
The search of the Cochrane Stroke Group Trials Register identified 19 potentially relevant new or ongoing trials, of which only one was completed and relevant (IST3 2012). Five trials are ongoing (DIAS‐3; DIAS‐4; DIAS‐J; EXTEND; WAKE‐UP 2011). Three trials are awaiting classification (FRALYSE; Lin 2006; TESPI): TESPI has recently been completed but not yet reported, and the other two are thought to have been completed but have not yet been published. The remainder of the publications were not relevant. The search of MEDLINE and EMBASE identified 3958 references, which included many additional publications to trials that were already in the review, and several that were relevant to another review (Wardlaw 2013), but none that was relevant to this review.
Included studies
We include 27 trials, involving 10,239 randomised participants, but data for only 10,187 participants were available for inclusion in the review (Abe 1981; ASK 1996; Atarashi 1985; ATLANTIS A 2000; ATLANTIS B 1999; AUST 2005; Chen 2000; DEDAS 2006; DIAS 2005; DIAS 2 2008; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; Haley 1993; IST3 2012; JTSG 1993; MAST‐E 1996; MAST‐I 1995; MELT 2007; Mori 1992; Morris 1995; NINDS 1995; Ohtomo 1985; PROACT 1998; PROACT 2 1999; Wang 2003). This review includes all possible available information about all trials in an effort to provide as complete a record as possible of the available data on thrombolysis for acute ischaemic stroke. The NINDS trial (NINDS 1995) was conducted in two consecutive parts, A and B, but published in one paper, so is included as one trial in this review. Although the USA Food and Drug Administration review of the primary analysis of the NINDS trial referred to an 'on‐treatment' analysis, the analysis was actually 'intention‐to‐treat' as no participants who had been entered into the trial were excluded from that analysis (NINDS 1995). Reasons for these comments and further analyses are provided in the Clinical Reviews submitted by Genentech to the USA Food and Drug administration in support of the license application for alteplase (www.fda.gov/cder/biologics/products/altegen061896.htm; see Clinical Review 2, pages 18 to 20).
The trials performed in the 1980s (Abe 1981; Atarashi 1985; Ohtomo 1985) were methodologically different to the trials performed from the 1990s onwards. The 1980s trials used very low doses of thrombolytic drug, given daily intravenously for several days, and started up to five or 14 days after the stroke. The trials from the 1990s onwards used a single large dose of thrombolytic drug (in the region of 80 mg to 100 mg rt‐PA or equivalent), given intravenously or intra‐arterially, in most trials, within three, six, nine or 24 hours of the stroke. The 1980s trials did not collect data on functional outcome and therefore only the 1990s‐onwards trials contribute to the analysis of death or dependency. All trials, however, contributed to analyses of intracranial haemorrhage and death by the end of follow‐up (although very few deaths or intracranial haemorrhages occurred in the trials in the 1980s). However, it is possible to see in the figures what effect the exclusion of these early trials would have on the overall results.
The MAST‐I trial (MAST‐I 1995), which tested intravenous streptokinase and oral aspirin given within six hours of stroke onset in a two‐by‐two factorial design, is the only trial to have tested for an interaction between thrombolytic and antithrombotic drugs in a randomised trial; the comparison of streptokinase plus aspirin versus aspirin from MAST‐I 1995 is included in this review (separated from the MAST‐I 1995 data in the absence of aspirin) because it represents the only available randomised evidence on this important interaction. As there was a significant adverse interaction between streptokinase and aspirin, which we felt was important to highlight, the data for the participants receiving streptokinase in the presence or absence of aspirin are presented separately (that is, streptokinase versus control separate from streptokinase with aspirin versus aspirin). It would not be methodologically appropriate to exclude MAST‐I 1995 participants allocated aspirin because in most other trials, some antithrombotic agents were used, and while it is possible to identify the proportion of participants in the trial that received antithrombotic treatment, it is not possible to identify and then exclude the individual participants.
Types and severities of strokes included
The selection of participants was based initially on clinical criteria to diagnose the stroke sub‐type (cortical versus lacunar versus posterior circulation):
eight trials randomised all types of ischaemic stroke: cortical, lacunar and posterior circulation (ATLANTIS A 2000; ATLANTIS B 1999; ECASS 3 2008; Haley 1993; IST3 2012; MAST‐I 1995; NINDS 1995; Wang 2003);
two trials included cortical and lacunar strokes (ASK 1996; Chen 2000);
five trials included only participants with symptoms of hemispheric cortical ischaemia (ECASS 1995; ECASS II 1998; EPITHET 2008; MAST‐E 1996; Morris 1995);
six trials included participants with angiographically proven occlusion of the internal carotid or middle cerebral artery (JTSG 1993; MELT 2007; Mori 1992; PROACT 1998; PROACT 2 1999) or vertebrobasilar arteries (AUST 2005);
three trials included presumed 'thrombotic' stroke of most severities and excluded presumed cardio‐embolic strokes (although it is not clear whether artery‐to‐artery embolism counted as 'embolic' in this context) (Abe 1981; Atarashi 1985; Ohtomo 1985);
three trials included participants with 'tissue at risk' as identified by MR DWI/PWI or CT perfusion imaging (DEDAS 2006; DIAS 2005; DIAS 2 2008).
Most trials used a stroke severity scale, such as the National Institutes of Health Stroke Scale (NIHSS) or Scandinavian Stroke Scale (SSS) or developed their own neurological stroke severity scale to measure the severity of the stroke at baseline.
All trials excluded people who were in a coma; most trials did not randomise many participants who were drowsy except one (MAST‐E 1996) in which 50% of the participants were drowsy or stuporous at randomisation.
Age of included participants
Only seven trials had no upper age limit at all and included also very elderly participants (Abe 1981; Atarashi 1985; EPITHET 2008; IST3 2012; MAST‐E 1996; MAST‐I 1995; Ohtomo 1985). EPITHET included 25 participants aged over 80 years and IST‐3 included 1617 participants aged over 80 years.
Seven trials had an upper age limit of 85 years (ASK 1996; AUST 2005; DEDAS 2006; DIAS 2005; DIAS 2 2008; PROACT 1998; PROACT 2 1999).
The NINDS trial (NINDS 1995) initial protocol stated an upper age limit of 80 years. However, this was removed after 188 participants had been recruited into Part A of the trial on 30 March 1992, and thereafter 69 participants over the age of 80 were randomised (the oldest participant was 90) (www.fda.gov/cber/products/altegen061896.htm; Clinical Review 2, page 27).
All the remaining trials, including all the other rt‐PA trials, (except EPITHET 2008; and IST3 2012), had an upper age limit of 80 years.
The upper age limit in two trials (Chen 2000; MELT 2007) was 75 years.
Visible infarction on the CT scan at randomisation
Three trials specified that the pre‐randomisation CT had to be normal (JTSG 1993; MELT 2007; Mori 1992).
One trial excluded people with early visible infarction (Wang 2003).
Six trials specified that the pre‐randomisation CT scan had to be normal or only show ischaemic changes in less than one‐third of the middle cerebral artery supply territory (ATLANTIS B 1999; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; Wang 2003).
Two trials excluded people with mass effect and midline shift on CT (PROACT 1998; PROACT 2 1999).
None of the other trials specified that people with a CT scan that showed an infarct (which was likely to be symptomatic) should be excluded, although individual doctors may have excluded these individuals in some centres depending on local opinion.
Three trials selected participants with 'tissue at risk' on the basis of DWI/PWI (DEDAS 2006; DIAS 2005), or MR DWI/PWI or CT with CT perfusion imaging (DIAS 2 2008).
Time to randomisation
The maximum time interval allowed between the onset of the stroke and the start of the treatment administration varied from within three hours to up to two weeks.
Two trials randomised participants within three hours (Haley 1993; NINDS 1995).
One trial randomised participants within four hours (ASK 1996).
One trial randomised participants between three hours and 4.5 hours (ECASS 3 2008).
One trial randomised participants between three hours and five hours (ATLANTIS B 1999).
14 trials randomised participants within six hours (ATLANTIS A 2000; Chen 2000; ECASS 1995; ECASS II 1998; IST3 2012; JTSG 1993; MAST‐E 1996; MAST‐I 1995; MELT 2007; Mori 1992; Morris 1995; PROACT 1998; PROACT 2 1999; Wang 2003). However, please note that in three studies (MELT 2007; PROACT 1998; PROACT 2 1999), the majority of the participants were actually randomised between three and six hours.
One trial randomised participants within three to six hours (EPITHET 2008).
Three trials randomised participants between three and nine hours (DEDAS 2006; DIAS 2005; DIAS 2 2008).
One trial randomised participants within 24 hours (AUST 2005).
Two trials randomised participants within five days (Atarashi 1985; Ohtomo 1985).
One trial randomised participants within two weeks (Abe 1981).
Please note that the latter three trials (Abe 1981; Atarashi 1985; Ohtomo 1985) do not contribute data to the analysis of early deaths or of death and dependency, as early deaths were not recorded and a functional outcome measure was not used in these trials. They do contribute data to the analyses of intracranial haemorrhages and deaths by the end of follow‐up.
Drug and dosage
Trials using intravenous rt‐PA contribute 7012 of the 10,187 participants, that is, 69% of the data in this review. Data and outcomes of all included substances are reported for completeness. However, rt‐PA data are also given as appropriate.
Four trials used streptokinase (ASK 1996; MAST‐E 1996; MAST‐I 1995, Morris 1995).
Twelve trials used recombinant tissue plasminogen activator (rt‐PA) (ATLANTIS A 2000; ATLANTIS B 1999; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; Haley 1993; IST3 2012; JTSG 1993; Mori 1992; NINDS 1995; Wang 2003).
Six used urokinase (UK) (Abe 1981; Atarashi 1985; AUST 2005; Chen 2000; MELT 2007; Ohtomo 1985).
Two used pro‐urokinase (pro‐UK) (PROACT 1998; PROACT 2 1999).
Three used desmoteplase (DEDAS 2006; DIAS 2005; DIAS 2 2008).
The mode of administration was intravenous in most trials.
-
In all except four of the above trials, the thrombolytic agent was administered intravenously.
In two studies (AUST 2005; MELT 2007) the thrombolytic agent was given intra‐arterially into the cerebral circulation.
Two studies used recombinant pro‐urokinase (rpro‐UK) given intra‐arterially into the cerebral circulation (PROACT 1998; PROACT 2 1999).
Please note that trials testing lumbrokinase did not meet the inclusion criteria for this review. Ongoing trials are testing other new thrombolytic agents such as microplasmin or tenecteplase (see Characteristics of studies awaiting classification and Characteristics of ongoing studies).
The doses were:
the streptokinase dose was 1.5 MU (as used to treat acute myocardial infarction) in four studies (ASK 1996; MAST‐E 1996; MAST‐I 1995; Morris 1995);
the rt‐PA dose was similar to that used to treat acute myocardial infarction at 1.1 mg/kg to a maximum of 100 mg in one study (ECASS 1995); about 20% less at 0.9 mg/kg to a maximum of 90 mg in eight studies (ATLANTIS A 2000; ATLANTIS B 1999; ECASS II 1998; ECASS 3 2008; EPITHET 2008; Haley 1993; IST3 2012; NINDS 1995); either 0.7 or 0.9 mg/kg in one study (Wang 2003); and about one‐third of 0.9 mg/kg in two studies (JTSG 1993; Mori 1992). All streptokinase and rt‐PA doses were administered by intravenous infusion through a peripheral arm vein, over one hour.
the urokinase dose in the Chinese UK trial (Chen 2000) was 1.5 or 1.0 MU intravenously over 30 minutes (considered to be similar to that used to treat acute myocardial infarction); in three studies (Abe 1981; Atarashi 1985; Ohtomo 1985) the urokinase dose was much lower than the equivalent for acute myocardial infarction and was administered intravenously once daily for seven days. The intra‐arterial urokinase dose in one study (AUST 2005) was up to 1.0 MU maximum and in another study (MELT 2007) was up to 60,000 IU;
the rpro‐UK dose was 6 mg in PROACT 1998 and 9 mg in PROACT 2 1999: in both trials it was given intra‐arterially, through a catheter with its tip embedded in the occluding thrombus;
the dose of desmoteplase was 62.5 μ/kg, 90 μ/kg or 125 μ/kg in one study (DIAS 2005), and 90 μ/kg or 125 μ/kg in two studies (DEDAS 2006; DIAS 2 2008).
Concomitant use of antithrombotic treatment
One trial (MAST‐I 1995) compared streptokinase versus control among participants who were either allocated to aspirin, or allocated to no aspirin, started within six hours of stroke onset, in a factorial randomisation (in the groups randomised to receive aspirin, it was continued for 10 days).
Antithrombotic use was not randomly assigned in any other trial and its permitted use varied:
in one study ASK 1996) all participants were to receive 300 mg aspirin starting within four hours of the streptokinase infusion and continued daily thereafter;
in one study (PROACT 1998) all participants were to receive 1000 u/hour intravenous heparin during the trial angiogram, reduced to 500 u/hour halfway through the trial;
in one study (PROACT 2 1999) all participants were to receive intravenous heparin 500 u/hour for four hours starting at the time of the angiogram infusion;
in one study (AUST 2005) all participants received 5000 IU heparin intra‐arterially followed by intravenous heparin to a target activated partial thromboplastin time (APTT) of 60 to 80 seconds for a minimum of two days followed by oral warfarin to a target international normalised ratio (INR) of 1.5 to 2.5 for six months;
in one study (MAST‐E 1996) aspirin and intravenous heparin were allowed to start at any time and continue for any time (about 25% of participants received aspirin or heparin within 24 hours and 75% within the first week of the stroke);
in three studies (ECASS 1995; ECASS II 1998; ECASS 3 2008) subcutaneous heparin was allowed within 24 hours of the stroke (and thereafter) and aspirin after 24 hours (in ECASS II 1998, about 20% of participants were taking aspirin at the time of their stroke and 54% of rt‐PA‐treated participants received subcutaneous heparin within the first 24 hours, but we are unsure of the corresponding numbers for ECASS 1995, or ECASS 3 2008, nor how many participants in either trial received aspirin or heparin after 24 hours);
in one study (Haley 1993) a few participants received antithrombotic drugs within 24 hours and thereafter;
in 10 studies (ATLANTIS A 2000; ATLANTIS B 1999; Chen 2000; DEDAS 2006; DIAS 2005; DIAS 2 2008; IST3 2012; MELT 2007; Mori 1992; NINDS 1995) no antithrombotic drugs were allowed within 24 hours but aspirin was allowed thereafter;
in three studies (Abe 1981; Atarashi 1985; Ohtomo 1985) antithrombotic drugs were not allowed during the seven days of treatment infusion, but could be used thereafter;
the antithrombotic drug use is not stated clearly three studies (EPITHET 2008; JTSG 1993; Morris 1995).
Follow‐up
Early outcome assessments were made at around seven to 10 days in most trials. Some trials also performed more frequent assessments in the first few hours and days after the trial treatment. In this review, outcome events occurring within the first seven to 10 days (whichever was the later date at which data were collected) have been used to determine the effect of thrombolytic therapy on early outcome.
The final outcome assessment was at:
about one month after the stroke (Abe 1981; Atarashi 1985; JTSG 1993; Mori 1992; Morris 1995; Ohtomo 1985);
three months after the stroke (ASK 1996; ATLANTIS A 2000; ATLANTIS B 1999; Chen 2000; DEDAS 2006; DIAS 2005; DIAS 2 2008; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; Haley 1993; MELT 2007; NINDS 1995; PROACT 1998; PROACT 2 1999; Wang 2003); and
six months after the stroke (AUST 2005; IST3 2012; MAST‐E 1996; MAST‐I 1995).
Please note that follow‐up at six months and one year have subsequently been reported for one study (NINDS 1995), but the three‐month outcome, the primary outcome originally reported, is used in this review. This also occurred in another study (IST3 2012) where the primary six‐month outcome originally reported is used, even if the 18‐month follow‐up, of a predefined vast majority of participating countries, has been subsequently reported.
Please note that because of the difficulty of blinding the biological effect of thrombolytic therapy, it is important to ensure that outcome assessment is blinded and objective. Follow‐up should therefore be performed by individuals unaware of the trial treatment allocation either because they have not been involved in the administration of the trial treatment, or in the care of the participant during at least the first few days. In one study (MAST‐I 1995) the six‐month follow‐up was by telephone by a trained observer blind to the treatment allocation. In another study (IST3 2012) the six‐month follow‐up was blinded and performed either by postal mail or telephone by a trained observer blind to the treatment allocation. Seven studies (ASK 1996; DEDAS 2006; DIAS 2005; DIAS 2 2008; EPITHET 2008; MAST‐E 1996; Wang 2003) did not specify who performed the follow‐up or that they should not have been involved in the trial treatment administration or participant care in the first 24 hours. In five studies (ATLANTIS A 2000; ATLANTIS B 1999; Chen 2000; ECASS 3 2008; NINDS 1995), follow‐up at all stages was done by a doctor who had not been involved in the randomisation or care of the participant in the first 24 hours. In four studies (ECASS 1995; ECASS II 1998; PROACT 1998; PROACT 2 1999), follow‐up was by a mixture of individuals; if possible, by someone who had not been involved in the participant's care within the first 24 hours but this may not always have been the case.
Assessment of functional outcome
The assessment of functional outcome was by:
the Barthel Scale in four studies (ASK 1996; JTSG 1993; Mori 1992; Morris 1995);
an undefined scale (no, mild, moderate or severe 'limitation') in one study (Haley 1993);
the Rankin Scale in two studies (MAST‐E 1996; MAST‐I 1995);
the modified Rankin Scale in 16 studies (ATLANTIS A 2000; ATLANTIS B 1999; AUST 2005; Chen 2000; DEDAS 2006; DIAS 2005; DIAS 2 2008; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; MELT 2007; NINDS 1995; PROACT 1998; PROACT 2 1999; Wang 2003);
the Oxford Handicap Scale (OHS), a commonly used variant of the modified Rankin score, was used in one study (IST3 2012);
it was not assessed in three studies (Abe 1981; Atarashi 1985; Ohtomo 1985).
Some trials used more than one scale to measure outcome; for example, six studies (ATLANTIS A 2000; ATLANTIS B 1999; DEDAS 2006; DIAS 2005; DIAS 2 2008; NINDS 1995) favoured a 'Global Outcome Statistic' which involved collecting Barthel, Rankin, Glasgow Outcome Score and NIHSS scores individually and then combining the four scores. Three trials (Abe 1981; Atarashi 1985; Ohtomo 1985) used the 'Global Improvement Rating', which measures change in neurological status and safety outcome as a composite surrogate for functional outcome.
There are differences in the primary outcome measure used between trials, in that some used a 'poor functional outcome' and some used a 'good outcome'. The following trials sought 'dependency' (that is, whether the participant was dependent or not in activities of daily living) as a measure of poor functional outcome: two studies (MAST‐E 1996; MAST‐I 1995) defined dependency as Rankin 3 or worse, and two studies (ASK 1996; Morris 1995) defined dependency as a Barthel score of 60 or worse. In one study (IST3 2012) 'alive and independent' (OHS 0 to 2; mRS 0 to 2) was the primary measure of outcome. The 'alive and favourable outcome' (mRS 0 to 1) and ordinal analysis were included in prespecified secondary outcome analyses.
Thirteen trials sought 'good functional outcome' (that is, whether the participant had made a complete or virtually complete recovery) defining 'good outcome' as mRS 0 or 1 (ATLANTIS A 2000; ATLANTIS B 1999; Chen 2000; DEDAS 2006; DIAS 2005; DIAS 2 2008; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; MELT 2007; NINDS 1995; Wang 2003).
For most trials, it has been possible to obtain data on participants in each individual Rankin (or Barthel) group, or data dichotomised on Rankin 0 to 2 versus 3 to 6, or 0 to 1 versus 2 to 6, so that dependency in this review refers to Rankin, (mRS or OHS) 3 to 5 (6 being dead) unless otherwise stated. There are only two trials for which the number of participants in individual Rankin groups is not so far available (and therefore the data shown are for Rankin 2 or worse) (ATLANTIS A 2000; PROACT 1998).
Excluded studies
We excluded two trials conducted prior to the availability of CT scanning (Meyer 1963; Meyer 1964) as there was no way of confirming that the stroke was ischaemic. One small trial of intra‐arterial streptokinase stopped prematurely after four participants had been randomised due to the impracticality of the intra‐arterial technique (Edinburgh 1991). A trial started in Hong Kong was abandoned after a few participants had been randomised because of concerns that streptokinase might cause too many haemorrhages (Hong Kong 1994) (two trials (ASK 1996; MAST‐E 1996) had both just stopped prematurely creating an adverse climate for the conduct of trials testing streptokinase). We excluded one trial (Naito 1984) after discussion with Professor T Abe (co‐investigator) as it was not possible to account for 11 of the 101 randomised participants (most of whom were in the control group). We excluded six trials conducted in China, two because of confounding (Xiang 1995; Yuan 1995), one because the duration of follow‐up was only three weeks (Pang 1993), two that evaluated oral lumbrokinase thrice daily for 21 days but within an unspecified time window and without clinical outcome assessment (Jin Urokinase metaanalysis 2000; Huang 2000), and one that assessed ahylysantifarctase but lacked clinical outcomes (Liu 1994). A further 73 trials have also been excluded due to a range of reasons given in the Characteristics of excluded studies table. Studies that were potentially relevant but were confounded are listed in the Characteristics of excluded studies table and the reason given.
Risk of bias in included studies
We have included 27 trials: six trials using intravenous thrombolytic therapy published prior to 1995, 17 trials from 1995 to 2012, and four trials using intra‐arterial thrombolytic therapy.
Allocation
Among the included studies 14 (52%) fulfilled criteria for high grade concealment. The concealment has successively improved over time with the development and utilisation of new randomisation methods, such as the use of a centralised computerised method with interactive interface for randomisation over the telephone or Internet.
Twelve trials used central telephone or Internet randomisation (ASK 1996; AUST 2005; DEDAS 2006; DIAS 2005; DIAS 2 2008; ECASS 3 2008; IST3 2012; MAST‐E 1996; MAST‐I 1995; MELT 2007; PROACT 1998; PROACT 2 1999). In five studies (AUST 2005; IST3 2012, MAST‐I 1995; MELT 2007; PROACT 2 1999), the allocated treatment was then given unblinded without a placebo. In one of the studies (IST3 2012) with the exception of 276 participants treated in the double‐blinded phase of the trial, the remaining participants (2759) were treated unblinded without placebo. In four other studies (ASK 1996; ECASS 3 2008; MAST‐E 1996; PROACT 1998) sealed prepacks of thrombolytic drug or identical‐appearing placebo were given according to the randomisation instructions.
In three trials randomisation was at the participating hospital by selection of a sealed, sequentially‐numbered, prepack (of active drug or identical appearing placebo) followed within two hours by a telephone call to the Central Trial Co‐ordinating Office to notify them of the participant and the number of the drug pack (ATLANTIS A 2000; ATLANTIS B 1999; NINDS 1995). In one study (NINDS 1995), the randomisation system, set up in an effort to reduce delays to treatment, led to 'out of order' trial treatment allocations in between 13 and 31 participants, which affected every subsequent participant until the error was detected, and led to participants appearing to cross between treatment allocations (more moved from rt‐PA to placebo than the other way round). Also in the interests of reducing delays to trial treatment administration, there were some participants who ultimately were not entered into the study after the pharmacy had prepared the trial pack (and therefore some discarded trial packs). Details of the randomisation are given at www.fda.gov/cber/products/altegen061896.htm; see Clinical Review 2, page 11‐12 and 18‐19.
In three trials, randomisation was by selection of a sequentially numbered, sealed drug prepack at the participating centre provided by the sponsor from a randomisation schedule drawn up centrally (ECASS 1995; ECASS II 1998; EPITHET 2008).
Of the remaining trials:
five trials used sealed drug prepacks of active drug or identical‐appearing placebo (Abe 1981; Atarashi 1985; JTSG 1993; Mori 1992; Ohtomo 1985);
one used sealed envelopes (Haley 1993);
one used sealed drug prepacks of active drug or normal saline (as placebo) (Chen 2000);
the method was not stated in two (Morris 1995; Wang 2003).
Please note that, therefore, only two of the rt‐PA trials (ECASS 3 2008: IST3 2012) recorded the participant details centrally over the telephone or Internet prior to starting trial treatment. In one of these trials (IST3 2012) a minimisation algorithm was used to balance the study arms for key prognostic variables like stroke severity before randomisation. Several later studies have made use of modern randomisation techniques and entering key prognostic variables into the IT system before randomisation, which allows balancing of the study arms ‐ as has been introduced in one trial (IST3 2012) .
Blinding
Five trials were single‐blind without a placebo (AUST 2005; MAST‐I 1995; MELT 2007; PROACT 2 1999; Wang 2003). In one trial (IST3 2012) the first 276 participants were treated in the double‐blinded phase of the trial and all 2759 remaining participants were included into the open phase of the trial. All participants in the study, irrespective of study phase, were blindly assessed by postal mail or telephone by a blinded and trained observer. In PROACT 2 1999, the control group underwent catheter placement but received no infusion. All the rest were double‐blind placebo‐controlled trials. However, it should also be noted that thrombolysis, due to its effects on the coagulation system at high doses, can be difficult to blind completely due to the obvious signs of bleeding (prolonged bleeding at venepuncture sites, easy bruising, gingival or conjunctival haemorrhages, etc). Thus, provision of an identical‐appearing placebo (in the syringe) may not fully blind investigators to treatment allocation. Furthermore, as thrombolytic agents are proteins, they froth when shaken in solution with water or saline, rather like egg white mixed with water and shaken. Normal saline is therefore not an identical‐looking placebo for a thrombolytic agent. Thus, in addition to the possibilities for failure of treatment allocation concealment inherent in the randomisation methods used as outlined above, it is possible that treatment allocation could be guessed accurately by the physicians caring for the participant in the acute phase because of these biological effects. Accordingly, methods for ensuring complete blinding of treatment allocation at late follow‐up are crucial. Only one study (MAST‐I 1995) used central telephone follow‐up by a blinded trained observer. Although seven other trials specified that follow‐up was to be by a physician not involved in the acute care of the participant, it is uncertain how completely this was achieved in practice. Other trials either did not specify who should do the follow‐up, or did not make it mandatory that follow‐up was by an independent physician, so in either case follow‐up may have been carried out by the acute phase physician who could have been influenced by their knowledge of events in the acute phase.
Incomplete outcome data
All available data are included. Data on six participants were missing from the ATLANTIS B 1999 trial publication and details have not been forthcoming from the investigators, and we have not yet received data on 46 participants from the Chinese UK Trial (Chen 2000) (these participants were randomised after the trial's six‐hour time limit and have not yet been supplied). More information is available for some trials than for others, either because the trial collaborators have published very actively on various aspects of their trial, or because in some cases further information is available from other sources (for example, reports on NINDS 1995 appear on the US Food and Drug Administration (FDA) website as part of the licence application process). The more frequent reporting or greater completeness of the data for some trials is merely a reflection that more information is available for those trials, and not intended to over‐ or under‐emphasise the actual results or quality of any particular trial (or trials) compared with others for which there is less detailed information available.
Selective reporting
We have avoided, as far as possible, any reporting bias by obtaining original data from the trial investigators where these have not been published. Only the intention‐to‐treat results are included here. In any trials where there have been exclusions, these were made prior to the breaking of the randomisation code. A strict intention‐to‐treat analysis was used in 18 studies (ASK 1996; ATLANTIS A 2000; ATLANTIS B 1999; AUST 2005; DEDAS 2006; DIAS 2005; DIAS 2 2008; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; IST3 2012; MAST‐E 1996; MAST‐I 1995; MELT 2007; PROACT 1998; PROACT 2 1999; Wang 2003), but not in any of the earlier trials. The administrative problems with randomisation in one study (NINDS 1995) led the FDA reviewer to describe the primary analysis as an 'on‐treatment analysis'. However, the primary analysis was undertaken without excluding any participants entered into the trial and was, therefore, an intention‐to‐treat analysis (www.fda.gov/cber/products/altegen061896.htm; see Clinical Review 2, page 20). For the earlier trials, with additional information from the principal investigators if necessary, we have attempted to find a final outcome for all randomised participants, rather than simply relying on the published data from which some randomised participants may have been excluded. Note that one trial (ECASS 1995) was published as intention‐to‐treat and as a 'target population' after about 20% of the randomised participants had been excluded, but only the intention‐to‐treat data have been included here.
Other potential sources of bias
Randomisation in two trials, ASK 1996 (in the over‐three‐hour group) and MAST‐E 1996, was stopped on the advice of their respective data monitoring committees after only about half of the originally intended number of participants had been randomised. One study (MAST‐I 1995) was suspended by its steering committee (in view of the stopping of MAST‐E 1996 and ASK 1996) to examine its interim results after randomising about one third of its originally intended number. Another study (MELT 2007) was discontinued on the advice of its data monitoring committee when rt‐PA was licensed in Japan in 2005. Another study (AUST 2005) was discontinued on the basis of very slow recruitment after 24 participants of a planned sample of 200 had been included. Four studies (ECASS 1995; ECASS II 1998; NINDS 1995; PROACT 2 1999) all reached their planned targets. One study (PROACT 1998) was stopped after completing two of its planned three dosage arms by the pharmaceutical provider. Another study (ATLANTIS A 2000) was stopped on publication of the NINDS 1995 trial, and continued in modified form as ATLANTIS B 1999, which in turn stopped in 1998 following a 'futility analysis' prompted by results from the ECASS II 1998 study. Examination of funnel plots for the main outcomes showed these to be symmetrical and therefore provided little evidence of publication bias.
Effects of interventions
See Data and analyses. Note that in each analysis, trials are grouped by thrombolytic drug and whether intravenous or intra‐arterial, with a subtotal odds ratio (OR) for that group. The overall OR for all trials appears at the bottom of each plot. Note that one study (MAST‐I 1995) appears twice in the analyses because the data in participants allocated aspirin have been entered separately from the participants allocated no aspirin. Also note that some outcomes have fewer trials contributing data than other outcomes. This is because not all trials collected data on all outcomes examined in this review, or if they did collect data on the particular outcome, it may not be available. If data were available for a particular outcome, then the trial appears listed in the relevant analysis. The 2012 systematic review and meta‐analysis of rt‐PA (Wardlaw 2012) conducted a comparison of the 11 earlier rt‐PA studies (ATLANTIS A 2000; ATLANTIS B 1999; ECASS 1995; ECASS 3 2008; ECASS II 1998; EPITHET 2008; Haley 1993; JTSG 1993; Mori 1992; NINDS 1995; Wang 2003) and IST3 2012 on its own, and analysed the effect of adding IST‐3 to the 11 earlier trials. That analysis is not repeated here.
Deaths from all causes within seven to 10 days
Data on deaths occurring within the first seven to 10 days were available for 13 trials (7458 participants; Analysis 1.1). Amongst the larger and more recently completed trials, data were not available for seven trials (ATLANTIS A 2000; ATLANTIS B 1999; DEDAS 2006; DIAS 2005; NINDS 1995; PROACT 1998; PROACT 2 1999). There was a significant excess of early deaths with thrombolysis: 11.5% of those allocated to thrombolytic therapy died compared with 7.4% of those allocated to control (OR 1.69, 95% confidence interval (CI) 1.44 to 1.98, P < 0.00001). In absolute terms, if confirmed, this is an increase of 40 (95% CI 30 to 55) early deaths per 1000 participants treated with thrombolysis. There was borderline significant heterogeneity (I² = 41%).
1.1. Analysis.
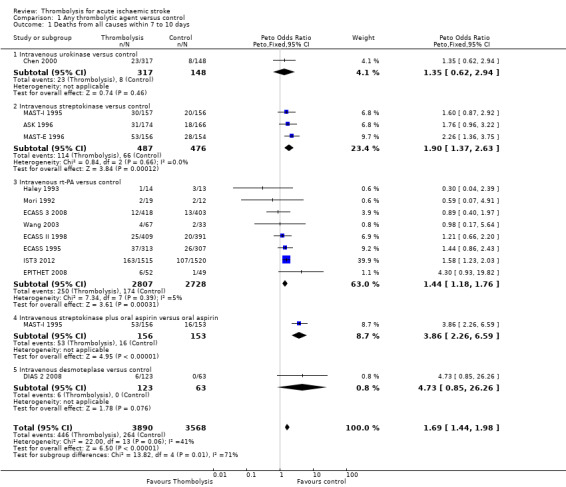
Comparison 1 Any thrombolytic agent versus control, Outcome 1 Deaths from all causes within 7 to 10 days.
Data on early deaths were available for eight trials using intravenous rt‐PA. The numerical (tabular) data on early deaths for the NINDS trial (NINDS 1995) have not been published, but the NINDS trial did publish a survival curve which suggested that fewer deaths occurred in the rt‐PA‐treated participants from 24 hours after treatment onwards. The tabular data available from the other rt‐PA trials showed a significant excess of early deaths: the OR was 1.44 (95% CI 1.18 to 1.76, P = 0.0003; 5535 participants) with no significant heterogeneity; the absolute effect was 25 more (95% CI 11 to 40 more) deaths per 1000 participants treated. In the three trials using streptokinase, there was also a significant excess of early deaths (OR 1.90, 95% CI 1.37 to 2.63; 963 participants).
We also performed an analysis of the data using a random‐effects model. This also shows a statistically significant excess of deaths with thrombolysis of similar magnitude to the fixed‐effect analysis (all trials: OR 1.68, 95% CI 1.30 to 2.16, P < 0.0001; just trials of rt‐PA: OR 1.44 , 95% CI 1.18 to 1.77, P = 0.0004).
Fatal intracranial haemorrhage within seven to 10 days
Data were available from 17 trials on fatal intracranial haemorrhage (9066 participants; Analysis 1.2). There are 10 trials for which this outcome is not currently available (Abe 1981; AUST 2005; DEDAS 2006; DIAS 2005; DIAS 2005; EPITHET 2008; JTSG 1993; Mori 1992; PROACT 2 1999; Wang 2003). This outcome may underestimate the frequency of intracranial haemorrhage since some of the participants who died without a post‐mortem examination or CT scan may have died of intracranial haemorrhage. There was a significant, approximate six‐fold increase in the rate of fatal intracranial haemorrhage with thrombolysis (4.19% of participants allocated to thrombolysis compared with 0.65% of those allocated to control, OR 4.53, 95% CI 3.47 to 5.91, P < 0.00001). There was no statistically significant heterogeneity (I² = 0%).
1.2. Analysis.
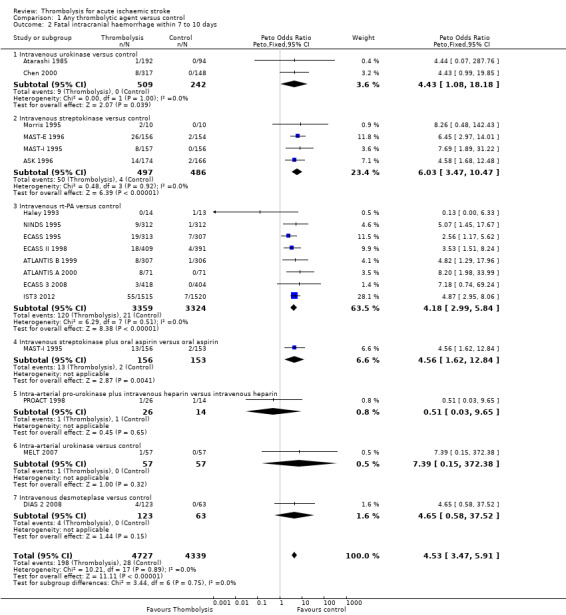
Comparison 1 Any thrombolytic agent versus control, Outcome 2 Fatal intracranial haemorrhage within 7 to 10 days.
In eight trials using rt‐PA, there were 30 (95% CI 20 to 40) extra fatal intracranial haemorrhages per 1000 participants treated (OR 4.18, 95% CI 2.99 to 5.84, P < 0.00001; 6683 participants) with no statistically significant heterogeneity between trials (I² = 0%).
The combination of streptokinase with aspirin in one study (MAST‐I 1995) significantly increased fatal intracranial haemorrhage (OR 4.56, 95% CI 1.62 to 12.84; 309 participants), and more participants died of cerebral causes without a CT scan or autopsy who may therefore also have had intracranial haemorrhage than in the group who received aspirin alone.
Deaths within the first seven to 10 days from causes other than fatal intracranial haemorrhage
We calculated the effect of thrombolysis on death from causes other than fatal intracranial haemorrhage for the 10 trials that provided data on both early death and fatal intracranial haemorrhage (7226 participants; Analysis 1.3). Note that, unfortunately, this excludes several large trials (ATLANTIS A 2000; ATLANTIS B 1999; DEDAS 2006; DIAS 2005; NINDS 1995; PROACT 1998; PROACT 2 1999), which did not provide data on early death. There were 264/3752 (7.0%) non‐intracranial haemorrhage deaths in the thrombolysis‐treated participants and 234/3474 (6.7%) in the control participants (OR 1.08, 95% CI 0.90 to 1.30, P = 0.39) with significant between‐trial heterogeneity (I² = 53%, P = 0.02). In comparison with Analysis 1.2 this suggests that most of the excess in early deaths of 42 per 1000 treated with thrombolysis is attributable to intracranial haemorrhage.
1.3. Analysis.
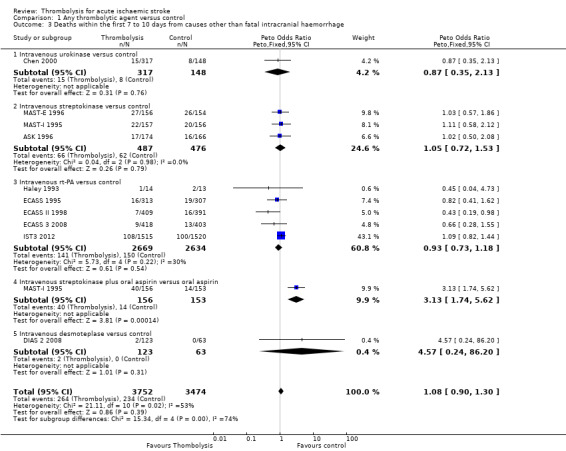
Comparison 1 Any thrombolytic agent versus control, Outcome 3 Deaths within the first 7 to 10 days from causes other than fatal intracranial haemorrhage.
In participants treated with rt‐PA (ECASS 1995; ECASS 3 2008; ECASS II 1998; Haley 1993; IST3 2012), 141/2669 (5.2%) died within the first seven to 10 days of causes other than intracranial haemorrhage, compared with 150/2634 (5.7%) in the control group, OR 0.93, 95% CI 0.73 to 1.18, P = 0.54, I² = 30%; 5303 participants),
Symptomatic (including fatal) intracranial haemorrhage within seven to 10 days
All trials provided data on intracranial haemorrhage and most provided them in a form that made it clear how many participants had suffered a neurological deterioration associated with the appearance of new haemorrhage in the brain on a CT or MR brain scan or at post‐mortem examination (10,186 participants; Analysis 1.4). There was a highly significant four‐fold increase in symptomatic intracranial haemorrhage with thrombolysis in 7.5% of those allocated to thrombolysis versus 1.7% of those allocated to control (OR 3.75, 95% CI 3.11 to 4.51, P < 0.00001) with no statistically significant between‐trial heterogeneity (P = 0.36). This represents an extra 60 (95% CI 50 to 65) symptomatic intracranial haemorrhages per 1000 participants treated.
1.4. Analysis.
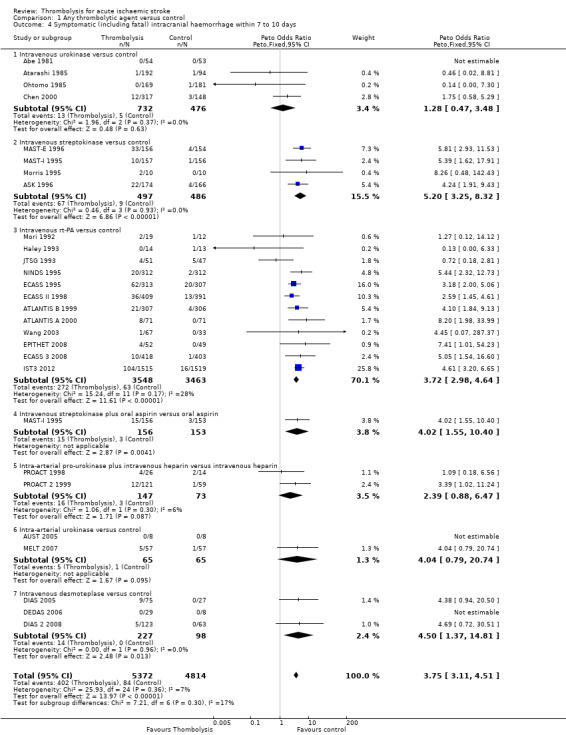
Comparison 1 Any thrombolytic agent versus control, Outcome 4 Symptomatic (including fatal) intracranial haemorrhage within 7 to 10 days.
In 12 trials using rt‐PA, there were 60 (95% CI 50 to 70) extra symptomatic intracranial haemorrhages per 1000 participants treated (OR 3.72, 95% CI 2.98 to 4.64, P < 0.00001; 7011 participants) with no heterogeneity between trials (I² = 28%, P = 0.17).
Excluding the trials that used lower doses of thrombolysis and had lower rates of fatal and symptomatic intracranial haemorrhage had little effect on the overall result as they contributed relatively few of the data to this analysis.
Symptomatic (including fatal) cerebral oedema
Six trials all testing rt‐PA provided data on symptomatic including fatal infarct swelling (ATLANTIS B 1999; ECASS 1995; ECASS II 1998; ECASS 3 2008; IST3 2012; NINDS 1995) (Analysis 1.5; 5961 participants). There was no overall reduction in symptomatic infarct swelling with thrombolysis: 10.2% of those allocated thrombolysis had symptomatic infarct swelling compared with 10.4% of those allocated control (OR 0.97, 95% CI 0.79 to 1.19, P = 0.75) with significant heterogeneity (I² = 71%, P = 0.004). Due to the heterogeneity we undertook an analysis according to a random‐effects model. This gave very similar results (OR 0.79, 95% CI 0.62 to 1.51, P = 0.88), and identical heterogeneity compared with the fixed‐effect model.
1.5. Analysis.
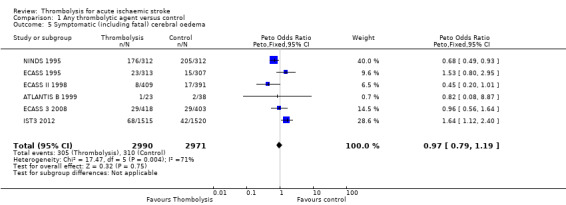
Comparison 1 Any thrombolytic agent versus control, Outcome 5 Symptomatic (including fatal) cerebral oedema.
Deaths from all causes during follow‐up
Data were available for all 27 trials (10,187 participants) (Analysis 1.8). There was a modest but significant increase in deaths within scheduled follow‐up, from 18.0% in controls to 19.4% in the participants allocated to thrombolysis (OR 1.18, 95% CI 1.06 to 1.30 , P < 0.002). In absolute terms, this represented an extra 15 (95% CI six fewer to 30 more) deaths at the end of follow‐up per 1000 participants treated with thrombolysis. There was heterogeneity between the trials (I² = 48%, P = 0.003) reflected in the fact that some trials (for example, ECASS II 1998; IST3 2012; MAST‐I 1995 (of participants allocated to the thrombolytic agent alone) and NINDS 1995) showed a non‐significant reduction and others (for example, ASK 1996, ATLANTIS A 2000 and MAST‐I 1995 (of participants allocated to the thrombolytic agent plus aspirin)) showed a significant increase in case fatality with thrombolysis.
1.8. Analysis.
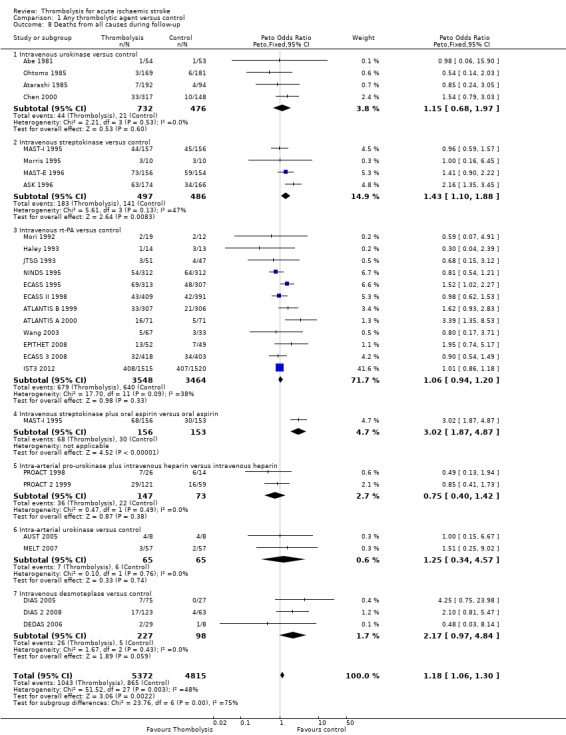
Comparison 1 Any thrombolytic agent versus control, Outcome 8 Deaths from all causes during follow‐up.
In the 12 trials using intravenous rt‐PA, there was no net effect on deaths (OR 1.06, 95% CI 0.94 to 1.20; 7012 participants) equivalent overall to seven more (two fewer to 25 more) deaths per 1000 participants treated. The heterogeneity of treatment effect among the trials of rt‐PA was not quite statistically significant (I² = 38%, P = 0.09).
In view of the statistically significant heterogeneity for all trials (I² = 48%), we performed an analysis of the data using a random‐effects model. This also shows a statistically significant excess of deaths with thrombolysis of similar magnitude to the fixed‐effect analysis (all trials: OR 1.26, 95% CI 1.04 to 1.52 P = 0.02).The results for just trials of rt‐PA (OR 1.12, 95% CI 0.90 to 1.38, P = 0.31) were also of similar magnitude as the fixed‐effect model and still without any statistical significance.
Deaths occurring between seven and 10 days and the end of follow‐up
We examined the number of deaths occurring between the first seven to 10 days and the end of follow‐up in the 13 trials that provided data for both early and late deaths (ASK 1996; Chen 2000; DIAS 2 2008; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; Haley 1993; IST3 2012; MAST‐E 1996; MAST‐I 1995; Mori 1992; Wang 2003). There were 425/3890 (10.9%) deaths in this period in the thrombolysis‐treated participants compared with 460/3568 (12.9%) in the control participants, a difference of 2 per 1000, OR 0.88 (95% CI 0.76, 1.02, P = 0.09; 7458 participants). There was significant heterogeneity (I² = 57%, P = 0.007) (Analysis 1.7). In eight trials testing rt‐PA, the corresponding OR was 0.84 (95% CI 0.71 to 0.99; 5535 participants) also indicating fewer deaths between seven and 10 days and the end of follow‐up. This analysis suggests that most of the deaths that occur following thrombolysis, including rt‐PA, occur in the first seven to 10 days, and that thereafter the number of deaths occurring in thrombolysis‐treated participants is very similar to that occurring in the control participants, or fewer than in the control group in trials testing rt‐PA. IST3 2012 provided data on death at six months, which suggested that the longer duration of follow‐up (six instead of three months) allowed for the deaths in the control group occurring after the first seven to 10 days (300/1520, 19.7%, versus 245/1515, 16.2% in rt‐PA treated participants) to overtake the excess of deaths due to fatal intracranial haemorrhage after rt‐PA occurring in the first seven to 10 days, leading to a net‐neutral overall effect on death at long‐term follow‐up with rt‐PA.
1.7. Analysis.
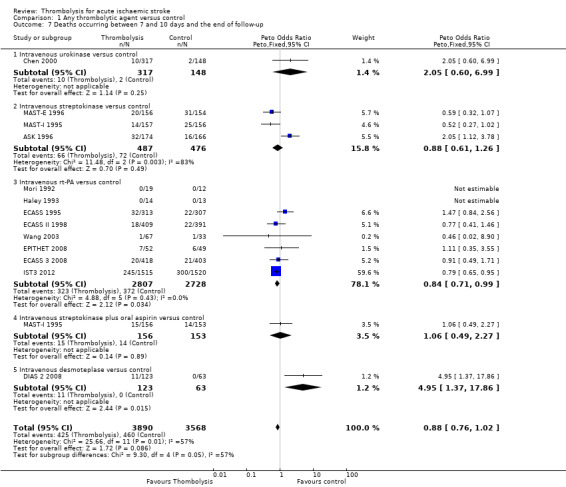
Comparison 1 Any thrombolytic agent versus control, Outcome 7 Deaths occurring between 7 and 10 days and the end of follow‐up.
Death or dependency at the end of follow‐up
Analysable data from 22 trials (including all recently completed and large trials) on functional outcome were available for 9318 participants (Analysis 1.6). Two further trials also assessed functional outcome but the data from one (Haley 1993) were incomplete (3/27 participants were alive but lost to follow‐up), and in the other (JTSG 1993) the Barthel Scores were not available.
1.6. Analysis.
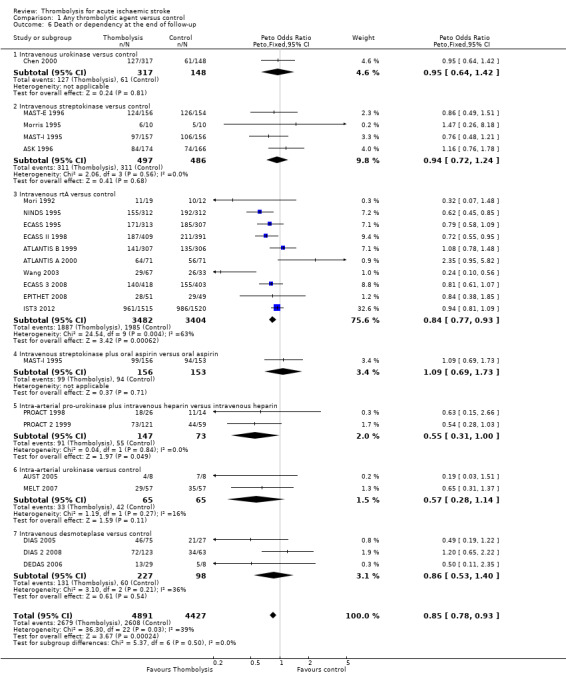
Comparison 1 Any thrombolytic agent versus control, Outcome 6 Death or dependency at the end of follow‐up.
There was a significant reduction in death or dependency with thrombolysis: 54.8% compared with 58.9% of those allocated to control (OR 0.85, 95% CI 0.78 to 0.93, P = 0.0002). This is equivalent to 41 (95% CI 20 to 60) fewer dead or dependent participants per 1000 treated. There was significant heterogeneity of treatment effect between the trials (I² = 39%, P = 0.03).
For the 10 trials using intravenous rt‐PA (6886 participants), the OR was 0.84 (95% CI 0.77 to 0.93, P = 0.0006), equivalent to 40 (95% CI 20 to 65) fewer participants being dead or dependent per 1000 treated. There was significant heterogeneity of treatment effect among the trials using rt‐PA (I² = 63%, P = 0.004).
In view of the statistically significant between‐trial heterogeneity, we performed an analysis of the data on death or dependency using a random‐effects model. This gives an OR of 0.83 (95% CI 0.73 to 0.95, P = 0.006) for all trials, and of 0.80 (95% CI 0.66 to 0.97, P = 0.03) for just trials of rt‐PA. (Note of caution: random‐effects analyses place undue weight on smaller studies and possibly should be avoided with combined outcomes. Death and dependency actually reflect two outcomes which may 'pull' in different directions. Small studies may have more extreme results and give less reliable estimates of true treatment effect than large studies).
If an alternative definition of 'poor outcome' (Rankin score 2 to 6) is used in this analysis, and the analysis is restricted to just the 21 trials with both definitions available, then the ORs are as follows:
mRS 2 to 6 for any thrombolytic drug versus control OR 0.76 (95% CI 0.70 to 0.84, P = 0.00001; 8824 participants) with significant between‐trial heterogeneity (I² = 45%, P = 0.02); for just rt‐PA trials, the OR was 0.79 (95% CI 0.71 to 0.88, P = 0.00001; 6887 participants), but also with significant heterogeneity (I² = 64%, P = 0.003) (Analysis 1.9);
mRS 3 to 6 for any thrombolytic drug versus control OR 0.85 (95% CI 0.78 to 0.93, P = 0.15 with non‐significant between‐trial heterogeneity (I² = 25%; 8824 participants); for 10 just rt‐PA trials, the OR was 0.85 (95% CI 0.77 to 0.94, P = 0.001; 6887 participants), and also with borderline significant heterogeneity (I² = 47%, P = 0.05) (Analysis 1.10).
1.9. Analysis.
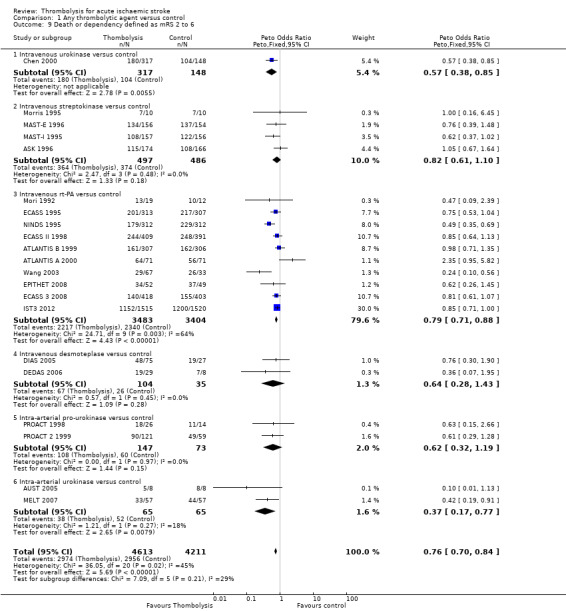
Comparison 1 Any thrombolytic agent versus control, Outcome 9 Death or dependency defined as mRS 2 to 6.
1.10. Analysis.
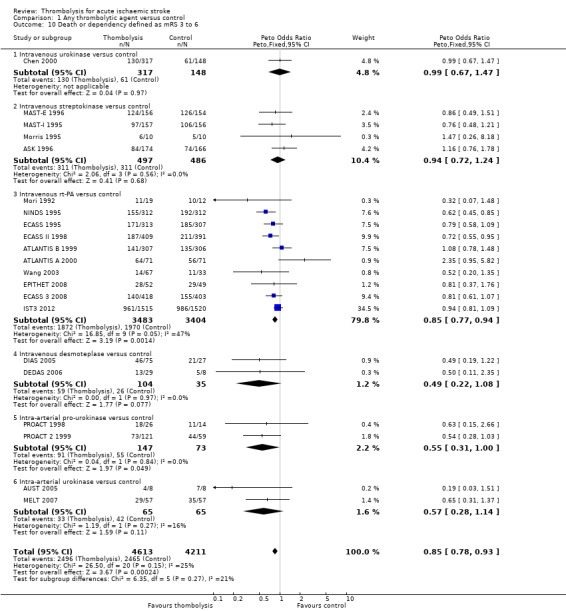
Comparison 1 Any thrombolytic agent versus control, Outcome 10 Death or dependency defined as mRS 3 to 6.
Although the confidence intervals for poor outcome defined by mRS 3 to 6 and 2 to 6 overlap for analyses of all thrombolytic drugs and of just rt‐PA, these data suggest that choosing mRS 2 to 6 as the primary outcome may provide a more positive trial result. Heterogeneity is present for poor outcome defined as mRS 2 to 6 as for poor outcome defined as mRS 3 to 6. This suggests that none of these dichotomous outcomes is specifically robust and that a more cautious estimate of overall thrombolysis and of rt‐PA effect, such as the ordinal shift analysis, is wise. Note that some individual trials 'wobble' from being positive to not positive in going between the mRS definitions (some go one way and some the other) but overall the trend is to more positive results with mRS 2 to 6. For example, ECASS II 1998 was neutral on its primary outcome of mRS 2 to 6, but positive on the alternative outcome of mRS 3 to 6; ECASS 3 2008 was positive on its primary outcome of mRS 2 to 6, but neutral on mRS 3 to 6; NINDS 1995 became less positive on mRS 3 to 6; Wang 2003 moves from very positive on mRS 2 to 6 to neutral on mRS 3 to 6, the latter trial illustrating the particular instability of small and highly positive studies, and IST3 2012 went from neutral on mRS 3 to 6 to positive on mRS 2 to 6 as well as on ordinal shift analysis.
Dependency at the end of follow‐up
We examined the effect of thrombolysis on dependency defined as mRS 3 to 5 in the 22 trials with analysable data (9318 participants; Analysis 1.11). There were 1649/4891 (33.7%) dependent participants amongst those treated with thrombolysis and 1761/4427 (39.8%) in the control group, an absolute reduction in dependent participants of 60 per 1000 participants treated with thrombolysis, OR 0.75 (95% CI 0.69 to 0.82, P < 0.00001), with borderline significant between‐trial heterogeneity (I² = 47%, P = 0.007) or between the groups treated with different thrombolytic drugs (I² = 55%, P = 0.04). This would suggest that amongst those who avoid early death (when most of the excess of deaths attributable to thrombolysis appear to occur), there is a highly significant and worthwhile reduction in the risk of being dependent with any thrombolytic treatment. Amongst those treated with rt‐PA, the reduction in dependency was similar, with an OR of 0.80 (95% CI 0.73 to 0.89, P < 0.0001; 6886 participants, 10 trials) but with between‐trial heterogeneity (I² = 53%, P = 0.02).
1.11. Analysis.
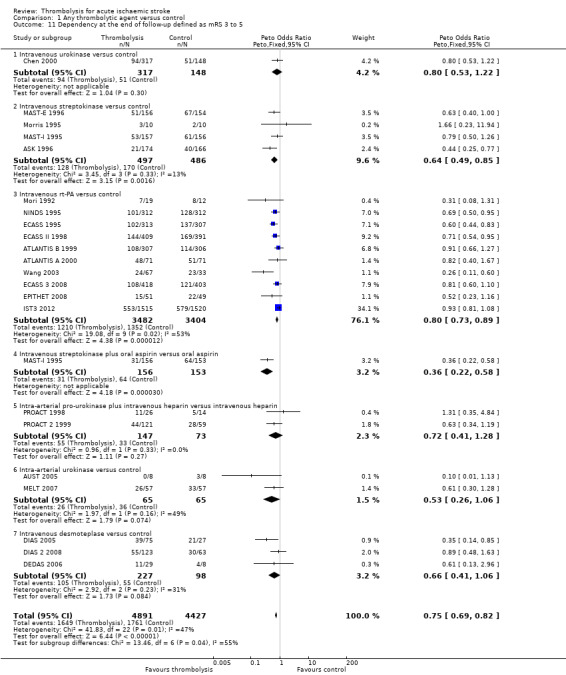
Comparison 1 Any thrombolytic agent versus control, Outcome 11 Dependency at the end of follow‐up defined as mRS 3 to 5.
Alive and independent at the end of follow‐up
We provide data on the number of participants who were alive and independent (mRS 0 to 2) (Analysis 1.12) and alive and with favourable outcome (mRS 0 to 1) (Analysis 1.13) at the end of follow‐up for trials testing rt‐PA up to six hours after stroke. For mRS 0 to 2, 1611/3483 participants given rt‐PA were alive and independent versus 1434/3404 (42%) allocated control, OR 1.17 (95% CI 1.06 to 1.29, P = 0.001; 10 trials, 6887 participants) with significant heterogeneity (I² = 47%, P = 0.05). For mRS 0 to 1, 1211/3483 participants allocated rt‐PA versus 998/3404 allocated control were alive with a favourable outcome (OR 1.29, 95% CI 1.16 to 1.43, P < 0.00001; 10 trials, 6887 participants) but with significant heterogeneity (I² = 57%, P = 0.01).
1.12. Analysis.
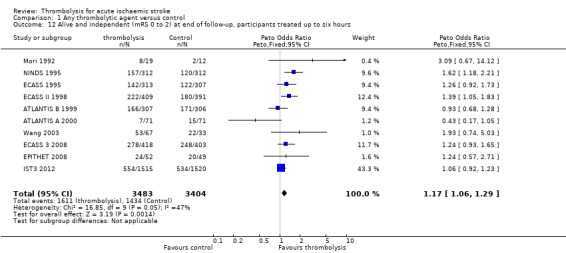
Comparison 1 Any thrombolytic agent versus control, Outcome 12 Alive and independent (mRS 0 to 2) at end of follow‐up, participants treated up to six hours.
1.13. Analysis.
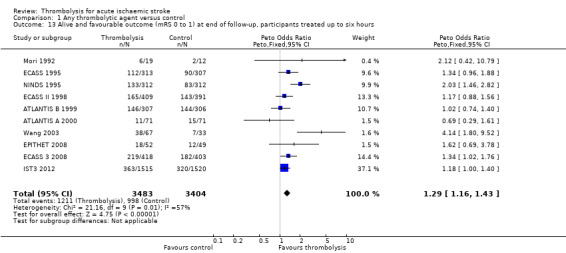
Comparison 1 Any thrombolytic agent versus control, Outcome 13 Alive and favourable outcome (mRS 0 to 1) at end of follow‐up, participants treated up to six hours.
Possible sources of between‐trial heterogeneity
To attempt to identify possible causes for the heterogeneity of the data on death (amongst all trials), functional outcome and symptomatic intracranial haemorrhage (SICH) in rt‐PA trials, we have ordered the trials by:
thrombolytic drug used (in all main outcome analyses);
concomitant antithrombotic drug usage;
time to treatment;
severity of stroke among participants randomised based on the case fatality in the control group (dichotomised on 0% to 19%, and 20% or more)
use of CT scanning versus MR diffusion‐ and perfusion‐weighted imaging (DWI/PWI) mismatch or CT perfusion prior to inclusion; and
CT scan ASPECTS score, which is a measure of the extent of acute ischaemic tissue changes.
We have also examined the effect of time to treatment and effect of imaging modality (CT versus MR DWI/PWI mismatch or CT perfusion) on:
death or dependency;
alive and independent;
death by the end of follow‐up; and
SICH.
There are obviously many other possible causes of heterogeneity, but it has not been possible to examine these systematically at the present time. These include, for example, the availability of data, the design of the trials, other aspects of the participant population apart from stroke severity, and the play of chance amongst what are still mainly relatively small trials.
Thrombolytic drug used
The indirect comparisons of the effects of each individual drug on death at the end of follow‐up (Analysis 1.8), death or dependency (Analysis 1.6), or SICH (Analysis 1.4) ‐ like all indirect comparisons ‐ are confounded by a number of factors, and hence not reliable. These trials differ in many respects apart from just the thrombolytic drug used, or its dose (for example the dose of streptokinase used in ASK 1996; MAST‐E 1996; and MAST‐I 1995 was similar to that used in myocardial infarction; a lower dose would perhaps be more relevant to a typical older stroke population). Although trends were observed, there were no statistically significant differences in symptomatic intracranial haemorrhage or death or dependency between trials using urokinase, streptokinase, rt‐PA or desmoteplase. There was significant between‐trial heterogeneity for death at the end of follow‐up (Analysis 1.8). Examination of this analysis shows that the MAST‐I streptokinase‐plus‐aspirin arm is a particular outlier with a large excess of deaths in the participants allocated streptokinase‐plus‐aspirin; the heterogeneity became non‐significant after removal of MAST‐I aspirin‐plus‐streptokinase‐allocated participants (I² dropped to 28%, P = 0.09). For all outcomes, including death at the end of follow‐up, there was considerable overlap between the 95% confidence intervals, clearly indicating that there are other differences between these trials in addition to the drug being tested. For example, the desmoteplase trials included participants as late as nine hours, whereas most other trials only included participants up to six hours. Any apparent differences between drugs may therefore be due to factors other than the drug in question.
Concomitant antithrombotic drug use
It is not possible to comment on the effect of aspirin use prior to the stroke; although some trials recorded prior aspirin use, we could not extract data from the publications in a comparable manner and none of the earlier trials balanced randomisation on prior aspirin use. In IST3 2012 treatment with antiplatelet drugs in the previous 48 hours was consistently collected and included among the pre‐randomisation variables. The interaction between thrombolytic drugs and antithrombotic drugs given simultaneously (or the latter very soon after the former) was only tested by random allocation in one study (MAST‐I 1995), which therefore provides the only truly valid evidence on this potential interaction. In this study there was a clinically important adverse interaction between aspirin and streptokinase when given simultaneously, resulting in a substantial increase in case fatality (early and late), which was not offset by a reduction in the number of dead or dependent participants by the end of follow‐up (28% of those allocated to streptokinase alone versus 43% of those allocated to streptokinase plus aspirin were dead by the end of follow‐up (P < 0.001), and 62% and 63% were dead or dependent respectively (versus 68% in the control group)). The actual cause of the increase in early and total deaths with streptokinase and aspirin appears largely to be due to cerebrovascular events. Aspirin with streptokinase significantly increased the number of deaths in hospital from all causes (OR 2.2, 95% CI 1.3 to 3.8), from neurological causes (OR 2.0, 95% CI 1.1 to 3.7), and intracranial haemorrhage on CT scan or at autopsy (OR 2.2, 95% CI 1.0 to 5.0) when compared with the group who received streptokinase alone. There was no difference in deaths from neurological causes without intracranial haemorrhage, but note also that more participants in the streptokinase plus aspirin group died of neurological causes without a CT scan or autopsy, so could also have had an intracranial haemorrhage, that is, the increase in intracranial haemorrhage with aspirin and streptokinase may be even greater (Ciccone 1998).
Information is also available on antithrombotic drug use (although not randomly allocated) in 20 other trials (ASK 1996; ATLANTIS A 2000; ATLANTIS B 1999; AUST 2005; Chen 2000; DEDAS 2006; DIAS 2005; DIAS 2 2008; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; Haley 1993; IST3 2012; MAST‐E 1996; MELT 2007; Mori 1992; NINDS 1995; PROACT 1998; PROACT 2 1999), and some further data in three other trials (Abe 1981; Atarashi 1985; Ohtomo 1985) (9674 participants; Analysis 1.14). The odds of death by the end of follow‐up were increased in line with the frequency, the amount, and proximity to the administration of thrombolysis of the concomitant antithrombotic drug use (OR 1.31 when all participants received antithrombotic drugs within 24 hours of thrombolysis; 1.27 when some participants received antithrombotic drugs within 24 hours; 1.13 when no participants received antithrombotic drugs within 24 hours but some thereafter; and 0.89 for no antithrombotic drugs within the first 10 to 14 days; I² = 50%, P = 0.002). Although these data are based on non‐randomised comparisons, they do support the evidence of a clinically significant adverse interaction between thrombolysis and antithrombotic drugs given concurrently found in the MAST‐I 1995 study and may go some way towards explaining the heterogeneity between the trials for case fatality
1.14. Analysis.
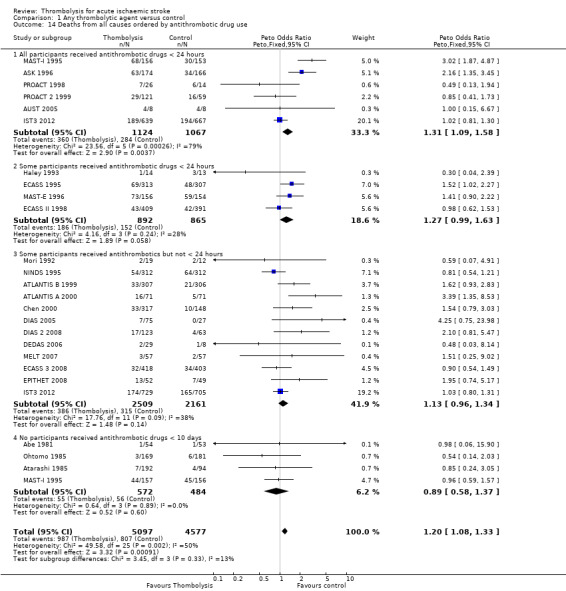
Comparison 1 Any thrombolytic agent versus control, Outcome 14 Deaths from all causes ordered by antithrombotic drug use.
Severity of stroke among randomised participants
There was no obvious statistically significant difference in the effect of thrombolysis on case fatality between trials with a case fatality rate less than 19% in the control group (OR 1.31, 95% CI 1.08 to 1.58; 17 trials, 4973 participants) and those with a case fatality rate greater than 20% in the control group (OR 1.05, 95% CI 0.92 to 1.19; 10 trials, 4905 participants; pooled I² = 28%, P = 0.09) (Analysis 1.15). However, this crude comparison may mask an important relationship between stroke severity and hazard with thrombolysis. Post‐hoc statistical correction methods are unlikely to be adequate, so combined individual patient data from published rt‐PA trials may be incorrect. Also, unfortunately, analysis based on an outcome in the control group is prone to bias due to regression to the mean (Sharp 1996). Nonetheless, the post‐hoc analyses of both the NINDS (Ingall 2003) and MAST‐I trials (Wardlaw 1999a) suggested that the risk reduction for death or dependency varied with stroke severity, being largest for participants with moderate stroke (NIHSS score around 10 to 15) and least in severe stroke (NINDS score around 18 to 30). However, these findings were based on small numbers in post‐hoc analysis and need to be verified prospectively in larger randomised trials. A first attempt has now been made in the IST3 2012 trial. In IST‐3 severity was well balanced between the study arms through minimisation and more than 30% of all participants had a NINDS score of 15 or more, Furthermore, case fatality was 20% or more in the control group. The participants with more severe stroke had at least the same benefit of thrombolytic treatment as those with less severe stroke.
1.15. Analysis.
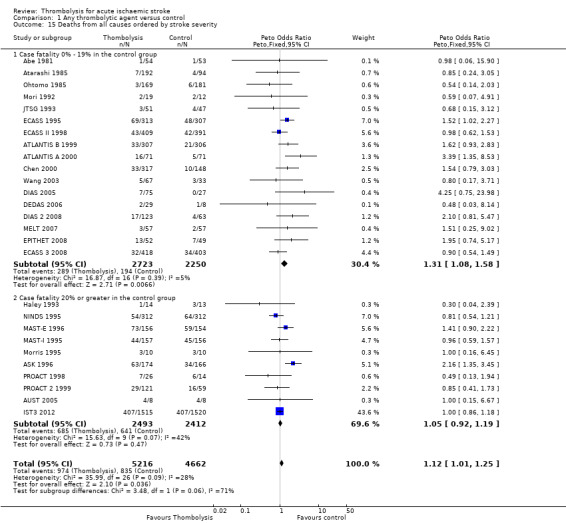
Comparison 1 Any thrombolytic agent versus control, Outcome 15 Deaths from all causes ordered by stroke severity.
Effect of time to treatment (randomisation): death or dependency at the end of follow‐up
There was a significant reduction in the number of dead or dependent participants allocated thrombolysis who were randomised within three hours (57.4% of those allocated to thrombolysis were dead or dependent compared with 67% of those allocated to control, OR 0.66, 95% CI 0.56 to 0.79, P = 0.00001; 10 trials, 2160 participants) with no statistically significant heterogeneity (P = 0.91) (Analysis 1.16). In absolute terms, this is equivalent to 95 (95% CI 55 to 136) fewer dead or dependent participants per 1000 treated with thrombolysis (all drugs combined). In trials testing rt‐PA, 59.3% of those allocated rt‐PA were dead or dependent compared with 68.3% % of those allocated to control, OR 0.65 (95% CI 0.54 to 0.80, P < 0.0001; 6 trials, 1779 participants) with no significant heterogeneity, equivalent to 90 per 1000 fewer (95% CI 46 to 135) dead or dependent participants with rt‐PA. Thus, heterogeneity present in the analysis of all trials, all time windows and drugs, is removed for participants randomised within three hours of stroke. This reinforces the previous finding of no heterogeneity for rt‐PA and is presumably due to the fact that the vast majority of the participants (82.4%) in this analysis were treated with rt‐PA, an effect of the inclusion of the IST3 2012 trial.
1.16. Analysis.
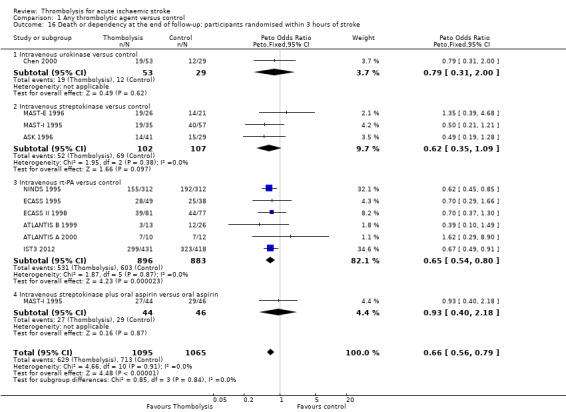
Comparison 1 Any thrombolytic agent versus control, Outcome 16 Death or dependency at the end of follow‐up: participants randomised within 3 hours of stroke.
To compare these data on the effects of treatment given within three hours with the effects when given after three hours, we examined only those trials that reported data for both time windows (ASK 1996; ATLANTIS A 2000; ATLANTIS B 1999; Chen 2000; ECASS 1995; ECASS II 1998; IST3 2012; MAST‐E 1996; MAST‐I 1995) to avoid confounding by other differences between trials (Analysis 1.17; 9 trials, 6941 participants). Although there appeared to be more reduction in death or dependency in participants treated within three hours, the difference was not significant: three hours, OR 0.69, 95% CI 0.55 to 0.85; between three and six hours OR 0.99, 95% CI 0.88 to 1.10, (I² = 28%, P = 0.13).
1.17. Analysis.
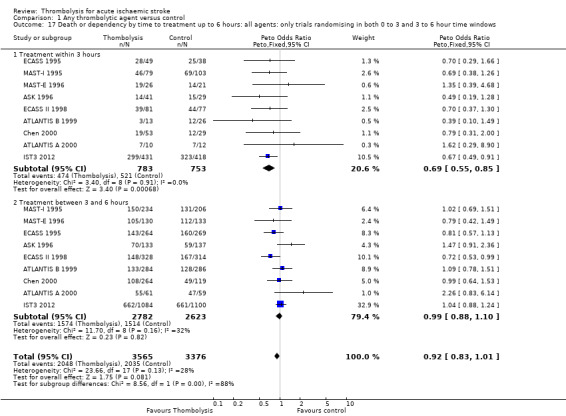
Comparison 1 Any thrombolytic agent versus control, Outcome 17 Death or dependency by time to treatment up to 6 hours: all agents: only trials randomising in both 0 to 3 and 3 to 6 hour time windows.
In trials using rt‐PA alone, amongst trials randomising in both less than three‐hour and three‐ to six‐hour time windows, the effect of treatment was not significantly different when given within three hours (OR 0.68, 95% CI 0.53 to 0.87; 5 trials, 1155 participants), or more than three hours after stroke (OR 0.97, 95% CI 0.85 to 1.09; 5 trials, 1449 participants), (I² = 45%, P = 0.06) (Analysis 1.18). Comparing all rt‐PA trials whether they randomised only under three hours or only between three and six hours or from zero to six hours made little difference (Analysis 1.19): within three hours OR 0.65, 95% CI 0.54 to 0.80; 6 trials, 1779 participants; between three and six hours OR 0.93, 95% CI 0.83 to 1.04; 7 trials, 4950 participants. This should not be interpreted to mean that time to treatment is unimportant, but rather that other factor(s) like stroke severity may have confounded the association between time and outcome, and cannot be corrected for in this tabular analysis.
1.18. Analysis.
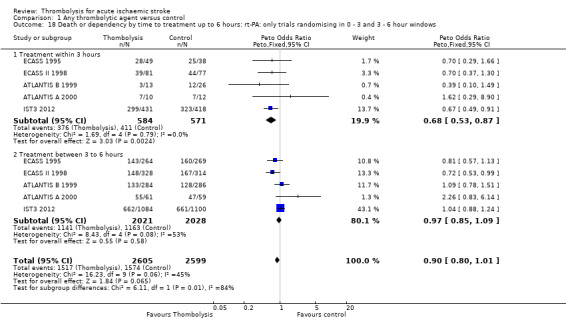
Comparison 1 Any thrombolytic agent versus control, Outcome 18 Death or dependency by time to treatment up to 6 hours: rt‐PA: only trials randomising in 0 ‐ 3 and 3 ‐ 6 hour windows.
1.19. Analysis.
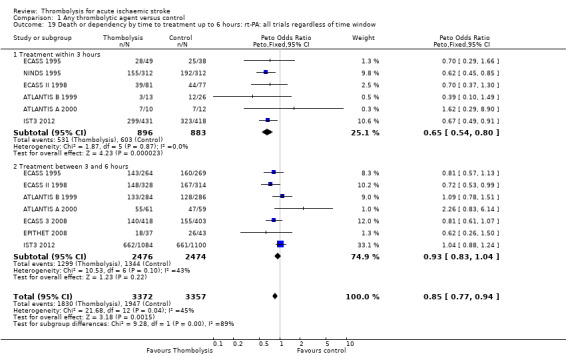
Comparison 1 Any thrombolytic agent versus control, Outcome 19 Death or dependency by time to treatment up to 6 hours: rt‐PA: all trials regardless of time window.
We also compared the outcome death or dependency at the end of follow‐up across all trials stratified by their latest time allowed to randomisation (Analysis 1.20). This provided data at up to three hours, up to 4.5 hours, up to six hours, up to nine hours and up to 24 hours. There was surprisingly little difference in ORs for each time point: zero to three hours, OR 0.62, 95% CI 0.45 to 0.85 (1 trial, 624 participants); zero to 4.5 hours, OR 0.93, 95% CI 0.66 to 1.32 (2 trials, 1161 participants); zero to six hours, OR 0.81, 95% CI 0.69 to 0.95 (15 trials, 6883 participants); zero to nine hours, OR 0.74, 95% CI 0.35 to 1.59 (3 trials, 325 participants); zero to 24 hours OR 0.14, 95% CI 0.01 to 1.76 (1 trial, 16 participants); I² = 35%, P = 0.05.
1.20. Analysis.
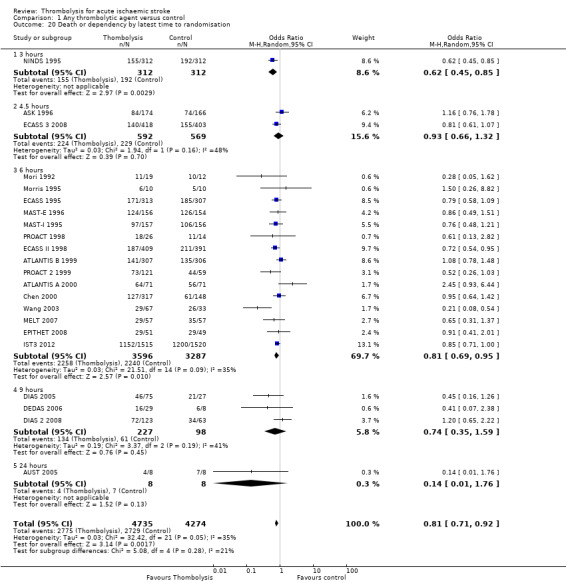
Comparison 1 Any thrombolytic agent versus control, Outcome 20 Death or dependency by latest time to randomisation.
An analysis of the effect of time on the proportion of participants who were alive and independent (Analysis 1.21; 8 trials, 6750 participants) or alive and with favourable outcome (Analysis 1.22; 6 trials, 1779 participants) for trials testing rt‐PA (other trials not assessed) suggested that earlier treatment increased the proportion with better outcomes than later treatment: for every 1000 participants given rt‐PA within three hours, 90 more would be alive and independent (P < 0.0001) with no heterogeneity, compared with 10 more if treated between three and six hours after stroke (P = 0.58).
1.21. Analysis.
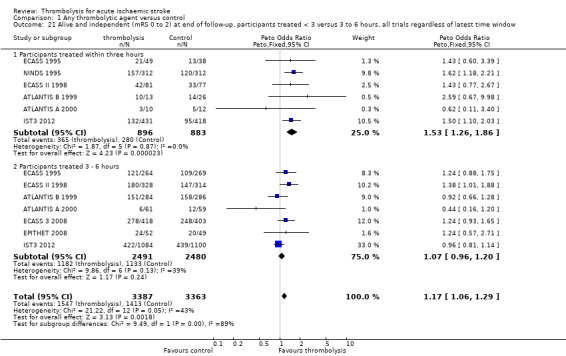
Comparison 1 Any thrombolytic agent versus control, Outcome 21 Alive and independent (mRS 0 to 2) at end of follow‐up, participants treated < 3 versus 3 to 6 hours, all trials regardless of latest time window.
1.22. Analysis.
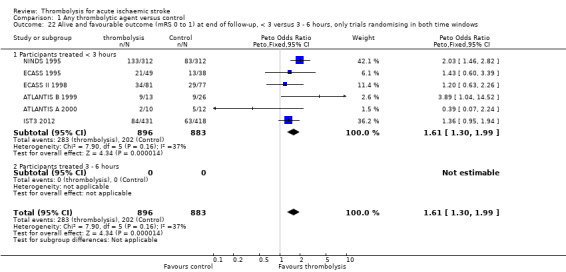
Comparison 1 Any thrombolytic agent versus control, Outcome 22 Alive and favourable outcome (mRS 0 to 1) at end of follow‐up, < 3 versus 3 ‐ 6 hours, only trials randomising in both time windows.
Effect of time to treatment (randomisation): death during follow‐up
Data on participants treated within three hours of stroke are available for 11 trials (Analysis 1.23). The NINDS 1995 trial contributes 29% of the data on all drugs (624/2187 participants). There was no excess of deaths during follow‐up with thrombolysis: 25.5 % of participants allocated to thrombolysis versus 25.8% of those allocated to control (OR 0.99 95% CI 0.82 to 1.21) but with statistically significant heterogeneity (I² = 65%, P = 0.0008). The main outlier was the MAST‐I 1995 streptokinase‐plus‐aspirin‐allocated participants who showed a significant excess of deaths even with treatment within three hours. In trials using rt‐PA, the equivalent figures were OR 0.91 (95% CI 0.73 to 1.13, P = 0.39; 7 trials, 1806 participants), with no statistically significant heterogeneity (P = 0.22 ) and 14 fewer per 1000 deaths (95% CI 26 fewer to 55 fewer).
1.23. Analysis.
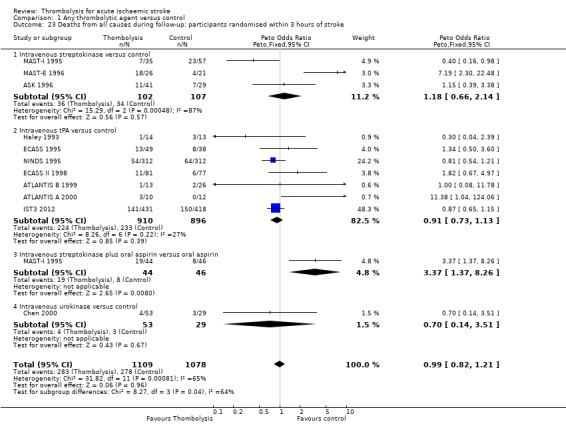
Comparison 1 Any thrombolytic agent versus control, Outcome 23 Deaths from all causes during follow‐up: participants randomised within 3 hours of stroke.
To compare treatment within three hours with treatment between three and six hours, we performed a similar analysis to those above for death and dependency (Analysis 1.24). Here there was a significant difference in treatment effect between those treated within three hours (OR 1.08, 95% CI 0.86 to 1.35; 9 trials, 1536 participants) and between three and six hours (OR 1.29, 95% CI 1.13 to 1.48; 9 trials, 5400 participants) after the stroke (I² = 63%, P = 0.0002) for all trials providing data in both time windows. For just trials testing intravenous rt‐PA (Analysis 1.25), there was a marginally significant excess of deaths for participants treated between three and six hours (OR 1.17, 95% CI 1.00 to 1.38; 5 trials, 4044 participants), compared with those treated within three hours (OR 0.97, 95% CI 0.75 to 1.26; 5 trials, 1155 participants. I² = 46%, P = 0.05). Including all rt‐PA data (i.e. all trials, all time windows) (Analysis 1.26) made little difference: within three hours: OR 0.91, 95% CI 0.73 to 1.13; 7 trials, 1806 participants; between three and six hours OR 1.16, 95% CI 1.00 to 1.35 (P = 0.07); 7 trials, 4966 participants.
1.24. Analysis.
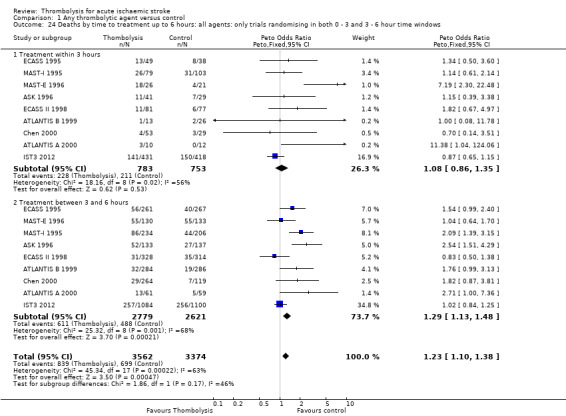
Comparison 1 Any thrombolytic agent versus control, Outcome 24 Deaths by time to treatment up to 6 hours: all agents: only trials randomising in both 0 ‐ 3 and 3 ‐ 6 hour time windows.
1.25. Analysis.
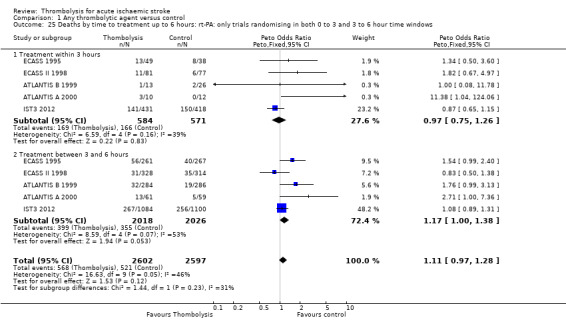
Comparison 1 Any thrombolytic agent versus control, Outcome 25 Deaths by time to treatment up to 6 hours: rt‐PA: only trials randomising in both 0 to 3 and 3 to 6 hour time windows.
1.26. Analysis.
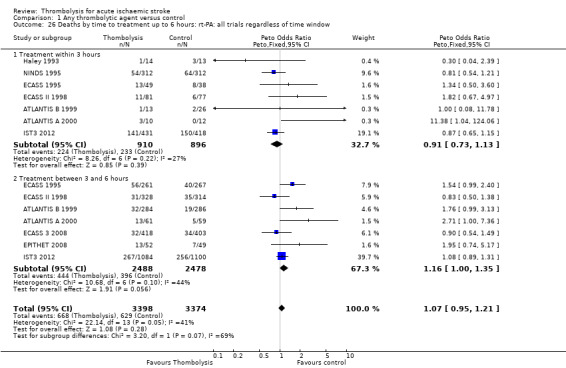
Comparison 1 Any thrombolytic agent versus control, Outcome 26 Deaths by time to treatment up to 6 hours: rt‐PA: all trials regardless of time window.
We also compared the outcome death at the end of follow‐up across all trials stratified by latest time to randomisation (Analysis 1.27). This provided data at up to three hours, up to 4.5 hours, up to six hours, up to nine hours and up to 24 hours. Although 'wobbly', there appeared to be an increase in ORs with increasing latest time to randomisation (and this was also just statistically significant, P = 0.04), but bear in mind that there are other major differences between these studies and very few data for later time windows: zero to three hours, OR 0.79 (95% CI 0.53 to 1.17; 2 trials, 651 participants); zero to 4.5 hours, OR 1.43 (95% CI 1.01 to 2.03; 2 trials, 1161 participants); zero to six hours, OR 1.12 (95% CI 0.99 to 1.26; 16 trials, 6886 participants ); zero to nine hours, OR 2.10 (95% CI 0.79 to 5.58; 3 trials, 325 participants); zero to 24 hours, OR 1.00 (95% CI 0.14 to 7.10; 1 trial, 16 participants; I² = 33%, P = 0.07).
1.27. Analysis.
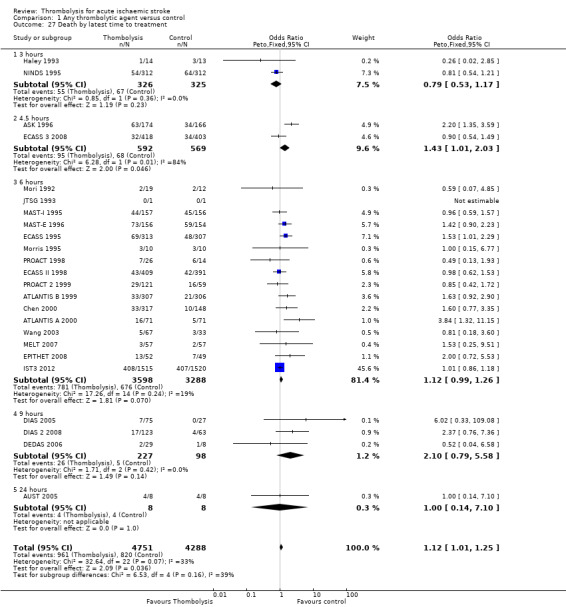
Comparison 1 Any thrombolytic agent versus control, Outcome 27 Death by latest time to treatment.
Effect of time to treatment (randomisation) ‐ symptomatic intracranial haemorrhage (SICH)
Data are available on SICH from trials randomising both in under three hours and between three‐ and six‐hour time windows for five rt‐PA trials (ATLANTIS B 1999; ATLANTIS A 2000; ECASS 1995; ECASS II 1998; IST3 2012) (Analysis 1.28). Within three hours, there was an excess of SICH with rt‐PA (OR 4.25, 95% CI 2.53 to 7.16; 1155 participants). This did not differ to the increase in SICH in participants randomised between three and six hours (OR 3.62 , 95% CI 2.76 to 4.76; 4013 participants; I² = 0%). When data from all rt‐PA trials were included (that is, those randomising only within three or only between three and six hours) (Analysis 1.29), there was little difference: within three hours OR 4.55 (95% CI 2.92 to 7.09; 6 trials, 1779 participants); between three and six hours OR 3.73 (95% CI 2.86 to 4.86; 7 trials, 4935 participants; I² = 0%).
1.28. Analysis.
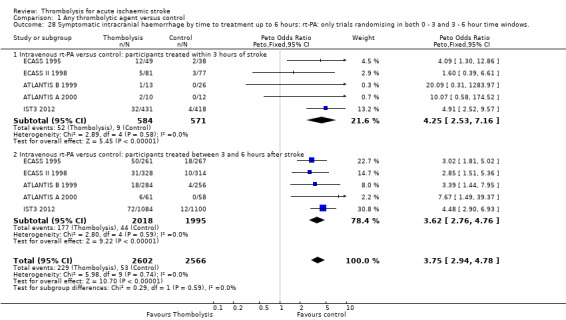
Comparison 1 Any thrombolytic agent versus control, Outcome 28 Symptomatic intracranial haemorrhage by time to treatment up to 6 hours: rt‐PA: only trials randomising in both 0 ‐ 3 and 3 ‐ 6 hour time windows..
1.29. Analysis.
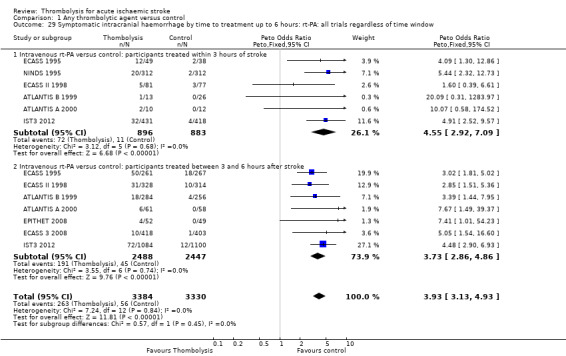
Comparison 1 Any thrombolytic agent versus control, Outcome 29 Symptomatic intracranial haemorrhage by time to treatment up to 6 hours: rt‐PA: all trials regardless of time window.
We also compared the outcome SICH across all trials stratified by latest time to randomisation (Analysis 1.30). There was surprisingly little difference in ORs for each time point: zero to three hours, OR 5.85 (95% CI 1.54 to 22.26; 2 trials, 651 participants); zero to 4.5 hours, OR 6.56 (95% CI 2.51 to 17.18; 2 trials, 1161 participants); zero to six hours, OR 4.20 (95% CI 3.21 to 5.50; 15 trials, 6951 participants); zero to nine hours, OR 6.82 (95% CI 0.88 to 52.78; 3 trials, 325 participants); zero to 24 hours, no SICH in AUST 2005; I² = 23, P = 0.17.
1.30. Analysis.
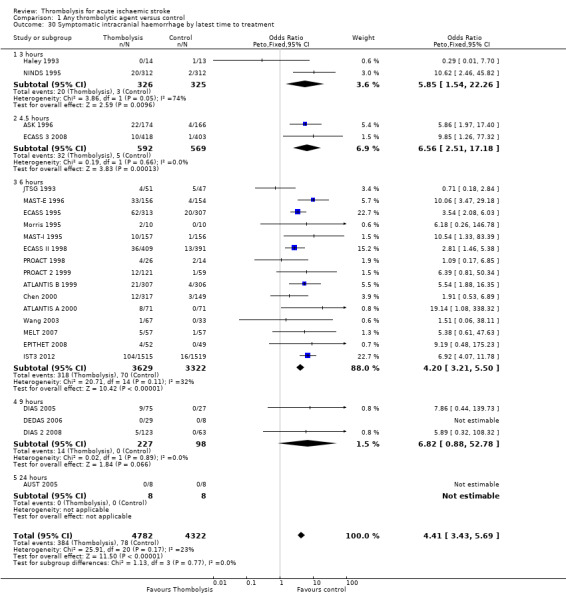
Comparison 1 Any thrombolytic agent versus control, Outcome 30 Symptomatic intracranial haemorrhage by latest time to treatment.
Effect of age under or over 80 years on death or dependency and alive and independent
We analysed the effect of age in just the rt‐PA trials. Three trials included participants aged over 80 years (EPITHET 2008; IST3 2012; NINDS 1995), most coming from IST3 2012. With treatment up to six hours, the effect of rtPA on reducing the proportion of participants who were dead or dependent aged over 80 years (OR 0.80, 95% CI 0.64 to 0.99, P = 0.04; 3 trials, 1696 participants) was the same as in participants aged up to and including 80 years (OR 0.85, 95% CI 0.76 to 0.95, P = 0.004; 10 trials, 5175 participants) (Analysis 1.31). For participants treated within three hours (Analysis 1.32), those aged over 80 (OR 0.56, 95% CI 0.40 to 0.78, P = 0.0007; 2 trials, 726 participants) did as well as those aged up to or including 80 years (0.66, 95% CI 0.52, 0.85, P = 0.001; 6 trials, 1039 participants) with no heterogeneity. There was similarly little difference between the proportions of those aged under or over 80 years who were alive and independent after rt‐PA whether treated up to six hours (Analysis 1.33; 10 trials, 6885 participants), within three hours (Analysis 1.34; 6 trials, 1779 participants) or between three and six hours (Analysis 1.35; 7 trials, 4971 participants).
1.31. Analysis.
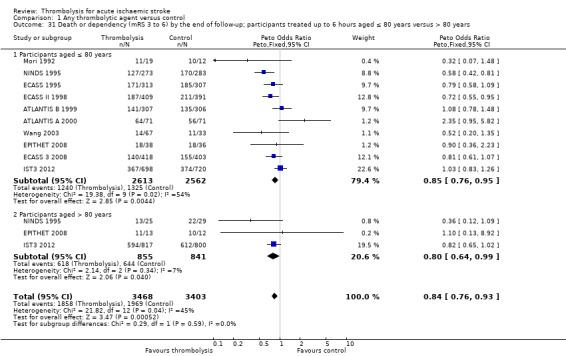
Comparison 1 Any thrombolytic agent versus control, Outcome 31 Death or dependency (mRS 3 to 6) by the end of follow‐up; participants treated up to 6 hours aged ≤ 80 years versus > 80 years.
1.32. Analysis.
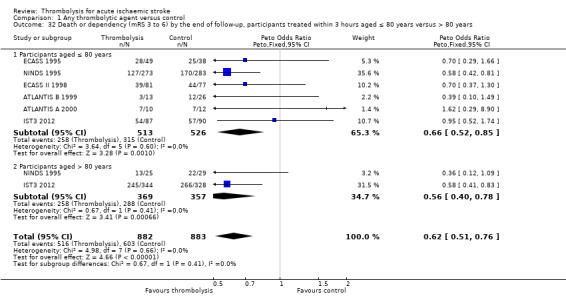
Comparison 1 Any thrombolytic agent versus control, Outcome 32 Death or dependency (mRS 3 to 6) by the end of follow‐up, participants treated within 3 hours aged ≤ 80 years versus > 80 years.
1.33. Analysis.
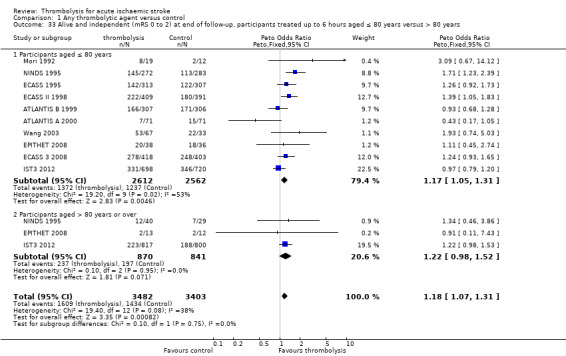
Comparison 1 Any thrombolytic agent versus control, Outcome 33 Alive and independent (mRS 0 to 2) at end of follow‐up, participants treated up to 6 hours aged ≤ 80 years versus > 80 years.
1.34. Analysis.
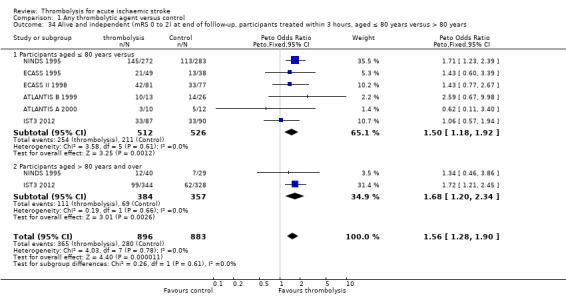
Comparison 1 Any thrombolytic agent versus control, Outcome 34 Alive and independent (mRS 0 to 2) at end of folllow‐up, participants treated within 3 hours, aged ≤ 80 years versus > 80 years.
1.35. Analysis.
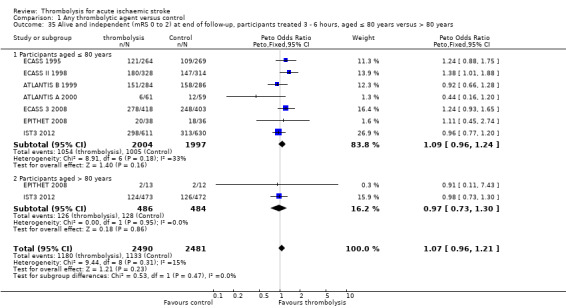
Comparison 1 Any thrombolytic agent versus control, Outcome 35 Alive and independent (mRS 0 to 2) at end of follow‐up, participants treated 3 ‐ 6 hours, aged ≤ 80 years versus > 80 years.
Selection using CT scanning or Magnetic Resonance diffusion and perfusion (DWI/PWI) mismatch
We restricted this analysis to just trials testing intravenous thrombolysis. Eleven trials contributed data on selection using CT scanning (ATLANTIS A 2000; ATLANTIS B 1999; Chen 2000; ECASS 1995; ECASS II 1998; ECASS 3 2008; IST3 2012; MAST‐E 1996; MAST‐I 1995; NINDS 1995; Wang 2003) and four trials contributed data on selection using DWI/PWI mismatch (DEDAS 2006; DIAS 2005; DIAS 2 2008; EPITHET 2008). We included the EPITHET trial in the DWI/PWI section, although participants were not actually selected for inclusion on the basis of the MR findings, but rather on the basis of plain CT. However EPITHET obtained MR DWI/PWI data prior to randomisation and provided data on mismatch findings in relation to thrombolysis effect. Amongst 8334 participants selected on the basis of plain CT, 19% allocated thrombolysis and 19.3% allocated control were dead at the end of follow‐up (OR 1.12, 95% CI 1.00 to 1.25; 15 trials); amongst 426 participants selected on the basis of DWI/PWI mismatch, 14% allocated thrombolysis and 8.2% allocated control were dead at the end of follow‐up (OR 2.05, 95% CI 1.02 to 4.15; 4 trials) (Analysis 1.36). Amongst 7843 participants selected on the basis of plain CT, 60.5% allocated thrombolysis and 66.3% allocated control were dead or dependent at the end of follow‐up (OR 0.81, 95% CI 0.73 to 0.89; 11 trials); amongst 425 participants selected on the basis of DWI/PWI mismatch, 58.6% allocated thrombolysis and 61.2% allocated control were dead or dependent at the end of follow‐up (OR 0.88, 95% CI 0.58 to 1.35; 4 trials) (Analysis 1.37). Amongst 8358 participants selected on the basis of plain CT, 8.1% allocated thrombolysis and 1.9% allocated control developed SICH (OR 4.38, 95% CI 3.38 to 5.69; 16 trials); amongst 426 participants selected on the basis of DWI/PWI mismatch, 6.4% allocated thrombolysis and 0% allocated control developed SICH (OR 7.51, 95% CI 1.40 to 40.35; 4 trials) (Analysis 1.38). These differences between selection by CT and by DWI/PWI mismatch were not statistically different.
1.36. Analysis.
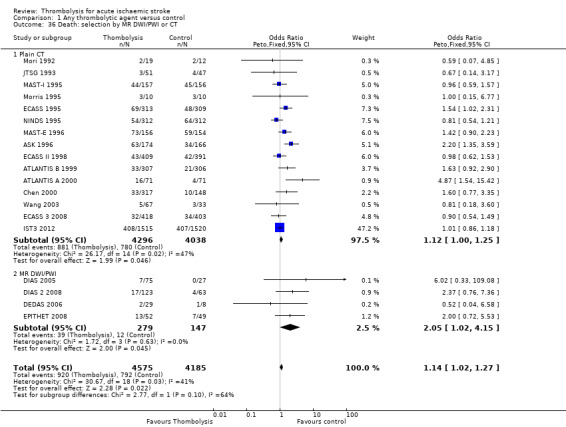
Comparison 1 Any thrombolytic agent versus control, Outcome 36 Death: selection by MR DWI/PWI or CT.
1.37. Analysis.
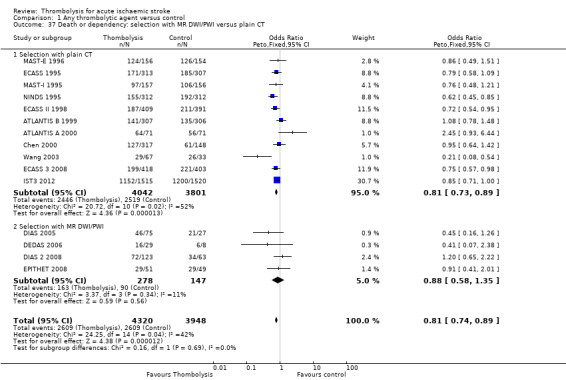
Comparison 1 Any thrombolytic agent versus control, Outcome 37 Death or dependency: selection with MR DWI/PWI versus plain CT.
1.38. Analysis.
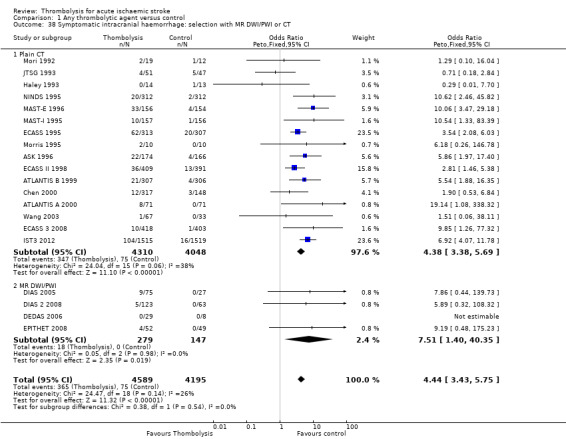
Comparison 1 Any thrombolytic agent versus control, Outcome 38 Symptomatic intracranial haemorrhage: selection with MR DWI/PWI or CT.
Four studies (ECASS II 1998; IST3 2012; NINDS 1995; PROACT 2 1999) assessed ischaemic tissue extent on plain CT according to the ASPECT score, 8 to 10 versus 0 to 7, and the probability of being alive and independent (mRS 0 to 1) by end of follow‐up after thrombolysis (Analysis 1.39). In all, 4567 participants provided data for this analysis. Among the 3317 participants with an ASPECT score indicating no or only a small area of ischaemic change (ASPECT 8 to 10), 38.9% of those allocated control versus 43.4% of those allocated thrombolysis had a favourable outcome (mRS 0 to 1) at the end of follow‐up, (OR 1.21, 95% CI 1.06 to 1.39, P = 0.006; 4 trials, I² = 79%, P = 0.002). For the 1250 participants with an ASPECT score indicative of more extensive ischaemia (ASPECT 0 to 7) 19.3% of participants allocated control versus 22.5% of participants allocated thrombolysis were alive and independent (OR 1.20, 95% CI 0.91 to 1.58, P = 0.19; 4 trials), with no heterogeneity (I² = 1%, P = 0.39). However, there was heterogeneity between the effect of rt‐PA in the participants with no or mild and extensive ischaemic change on CT (I² = 60, P = 0.01). In this relatively small sample tested for the effect on outcome by grade of CT‐visible infarction, there were several types of drugs and different administration (pro‐urokinase intra‐arterially and rt‐PA intravenously) which may account for the between‐group heterogeneity.
1.39. Analysis.
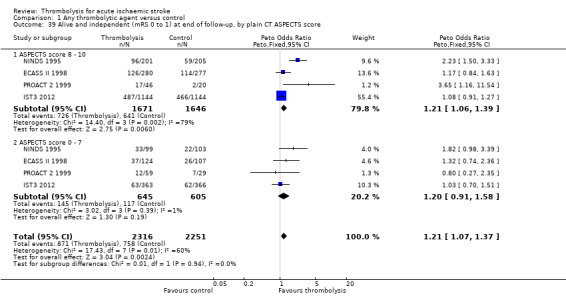
Comparison 1 Any thrombolytic agent versus control, Outcome 39 Alive and independent (mRS 0 to 1) at end of follow‐up, by plain CT ASPECTS score.
Trials testing intra‐arterial thrombolysis
Amongst participants allocated to thrombolysis, 56.6% were dead or dependent at the end of follow‐up compared with 70.3% of those allocated to control, OR 0.49 (95% CI 0.31 to 0.79; 4 trials, 350 participants) (Analysis 1.40). This can be compared crudely with analysis of death or dependency for the combined intravenous thrombolysis trials which gave an OR of 0.79 (95% CI 0.71 to 0.89). These are not direct comparisons, and there are many other differences between the trials apart from the route of administration; this analysis should therefore be regarded with extreme caution. A separate Cochrane review presents the evidence, at present very limited, on direct randomised comparisons of intravenous with intra‐arterial thrombolysis (Wardlaw 2013).
1.40. Analysis.
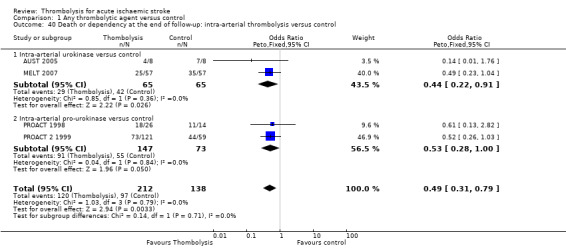
Comparison 1 Any thrombolytic agent versus control, Outcome 40 Death or dependency at the end of follow‐up: intra‐arterial thrombolysis versus control.
Discussion
Summary of main results
There is strong evidence from 27 trials in 10,187 participants on the immediate hazards and the apparent net benefit of thrombolytic therapy given up to within three hours of acute ischaemic stroke, with overall benefit suggested up to six hours, for people aged over or under 80 years, and with different stroke severities. Overall, thrombolytic therapy was associated with a significant excess of deaths within the first seven to 10 days, symptomatic and fatal intracranial haemorrhages and (for all drugs) deaths by the end of follow‐up. Most of the excess of deaths with thrombolysis occurred early and was explained by fatal intracranial haemorrhage. However, dependency was reduced in survivors so overall there was a significant net benefit. For every 1000 people treated with thrombolysis, 41 avoided death or dependency. Treatment within three hours resulted in 95/1000 fewer dead or dependent people. Trials using intravenous recombinant tissue plasminogen activator (rt‐PA) contributed the most data to this review, and rt‐PA appeared more favourable. Nevertheless, it was associated with an excess of early deaths, virtually all attributable to fatal intracranial haemorrhage (ICH), and a significant excess of symptomatic intracranial haemorrhage (SICH), but a neutral effect on deaths by the end of follow‐up, and significantly more people avoiding dependent survival. If the factors associated with early fatal ICH can be identified (Whiteley 2012), then it may become easier to identify those who are at greatest risk of harm and clarify the risk‐to‐benefit ratio for thrombolysis. There was no clear increase in hazard (ICH or death) with increasing time up to six hours after stroke, although there was some evidence of decreasing benefit (reduction in death and dependency). Therefore, increasing time to treatment may reduce benefit more than it increases the hazard of thrombolysis.
IST3 2012 has provided substantial new data since the last update, and includes a wide range of participants that has helped to answer questions about how to select patients (to maximise benefit and minimise hazard), by using the variables age, time from onset, stroke severity, stroke subtype, role of imaging findings and co‐morbidities. However, questions remain about whether hazard can be minimised by novel thrombolytic drugs, different doses and routes or speed of administration (Wardlaw 2013) or strategies to improve access of rt‐PA to the occluding thrombus (e.g. ultrasound or glyceryl trinitrate (GTN)): some trials addressing these questions are ongoing or are in the planning stages.
There is now good data on the effect of thrombolytic therapy in the elderly, in whom stroke is most common. Prior to IST3 2012, only EPITHET 2008, MAST‐E 1996, MAST‐I 1995, and NINDS 1995 did not have an upper age limit but they included few participants (69 in NINDS and 25 in EPITHET) older than 80 years. People over 80 constitute a significant and increasing proportion of patients with stroke. The European license has an upper age limit of 80 years and has further strict selection criteria compared with the USA, where the license is based mainly on the NINDS 1995 trial and does not have an upper age limit. Among other limitations in Europe, an upper limit of stroke severity has also been introduced, as has a contraindication in people with the combined occurrence of diabetes and previous stroke. Hopefully these new data showing similar benefits of rt‐PA in older as in younger people, particularly if treated within three hours of stroke, will lead to a relaxation of the licence.
Despite the large increase in available data with the inclusion of IST3 2012 (and near doubling of data for rt‐PA) there is still significant between‐trial heterogeneity for death or dependency at the end of follow‐up both for trials testing rt‐PA, and for all thrombolysis trials together. For rt‐PA, there is heterogeneity for death or dependency whether defined as modified Rankin Score (mRS) 3 to 6 or 2 to 6. This indicates that more data are needed to provide more robust results. The need for more data from new trials is also supported by the fact that the data are relatively unstable. For example, changing the definition of death or dependency from mRS 3 to 6 to 2 to 6 increases the heterogeneity for all thrombolysis trials (and is still present for just rt‐PA trials) (Wardlaw 2000). In this latter analysis, although the overall conclusion of the trials combined was not materially altered, some individual trials 'wobbled' between being statistically significantly positive to statistically non‐significant on moving from Rankin score 2 to 6 to 3 to 6, and vice versa (ECASS II 1998; ECASS 3 2008; PROACT 2 1999; Wardlaw 2000), with the most recent victim of this play of chance being IST3 2012. The fact that a neutral trial can become positive, or vice versa, or heterogeneity can be apparently removed by such a small alteration in the endpoint analysed simply emphasises the instability of the data and also advocates for alternative more robust ways of analysing data. The heterogeneity could have arisen from many sources, including differences in the design of the trials, in the type of participants included, in the availability of data to contribute to the present analysis (not all trials contributed data to all outcome analyses), and in the fact that these can only be considered as small trials for a condition as heterogeneous as ischaemic stroke. Individual data will be able to examine whether factors such as sex, blood glucose, etc. influence the effect of thrombolysis (Kent 2005a; Kent 2005b; Kent 2007; Mann 2005; Sandercock 2006). Comparisons of different thrombolysis drugs, doses and routes of administration are addressed in a separate Cochrane review (Wardlaw 2013).
There was a suggestion that the presence of a visible recent infarct on the CT scan prior to randomisation may be related to increased hazard (risk of ICH and death) but this was based on a post‐hoc analysis of the CT scans in ECASS 1995, in which the baseline CT scans were not read blind to follow‐up CT scans. Some trials had CT‐visible infarction exclusion criteria and some, including NINDS 1995, did not. The reported rate of CT‐visible infarction varied between trials, either reflecting differences in participant selection, observer sensitivity, or definition of visible infarction signs. We have now been able to include data on CT‐visible infarct extent categorised using the ASPECTS score from secondary analyses of CT scans in several trials. We did not find any evidence of an interaction between visible infarct extent and rt‐PA on death or dependency. Other possible risk factors identifiable on the CT scan that might interact with rt‐PA (such as evidence of small vessel disease), are being addressed in IST3 2012. Whether or not other, more advanced imaging modalities, such as CT perfusion or magnetic resonance (MR) diffusion with perfusion, would improve patient selection or allow an extension of time window to treatment is the subject of ongoing trials (EXTEND). At present, the modest data available in an indirect comparison do not suggest that selection on the basis of MR diffusion‐ or perfusion‐weighted imaging (DWI/PWI) leads to better effect of thrombolysis than for those selected on the basis of plain CT (Analysis 1.36; Analysis 1.37).
Overall completeness and applicability of evidence
The trials included in this review are small in comparison with the thrombolytic therapy in myocardial infarction trials. Nonetheless, this version of the review, with the addition of IST3 2012, includes a wider range of participants, with many more aged over 80 years, than previous versions. This is an effect of the principal methodology of IST3 2012 with the application of the uncertainty principle, which states that when there is a clear indication for treatment the person should be treated, and when there clearly is a contraindicated the person should not be treated; only where the tested treatment is promising but unproven could the participant be randomised. This approach provides the chance to test wider treatment criteria. There are substantially more data with the inclusion of 3035 participants from IST3 2012. However, not all trials contributed to all analyses, some analyses only include five or so trials and there were imbalances in stroke severity and age between treatment groups in some earlier trials. There remains significant heterogeneity for some outcomes and lack of a complete picture of the sources of heterogeneity, meaning that there is scope for more trials. This is particularly the case for mild strokes. Although there is a lack of information on concomitant aspirin usage, it seems fairly clear from MAST‐I 1995 and the non‐random comparisons in Analysis 1.14 that aspirin (or other antithrombotic drugs) given at the same time as thrombolysis is hazardous. The adverse effect of aspirin together with rt‐PA was confirmed in one recent trial that stopped prematurely due to excess bleeding with aspirin and rt‐PA combined (Zinkstok 2008). We have not been able to identify clear reasons why some people do poorly with thrombolysis. For example, the absence of any apparent time dependence of SICH with thrombolysis suggests that some other non‐time‐dependent factors may increase haemorrhage risk ‐ i.e., not the presence of acute ischaemic change or other time‐dependent factors. In contrast, the benefit of thrombolysis declines with time, fewer patients being alive and independent the later the treatment. The independent data meta‐analysis of all rt‐PA trials may be better able to identify factors influencing hazard.
The time window beyond which there is unlikely to be any benefit (or too much hazard) with thrombolytic therapy is unclear. The modifiers of the adverse effects of thrombolytic mode of action remain undetermined. There is a clear time dependency, with fewer participants treated within three hours of stroke being dead or dependent, than participants treated between three and six hours, but the latest time window remains undetermined. Other trials that tested other thrombolytic regimens beyond six hours suggest that the benefit may extend to nine hours or even longer in selected people. Although these trials were themselves not positive, when combined the overall result suggests that thrombolysis reduces death or dependency even at these later times. Thus, the time window for benefit probably extends to, and even beyond, six hours in selected people. However, this should not encourage complacency about the need for speedy treatment in ischaemic stroke. It simply underlines the need for more data so as to be able to provide individually‐tailored treatment accounting for age, sex, stroke severity, prior aspirin use, brain scan appearances, etc., to name but a few factors in addition to time, which are likely to affect thrombolysis effect.
There is little information on which thrombolytic drug might have most benefit and least hazard, and there is little information on which dosage of drug has least hazard and most benefit (Wardlaw 2013). Direct randomised comparisons would be required (Dundar 2003). The Chinese UK trial (Chen 2000) had two doses of urokinase, but was underpowered to detect any difference between them. Similarly, the DEDAS 2006, DIAS 2005, and DIAS 2 2008 trials were together underpowered to detect a difference between doses of desmoteplase. Note that further details on direct randomised comparisons of drug or dose are included in a separate Cochrane Review (Wardlaw 2013), for which there are few additional data since its original publication.
There is limited information about the effect of thrombolysis on survival in a longer time frame, as most of the trials (all of the recent rt‐PA trials) performed the follow‐up at three months. NINDS 1995 published data on functional outcome at six months and one year which indicate that the effect of rt‐PA was sustained beyond three months. IST3 2012 published data on functional outcome and death at 18 months which also indicate long‐term benefits, but there are few other data on whether the benefit of thrombolysis is sustained (or even increases) at one year. This information is important for understanding the impact of thrombolytic treatment on health economics.
It is difficult to assess the cost effectiveness of thrombolytic treatment. A review for the UK National Health Service Health Technology Assessment (HTA) Programme on the cost effectiveness of thrombolytic treatment for acute ischaemic stroke needs to be updated (Sandercock 2002). One trial has a prospective substudy ongoing for the measured and modelled evaluation of cost effectiveness (IST3 2012).
Quality of the evidence
The overall quality of evidence, particularly for the drug with the majority of data, rt‐PA, is good. The concerns about quality in earlier trials are largely overcome. The recent trials had good allocation concealment, central telephone randomisation, central blinded follow‐up, and very few losses to follow‐up.
Potential biases in the review process
This review is the result of an ongoing process involving the collaborative effort of many researchers worldwide and the principal investigators of many of the thrombolysis trials. At present, this review represents all of the evidence from the randomised controlled studies on the effects of thrombolytic therapy on acute ischaemic stroke. Comparisons of trials using different thrombolytic drugs should be treated with caution as these comparisons are indirect; available data on direct comparisons are presented in the companion review (Wardlaw 2013). We have tried to include all available tabular data and have checked the accuracy of it rigorously. We have tried not to miss any relevant completed trials. We can only apologise if we have overlooked some available data on an outcome in an included trial or have overlooked a trial completely.
Agreements and disagreements with other studies or reviews
A more detailed individual patient data meta‐analysis of the streptokinase trials, using data from MAST‐E 1996, MAST‐I 1995 and ASK 1996, has been completed (TAS‐PP 1999); urokinase trials (Jin Urokinase metaanalysis 2000) have been completed, and three rounds of a meta‐analysis of individual data from the rt‐PA trials have been published (rt‐PA pooled analysis 2004; rt‐PA pooled analysis 2008; TTAS) with a third completed and accepted for publication (in press). The analyses based on tabular data in the present review are consistent with these individual patient data analyses.
The SICH, death and functional outcome rates in the large registry of people treated open‐label with rt‐PA (SITS‐MOST) are also similar to those found in the randomised trials. However, patient surveys are ultimately voluntary and therefore are inevitably prone to potential bias through incomplete data.
Authors' conclusions
Implications for practice.
Taken overall, in people given thrombolysis in the acute phase of ischaemic stroke, there appears to be a net benefit of a significant reduction in the proportion who are dead or dependent at the end of follow‐up across all drugs and time windows.
Faster treatment is more beneficial. People treated within three hours of stroke are less likely to be dead or dependent than those treated after three hours, although some may still derive benefit if treated up to six hours.
There is, overall, proof of an excess risk of symptomatic and fatal intracranial haemorrhage and early death from all causes with thrombolytic therapy. Evidence on risk factors, however, is incomplete.
More data are available for recombinant tissue plasminogen activator (rt‐PA) than for other drugs; with rt‐PA, there was no net effect on death from all causes at long‐term follow‐up.
People aged over 80 derive as much benefit from rt‐PA as do those aged under 80 years, especially if treated within three hours of stroke.
Despite the overall net benefit, the available data do not provide sufficient evidence to determine the duration of the therapeutic time window, the clinical or radiological features which identify those most likely to benefit (or be harmed) including whether or not people with mild stroke benefit or not, or the optimum agent (or dose or route of administration).
The data indicate that antithrombotic treatment should be avoided until at least 24 hours after thrombolytic treatment.
In the light of these considerations, current evidence supports configuration of stroke services so as to be able to treat as many people as possible as fast as possible with the licensed drug rt‐PA, including those aged over 80. There is no evidence to withhold rt‐PA on the basis of age, early CT ischaemic changes, or severity of stroke if it can be administered within 4.5 hours and preferably within three hours. While the data suggest that some people may benefit even up to six hours, change in clinical practice should await results of further trials to determine the latest time window for benefit.
Implications for research.
These data leave some uncertainties, which suggest that further large‐scale randomised trials testing aspects of delivery of thrombolytic therapy in people with acute ischaemic stroke are needed:
To identify means of minimising the hazard without reducing the benefit, e.g. lower dose, avoiding people with specific characteristics (yet to be determined) or combinations of characteristics (e.g. elderly, severe stroke and some imaging feature), slower administration of the rt‐PA bolus, different drug with lower haemorrhage risk, etc;
To provide data on the latest time window for treatment in which people and by what means of selection;
To provide data on benefits or harms of thrombolysis in mild stroke;
To provide randomised data on quality of life and cost effectiveness.
In future trials it would be helpful if data could be collected in such a way as to be compatible with the simple and fundamental effect parameters used in this review (e.g. early and late death, fatal intracranial haemorrhage, functional outcome). This would help to address the problem in the present review of between‐trial heterogeneity (which may be exacerbated by missing data), and facilitate future meta‐analyses.
Feedback
Concomitant use of antithrombotic treatment
Summary
Category: Methodological qualities of included studies
This review (The Cochrane Library Issue 1,1997) states that ".. in the NINDS (1995), Mori et al 1992, and Haley et al 1993 no antithrombotic drugs were allowed within 24 hours .". However, Haley et al published that "Intravenous heparin was prohibited until at least 30 minutes after the infusion was complete .." thereby suggesting that heparin may have been given much earlier than 24 hours.
Reply
Wardlaw et al have tried to contact Haley et al but without success. Hence, they have changed the text to ".. in Haley et al (1993) a few patients received antithrombotic drugs within 24 hours ..".
Contributors
Comment by Ryan CW, April 1997 Response by Wardlaw J et al, December 1997
Feedback, 21 March 2015
Summary
Comment: We commented on this Cochrane review and the overall evidence reported for alteplase use at 3‐4.5 hours after stroke (BMJ. 2015 Mar 17;350:h1075) and provide specific feedback for improving this Cochrane review. In the Risk of bias in included studies section, it would be helpful: 1.) to include a risk of bias summary table 2.) in the paragraph on incomplete outcome data, to describe the completeness of the available data explicitly rather than stating all available data are included. Describe the risk of bias from incomplete data, including a trial‐specific understanding of how prevalent or nonprevalent this risk is 3.) in the paragraph on selective reporting, to define what you mean by "strict intention‐to‐treat analysis". Does this mean that outcomes were assumed and assigned for all randomized patients, regardless of follow‐up data availability? And if missing data was imputed, how was it done, and was it done in a similar way in each trial? Also, presenting sensitivity analyses and including an available case analysis would be useful. Regardless of how you describe your methodology for approaching intention‐to‐treat analysis, comment on whether the data is available or the intention‐to‐treat analyses were available from the trials to determine if absence of intention‐to‐treat analyses represents a risk of bias in the data at a trial‐specific level. 4.) to add a discussion on baseline differences: What relevant factors, if any, were imbalanced between groups in each trial? This is an important risk of bias criterion not addressed in the review. In the Results section and subsequent discussions and conclusions: 1.) While there are many analyses that can be grouped by time in many ways (0‐3 hours, 0‐4.5 hours, 0‐6 hours, 3‐6 hours, 3‐4.5 hours, 4.5‐6 hours), providing data for many overlapping combinations is confusing, so timeframes that do not represent clinically useful and methodologically sound groups should be dropped from the results reporting. 2.) Overall timeframes such as 0‐4.5 hours and 0‐6 hours are not useful for clinical decision making or overall data interpretation because: a. Clinical practice patterns and guidelines have “chunked” practice into 0‐3 hours, 3‐4.5 hours, and 4.5‐6 hours for practical implementation. b. The data in the existing Cochrane review have such different results in subgroup analyses for 0‐3 hours and for 3‐6 hours that lumping these two together for an overall analysis does not provide a summary statistic that is representative of a clinically representative grouping of patients. The 'average' of these two groupings does not represent the 'whole'. c. If data for 3‐4.5 hours and 4.5‐6 hours could be separated this may be more clear than analyses for 3‐6 hours. 3.) Addressing the impact of risk of bias assessments could be important for results interpretation and discussion. The changes above could be applied relatively quickly and are important considering the Cochrane review could be expected to be the most valid, thorough and current systematic review for this subject. Meanwhile, if Cochrane reviewers could obtain the full data, address population subgroupings considered most clinically relevant (age < 80 years vs. age > 80 years, timeframes 0‐3 hours vs. 3‐4.5 hours), and provide an updated individual patient data meta‐analysis, it would immensely help policy‐makers view this in full detail. A protocol for such an analysis being shared a priori with an opportunity for clinicians, researchers, and content domain experts to comment on which subgroupings are most clinically relevant and which approaches to the data interpretation and analysis are most likely to be informative or potentially introducing a risk of bias would allow for a meta‐analysis with wider acceptance and greater confidence in its results.
Reply
We thank Brian Alper and colleagues for their interest in our review, and for their comments. Our responses (in bold) are listed below point by point. Many of the points raised cannot be answered by the tabular data of the review (for example the effects in different time splits). However, these were answered by the Stroke Thrombolysis Trialists' Collaboration (STTC) publication in the Lancet in 2014, which is referenced in the review.
Comment: We commented on this Cochrane review and the overall evidence reported for alteplase use at 3‐4.5 hours after stroke (BMJ 2015 Mar 17;350:h1075) and provide specific feedback for improving this Cochrane review.
Response: Thank you for your interest in the review and for taking the time to submit this critique.
In the Risk of bias in included studies section, it would be helpful:
1) to include a risk of bias summary table.
Response: Thank you. We have provided 'Risk of bias' tables for the trials included since the last update, and plan to extend to all trials in the next revision. However, we clearly state the major issues related to bias in the text and state in the Methods in 'Assessment of risk of bias':
"We assessed risk of bias as specified in the Cochrane Handbook for Systematic Reviews of Interventions, Version 5.1.0 (March 2011), Chapter 8 (Higgins 2011). We assessed whether the method of randomisation would allow allocation concealment, the adequacy of efforts to blind treatment administration and outcome assessment. For each included trial we collected information about:
the method of randomisation (including information on allocation concealment);
blinding of treatment administration;
blinding of outcome assessment; and
whether an intention‐to‐treat analysis was done, or could possibly be done.
We provide detailed 'Risk of bias' tables for the trial included since the last update."
2) to describe, in the paragraph on incomplete outcome data, the completeness of the available data explicitly rather than stating all available data are included. Describe the risk of bias from incomplete data, including a trial‐specific understanding of how prevalent or nonprevalent this risk is.
Response: We do report the following about incomplete outcome data:
"Incomplete outcome data (attrition bias)
All available data are included. Data on six participants were missing from the ATLANTIS B 1999 trial publication and details have not been forthcoming from the investigators, and we have not yet received data on 46 participants from the Chinese UK Trial (Chen 2000) (these participants were randomised after the trial's six‐hour time limit and have not yet been supplied). More information is available for some trials than for others, either because the trial collaborators have published very actively on various aspects of their trial, or because in some cases further information is available from other sources (for example, reports on NINDS 1995 appear on the US Food and Drug Administration (FDA) website as part of the licence application process). The more frequent reporting or greater completeness of the data for some trials is merely a reflection that more information is available for those trials, and not intended to over‐ or under‐emphasise the actual results or quality of any particular trial (or trials) compared with others for which there is less detailed information available.
There was more detail about each trial in the text in earlier versions and in drafts of the present version. However, space constraints imposed by the Cochrane Database of Systematic Reviews (CDSR), the large volume of important information that now has to be included, and the historical nature of some of the trials (which, note, are also relatively small), meant that these details have not been retained in the text. Note we state clearly in Methods, 'Dealing with missing data':
"We contacted trial investigators to obtain all unpublished missing data where possible. Where data were still missing or had not been collected in the original trial, then that trial did not contribute to the relevant outcome. We clarified missing or unclear data with the principal investigator. The outcomes in two studies were very clearly described in the original texts and verification with the principal investigators was not necessary (Haley 1993; Morris 1995)."
Also see Methods 'Assessment of reporting biases':
"We have endeavoured to include data from all trials on all prespecified outcomes, obtained from secondary publications or the trial investigators if unpublished. We assessed the likelihood of missing trials using a funnel plot."
The risk of bias from incomplete data is dependent on whether or not the trials used blinded outcome assessment. We will therefore also refer to Results, from the five sections on five different important sources of bias:
"Blinding (performance bias and detection bias)
Five trials were single‐blind without a placebo (AUST 2005; MAST‐I 1995; MELT 2007; PROACT 2 1999; Wang 2003). In one trial (IST3 2012) the first 276 participants were treated in the double‐blinded phase of the trial and all 2759 remaining participants were included into the open phase of the trial. All participants in the study, irrespective of study phase, were blindly assessed by postal mail or telephone by a blinded and trained observer. In PROACT 2 1999, the control group underwent catheter placement but received no infusion. All the rest were double‐blind placebo‐controlled trials. However, it should also be noted that thrombolysis, due to its effects on the coagulation system at high doses, can be difficult to blind completely due to the obvious signs of bleeding (prolonged bleeding at venepuncture sites, easy bruising, gingival or conjunctival haemorrhages, etc). Thus, provision of an identical‐appearing placebo (in the syringe) may not fully blind investigators to treatment allocation. Furthermore, as thrombolytic agents are proteins, they froth when shaken in solution with water or saline, rather like egg white mixed with water and shaken. Normal saline is therefore not an identical‐looking placebo for a thrombolytic agent. Thus, in addition to the possibilities for failure of treatment allocation concealment inherent in the randomisation methods used as outlined above, it is possible that treatment allocation could be guessed accurately by the physicians caring for the participant in the acute phase because of these biological effects. Accordingly, methods for ensuring complete blinding of treatment allocation at late follow‐up are crucial. Only one study (MAST‐I 1995) used central telephone follow‐up by a blinded trained observer. Although seven other trials specified that follow‐up was to be by a physician not involved in the acute care of the participant, it is uncertain how completely this was achieved in practice. Other trials either did not specify who should do the follow‐up, or did not make it mandatory that follow‐up was by an independent physician, so in either case follow‐up may have been carried out by the acute phase physician who could have been influenced by their knowledge of events in the acute phase."
Also, the risk of bias from incomplete data is dependent on other sources of bias. We have, therefore, copied from the three other sections on different important sources of bias in the Results section, here for avoidance of doubt:
"Allocation (selection bias)
Among the included studies 14 (52%) fulfilled criteria for high grade concealment. The concealment has successively improved over time with the development and utilisation of new randomisation methods, such as the use of a centralised computerised method with interactive interface for randomisation over the telephone or Internet.
Twelve trials used central telephone or Internet randomisation (ASK 1996; AUST 2005; DEDAS 2006; DIAS 2005; DIAS 2 2008; ECASS 3 2008; IST3 2012; MAST‐E 1996; MAST‐I 1995; MELT 2007; PROACT 1998; PROACT 2 1999). In five studies (AUST 2005; IST3 2012, MAST‐I 1995; MELT 2007; PROACT 2 1999), the allocated treatment was then given unblinded without a placebo. In one of the studies (IST3 2012) with the exception of 276 participants treated in the double‐blinded phase of the trial, the remaining participants (2759) were treated unblinded without placebo. In four other studies (ASK 1996; ECASS 3 2008; MAST‐E 1996; PROACT 1998) sealed prepacks of thrombolytic drug or identical‐appearing placebo were given according to the randomisation instructions.
In three trials randomisation was at the participating hospital by selection of a sealed, sequentially‐numbered, prepack (of active drug or identical appearing placebo) followed within two hours by a telephone call to the Central Trial Co‐ordinating Office to notify them of the participant and the number of the drug pack (ATLANTIS A 2000; ATLANTIS B 1999; NINDS 1995). In one study (NINDS 1995), the randomisation system, set up in an effort to reduce delays to treatment, led to 'out of order' trial treatment allocations in between 13 and 31 participants, which affected every subsequent participant until the error was detected, and led to participants appearing to cross between treatment allocations (more moved from rt‐PA to placebo than the other way round). Also in the interests of reducing delays to trial treatment administration, there were some participants who ultimately were not entered into the study after the pharmacy had prepared the trial pack (and therefore some discarded trial packs). Details of the randomisation are given at www.fda.gov/cber/products/altegen061896.htm; see Clinical Review 2, page 11‐12 and 18‐19.
In three trials, randomisation was by selection of a sequentially numbered, sealed drug prepack at the participating centre provided by the sponsor from a randomisation schedule drawn up centrally (ECASS 1995; ECASS II 1998; EPITHET 2008).
Of the remaining trials:
five trials used sealed drug prepacks of active drug or identical‐appearing placebo (Abe 1981; Atarashi 1985; JTSG 1993; Mori 1992; Ohtomo 1985);
one used sealed envelopes (Haley 1993);
one used sealed drug prepacks of active drug or normal saline (as placebo) (Chen 2000);
the method was not stated in two (Morris 1995; Wang 2003).
Please note that, therefore, only two of the rt‐PA trials (ECASS 3 2008: IST3 2012) recorded the participant details centrally over the telephone or Internet prior to starting trial treatment. In one of these trials (IST3 2012) a minimisation algorithm was used to balance the study arms for key prognostic variables like stroke severity before randomisation. Several later studies have made use of modern randomisation techniques and entering key prognostic variables into the IT system before randomisation, which allows balancing of the study arms ‐ as has been introduced in one trial (IST3 2012).
Selective reporting (reporting bias)
We have avoided, as far as possible, any reporting bias by obtaining original data from the trial investigators where these have not been published. Only the intention‐to‐treat results are included here. In any trials where there have been exclusions, these were made prior to the breaking of the randomisation code. A strict intention‐to‐treat analysis was used in 18 studies (ASK 1996; ATLANTIS A 2000; ATLANTIS B 1999; AUST 2005; DEDAS 2006; DIAS 2005; DIAS 2 2008; ECASS 1995; ECASS II 1998; ECASS 3 2008; EPITHET 2008; IST3 2012; MAST‐E 1996; MAST‐I 1995; MELT 2007; PROACT 1998; PROACT 2 1999; Wang 2003), but not in any of the earlier trials. The administrative problems with randomisation in one study (NINDS 1995) led the FDA reviewer to describe the primary analysis as an 'on‐treatment analysis'. However, the primary analysis was undertaken without excluding any participants entered into the trial and was, therefore, an intention‐to‐treat analysis (www.fda.gov/cber/products/altegen061896.htm; see Clinical Review 2, page 20). For the earlier trials, with additional information from the principal investigators if necessary, we have attempted to find a final outcome for all randomised participants, rather than simply relying on the published data from which some randomised participants may have been excluded. Note that one trial (ECASS 1995) was published as intention‐to‐treat and as a 'target population' after about 20% of the randomised participants had been excluded, but only the intention‐to‐treat data have been included here.
Other potential sources of bias
Randomisation in two trials, ASK 1996 (in the over‐three‐hour group) and MAST‐E 1996, was stopped on the advice of their respective data monitoring committees after only about half of the originally intended number of participants had been randomised. One study (MAST‐I 1995) was suspended by its steering committee (in view of the stopping of MAST‐E 1996 and ASK 1996) to examine its interim results after randomising about one third of its originally intended number. Another study (MELT 2007) was discontinued on the advice of its data monitoring committee when rt‐PA was licensed in Japan in 2005. Another study (AUST 2005) was discontinued on the basis of very slow recruitment after 24 participants of a planned sample of 200 had been included. Four studies (ECASS 1995; ECASS II 1998; NINDS 1995; PROACT 2 1999) all reached their planned targets. One study (PROACT 1998) was stopped after completing two of its planned three dosage arms by the pharmaceutical provider. Another study (ATLANTIS A 2000) was stopped on publication of the NINDS 1995 trial, and continued in modified form as ATLANTIS B 1999, which in turn stopped in 1998 following a 'futility analysis' prompted by results from the ECASS II 1998 study. Examination of funnel plots for the main outcomes showed these to be symmetrical and therefore provided little evidence of publication bias."
3) to define, in the paragraph on selective reporting, what you mean by "strict intention‐to‐treat analysis". Does this mean that outcomes were assumed and assigned for all randomised patients, regardless of follow‐up data availability? And if missing data was imputed, how was it done, and was it done in a similar way in each trial? Also, presenting sensitivity analyses and including an available case analysis would be useful. Regardless of how you describe your methodology for approaching intention‐to‐treat analysis, comment on whether the data is available or the intention‐to‐treat analyses were available from the trials to determine if absence of intention‐to‐treat analyses represents a risk of bias in the data at a trial‐specific level.
Response: 'Intention to treat' means analysed by treatment group to which the subject was assigned at randomisation, rather than by the treatment that they actually got. This is a standard term. All analyses were done as intention‐to‐treat analyses, and there were no assumptions, imputations or other fiddling with the data. The data are as published in the individual trials in almost all cases and can be extracted should you wish to repeat these analyses yourselves. In a few trials, additional unpublished data were sought and included but these were mostly for secondary outcomes. Also see above.
4) to add a discussion on baseline differences: What relevant factors, if any, were imbalanced between groups in each trial? This is an important risk of bias criterion not addressed in the review.
Response: A detailed discussion on baseline imbalance was included in previous versions of this review over the last 15 years but has mostly been removed from the current version due mainly to space constraints and because there has been an extensive debate and reanalysis (e.g. of the NINDS trial) all of which is referenced, plus the STTC individual patient data meta‐analysis (Lancet 2014, see references), all of which overcome concerns regarding baseline imbalance. Also see sections on bias from Results copied above.
In the Results section and subsequent discussions and conclusions:
1) While there are many analyses that can be grouped by time in many ways (0‐3 hours, 0‐4.5 hours, 0‐6 hours, 3‐6 hours, 3‐4.5 hours, 4.5‐6 hours), providing data for many overlapping combinations is confusing, so timeframes that do not represent clinically useful and methodologically sound groups should be dropped from the results reporting.
Response: With respect, a) many would disagree on what constitutes a clinically meaningful time window, and b) tabular data on 3‐4.5 or 4.5‐6 hours have not been published from most trials. Only ECASS‐3 examined 3‐4.5 hours and IST‐3 provided 3‐4.5 hour data in a prespecified subgroup analysis. There is substantial interest in 0‐3 hours and 3‐6 hours and these times are available. Therefore we provide these times and refer the reader to the STTC analysis (Lancet 2014). The several time subgroups are clearly labelled and their presence explained in the text. The separate analyses are retained for trials that randomised in both 0‐3 and 3‐6 hour time windows to deal with the difference in stroke severity seen in early vs later presenting patients, which was not otherwise possible with tabular data.
2) Overall timeframes such as 0‐4.5 hours and 0‐6 hours are not useful for clinical decision making or overall data interpretation because:
a. Clinical practice patterns and guidelines have “chunked” practice into 0‐3 hours, 3‐4.5 hours, and 4.5‐6 hours for practical implementation.
b. The data in the existing Cochrane review have such different results in subgroup analyses for 0‐3 hours and for 3‐6 hours that lumping these two together for an overall analysis does not provide a summary statistic that is representative of a clinically representative grouping of patients. The “average” of these two groupings does not represent the “whole”.
c. If data for 3‐4.5 hours and 4.5‐6 hours could be separated this may be more clear than analyses for 3‐6 hours.
Response: See above. As only 2 trials provide 3‐4.5 hr data, there is little point in creating separate tables, because the sample would be too small. The STTC analysis addresses this issue adequately, clearly showing that the latest time window for alteplase (rt‐PA) is around 5 hrs or just after. Please note a methodological point however: the lack of patients randomised beyond six hours means that it is not possible to determine the very latest time window at present – for now we can only be confident of some benefit out to 5 hours – a larger sample, as is seen where all thrombolytic drugs (10,000 patients) are considered, suggests a definite benefit out to six hours. Confirmation of this point will be possible with the results of more later time window trials. Note that the recent positive (small) thrombectomy trials had time windows out to 8 hours.
3) Addressing the impact of risk of bias assessments could be important for results interpretation and discussion.
Response: We have above copied from the five sections on different important sources of bias, from Results. For avoidance of doubt, please see also the three sections on these points in the Discussion:
"Overall completeness and applicability of evidence
The trials included in this review are small in comparison with the thrombolytic therapy in myocardial infarction trials. Nonetheless, this version of the review, with the addition of IST3 2012, includes a wider range of participants, with many more aged over 80 years, than previous versions. This is an effect of the principal methodology of IST3 2012 with the application of the uncertainty principle, which states that when there is a clear indication for treatment the person should be treated, and when there clearly is a contraindicated the person should not be treated; only where the tested treatment is promising but unproven could the participant be randomised. This approach provides the chance to test wider treatment criteria. There are substantially more data with the inclusion of 3035 participants from IST3 2012. However, not all trials contributed to all analyses, some analyses only include five or so trials and there were imbalances in stroke severity and age between treatment groups in some earlier trials. There remains significant heterogeneity for some outcomes and lack of a complete picture of the sources of heterogeneity, meaning that there is scope for more trials. This is particularly the case for mild strokes. Although there is a lack of information on concomitant aspirin usage, it seems fairly clear from MAST‐I 1995 and the non‐random comparisons in Analysis 1.14 that aspirin (or other antithrombotic drugs) given at the same time as thrombolysis is hazardous. The adverse effect of aspirin together with rt‐PA was confirmed in one recent trial that stopped prematurely due to excess bleeding with aspirin and rt‐PA combined (Zinkstok 2008). We have not been able to identify clear reasons why some people do poorly with thrombolysis. For example, the absence of any apparent time dependence of SICH with thrombolysis suggests that some other non‐time‐dependent factors may increase haemorrhage risk ‐ i.e., not the presence of acute ischaemic change or other time‐dependent factors. In contrast, the benefit of thrombolysis declines with time, fewer patients being alive and independent the later the treatment. The independent data meta‐analysis of all rt‐PA trials may be better able to identify factors influencing hazard.
The time window beyond which there is unlikely to be any benefit (or too much hazard) with thrombolytic therapy is unclear. The modifiers of the adverse effects of thrombolytic mode of action remain undetermined. There is a clear time dependency, with fewer participants treated within three hours of stroke being dead or dependent, than participants treated between three and six hours, but the latest time window remains undetermined. Other trials that tested other thrombolytic regimens beyond six hours suggest that the benefit may extend to nine hours or even longer in selected people. Although these trials were themselves not positive, when combined the overall result suggests that thrombolysis reduces death or dependency even at these later times. Thus, the time window for benefit probably extends to, and even beyond, six hours in selected people. However, this should not encourage complacency about the need for speedy treatment in ischaemic stroke. It simply underlines the need for more data so as to be able to provide individually‐tailored treatment accounting for age, sex, stroke severity, prior aspirin use, brain scan appearances, etc., to name but a few factors in addition to time, which are likely to affect thrombolysis effect.
There is little information on which thrombolytic drug might have most benefit and least hazard, and there is little information on which dosage of drug has least hazard and most benefit (Wardlaw 2013). Direct randomised comparisons would be required (Dundar 2003). The Chinese UK trial (Chen 2000) had two doses of urokinase, but was underpowered to detect any difference between them. Similarly, the DEDAS 2006, DIAS 2005, and DIAS 2 2008 trials were together underpowered to detect a difference between doses of desmoteplase. Note that further details on direct randomised comparisons of drug or dose are included in a separate Cochrane Review (Wardlaw 2013), for which there are few additional data since its original publication.
There is limited information about the effect of thrombolysis on survival in a longer time frame, as most of the trials (all of the recent rt‐PA trials) performed the follow‐up at three months. NINDS 1995 published data on functional outcome at six months and one year which indicate that the effect of rt‐PA was sustained beyond three months. IST3 2012 published data on functional outcome and death at 18 months which also indicate long‐term benefits, but there are few other data on whether the benefit of thrombolysis is sustained (or even increases) at one year. This information is important for understanding the impact of thrombolytic treatment on health economics.
It is difficult to assess the cost effectiveness of thrombolytic treatment. A review for the UK National Health Service Health Technology Assessment (HTA) Programme on the cost effectiveness of thrombolytic treatment for acute ischaemic stroke needs to be updated (Sandercock 2002). One trial has a prospective substudy ongoing for the measured and modelled evaluation of cost effectiveness (IST3 2012).
Quality of the evidence
The overall quality of evidence, particularly for the drug with the majority of data, rt‐PA, is good. The concerns about quality in earlier trials are largely overcome. The recent trials had good allocation concealment, central telephone randomisation, central blinded follow‐up, and very few losses to follow‐up.
Potential biases in the review process
This review is the result of an ongoing process involving the collaborative effort of many researchers worldwide and the principal investigators of many of the thrombolysis trials. At present, this review represents all of the evidence from the randomised controlled studies on the effects of thrombolytic therapy on acute ischaemic stroke. Comparisons of trials using different thrombolytic drugs should be treated with caution as these comparisons are indirect; available data on direct comparisons are presented in the companion review (Wardlaw 2013). We have tried to include all available tabular data and have checked the accuracy of it rigorously. We have tried not to miss any relevant completed trials. We can only apologise if we have overlooked some available data on an outcome in an included trial or have overlooked a trial completely."
The changes above could be applied relatively quickly and are important considering the Cochrane review could be expected to be the most valid, thorough and current systematic review for this subject.
Response: Thank you but please take note of the above points, particularly that of space constraints. A large amount of text had to be culled from the present version at a late stage. Perhaps the CDSR could consider increasing their space – an electronic publishing medium should not impose text space constraints. The information you refer to is not ‘missing’ from the review – information on bias is given in the text and the published trial reports do not contain tabular data on the specific time window you request. Earlier versions provide detailed descriptions and discussion of baseline imbalance. However these issues concern only two trials (NINDS and ECASS III) of only about 1400 patients (of 7000 patients treated with alteplase or 10,000 patients treated with any thrombolytic drug). Disputes over this have already been settled, and the STTC individual patient data meta‐analysis has already overcome concerns over this.
Meanwhile, if the Cochrane reviewers could obtain the full data, address population subgroupings considered most clinically relevant (age < 80 years vs. age > 80 years, timeframes 0‐3 hours vs. 3‐4.5 hours), and provide an updated individual patient data meta‐analysis, it would immensely help policy‐makers view this in full detail. A protocol for such an analysis being shared a priori with an opportunity for clinicians, researchers, and content domain experts to comment on which subgroupings are most clinically relevant and which approaches to the data interpretation and analysis are most likely to be informative or potentially introducing a risk of bias would allow for a meta‐analysis with wider acceptance and greater confidence in its results.
Response: The STTC has already done exactly what you suggest and published both the protocol and the results in 2014. For the Cochrane review, all available data are in the data tables, and the primary and secondary trials publications are all listed in the review so that interested readers can go and replicate this 25 years' worth of analysis should they wish.
Contributors
Commentors: Brian S Alper, Meghan Malone‐Moses, James S McLellan, Kameshwar Prasad, Eric Manheimer
Responders: Joanna Wardlaw, Eivind Berge
Feedback, 11 November 2015
Summary
We previously provided feedback (dated March 21, 2015) and your many responses to numerous concerns did not address a highly specific concern regarding the baseline difference in history of prior stroke in ECASS III, and how adjusting for this may negate any evidence of efficacy for alteplase 3 to 4.5 hours after stroke onset.
The ECASS III trial (Hacke et al. New England Journal of Medicine 2008 25;359(13):1317‐29) is the only trial supporting efficacy of alteplase 3 to 4.5 hours after stroke. A potentially serious risk of bias in ECASS III was the baseline difference in history of prior stroke, a finding that was present in 7.7% of alteplase patients and 14.1% of placebo patients.
Detailed discussions on baseline imbalances that were included in previous versions of this review (2000, 2003, or 2009) do not adequately cover the baseline difference in history of prior stroke in ECASS III and its specific influence on conclusions regarding efficacy 3 to 4.5 hours after stroke, either in ECASS III alone or in analysis of all available data for this timeframe.
This baseline difference was erroneously reported in ECASS III as non‐significant in the original trial report in 2008 and not corrected until 2011, so none of this should be considered "settled" or referred to historically. In the 2008 ECASS III report, the adjusted analyses to show the primary outcome was robust when adjusting for baseline differences did not adjust for the history of prior stroke. The P value for this baseline difference in history of stroke, a potentially confounding variable, was corrected online in 2011 (from 0.03 to 0.003) without a correction notice, and this is only known based on a letter published in another journal in 2012 (Shy. Journal of Emergency Medicine 2014;46(3):385‐6. Epub 2012 November 8). This letter also reported a reanalysis of ECASS III based on available summary data and limited to the 89% of patients without a prior stroke, which found no significant effect on the primary outcome.
Thus, this specific key baseline difference appears to be the primary contributor to the "statistically significant benefit" reported in ECASS III and conclusions of efficacy derived from this data or any other data should require substantial transparency and scrutiny to be accepted.
The ECASS III trial data is reported to be available at clinicalstudydatarequest.com and a reanalysis using the original data could be conducted independently. Until such reanalysis is done, it is most prudent to acknowledge this specific risk of bias, recognize its potential effect on the overall body of evidence specific to use of alteplase 3 to 4.5 hours after stroke, and not make conclusions about alteplase use 3 to 4.5 hours after stroke that extrapolate from data outside this timeframe or analyses that have not accounted for such risk of bias.
Reply
We thank Dr Alpers and colleagues for their continuing interest in the review of thrombolysis in acute ischaemic stroke. We are sorry that our responses did not address one highly specific concern.
To summarise, in ECASS 3, there was a difference in history of stroke (7.7% in the patients allocated to alteplase and 14.1% in the patients allocated to placebo). You are concerned that this difference in baseline characteristic is enough to account for the apparent benefit of rt‐PA on mRS 0 to 1 at 90 days that was seen in ECASS 3, which only randomised patients between 3 to 4.5 hours after stroke. You cite a letter by Shy published in the Journal of Emergency Medicine 2014 in support of this concern. Shy refers to an adjusted analysis done by Bluhmki et al [1] restricted to the 89% of patients without a prior stroke in ECASS 3, which produced an odds ratio for the effect of rt‐PA on mRS 0 to 1 of 1.19, 95% CI 0.89 to 1.59. You interpret this to mean that there is no benefit of rt‐PA within the 3 to 4.5 hour time window.
We believe that the loss of significance when analysis is restricted to 89% of the data is more likely due to the diminished sample. Bluhmki et al [1] also performed a fully adjusted analysis including prior stroke, and also reported several additional outcomes and subgroup analyses beyond those provided in the original NEJM primary report [2]. In the fully adjusted model of the effect of rt‐PA on mRS 0 to 1 at 90 days, Bluhmki et al [1] adjusted for NIHSS, age, weight, onset to treatment time, diastolic and systolic blood pressure, dose in mg/kg body weight, smoking, previous stroke, previous diabetes, atrial fibrillation, hypertension, previous use of antithrombotic drugs, and sex, giving an OR for mRS 0 to 1 at 90 days after stroke of 1.43 (95% CI 1.02, 2.00, P value = 0.040), which was no different to the OR in the model that included about half of those variables but not prior stroke (OR 1.42, 95% CI 1.02‐1.98, P value = 0.037) and the analysis that was completely unadjusted (OR 1.34, 95% CI 1.02‐1.76, P value = 0.038): see Figure 3 in 1. This would suggest that the effect of prior stroke is unlikely to be sufficient to negate the positive result of ECASS 3 on mRS 0 to 1.
We note two further points.
None of the trials were especially large. As we explain in the review, a consequence is that several trials go from positive to neutral, or neutral to positive, simply by switching between mRS 0 to 1 and mRS 0 to 2, or between mRS 3 to 6 and mRS 2 to 6 as the outcome. Figures 1.9, 1.10, 1.12, 1.13, etc, explore this effect in the Review and it is described in the Results and discussed in detail in the Discussion. This illustrates that the data should be viewed in totality rather than focusing too closely on individual trials, which of course is the purpose of Cochrane reviews.
You are concerned that the imbalance in ECASS 3 is important because ECASS 3 was the only trial that randomised in the 3 to 4.5 hour time window. However, several other trials also randomised in that time window although not exclusively. Unfortunately, as explained in a previous response, not enough of those trials published tabular data on the 3 to 4.5 hour window to make a tabular meta‐analysis feasible, therefore we were not able to provide the 3 to 4.5 hour time window separately. However, as stated in the Review, there is now an individual patient data (IPD) meta‐analysis which includes ECASS 3. This IPD, performed by independent statisticians, has published a very detailed analysis of the effect of rt‐PA by time, with adjustments, and clearly showed that there is benefit for rt‐PA between 3 and 4.5 hours after stroke [3], indeed up to between 4.5 and 5 hours after stroke, when considering all the data.
The Cochrane Review has several sections on risk of bias focusing on the main messages that apply to multiple trials. For example, about half the trials had various baseline imbalances, which were inevitable as they did not use central randomisation with minimisation, and some were of similar size or worse than the prior stroke imbalance in ECASS 3 (like those in NINDS). We believe that these major messages are more relevant than to focus on only one trial. Indeed, Cochrane editorial policy means that the limited space precludes providing very detailed descriptions of all variables in all trials.
References
Bluhmki E, Chamorro A, Davalos A, et al. Additional outcomes and subgroup analyses of ECASS III: a stroke treatment study with alteplase given 3–4·5 h after onset of acute ischaemic stroke. Lancet Neurology 2009;8:1095‐102.
Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. New England Journal of Medicine 2008;359:1317‐29.
Emberson J, Lees KR, Lyden P et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta‐analysis of individual patient data from randomised trials. Lancet 2014;384:1929‐35.
Contributors
Feedback: Brian S Alper, Meghan Malone‐Moses, James S McLellan, Kameshwar Prasad, Eric Manheimer
Responders: Joanna Wardlaw, Eivind Berge
What's new
| Date | Event | Description |
|---|---|---|
| 5 January 2016 | Feedback has been incorporated | See Feedback 3 |
History
Protocol first published: Issue 1, 1995 Review first published: Issue 1, 1995
| Date | Event | Description |
|---|---|---|
| 12 May 2015 | Feedback has been incorporated | See Feedback 2 |
| 2 April 2014 | New search has been performed | We have updated the searches to November 2013 with further handsearch to March 2014. The IST‐3 study was the only new, completed trial since the previous update of the review in 2009. The IST‐3 trial adds 3035 new participants, increasing the total number of participants to 10,187, of which the rt‐PA‐data increased by 43%. We have added new information from previously published trials and several new analyses of new and existing outcomes. |
| 2 April 2014 | New citation required and conclusions have changed | Substantially more information is now available on essential outcomes in subgroups of patients, e.g. by age, stroke severity, time to treatment and essential outcomes like early and late case fatality. Markers of risk for early intracerebral haemorrhage are still unidentified. |
| 26 June 2008 | Amended | Converted to new review format. |
| 14 March 2003 | New search has been performed | Additional data are included from rt‐PA trials broken into under three and three to six‐hour time windows. Information about the organisation of the NINDS trial randomisation method and resulting problems have come to light. A new trial from China of intravenous urokinase within six hours is included. A plain language summary has been added. |
Acknowledgements
Although supported in part by a grant from the NHS Executive, the opinions and views expressed in this update do not necessarily reflect those of the NHS Executive.
We thank Prof Take Yamaguchi, now Emeritus Professor of Neurology, Cardiovascular Division, National Cardiovascular Centre, Osaka, Japan (author from the first version in 1995 to the third version in 2003) and Prof Werner Hacke, Professor of Neurology, University of Heidelberg, Germany (author on the first version in 1995) for their support on previous versions of this review.
We are very grateful to the principal investigators of all the trials who provided additional unpublished information, particularly Prof E Mori, Prof T Abe, Prof E Ohtomo, Prof L Candelise, Prof W Hacke, Prof M Kaste, Dr D Meier, Dr J Marler, Prof G Donnan, Dr M Hommel, Prof T Furlan, Dr G Albers, Dr W Clarke, and Prof Q Chen.
We would like to thank Dr Yasir Al‐Rawi for providing additional unpublished details of the DEDAS 2006, DIAS 2005, and DIAS 2 2008 trials on behalf of Paion GMBH. We would also like to thank Dr Mei‐Chiun Tseng for translating Chinese papers, Dr Kyoshi Miyagawa for translating the Japanese language papers, and Dr Kazuo Minematsu and Dr Masahiro Yasaka of the National Cardiovascular Centre, Osaka, Japan for assistance in journal searching and translation, Dr David Moher of the Ottawa Stroke Trials Registry, Dr Ming Liu and Dr Mei‐Chiun Tseng for the information on the Chinese trials, Dr Hans‐Goran Hardemark for pointing out discrepancies in the Rankin dichotomisation point for some trials, and Hazel Fraser and Brenda Thomas of the Cochrane Stroke Group for trial lists. We would also like to thank Brenda Thomas for assistance with literature searching. We would like to thank Ms Kirsten Shuler for entering references and data. We also thank the Stroke Association of the United Kingdom, and the UK Medical Research Council for previous financial support for this review. The Norwegian Research Council supported Dr Eivind Berge. The Swedish Heart‐Lungfund, AFA Insurances and Karolinska Institutet supported Dr Veronica Murray. Anyone aware of any thrombolysis trials not mentioned herein, or inaccuracies, should please contact us with the relevant information.
Appendices
Appendix 1. MEDLINE search strategy
MEDLINE (Ovid) 1966 to November 2013
1. cerebrovascular disorders/ or basal ganglia cerebrovascular disease/ or exp brain ischemia/ or carotid artery diseases/ or carotid artery thrombosis/ or intracranial arterial diseases/ or cerebral arterial diseases/ or exp "intracranial embolism and thrombosis"/ or exp stroke/
2. (isch?emi$ adj6 (stroke$ or apoplex$ or cerebral vasc$ or cerebrovasc$ or cva)).tw.
3. ((brain or cerebr$ or cerebell$ or vertebrobasil$ or hemispher$ or intracran$ or intracerebral or infratentorial or supratentorial or middle cerebr$ or mca$ or anterior circulation) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$ or hypoxi$)).tw.
4. 1 or 2 or 3
5. thrombolytic therapy/
6. fibrinolytic agents/ or plasmin/ or plasminogen/ or tissue plasminogen activator/ or exp plasminogen activators/ or urokinase‐type plasminogen activator/
7. fibrinolysis/
8. (thromboly$ or fibrinoly$ or recanalis$ or recanaliz$).tw.
9. ((clot$ or thrombus) adj5 (lyse or lysis or dissolve$ or dissolution)).tw.
10. (tPA or t‐PA or rtPA or rt‐PA or plasminogen or plasmin or alteplase or actilyse).tw.
11. (anistreplase or streptodornase or streptokinase or urokinase or pro?urokinase or rpro?uk or lumbrokinase or duteplase or lanoteplase or pamiteplase or reteplase or saruplase or staphylokinase or streptase or tenecteplase or desmoteplase or retevase).tw.
12. 5 or 6 or 7 or 8 or 9 or 10 or 11
13. Randomized Controlled Trials as Topic/
14. random allocation/
15. Controlled Clinical Trials as Topic/
16. control groups/
17. clinical trials as topic/ or clinical trials, phase i as topic/ or clinical trials, phase ii as topic/ or clinical trials, phase iii as topic/ or clinical trials, phase iv as topic/
18. double‐blind method/
19. single‐blind method/
20. Placebos/
21. placebo effect/
22. Drug Evaluation/
23. Research Design/
24. randomized controlled trial.pt.
25. controlled clinical trial.pt.
26. (clinical trial or clinical trial phase i or clinical trial phase ii or clinical trial phase iii or clinical trial phase iv).pt.
27. (random$ or RCT or RCTs or quasi‐random$ or quasi random$ or pseudo‐random$ or pseudo random$).tw.
28. (controlled adj5 (trial$ or stud$)).tw.
29. (clinical$ adj5 trial$).tw.
30. ((control or treatment or experiment$ or intervention) adj5 (group$ or subject$ or patient$)).tw.
31. ((control or experiment$ or conservative) adj5 (treatment or therapy or procedure or manage$)).tw.
32. ((singl$ or doubl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).tw.
33. placebo$.tw.
34. (assign$ or allocat$).tw.
35. controls.tw.
36. trial.ti.
37. or/13‐36
38. 4 and 12 and 37
39. exp animals/ not humans.sh.
40. 38 not 39
Appendix 2. EMBASE search strategy
EMBASE (Ovid) 1980 to November 2013
1. cerebrovascular disease/ or cerebral artery disease/ or cerebrovascular accident/ or stroke/ or vertebrobasilar insufficiency/ or carotid artery disease/ or exp carotid artery obstruction/ or exp brain infarction/ or exp brain ischemia/ or exp occlusive cerebrovascular disease/ or stroke patient/ or stroke unit/
2. (isch?emi$ adj6 (stroke$ or apoplex$ or cerebral vasc$ or cerebrovasc$ or cva or attack$)).tw.
3. ((brain or cerebr$ or cerebell$ or vertebrobasil$ or hemispher$ or intracran$ or intracerebral or infratentorial or supratentorial or middle cerebr$ or mca$ or anterior circulation) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$ or hypoxi$)).tw.
4. 1 or 2 or 3
5. fibrinolytic therapy/
6. fibrinolytic agent/ or plasmin/ or plasminogen/ or exp plasminogen activator/
7. blood clot lysis/
8. fibrinolysis/
9. (thromboly$ or fibrinoly$ or recanalis$ or recanaliz$).tw.
10. ((clot$ or thrombus) adj5 (lyse or lysis or dissolve$ or dissolution)).tw.
11. (tPA or t‐PA or rtPA or rt‐PA or plasminogen or plasmin or alteplase or actilyse).tw.
12. (anistreplase or streptodornase or streptokinase or urokinase or pro?urokinase or rpro?uk or lumbrokinase or duteplase or lanoteplase or pamiteplase or reteplase or saruplase or staphylokinase or streptase or tenecteplase or desmoteplase or retevase).tw.
13. 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12
14. Randomized Controlled Trial/
15. Randomization/
16. Controlled Study/
17. control group/
18. clinical trial/ or phase 1 clinical trial/ or phase 2 clinical trial/ or phase 3 clinical trial/ or phase 4 clinical trial/ or controlled clinical trial/
19. Double Blind Procedure/
20. Single Blind Procedure/ or triple blind procedure/
21. placebo/
22. drug comparison/ or drug dose comparison/
23. "types of study"/
24. (random$ or RCT or RCTs or quasi‐random$ or quasi random$ or pseudo‐random$ or pseudo random$).tw
25. (controlled adj5 (trial$ or stud$)).tw.
26. (clinical$ adj5 trial$).tw.
27. ((control or treatment or experiment$ or intervention) adj5 (group$ or subject$ or patient$)).tw.
28. ((control or experiment$ or conservative) adj5 (treatment or therapy or procedure or manage$)).tw.
29. ((singl$ or doubl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).tw.
30. placebo$.tw.
31. (assign$ or allocat$).tw.
32. trial.ti.
33. or/14‐32
34. 4 and 13 and 33
35. (ANIMAL/ or NONHUMAN/ or ANIMAL EXPERIMENT/) and HUMAN/
36. ANIMAL/ or NONHUMAN/ or ANIMAL EXPERIMENT/
37. 36 not 35
38. 34 not 37
Data and analyses
Comparison 1. Any thrombolytic agent versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Deaths from all causes within 7 to 10 days | 13 | 7458 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.69 [1.44, 1.98] |
| 1.1 Intravenous urokinase versus control | 1 | 465 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.35 [0.62, 2.94] |
| 1.2 Intravenous streptokinase versus control | 3 | 963 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.90 [1.37, 2.63] |
| 1.3 Intravenous rt‐PA versus control | 8 | 5535 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.44 [1.18, 1.76] |
| 1.4 Intravenous streptokinase plus oral aspirin versus oral aspirin | 1 | 309 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.86 [2.26, 6.59] |
| 1.5 Intravenous desmoteplase versus control | 1 | 186 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.73 [0.85, 26.26] |
| 2 Fatal intracranial haemorrhage within 7 to 10 days | 17 | 9066 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.53 [3.47, 5.91] |
| 2.1 Intravenous urokinase versus control | 2 | 751 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.43 [1.08, 18.18] |
| 2.2 Intravenous streptokinase versus control | 4 | 983 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 6.03 [3.47, 10.47] |
| 2.3 Intravenous rt‐PA versus control | 8 | 6683 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.18 [2.99, 5.84] |
| 2.4 Intravenous streptokinase plus oral aspirin versus oral aspirin | 1 | 309 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.56 [1.62, 12.84] |
| 2.5 Intra‐arterial pro‐urokinase plus intravenous heparin versus intravenous heparin | 1 | 40 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.51 [0.03, 9.65] |
| 2.6 Intra‐arterial urokinase versus control | 1 | 114 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.39 [0.15, 372.38] |
| 2.7 Intravenous desmoteplase versus control | 1 | 186 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.65 [0.58, 37.52] |
| 3 Deaths within the first 7 to 10 days from causes other than fatal intracranial haemorrhage | 10 | 7226 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.08 [0.90, 1.30] |
| 3.1 Intravenous urokinase versus control | 1 | 465 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.87 [0.35, 2.13] |
| 3.2 Intravenous streptokinase versus control | 3 | 963 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.05 [0.72, 1.53] |
| 3.3 Intravenous rt‐PA versus control | 5 | 5303 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.93 [0.73, 1.18] |
| 3.4 Intravenous streptokinase plus oral aspirin versus oral aspirin | 1 | 309 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.13 [1.74, 5.62] |
| 3.5 Intravenous desmoteplase versus control | 1 | 186 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.57 [0.24, 86.20] |
| 4 Symptomatic (including fatal) intracranial haemorrhage within 7 to 10 days | 27 | 10186 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.75 [3.11, 4.51] |
| 4.1 Intravenous urokinase versus control | 4 | 1208 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.28 [0.47, 3.48] |
| 4.2 Intravenous streptokinase versus control | 4 | 983 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 5.20 [3.25, 8.32] |
| 4.3 Intravenous rt‐PA versus control | 12 | 7011 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.72 [2.98, 4.64] |
| 4.4 Intravenous streptokinase plus oral aspirin versus oral aspirin | 1 | 309 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.02 [1.55, 10.40] |
| 4.5 Intra‐arterial pro‐urokinase plus intravenous heparin versus intravenous heparin | 2 | 220 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.39 [0.88, 6.47] |
| 4.6 Intra‐arterial urokinase versus control | 2 | 130 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.04 [0.79, 20.74] |
| 4.7 Intravenous desmoteplase versus control | 3 | 325 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.50 [1.37, 14.81] |
| 5 Symptomatic (including fatal) cerebral oedema | 6 | 5961 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.97 [0.79, 1.19] |
| 6 Death or dependency at the end of follow‐up | 22 | 9318 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.85 [0.78, 0.93] |
| 6.1 Intravenous urokinase versus control | 1 | 465 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.95 [0.64, 1.42] |
| 6.2 Intravenous streptokinase versus control | 4 | 983 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.94 [0.72, 1.24] |
| 6.3 Intravenous rtA versus control | 10 | 6886 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.84 [0.77, 0.93] |
| 6.4 Intravenous streptokinase plus oral aspirin versus oral aspirin | 1 | 309 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.09 [0.69, 1.73] |
| 6.5 Intra‐arterial pro‐urokinase plus intravenous heparin versus intravenous heparin | 2 | 220 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.55 [0.31, 1.00] |
| 6.6 Intra‐arterial urokinase versus control | 2 | 130 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.57 [0.28, 1.14] |
| 6.7 Intravenous desmoteplase versus control | 3 | 325 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.86 [0.53, 1.40] |
| 7 Deaths occurring between 7 and 10 days and the end of follow‐up | 13 | 7458 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.88 [0.76, 1.02] |
| 7.1 Intravenous urokinase versus control | 1 | 465 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.05 [0.60, 6.99] |
| 7.2 Intravenous streptokinase versus control | 3 | 963 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.88 [0.61, 1.26] |
| 7.3 Intravenous rt‐PA versus control | 8 | 5535 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.84 [0.71, 0.99] |
| 7.4 Intravenous streptokinase plus oral aspirin versus control | 1 | 309 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.06 [0.49, 2.27] |
| 7.5 Intravenous desmoteplase versus control | 1 | 186 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.95 [1.37, 17.86] |
| 8 Deaths from all causes during follow‐up | 27 | 10187 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.18 [1.06, 1.30] |
| 8.1 Intravenous urokinase versus control | 4 | 1208 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.15 [0.68, 1.97] |
| 8.2 Intravenous streptokinase versus control | 4 | 983 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.43 [1.10, 1.88] |
| 8.3 Intravenous rt‐PA versus control | 12 | 7012 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.06 [0.94, 1.20] |
| 8.4 Intravenous streptokinase plus oral aspirin versus oral aspirin | 1 | 309 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.02 [1.87, 4.87] |
| 8.5 Intra‐arterial pro‐urokinase plus intravenous heparin versus intravenous heparin | 2 | 220 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.75 [0.40, 1.42] |
| 8.6 Intra‐arterial urokinase versus control | 2 | 130 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.25 [0.34, 4.57] |
| 8.7 Intravenous desmoteplase versus control | 3 | 325 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.17 [0.97, 4.84] |
| 9 Death or dependency defined as mRS 2 to 6 | 21 | 8824 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.76 [0.70, 0.84] |
| 9.1 Intravenous urokinase versus control | 1 | 465 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.57 [0.38, 0.85] |
| 9.2 Intravenous streptokinase versus control | 4 | 983 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.82 [0.61, 1.10] |
| 9.3 Intravenous rt‐PA versus control | 10 | 6887 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.79 [0.71, 0.88] |
| 9.4 Intravenous desmoteplase versus control | 2 | 139 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.64 [0.28, 1.43] |
| 9.5 Intra‐arterial pro‐urokinase versus control | 2 | 220 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.62 [0.32, 1.19] |
| 9.6 Intra‐arterial urokinase versus control | 2 | 130 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.37 [0.17, 0.77] |
| 10 Death or dependency defined as mRS 3 to 6 | 21 | 8824 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.85 [0.78, 0.93] |
| 10.1 Intravenous urokinase versus control | 1 | 465 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.99 [0.67, 1.47] |
| 10.2 Intravenous streptokinase versus control | 4 | 983 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.94 [0.72, 1.24] |
| 10.3 Intravenous rt‐PA versus control | 10 | 6887 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.85 [0.77, 0.94] |
| 10.4 Intravenous desmoteplase versus control | 2 | 139 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.49 [0.22, 1.08] |
| 10.5 Intra‐arterial pro‐urokinase versus control | 2 | 220 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.55 [0.31, 1.00] |
| 10.6 Intra‐arterial urokinase versus control | 2 | 130 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.57 [0.28, 1.14] |
| 11 Dependency at the end of follow‐up defined as mRS 3 to 5 | 22 | 9318 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.75 [0.69, 0.82] |
| 11.1 Intravenous urokinase versus control | 1 | 465 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.80 [0.53, 1.22] |
| 11.2 Intravenous streptokinase versus control | 4 | 983 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.64 [0.49, 0.85] |
| 11.3 Intravenous rt‐PA versus control | 10 | 6886 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.80 [0.73, 0.89] |
| 11.4 Intravenous streptokinase plus oral aspirin versus oral aspirin | 1 | 309 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.36 [0.22, 0.58] |
| 11.5 Intra‐arterial pro‐urokinase plus intravenous heparin versus intravenous heparin | 2 | 220 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.72 [0.41, 1.28] |
| 11.6 Intra‐arterial urokinase versus control | 2 | 130 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.53 [0.26, 1.06] |
| 11.7 Intravenous desmoteplase versus control | 3 | 325 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.66 [0.41, 1.06] |
| 12 Alive and independent (mRS 0 to 2) at end of follow‐up, participants treated up to six hours | 10 | 6887 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.17 [1.06, 1.29] |
| 13 Alive and favourable outcome (mRS 0 to 1) at end of follow‐up, participants treated up to six hours | 10 | 6887 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.29 [1.16, 1.43] |
| 14 Deaths from all causes ordered by antithrombotic drug use | 24 | 9674 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.20 [1.08, 1.33] |
| 14.1 All participants received antithrombotic drugs < 24 hours | 6 | 2191 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.31 [1.09, 1.58] |
| 14.2 Some participants received antithrombotic drugs < 24 hours | 4 | 1757 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.27 [0.99, 1.63] |
| 14.3 Some participants received antithrombotics but not < 24 hours | 12 | 4670 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.13 [0.96, 1.34] |
| 14.4 No participants received antithrombotic drugs < 10 days | 4 | 1056 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.89 [0.58, 1.37] |
| 15 Deaths from all causes ordered by stroke severity | 27 | 9878 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.12 [1.01, 1.25] |
| 15.1 Case fatality 0% ‐ 19% in the control group | 17 | 4973 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.31 [1.08, 1.58] |
| 15.2 Case fatality 20% or greater in the control group | 10 | 4905 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.05 [0.92, 1.19] |
| 16 Death or dependency at the end of follow‐up: participants randomised within 3 hours of stroke | 10 | 2160 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.66 [0.56, 0.79] |
| 16.1 Intravenous urokinase versus control | 1 | 82 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.79 [0.31, 2.00] |
| 16.2 Intravenous streptokinase versus control | 3 | 209 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.62 [0.35, 1.09] |
| 16.3 Intravenous rt‐PA versus control | 6 | 1779 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.65 [0.54, 0.80] |
| 16.4 Intravenous streptokinase plus oral aspirin versus oral aspirin | 1 | 90 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.93 [0.40, 2.18] |
| 17 Death or dependency by time to treatment up to 6 hours: all agents: only trials randomising in both 0 to 3 and 3 to 6 hour time windows | 9 | 6941 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.92 [0.83, 1.01] |
| 17.1 Treatment within 3 hours | 9 | 1536 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.69 [0.55, 0.85] |
| 17.2 Treatment between 3 and 6 hours | 9 | 5405 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.99 [0.88, 1.10] |
| 18 Death or dependency by time to treatment up to 6 hours: rt‐PA: only trials randomising in 0 ‐ 3 and 3 ‐ 6 hour windows | 5 | 5204 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.90 [0.80, 1.01] |
| 18.1 Treatment within 3 hours | 5 | 1155 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.68 [0.53, 0.87] |
| 18.2 Treatment between 3 to 6 hours | 5 | 4049 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.97 [0.85, 1.09] |
| 19 Death or dependency by time to treatment up to 6 hours: rt‐PA: all trials regardless of time window | 8 | 6729 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.85 [0.77, 0.94] |
| 19.1 Treatment within 3 hours | 6 | 1779 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.65 [0.54, 0.80] |
| 19.2 Treatment between 3 and 6 hours | 7 | 4950 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.93 [0.83, 1.04] |
| 20 Death or dependency by latest time to randomisation | 22 | 9009 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.71, 0.92] |
| 20.1 3 hours | 1 | 624 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.45, 0.85] |
| 20.2 4.5 hours | 2 | 1161 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.66, 1.32] |
| 20.3 6 hours | 15 | 6883 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.69, 0.95] |
| 20.4 9 hours | 3 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.35, 1.59] |
| 20.5 24 hours | 1 | 16 | Odds Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 1.76] |
| 21 Alive and independent (mRS 0 to 2) at end of follow‐up, participants treated < 3 versus 3 to 6 hours, all trials regardless of latest time window | 8 | 6750 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.17 [1.06, 1.29] |
| 21.1 Participants treated within three hours | 6 | 1779 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.53 [1.26, 1.86] |
| 21.2 Participants treated 3 ‐ 6 hours | 7 | 4971 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.07 [0.96, 1.20] |
| 22 Alive and favourable outcome (mRS 0 to 1) at end of follow‐up, < 3 versus 3 ‐ 6 hours, only trials randomising in both time windows | 6 | 1779 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.61 [1.30, 1.99] |
| 22.1 Participants treated < 3 hours | 6 | 1779 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.61 [1.30, 1.99] |
| 22.2 Participants treated 3 ‐ 6 hours | 0 | 0 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 23 Deaths from all causes during follow‐up: participants randomised within 3 hours of stroke | 11 | 2187 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.99 [0.82, 1.21] |
| 23.1 Intravenous streptokinase versus control | 3 | 209 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.18 [0.66, 2.14] |
| 23.2 Intravenous tPA versus control | 7 | 1806 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.91 [0.73, 1.13] |
| 23.3 Intravenous streptokinase plus oral aspirin versus oral aspirin | 1 | 90 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.37 [1.37, 8.26] |
| 23.4 Intravenous urokinase versus control | 1 | 82 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.70 [0.14, 3.51] |
| 24 Deaths by time to treatment up to 6 hours: all agents: only trials randomising in both 0 ‐ 3 and 3 ‐ 6 hour time windows | 9 | 6936 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.23 [1.10, 1.38] |
| 24.1 Treatment within 3 hours | 9 | 1536 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.08 [0.86, 1.35] |
| 24.2 Treatment between 3 and 6 hours | 9 | 5400 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.29 [1.13, 1.48] |
| 25 Deaths by time to treatment up to 6 hours: rt‐PA: only trials randomising in both 0 to 3 and 3 to 6 hour time windows | 5 | 5199 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.11 [0.97, 1.28] |
| 25.1 Treatment within 3 hours | 5 | 1155 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.97 [0.75, 1.26] |
| 25.2 Treatment between 3 and 6 hours | 5 | 4044 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.17 [1.00, 1.38] |
| 26 Deaths by time to treatment up to 6 hours: rt‐PA: all trials regardless of time window | 9 | 6772 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.07 [0.95, 1.21] |
| 26.1 Treatment within 3 hours | 7 | 1806 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.91 [0.73, 1.13] |
| 26.2 Treatment between 3 and 6 hours | 7 | 4966 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.16 [1.00, 1.35] |
| 27 Death by latest time to treatment | 24 | 9039 | Odds Ratio (Peto, Fixed, 95% CI) | 1.12 [1.01, 1.25] |
| 27.1 3 hours | 2 | 651 | Odds Ratio (Peto, Fixed, 95% CI) | 0.79 [0.53, 1.17] |
| 27.2 4.5 hours | 2 | 1161 | Odds Ratio (Peto, Fixed, 95% CI) | 1.43 [1.01, 2.03] |
| 27.3 6 hours | 16 | 6886 | Odds Ratio (Peto, Fixed, 95% CI) | 1.12 [0.99, 1.26] |
| 27.4 9 hours | 3 | 325 | Odds Ratio (Peto, Fixed, 95% CI) | 2.10 [0.79, 5.58] |
| 27.5 24 hours | 1 | 16 | Odds Ratio (Peto, Fixed, 95% CI) | 1.0 [0.14, 7.10] |
| 28 Symptomatic intracranial haemorrhage by time to treatment up to 6 hours: rt‐PA: only trials randomising in both 0 ‐ 3 and 3 ‐ 6 hour time windows. | 5 | 5168 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.75 [2.94, 4.78] |
| 28.1 Intravenous rt‐PA versus control: participants treated within 3 hours of stroke | 5 | 1155 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.25 [2.53, 7.16] |
| 28.2 Intravenous rt‐PA versus control: participants treated between 3 and 6 hours after stroke | 5 | 4013 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.62 [2.76, 4.76] |
| 29 Symptomatic intracranial haemorrhage by time to treatment up to 6 hours: rt‐PA: all trials regardless of time window | 8 | 6714 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.93 [3.13, 4.93] |
| 29.1 Intravenous rt‐PA versus control: participants treated within 3 hours of stroke | 6 | 1779 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.55 [2.92, 7.09] |
| 29.2 Intravenous rt‐PA versus control: participants treated between 3 and 6 hours after stroke | 7 | 4935 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.73 [2.86, 4.86] |
| 30 Symptomatic intracranial haemorrhage by latest time to treatment | 23 | 9104 | Odds Ratio (Peto, Fixed, 95% CI) | 4.41 [3.43, 5.69] |
| 30.1 3 hours | 2 | 651 | Odds Ratio (Peto, Fixed, 95% CI) | 5.85 [1.54, 22.26] |
| 30.2 4.5 hours | 2 | 1161 | Odds Ratio (Peto, Fixed, 95% CI) | 6.56 [2.51, 17.18] |
| 30.3 6 hours | 15 | 6951 | Odds Ratio (Peto, Fixed, 95% CI) | 4.20 [3.21, 5.50] |
| 30.4 9 hours | 3 | 325 | Odds Ratio (Peto, Fixed, 95% CI) | 6.82 [0.88, 52.78] |
| 30.5 24 hours | 1 | 16 | Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 31 Death or dependency (mRS 3 to 6) by the end of follow‐up; participants treated up to 6 hours aged ≤ 80 years versus > 80 years | 10 | 6871 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.84 [0.76, 0.93] |
| 31.1 Participants aged ≤ 80 years | 10 | 5175 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.85 [0.76, 0.95] |
| 31.2 Participants aged > 80 years | 3 | 1696 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.80 [0.64, 0.99] |
| 32 Death or dependency (mRS 3 to 6) by the end of follow‐up, participants treated within 3 hours aged ≤ 80 years versus > 80 years | 6 | 1765 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.62 [0.51, 0.76] |
| 32.1 Participants aged ≤ 80 years | 6 | 1039 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.66 [0.52, 0.85] |
| 32.2 Participants aged > 80 years | 2 | 726 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.56 [0.40, 0.78] |
| 33 Alive and independent (mRS 0 to 2) at end of follow‐up, participants treated up to 6 hours aged ≤ 80 years versus > 80 years | 10 | 6885 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.18 [1.07, 1.31] |
| 33.1 Participants aged ≤ 80 years | 10 | 5174 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.17 [1.05, 1.31] |
| 33.2 Participants aged > 80 years or over | 3 | 1711 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.22 [0.98, 1.52] |
| 34 Alive and independent (mRS 0 to 2) at end of folllow‐up, participants treated within 3 hours, aged ≤ 80 years versus > 80 years | 6 | 1779 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.56 [1.28, 1.90] |
| 34.1 Participants aged ≤ 80 years versus | 6 | 1038 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.50 [1.18, 1.92] |
| 34.2 Participants aged > 80 years and over | 2 | 741 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.68 [1.20, 2.34] |
| 35 Alive and independent (mRS 0 to 2) at end of follow‐up, participants treated 3 ‐ 6 hours, aged ≤ 80 years versus > 80 years | 7 | 4971 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.07 [0.96, 1.21] |
| 35.1 Participants aged ≤ 80 years | 7 | 4001 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.09 [0.96, 1.24] |
| 35.2 Participants aged > 80 years | 2 | 970 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.97 [0.73, 1.30] |
| 36 Death: selection by MR DWI/PWI or CT | 19 | 8760 | Odds Ratio (Peto, Fixed, 95% CI) | 1.14 [1.02, 1.27] |
| 36.1 Plain CT | 15 | 8334 | Odds Ratio (Peto, Fixed, 95% CI) | 1.12 [1.00, 1.25] |
| 36.2 MR DWI/PWI | 4 | 426 | Odds Ratio (Peto, Fixed, 95% CI) | 2.05 [1.02, 4.15] |
| 37 Death or dependency: selection with MR DWI/PWI versus plain CT | 15 | 8268 | Odds Ratio (Peto, Fixed, 95% CI) | 0.81 [0.74, 0.89] |
| 37.1 Selection with plain CT | 11 | 7843 | Odds Ratio (Peto, Fixed, 95% CI) | 0.81 [0.73, 0.89] |
| 37.2 Selection with MR DWI/PWI | 4 | 425 | Odds Ratio (Peto, Fixed, 95% CI) | 0.88 [0.58, 1.35] |
| 38 Symptomatic intracranial haemorrhage: selection with MR DWI/PWI or CT | 20 | 8784 | Odds Ratio (Peto, Fixed, 95% CI) | 4.44 [3.43, 5.75] |
| 38.1 Plain CT | 16 | 8358 | Odds Ratio (Peto, Fixed, 95% CI) | 4.38 [3.38, 5.69] |
| 38.2 MR DWI/PWI | 4 | 426 | Odds Ratio (Peto, Fixed, 95% CI) | 7.51 [1.40, 40.35] |
| 39 Alive and independent (mRS 0 to 1) at end of follow‐up, by plain CT ASPECTS score | 4 | 4567 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.21 [1.07, 1.37] |
| 39.1 ASPECTS score 8 ‐ 10 | 4 | 3317 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.21 [1.06, 1.39] |
| 39.2 ASPECTS score 0 ‐ 7 | 4 | 1250 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.20 [0.91, 1.58] |
| 40 Death or dependency at the end of follow‐up: intra‐arterial thrombolysis versus control | 4 | 350 | Odds Ratio (Peto, Fixed, 95% CI) | 0.49 [0.31, 0.79] |
| 40.1 Intra‐arterial urokinase versus control | 2 | 130 | Odds Ratio (Peto, Fixed, 95% CI) | 0.44 [0.22, 0.91] |
| 40.2 Intra‐arterial pro‐urokinase versus control | 2 | 220 | Odds Ratio (Peto, Fixed, 95% CI) | 0.53 [0.28, 1.00] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Abe 1981.
| Methods | Double‐blind RCT Sealed drug prepacks Not intention‐to‐treat | |
| Participants | All grades of presumed thrombotic stroke < 2 weeks, pre‐entry CT, age 18 years and over Excluded: presumed embolic stroke, severe neurological deficit | |
| Interventions | Tissue‐cultured urokinase (Abbott Labs, USA) 60,000 U/day i.v. for 7 days versus identical‐looking placebo | |
| Outcomes | Global Improvement Rating and safety assessment at 4 weeks after treatment start (including follow‐up CT if neurologically deteriorated) | |
| Notes | Further information from Prof Abe: 11 participants (6 treatment and 5 placebo) who received non‐trial drugs outside the trial protocol were excluded from analysis prior to breaking the randomisation code plus 2 who underwent superficial temporal artery‐middle cerebral artery bypass surgery Published in Japanese | |
ASK 1996.
| Methods | Double‐blind, central telephone randomisation RCT Sealed drug prepacks of streptokinase or identical‐looking placebo Intention‐to‐treat | |
| Participants | Any acute ischaemic stroke (though not very mild or rapidly recovering) that could be randomised and treatment start within 4 hours of clearly defined symptom onset; age > 18 years and < 85 years; CT scan mandatory to exclude cerebral haemorrhage pre‐randomisation Excluded: people with recent trauma or surgery, stroke within the last 3 months or at any time if in the same hemisphere as the presenting stroke, pregnancy, any anticoagulants given within the previous 48 hours (except aspirin), uncontrolled hypertension (systolic BP > 200 mmHg, diastolic > 120 mmHg) | |
| Interventions | Streptokinase (Hoechst, Australia) 1.5 MU in 100 ml of normal saline i.v. over 1 hour versus identical‐looking placebo (prepared by Berhingwerke, Germany, in association with Hoechst) Infusion to be stopped and i.v. Haemaccel given (plasma expander) should the BP fall below 100 mmHg systolic (or by more than 20 mmHg from the initial systolic BP) during the infusion This procedure to be repeated should hypotension occur again, but the infusion to be completed within 3 hours or abandoned Aspirin 100 mg to be given orally within 4 hours of the trial treatment infusion and thereafter daily for the duration of the study No other anticoagulants to be given within 48 hours of the trial treatment | |
| Outcomes | Death and dependency at 3 months assessed by the Barthel Index, i.e. favourable outcome = alive and Barthel Score > 60, unfavourable = dead or Barthel Score < 60 CT scan at 7 to 10 days (or sooner if clinically indicated) to look for cerebral haemorrhage | |
| Notes | Terminated prematurely after randomisation of 340 (of the intended 600) participants, initially just the 3 to 4 hour randomisation (because of significant excess early mortality in the streptokinase group) and then all randomisation because the randomisation rate within 3 hours was too slow to be viable Apparent excess of problems with hypotension during the streptokinase infusion compared with MAST‐I 1995 and MAST‐E 1996 (20% in ASK versus only a few % in the 2 MAST trials), and note routine use of aspirin within 4 hours of trial treatment | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
Atarashi 1985.
| Methods | Double‐blind RCT Sealed prepacked drug or placebo Not intention‐to‐treat | |
| Participants | Presumed cerebral arterial thrombosis (angiography where possible) < 5 days, aged 18 years and up, entry CT Excluded: presumed embolic stroke, severe neurological deficit | |
| Interventions | High‐dose UK (240,000 u/day i.v. for 7 days) versus low dose (60,000 u/day i.v. for 7 days) versus identical‐looking placebo The high‐ and low‐dose participants were analysed as 1 group for the purpose of this review, i.e. the comparison is 'any UK treatment versus placebo' | |
| Outcomes | Clinical improvement (Final Global Improvement Rating) at 4 weeks from start of treatment, safety (absence of side effects), including follow‐up CT if neurological deterioration | |
| Notes | Further information provided by Prof Ohtomo, co‐investigator 6 participants (4 treated low‐dose, 2 placebo) were excluded from analysis prior to breaking the randomisation code as the diagnosis thought not to be stroke | |
ATLANTIS A 2000.
| Methods | Double‐blind, placebo‐controlled RCT; August 1991 to November 1993 Randomisation was by selection of a numbered treatment pack held in the participating centre (of tPA or identical‐looking placebo); the numbered pack had been identified by telephoning the trial co‐ordinating centre in advance of the arrival of the next participant to be randomised Once administered, the trial co‐ordinating centre was telephoned to inform them of the participant's details and identify the next treatment pack to be given to the next participant to be randomised | |
| Participants | Any acute ischaemic stroke that could be randomised and start treatment within 6 hours of clearly‐defined stroke onset Participant selection criteria believed to be otherwise similar to those in the NINDS 1995 trial CT scan mandatory prior to randomisation to exclude cerebral haemorrhage, but no exclusion criteria based on visible infarction Age: 18 ‐ 80 years | |
| Interventions | Alteplase (tPA ‐ Activase, Genentech, South San Francisco) 0.9 mg/kg body weight to a maximum of 90 mg, in 100 ml normal saline, the initial 10% given as a bolus and the rest over an hour i.v. Blood pressure control as for NINDS 1995 Aspirin and anticoagulants to be avoided for the first 24 hours, but could be used thereafter | |
| Outcomes | NIH Stroke Scale improvement at 30 days Death and dependency at 90 days (modified Rankin 2 to 6) Barthel NIH Stroke Scale Cerebral haemorrhage on repeat CT The final follow‐up assessment was performed by a neurologist who had not cared for the participant at randomisation or during the first few days after treatment | |
| Notes | Stopped in November 1993, protocol modified and continued as ATLANTIS B 1999 in the same centres 42 active centres, 142 participants randomised | |
ATLANTIS B 1999.
| Methods | Double‐blind, placebo‐controlled RCT Randomisation was by selection of a numbered treatment pack held in the participating centre (of tPA or identical‐looking placebo); the numbered pack had been identified by telephoning the trial co‐ordinating centre in advance of the arrival of the next participant to be randomised Once administered, the trial co‐ordinating centre was telephoned to inform them of the participant's details and identify the next treatment pack to be given to the next participant to be randomised | |
| Participants | December 1993 to January 1996: 0 to 5‐hour time window February 1996 to end of trial: 3 to 5‐hour time window and CT exclusion criteria introduced (visible infarction in > ⅓ of the MCA territory excluded) Within the above time periods, anyone with acute ischaemic stroke with clearly defined symptom onset who could be treated within the specified time period Age 18 ‐ 80 years CT pre‐randomisation mandatory to exclude cerebral haemorrhage only until February 1996, but thereafter also to exclude visible infarction in more than ⅓ of the MCA territory 120 centres active | |
| Interventions | Alteplase (tPA ‐ Activase, Genentech, South San Francisco) 0.9 mg/kg to maximum dose of 90 mg in 100 ml normal saline or identical‐looking placebo, the first 10% as a bolus and the rest infused over an hour i.v. Aspirin to be avoided within the first 24 hours, but could be used thereafter | |
| Outcomes | NIHSS modified Rankin Scale (death or dependency = 2 to 6) BI at 90 days Death Cerebral haemorrhage (symptomatic and fatal) The final follow‐up assessment was performed by a neurologist who had not cared for the participant at randomisation or during the first few days after treatment | |
| Notes | Stopped in mid‐1998 following a futility analysis, prior to the publication of ECASS II 1998 Data on all 619 participants randomised has not yet been presented, only on 547 randomised between 3 and 5 hours | |
AUST 2005.
| Methods | PROBE: Prospective Randomised Open controlled study with Blinded Endpoint assessment; RCT Location: multi‐centre, 7 centres in Australia and New Zealand, ongoing January 1996 to May 2003 Method of randomisation: telephone to a central office (for 2 participants by coin tossing when system was unavailable, after consent from the Steering Committee) Analysis: intention‐to‐treat (LOCF) | |
| Participants | Eligibility: people with clinical presentation of a posterior lesion and where CT and angiography supported an ischaemic stroke with a posterior circulation vascular occlusion and where the participant was judged suitable for long‐term anticoagulation Age limits: 18 ‐ 85 years NIHSS: not defined Time window: 0 to 24 hours Participants: ‐ Treatment group: male: 7 (88%), mean age: 64.2 (± 11.1) years, mean delay (range): 710 (345 to 1305) minutes, NIHSS median (range): 23 (7 to 29), GCS: 6 (3 to 15) ‐ Control group: male: three (38%), mean age: 63.7 (± 12.3) years, mean delay (range): 749 (205 to 1350) minutes, NIHSS median (range): 18 (5 to 29); GCS median (range): 12 (3 to 15) Baseline angiography findings available in 14 participants, 7 in treatment group, 7 in control group: basilar artery, unilateral vertebral artery/posterior inferior cerebellar artery, and unilateral PCA were included Number of randomised participants: 16 (8 in treatment group, 8 in control group) | |
| Interventions | Treatment group: UK i.a. with increments of 100,000 IU to max 1,000,000 IU Control group: no thrombolysis All participants were anticoagulated acutely (5,000 IU heparin i.a. followed by i.v. heparin to a target APTT 60 to 80 for minimum 2 days, and then oral warfarin to a target INR of 1.5 to 2.5 for 6 months | |
| Outcomes | Clinical at 6 months: mRS, BI, NIHSS assessed by a research nurse or neurologist blinded to treatment allocation and not involved in the participant's initial care, and determined by an independent outcomes committee blinded to all documentation at 6 months Primary efficacy endpoint was the combined morbidity (BI and mRS) and mortality at 6 months Pre‐specified secondary endpoints were arterial re‐canalisation at day 7 to 10 after treatment, neurological impairment at 6 months, safety and tolerability, and cost effectiveness | |
| Notes | Study groups were unbalanced with more males and more severe strokes in the treatment group The trial was prematurely terminated in June 2003 due to slow recruitment, and withdrawal of sale of UK Given the small numbers of recruited participants only the primary endpoint and safety are presented | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
Chen 2000.
| Methods | Double‐blind, placebo‐controlled RCT Randomisation of a numbered treatment pack (UK or normal saline) held in the participating centre Blinded follow‐up | |
| Participants | People aged 35 to 75 years with clinical diagnosis of carotid distribution ischaemic stroke (including lacunar), within 6 hours of stroke No visible infarct on CT | |
| Interventions | UK (Roxin, Guangdong Techpool Biochem Pharma Co Ltd, China) Group A: 1.5 MU Group B: 1.0 MU Group C: placebo (normal saline) i.v. over 30 minutes | |
| Outcomes | European Stroke Scale serially up to 90 days Barthel Index and modified Rankin Score at 90 days Assessment was by a doctor not involved in administering the trial drug | |
| Notes | All participants also received low molecular weight dextran 500 ml i.v. per day for 10 days and 300 mg aspirin (starting 24 hours after thrombolysis) for 10 days | |
DEDAS 2006.
| Methods | Double‐blind, randomised, placebo‐controlled body‐weight‐adjusted dose‐escalation study; RCT Location: 21 centres in the USA and 4 centres in Germany, between March 2003 and October 2004 Method of randomisation: interactive voice response system Analysis: intention‐to‐treat, analysis of target population defined before unblinding by the core laboratory to consist only of participants with mismatch and no isolated ICA occlusion In addition to the ITT analysis, a target population was defined before unblinding and included only those who had had mismatch and no isolated ICA occlusion (as determined by the core laboratory) MRI at screening, 4 to 8 hours post‐treatment, and at 30 days CT was performed at 24 hours | |
| Participants | Eligibility: principally according to NINDS 1995 criteria, but with NIHSS limits 4 to 20 and MRI evidence of DWI/PWI mismatch (20% mismatch with a perfusion deficit, with or without DWI lesion of > 2 cm in diameter and involving the cerebral cortex) Participants: age limits: 18 ‐ 85 years NIHSS limits: 4 to 20 Time window: 3 to 9 hours Summary of participant characteristics: ‐ Treatment groups: N = 37, male 55%, median age (range): 73 (42 to 85) years, NIHSS: 9 (4 to 19), time after onset to treatment: 449 (222 to 568) minutes, DWI lesion: 22.2 (1.6 to 78.1) ml ‐ Control group: N = 8, male 63%, median age (range): 71.5 (42 to 85) years, NIHSS 12 (6 to 18), time after onset to treatment: 443 (220 to 516) minutes, DWI lesion volume: 35.1 (1.5 to 68.6) ml Number randomised: 37 (29 in the treatment groups; 8 in the control group) | |
| Interventions | Desmoteplase i.v. administered as a bolus over 1 to 2 minutes 2 doses: 90 µg/kg (N = 14) and 125 µg/kg (N = 15) versus placebo (N = 8) Each dose tier included 15 treatment participants and 4 controls | |
| Outcomes | Primary safety endpoint: rate of SICH as defined by CT and according to NINDS 1995 criteria Co‐primary efficacy endpoint: reperfusion at 4 to 8 hours (≥ 30% reduction of MTT volume or ≥ 2 points improvement on MR‐adjusted TIMI); clinical outcome at 90 days (good outcome composite of: NIHSS ≥ 8 points improvement or a score of 0 to 1, mRS 0 to 2, BI ≥ 75) | |
| Notes | In 12 randomised participants the MRI‐entry criteria had been violated: 6 had an isolated ICA occlusion; another 6 had either no mismatch or no perfusion deficit 6 of the violations occurred in the 90 µg/kg dose group, 4 in the 125 µg/kg group, and 2 in the placebo group The target population in total was 25 participants (placebo N = 6, 90 µg/kg N = 8, 125 µg/kg N =11) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
DIAS 2 2008.
| Methods | Double‐blind, randomised, controlled prospective single bolus study investigating the efficacy and safety of 2 doses versus placebo in participants with ≥ 20% mismatch as detected by MRI or perfusion CT Method of randomisation: interactive voice response system Imaging: MR (DWI/PWI) or CT perfusion Analysis: intention‐to‐treat | |
| Participants | Eligibility: principally according to NINDS 1995 criteria, but with NIHSS limits 4 to 20 and MRI evidence of DWI/PWI mismatch with a distinct penumbra ≥ 20% on MRI or perfusion CT Age limits: 18 ‐ 85 years NIHSS: 4 to 20 Time window: 3 to 9 hours Participants: ‐ Treatment groups: 90 µg/kg bodyweight N = 57, 125 µg/kg bodyweight N = 66 ‐ Control group: N = 63 Number randomised N =186 (treatment group N = 123, control group N = 63) | |
| Interventions | Desmoteplase i.v. administered as a single bolus over 1 to 2 minutes; 2 doses: 90 µg/kg and 125 µg/kg versus placebo | |
| Outcomes | Primary outcome included: clinical improvement day 90 defined by the composite endpoint: improvement in NIHSS ≥ 8 points or NIHSS 0 to 1, BI ≥ 75, and mRS 0 to 2 Primary safety endpoints: rate of SICH at 72 hours, and all‐cause mortality | |
| Notes | Trial information based on press release of June 2007 after report of the trial at the 16th European Stroke Conference, Glasgow 2007 There was increased mortality (14 deaths) in the 125 µg/kg bodyweight versus 90 µg/kg and placebo (among the 14 deaths, 10 were considered by the investigators to be unrelated to treatment, 9 were non‐neurological, and 9 occurred more than 10 days after administration of the drug) Further information is sought The trial was halted on 25 October 2006 by the Data and Safety Committee (DCM) for apparent safety concerns which were not disclosed by the DCM; after 5 days the trial was allowed to resume enrolment without modification of the protocol and with remaining blinded data | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
DIAS 2005.
| Methods | Double‐blind, randomised placebo‐controlled, dose‐finding, phase II trial Location: 44 centres in 12 countries, trial ongoing between January 2001and October 2003 Method of randomisation: interactive voice response system collected participant's date of birth, weight, and NIHSS score and participants were randomised to a dose of desmoteplase or placebo The trial was undertaken in 2 parts: ‐ Part 1: 1st period: participants were allocated into 4 groups, 3 with fixed treatment doses, and 1 placebo‐control group; 2nd period (from participant number 31): 1 fixed treatment dose versus placebo ‐ Part 2: after participant 47, the trial continued as a dose escalation trial with 3 body weight‐adjusted treatment doses versus placebo Stratification according to age: ≤ 75 years versus > 75 years Imaging inclusion method: MRI, DWI/PWI mismatch, perfusion abnormality > 2 cm in diameter involving hemispheric grey matter; DWI/PWI mismatch ≥ 20% MRI at screening, 4 to 8 hours post‐treatment, and at 30 days CT was performed at 24 hours Analyses: included an intention‐to‐treat sample (deceased participants were given worst possible score for all outcomes; further missing data were analysed as LOCF observations); and per‐protocol (104 randomised, 102 included in analyses) | |
| Participants | Eligibility: people with ischaemic stroke and MRI evidence of mismatch involving hemispheric grey matter NIHSS limits for first 9 participants: 8 to 20, to increase recruitment widened limits from participant number 10: 4 to 20 and a prolonged time window from 3 to 6 to 3 to 9 hours After the first 5 participants a 30‐minute MRI‐to‐treatment time requirement was applied Further, the upper limit of the DWI lesion at baseline was reduced from ⅔ to ⅓ of the MCA territory In addition, participants on any platelet inhibitor could be excluded at the discretion of the treating physician Patients: age limits 18 years ‐ 85 years NIHSS: 4 to 20 Time window: 3 to 9 hours (first 9 participants: 3 to 6 hours, from participant number 10: 3 to 9 hours) Summary from both parts of trial: ‐ Treatment groups: N = 75; male 55%, age (median) 68 years, NIHSS 12, time from onset 324 minutes, DWI lesion volume ml 17.76 ‐ Control group: N = 27; male 52%, (median) age 68 years, NIHSS 12, time from onset 325 minutes, DWI lesion volume ml 20.40 Number randomised:104 (Part 1: 47, Part 2: 57) Of the 104 randomised participants, 2 control participants received no trial medication and were excluded from all analyses leaving for both parts 102 participants per protocol: treatment group 75; control group 27 | |
| Interventions | Desmoteplase i.v. administered as a bolus over 1 to 2 minutes Each dose tier included 15 treatment participants and 4 control participants Doses: ‐ Part 1, 1st period: 25 mg, 37.5 mg, 50 mg versus placebo; 2nd period: 25 mg versus control ‐ Part 2, dose titration from 62.5 µg/kg, 90 µg/kg to 125 µg/kg versus control Participants < 66 kg were administered 80% of the dose; participants ≥ 66 kg received 100% | |
| Outcomes | Primary safety endpoint: SICH according to NINDS 1995 criteria, CT at 72 hours Primary clinical endpoint: combined NIHSS (0 to 1), mRS (0 to 2), and BI (≥ 8 points improvement or a score of ≥ 75) at 90 days Co‐primary efficacy endpoints: reperfusion at 4 to 8 hours defined as ≥ 30% reduction of MTT volume or ≥ 2 points improvement on MRI‐adapted TIMI grading Other endpoint: change in infarct volume on DWI from baseline to day 90 | |
| Notes | The trial with 3 doses versus control was discontinued by the Data Monitoring Committee (DMC) after the 30th participant and occurrence of 3 SICH in the 37.5 mg group and 1 in the 50 mg group After the 47th participant, excess SICH in the treatment group (25 mg) prompted a halt by the DMC, interim analysis, and subsequent amendment In Part 2 of the trial, a placebo‐controlled bodyweight‐adjusted dose‐escalation design starting at a dose of 62.5 µg/kg, followed by 90 µg/kg and 125 µg/kg was applied Further, in association with the interim analysis of Part 1, the upper limit of blood sugar was reduced from 22 to 11 mmol/L (in Part 1 blood glucose > 10 mmol/L was associated with an increased risk of ICH) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
ECASS 1995.
| Methods | Double‐blind RCT Sealed drug prepacks (no central telephone randomisation) Only intention‐to‐treat data used in this review | |
| Participants | People with acute MCA territory ischaemic stroke who could be randomised and start treatment within 6 hours of symptom onset Pre‐entry CT to exclude cerebral haemorrhage and people whose infarct was already visible; age > 18 years and < 80 years Excluded: people with mild strokes or whose symptoms were rapidly improving, in coma, BP > 110 diastolic and > 200 systolic; recent trauma or surgery, pregnancy, weight > 100 kg (because of dose limit) | |
| Interventions | Actilyse (recombinant tissue Plasminogen Activator, tPA) 1.1 mg/kg body weight up to a maximum of 100 mg (Boehringer Ingelheim, Germany), or identical‐looking placebo i.v. The first 10% of the total dose was given as a bolus followed by infusion of the remainder over 60 minutes All anticoagulants and aspirin to be avoided in the first 24 hours (subcutaneous heparin allowed); thereafter, the use of these drugs was at the discretion of the attending physician | |
| Outcomes | Primary: BI and mRS scores at 90 days Secondary: case fatality at 30 days, various combined stroke scores at 90 days, duration of hospital stay Rankin 2 or worse = disabled on this modified scale: 0 = no symptoms, 6 = dead | |
| Notes | Trial sponsored by Boehringer‐Ingelhei Results presented as ITT analysis and 'Target Population', i.e. after exclusion of protocol violations most of which were due to visibility of early signs of cerebral infarction on the randomisation CT on review by the central CT monitoring committee In this review, only the ITT data have been used The published data were supplemented by additional information from the principal investigators, and with data presented at the meeting organised by the sponsors to present the results held in Barcelona in March 1995 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
ECASS 3 2008.
| Methods | Double‐blind, randomised, placebo‐controlled, parallel group efficacy and safety trial Location and time: 130 centres in 19 European countries, July 2003 to November 2007 Method of randomisation: interactive voice randomisation system Treatment packs of alteplase or matched placebo, in blocks of 4 at each centre and having been generated randomly centrally Investigators were blinded to the size of the blocks Blinded assessment at the time of enrolment at 1, 2 and 24 hours, and on days 7, 30 and 90 Analysis: intention‐to‐treat (worst possible outcome in cases who were known to be alive) | |
| Participants | Eligibility: with the exception of the time window, the study drug was to be used in accordance with the current European licensing Age: 18 ‐ 80 years NIHSS: 4 to 25 Time window: 3 to 4 hours (228 participants), and 3 to 4.5 hours (from participant 229 to 821) Imaging: CT or MR pre‐randomisation to exclude ICH and major ischaemic infarction, repeated 22 to 36 hours after treatment Summary of participants enrolled: ‐ Treatment group: age ± SD: 64.9 ± 12.2 years; male: 63%, NIHSS (mean): 10.7± 5.6; time from onset (median): 3 hours 59 minutes, 3 to ≤ 3.5 hours: 9.6%, 3.5 to ≤ 4 hours: 45.7%, 4 to ≤ 4.5 hours: 41.6% ‐ Control group: age ± SD. 65.6 ± 11.0 years; male: 57%; NIHSS (mean): 11.6 ± 5.9; time from onset (median): 3 hours 58 minutes, 3 to ≤ 3.5 hours: 10.4%%, 3.5 to ≤ 4 hours: 47.9%, 4 to ≤ 4.5 hours: 36.7% Number randomised: total N = 821; treatment group N = 418 of which 375 received treatment; control group N = 403, of which 355 received placebo | |
| Interventions | Actilyse (rt‐PA) 0.9 mg/kg to a maximum dose of 90 mg (Boehringer Ingelheim, Germany) or identical‐looking placebo with 10% given as an i.v. bolus and the remainder as a continuous infusion over 60 minutes | |
| Outcomes | The primary efficacy endpoint: disability day 90 assessed by means of a dichotomised mRS (0 to 1) or unfavourable outcome (mRS 2 to 6) Secondary endpoint: composite global outcome: mRS 0 to 1, BI ≥ 95, NIHSS 0 to 1, and GCS = 1 Further functional endpoints: NIHSS improvement > 8, mRS 0 to 2 or 3 to 6, and BI ≥ 95 Safety endpoints: overall mortality at day 90, any ICH, SICH*, symptomatic brain oedema** * SICH = any apparent extravascular blood in the brain/within the cranium associated with a clinical deterioration > 4 on NIHSS, or that led to death and that was identified as the predominant cause of the neurologic deterioration To allow comparison with published data we also performed a post‐hoc analysis of rates of SICH according to definitions used in other trials **Symptomatic oedema = brain oedema with a mass effect as predominant cause of clinical deterioration | |
| Notes | Monitoring and data management by the manufacturer, Boehringer Ingelheim Subcutaneous heparin (< 10,000 IU) or equivalent doses of low molecular weight heparin was permitted ≤ 24 hours | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
ECASS II 1998.
| Methods | Double‐blind, placebo‐controlled RCT Randomisation by sequential numbered packs at each centre (the allocation having been generated randomly centrally) | |
| Participants | Acute hemispheric ischaemic stroke, people aged 18 ‐ 80 years, within 6 hours of onset, CT having excluded intracranial haemorrhage and visible infarction in more than ⅓ of the MCA territory | |
| Interventions | Actilyse (rt‐PA) 0.9 mg/kg to a maximum dose of 90 mg (Boehringer Ingelheim, Germany), or identical‐looking placebo, 10% given as a bolus and the rest infused over 1 hour, to be started within 6 hours of stroke onset Aspirin and anticoagulants (apart from subcutaneous heparin) to be avoided in the first 24 hours; thereafter the use of these drugs was at the discretion of the attending physician | |
| Outcomes | Death or dependency at 90 days defined as modified Rankin 2 to 6 Intracranial haemorrhage Death Various other scores at 90 days | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
EPITHET 2008.
| Methods | Double‐blind, randomised, prospective, phase II placebo‐controlled study Location: 15 centres in Australia, New Zealand, Belgium and the UK during 2001 to 2007 Method of randomisation: computerised randomisation giving out the number of a treatment pack The packs were centrally prepared in blocks for each centre (4 treatment packs per block) Analysis: intention‐to‐treat, and of primary outcome measure: per‐protocol | |
| Participants | Acute hemispheric ischaemic stroke The participants were randomised on the basis of plain CT Age: ≥ 18 years NIHSS: ≥ 4; premorbid mRS ≤ 2 Time window: 3 to 6 hours Baseline CT (in one centre MRI) to exclude: haemorrhage and early ischaemic changes ≥ ⅓ of the MCA territory; CT was repeated in clinical deterioration possibly due to a haemorrhagic transformation MRI was performed before start of treatment, at day 3 to 5 (DWI, PWI/concentration‐time curves after gadolinium, and MRA (time of flight or phase contrast)) At day 90 (T2‐weighted images to measure final infarct volume) To standardise image analysis, all MRI were centrally read Number randomised: 101 (treatment group: 52; control group: 49) | |
| Interventions | Alteplase (rt‐PA) 0.9 mg/kg i.v. up to a maximum of 90 mg, 10% given as a bolus, the remainder as infusion over 1 hour, or placebo | |
| Outcomes | MRI definitions and outcome measures: mismatch, PWI‐DWI volume > 1 to 2, and PWI‐DWI volume ≥ 10 ml; infarct growth, 4 measurements, i.e. expansion between baseline and day 90 T2‐weighted lesion; reperfusion > 90% reduction between baseline and day 3 PWI volumes; recanalisation: improvement of TIMI from baseline to day 3 to 5 by ≥ 2 points; SICH according to SITS‐MOST criteria; target mismatch, mismatch excluding 'malignant profile'; 'malignant profile': DWI volume ≥100 ml, PWI volume ≥ 100 ml, or both with PWI defined as Tmax delay ≥ 8 seconds Clinical: NIHSS at day 3 to 5 and day 90; mRS day 90 Good neurological outcome: NIHSS 0 to 1 or improvement ≥ 8 from baseline Good functional outcome: mRS 0 to 2 Primary outcome measure: infarct growth Secondary outcome measures included: difference in mismatch participants in reperfusion; good neurological and functional outcome between the treatment and control group; difference in DWI lesion volumes in the treatment group between participants with and without SICH; difference in infarct growth in non‐mismatch participants between the treatment arms; difference in infarct growth, good neurological and functional outcomes in the treatment group between participants with and without mismatch; difference in infarct growth, good neurological and functional outcomes in the treatment group between participants with target mismatch and those with 'malignant profile' | |
| Notes | The paucity of participants without mismatch precluded comparisons of the effect of rt‐PA in the presence versus absence of mismatch | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
Haley 1993.
| Methods | Double‐blind, randomised, blinded, placebo‐controlled RCT Sealed envelope treatment allocation, opened by pharmacist at randomisation and appropriate infusion made up Other study personnel remained blinded Not intention‐to‐treat | |
| Participants | Ischaemic stroke < 90 or < 180 minutes from onset, 18 ‐ 80 years, pre‐entry CT Excluded: TIA, very mild and very severe neurological deficits | |
| Interventions | Alteplase (tPA, Genentech) 0.85 mg/kg or identically appearing placebo i.v. over 60 minutes Early (0 to 90 minutes) versus late (91 to 180 minutes) treatment versus placebo, although in this review the early and late groups have been analysed together, i.e. the comparison is 'any tPA versus placebo' | |
| Outcomes | Clinical improvement using NIH Stroke Scale at 24 hours, 2 + 7 days, and 3 months Follow‐up CT at 24 hours, 7 days and 3 months for infarct volume and haemorrhagic transformation | |
| Notes | Pilot for larger NINDS 1995 trial | |
IST3 2012.
| Methods | Randomised, open‐label, controlled trial with blinded outcome assessments (PROBE) | |
| Participants | Participants (n = 3035) with acute ischaemic stroke < 6 hours, no age limit Protocol at www.dcn.ed.ac.uk/IST3 or www.ist3.com | |
| Interventions | I.v. rt‐PA 0.9 mg/kg to maximum 90 mg versus placebo | |
| Outcomes | Death at 7 days, SICH, death or dependency at 6 months | |
| Notes | Moving from start‐up to extended pilot phase in July 2002, trial ended 31 January 2012 and the main publication and a meta‐analysis on rt‐PA trials (11 earlier included plus IST‐3) were both published in the Lancet 2012;379:2352‐63 and 2012;379: 2364‐72, respectively. The IST‐3 trial protocol and the Statistical Analysis Plan had been published before the breaking of the code (all references available in reference list) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised after core data entry was complete via the Internet or telephone, stratified by country and minimised on key prognostic variables |
| Allocation concealment (selection bias) | Low risk | Central web‐ or telephone‐based randomisation ensured allocation concealment |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Treatment was given open‐label, therefore participants and personnel knew whether rt‐PA or control was used. However, all participants were to receive best medical care and there was no evidence of differences in care between treatment groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Treatment was given open‐label, therefore participants and personnel knew whether rt‐PA or control was used |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Symptomatic and fatal ICH and all 6‐month outcomes were adequately blinded to treatment. Haemorrhage was assessed centrally by scan adjudicators blind to all clinical details; 6‐month outcomes were assessed centrally by staff blinded to treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Very few data were lost to follow‐up; no difference between trial groups |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported; no difference in reporting between groups |
| Other bias | Low risk | No other biases evident |
JTSG 1993.
| Methods | Double‐blind RCT Sealed identical prepacks of drug or placebo Not intention‐to‐treat | |
| Participants | Thromboembolic stroke < 6 hours, aged 18 ‐ 80 years, pre‐entry CT and angiography Excluded: haemorrhagic stroke or patent cerebral arteries at angiography | |
| Interventions | Duteplase 20 MIU versus identical‐looking placebo i.v. over 60 minutes Duteplase supplied by the Sumimoto Corporation, Japan | |
| Outcomes | Reperfusion (immediate post‐infusion angiography), clinical improvement using Hemispheric Stroke Scale (HSS) at 4 weeks after stroke, haemorrhagic transformation on follow‐up CT, death | |
| Notes | 112 participants recruited, 14 excluded for protocol violations before breaking the randomisation code, analysis is of 98 patients who completed the study Further information provided by Prof Yamaguchi | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
MAST‐E 1996.
| Methods | Double‐blind, randomised controlled trial Central telephone randomisation Sealed prepacks of drug (streptokinase) or identical placebo Intention‐to‐treat | |
| Participants | People with symptoms of large acute ischaemic stroke in the MCA territory who could be randomised and start treatment within 6 hours of symptom onset; age > 18 years but no upper limit; CT scan mandatory prior to randomisation to exclude cerebral haemorrhage Exclusions: people with mild neurological deficit (MAST Unified Scale > 55), or who were improving rapidly when assessed; previous disabling stroke; pregnancy; systolic BP > 220, diastolic BP > 110; oral anticoagulants (not aspirin); recent trauma, surgery, peptic ulcer disease, etc | |
| Interventions | Streptokinase 1.5 MU in 100 ml normal saline i.v. over 1 hour versus identical‐looking placebo Heparin and aspirin use were allowed in the first 24 hours (as well as later) at the discretion of the attending physician provided that the dose, route and time of administration were recorded | |
| Outcomes | Death and disability (MAST unified scale, Rankin and Barthel scales) at 10 days and 6 months after randomisation (disability = Rankin 3 or worse); cerebral haemorrhage within the first 10 days; other adverse events (hypotension, systemic haemorrhage) Death and disability at 1 year is being collected | |
| Notes | Trial terminated in September 1994 after 310 participants had been randomised on the advice of the Data Monitoring Committee due to an excess of cerebral haemorrhages and associated early mortality in the streptokinase‐treated group Original sample size was to have been 600 participants The results included here are only for the first 270 participants (published in the Lancet in January 1995); the full results on the 310 participants cannot be included until the trial has been published in full Note the frequent use of heparin (25%) and aspirin (13.5%) within 24 hours of trial treatment and during the first 2 weeks (65% and 25% respectively) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
MAST‐I 1995.
| Methods | Randomised controlled trial with 2 x 2 factorial design Central telephone randomisation Intention‐to‐treat Control group did not receive a placebo but 6‐month follow‐up was by blinded investigators | |
| Participants | All people with acute ischaemic strokes that could be randomised and start treatment within 6 hours from symptom onset; age > 18 years, but no upper limit; pre‐entry CT mandatory to exclude cerebral haemorrhage Exclusions: rapidly improving symptoms likely to be a TIA; recent trauma or surgery; oral anticoagulant treatment (not aspirin); aspirin or streptokinase not either definitely indicated or definitely contraindicated; streptokinase in the past year, etc | |
| Interventions | Streptokinase 1.5 MU i.v. over 1 hour immediately after randomisation, or aspirin 300 mg oral started immediately and continued for 10 days (or via nasogastric tube, per rectum, or i.v. in Italy), or both, or neither Other anticoagulants to be avoided within the first 24 hours, but could be used thereafter Aspirin use encouraged after 10 days or at hospital discharge (whichever came first) | |
| Outcomes | Death within the first 10 days; cerebral haemorrhage; death and disability at 6 months (Rankin scale: disabled = Rankin 3 or more) 1‐year follow‐up is also being performed | |
| Notes | Trial suspended after 622 participants randomised because of slow randomisation rate, partly due to increasing confusion about thrombolysis in stroke from ASK 1996 and MAST‐E 1996 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
MELT 2007.
| Methods | PROBE: Prospective Randomised Open control study with Blinded Endpoint assessment; RCT Location: multi‐centre, 57 centres in Japan; ongoing January 2002 to October 2005 Method of randomisation: Internet‐based in 2 steps: ‐ Step 1: pre‐registration of all eligible people to angiography ‐ Step 2: allocation to treatment or control Intention‐to‐treat | |
| Participants | Eligibility and randomisation: ‐ Step 1: all eligible people 20 ‐ 75 years with clinical presentation of an MCA lesion possibly embolic, NIHSS 5 to 22, CT normal or with only subtle early ischaemic changes where angiography revealed complete occlusion of the horizontal M1 segment or the M2 division of the MCA proceed to randomisation ‐ Step 2: treatment allocation Age limits: 20 ‐ 75 years NIHSS > 5 Time window: 0 to 6 hours Participants: ‐ Treatment group: male: 64.9%, age mean ± SD: 66.9 years (± 9.3), NIHSS: 14 (IQR 8), time from onset to allocation minimum 68 ± 61, occlusion location MCA M1: 68%, M2: 32% ‐ Control group: male: 64.9%, age mean ± SD: 67.3 ± 8.5, NIHSS: 14 (IQR 7), time from onset to allocation minimum: 206 ± 54, occlusion location MCA M1: 74%, M2: 26% Previous lacunar infarcts allowed if no sequels Number randomised: 114 (treatment group 57; control group 57) | |
| Interventions | UK i.a. infusion (treatment group) or conventional treatment (control group) After allocation treatment group immediate i.v. heparin (5000 IU) infusion An infusion catheter with a single end hole was then passed through the clot and positioned on the distal side of the thrombus if possible More proximal regional infusion than inside the clot was prohibited Mechanical disruption of clots was permitted with the guidewire Repeat CT at maximum intervals of 2 hours was required and only in the absence or with subtle early ischaemic signs was the next step accepted: i.a. infusion of UK (120,000 IU over 5 minutes) This was repeated until a total dose of 600,000 IU; 2 hours had passed; or complete re‐canalisation was achieved Control group: no specific treatment, osmotic diuretics in participants manifesting high intracranial pressure Neither group was allowed fibrinolytic therapy or antithrombotics (heparin, aspirin, ticlopidine) for 24 hours after fibrinolysis in the treatment group | |
| Outcomes | Clinical outcome: NIHSS, mRS, BI at 7, 30 and 90 days after symptom onset (by blinded physician) Follow‐up CT was scheduled at 24 hours, 7 and 90 days after symptom onset SICH was defined according to NINDS 1995 but also included participants with < 4 points on NIHSS in case of 'apparent signs considered as symptomatic' Follow‐up angiography only in the treatment group and re‐canalisation was evaluated by a blinded Film Reading Committet as: no, partial (≥ 50%), or complete re‐canalisation | |
| Notes | The trial was prematurely stopped upon advice from the Independent Monitoring Committee when i.v. rt‐PA was approved for ischaemic stroke in October 2005 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
Mori 1992.
| Methods | Double‐blind RCT Identical coded drug prepacks Not intention‐to‐treat | |
| Participants | Ischaemic stroke in carotid territory < 6 hours from onset, aged < 80 years, pre‐entry CT (to exclude haemorrhage) and angiography (to confirm arterial occlusion) Excluded: people in deep coma | |
| Interventions | Duteplase (Sumitomo Pharmaceuticals, Tokyo) 20 or 30 MIU versus identical placebo i.v. over 1 hour The 20 and 30 MIU groups were analysed as 1 group in this review, i.e. the comparison is 'any duteplase versus placebo' | |
| Outcomes | Reperfusion at 60 minutes after start of infusion, clinical status at 30 days using modified Hemispheric Stroke Scale (HSS) CT at 1, 2, 7 and 30 days for haemorrhagic transformation | |
| Notes | Additional information from Prof Mori Unbalanced numbers due to accidental loss of a 20 MIU dose pack and substitution with placebo No participants excluded, therefore an ITT analysis was possible | |
Morris 1995.
| Methods | Double‐blind, randomised trial of streptokinase 1.5 MU or placebo Randomisation method not stated (sequentially number drug prepacks probably) Intention‐to‐treat Note saline is not an identical placebo for streptokinase | |
| Participants | Anterior circulation acute ischaemic stroke that could be randomised and start treatment within 6 hours of onset (CT mandatory to exclude any haemorrhage), aged 40 ‐ 80 years, no previous stroke | |
| Interventions | Streptokinase 1.5 MU i.v. over 1 hour versus saline placebo | |
| Outcomes | Death at 3 months, neurological improvement at 3 months, ICH | |
| Notes | No additional information forthcoming from the authors | |
NINDS 1995.
| Methods | Randomised, placebo‐controlled, blinded trial On‐treatment analysis, not intention‐to‐treat Randomisation was at the participating hospital by sequentially numbered drug (or identical‐looking placebo) prepacks, followed within 2 hours by a telephone call to the Trial Co‐ordinating centre to notify them that an individual had been randomised | |
| Participants | People with ischaemic stroke (CT mandatory to exclude ICH) with a clearly defined time of symptom onset who could receive the trial treatment within 180 minutes of symptom onset; age > 18 and < 80 years initially (80‐year age limit removed on 30 March 1992 after 188 participants had been entered in Part A; a neurological deficit measurable on the NIH stroke scale (i.e. not very mild and not improving rapidly at the time of assessment) ‐ thus cortical and lacunar strokes were eligible. People with previous stroke or head trauma within 3 months, pregnancy/lactation, abdominal surgery, heparin within 48 hours or deranged clotting factors/platelets, systolic BP > 180 or diastolic BP > 110 (+ various other features) were excluded | |
| Interventions | Alteplase (rt‐PA, Activase, Genentech, South San Francisco) in a dose of 0.9 mg per kg body weight (maximum dose 90 mg) or identical‐looking placebo (prepared by Genentech) given i.v. The first 10% of the dose was given as a bolus followed by the remainder as a constant infusion over 60 minutes No antiplatelet or anticoagulants were to be given during the first 24 hours after randomisation, and BP had to be kept within prespecified limits (< 180 systolic and < 110 diastolic) The participants randomised within the first 90 minutes after the stroke were analysed separately from those randomised between 91 and 180 minutes after the stroke in the publication but have been put together in this review | |
| Outcomes | Assessment was by a physician who had not been involved in the randomisation or treatment administration using the following: NIH stroke scale at 2 hours after start of treatment, and at 24 hours, 7 to 10 days and 3 months after onset of the stroke Glasgow outcome score at 3 months Barthel Index at 7 to 10 days and 3 months modified Rankin score (0 = no symptoms, 5 = severe disability) at 7 to 10 days and 3 months CT scan at 24 hours, 7 to 10 days and 3 months Rankin 2 or worse was the cut‐off for disability in the analysis The data cannot be subdivided further at this point as the results have not yet been published by individual Rankin group | |
| Notes | The trial was conducted in 2 phases: the first (291 participants) was to assess the effect of tPA on outcome at 24 hours after the stroke; the second (333 participants) was to assess the effect of tPA on outcome at 3 months The investigators remained blinded to the results of Part 1 until the end of Part 2 The protocols were identical for the 2 parts except for their primary hypotheses, i.e. participants in Part 1 were followed up at the same time points as in Part 2 Additional information to that published is being sought from the principal investigators, but has not yet been received | |
Ohtomo 1985.
| Methods | Double‐blind, randomised trial Identical prepacked drugs Not intention‐to‐treat | |
| Participants | Presumed 'non‐embolic' ischaemic stroke < 5 days, no age limit, pre‐entry CT | |
| Interventions | UK 60,000 U/day i.v. over 1 hour for 7 days versus identical‐looking placebo UK supplied by Abbott Labs, USA | |
| Outcomes | Clinical status (Global Improvement and Severity Ratings) and safety (absence of side effects) at 4 weeks from start of treatment Follow‐up CT to assess haemorrhagic transformation | |
| Notes | Additional information from Prof Ohtotmo All cases accounted for including those who 'dropped out'; therefore, an intention‐to‐treat analysis possible | |
PROACT 1998.
| Methods | Double‐blind, placebo‐controlled trial Centralised randomisation Follow‐up blinded to treatment allocation by an independent neurologist who was blinded to the angiogram findings and in‐hospital course | |
| Participants | People aged 18 ‐ 85 years without prior stroke, within 6 hours of onset of symptoms indicative of an MCA occlusion, NIHSS score > 4 and < 30, and who had an MCA occlusion (complete or major branch) on conventional intra‐arterial angiography | |
| Interventions | Recombinant prourokinase 6 mg or saline placebo i.a. through the angiogram catheter with the tip in the thrombus; heparin was also administered in a high dose to the first 16 participants, and thereafter in a lower dose, so the study was confounded Funding and drug supplied by Abbott Laboratories | |
| Outcomes | Recanalisation of the MCA at 120 minutes after treatment infusion NIHSS, Barthel, Rankin, at 7, 30 and 90 days after treatment Haemorrhagic transformation of the infarct Extracranial bleeding Death | |
| Notes | This trial was confounded by heparin (the dose of which was altered half way through the trial, and was testing i.a. not i.v. thrombolysis It has been included in the present data tables, though in a separate subgroup, until further information on i.a. thrombolysis becomes available to make a separate review worthwhile | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
PROACT 2 1999.
| Methods | Randomised controlled trial Central telephone randomisation No placebo Follow‐up assessment by a neurologist blinded to the angiogram result, treatment allocation, and to the in‐hospital course, who was not involved in the care of the participant during the acute treatment phase Analysis was by intention‐to‐treat | |
| Participants | People with acute ischaemic stroke, aged 18 ‐ 85 years, with NIH stroke scale ≥ 4 but < 30, without visible infarction in > ⅓ of the MCA territory on baseline CT scan, with angiography‐proven MCA main stem (M1) or major branch (M2) occlusion, within 6 hours of onset of stroke symptoms Randomisation stratified by stroke severity according to the baseline NIH Stroke Scale, and 2 participants allocated to active treatment for every 1 allocated control | |
| Interventions | Pro‐urokinase (Abbott Laboratories) 9 mg given i.a. through the angiography catheter with the tip embedded in the thrombus No placebo Heparin 2000 IU bolus i.v. at time of angiography followed by 500 IU/hour i.v. for 4 hours, to both treatment groups | |
| Outcomes | Rankin ≤ 2 (i.e. independent) at 90 days after treatment, Rankin < 2 at 90 days, total deaths at 90 days, SICH, recanalisation (according to the TIMI classification for coronary occlusions) as documented on angiography at 2 hours after the start of the trial infusion | |
| Notes | During the trial 12,323 people were screened, of whom 476 underwent angiography and only 180 were randomised | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | |
Wang 2003.
| Methods | RCT Location: Beijing Emergency Medical Centre and Statistical Department, Peking University, Beijing, China Method of randomisation: unclear 3 parallel groups: 2 doses and 1 control Single‐centre trial Analysis: intention‐to‐treat | |
| Participants | Eligibility: those eligible were divided into 3 groups Age limit: 35 ‐ 80 years NIHSS: not defined Time window: 0 to 6 hours Participants: Chinese study population; BP 180/100 mmHg Treatment group A: rt‐PA 0.9 mg/kg, N = 34 Treatment group B: rt‐PA 0.7 mg/kg, N = 33 Treatment group C: control, N = 33 CT: to rule out ICH or low‐density lesion but not early ischaemic changes Number randomised: 100 (treatment group 67; control group 33) | |
| Interventions | Actilyse (t‐PA) 0.9 mg/kg to a maximum dose of 90 mg (Boehringer Ingelheim Germany) Treatment group A: 0.9 mg/kg Treatment group B: 0.7 mg/kg In both groups 8 mg rt‐PA was injected i.v. as a bolus and the rest was given over 60 minutes to a maximum dose of 90 mg Control group: no thrombolytic therapy | |
| Outcomes | Clinical: CSS (Chinese Stroke Scale) BI at 24 hours and 90 days Mortality at 30 days CT: 12 and 24 hours, 7 and 30 days Primary outcome: neurological and functional status, mortality, safety (ICH) | |
| Notes | Same group of participants seem to have been included in Wang 2006 and Zeng 2006 but with more added; awaiting further assessment of these later publications | |
APTT = activated partial thromboplastin time BI: Barthel Index BP: blood pressure CT: computed tomography DWI: diffusion weighted imaging GCS: Glasgow Coma Score i.a.: intra‐arterial ICA: internal carotid artery ICH: intracranial haemorrhage INR: measure of anticoagulation in blood used to monitor warfarin dose IQR: interquartile range ITT: intention‐to‐treat i.v.: intravenous LOCF: last observation carried forward M1, M2: different segments of the middle cerebral artery MCA: middle cerebral artery MRI: magnetic resonance imaging mRS: modified Rankin Scale MTT: mean transit time MU: mega units NIH: National Institutes of Health NIHSS: National Institutes of Health Stroke Scale PCA: posterior cerebral artery PWI: perfusion imaging RCT: randomised controlled trial rPRO‐UK: recombinant pro‐urokinase rt‐PA: recombinant tissue plasminogen activator SICH: symptomatic intracranial haemorrhage SITS‐MOST: Safe Implementation of Thrombolysis in Stroke Monitoring Study TIA: transient ischaemic attack TIMI: thrombolysis in myocardial infarction, and was an angiographic scale developed to quantify arterial patency in the coronary arteries, now adapted to the intracranial arteries for stroke Tmax: a method of quantifying a cerebral perfusion abnormality tPA: tissue plasminogen activator UK: urokinase
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bao 2003 | Not randomised; no relevant outcomes |
| Boehringer Mannheim 1994 | Study never started |
| CLEAR 2008 | Low versus high dose of rt‐PA |
| Davalos 2003 | Not a RCT: observational study of DWI/PWI mismatch |
| Defuse 2001 | Not a RCT: all participants received rt‐PA |
| DeWinter 1999 | Not relevant |
| Ding 2006 | Not clearly randomised; comparison of lumbrokinase with control; latest outcome 28 days |
| Ducrocq 2005 | Urokinase i.a. versus i.v. |
| EAST II [pers comm] 1999 | EAST = pilot, open‐dose escalation trial of a modified i.v. t‐PA EAST II = RCT of same agent but stopped prematurely at very early stage due to unexpectedly high SICH rate |
| Edinburgh 1991 | This study terminated prematurely because of the impracticality of i.a. thrombolytic treatment 4 participants were randomised between streptokinase (250,000 MU into the occluded cerebral artery) or placebo during the year that the trial ran (1991). It had been intended to randomise at least 10 participants; however, only 3 received streptokinase, 1 placebo. 1 (streptokinase) died within a week of the stroke of a massive cerebral infarct; 6‐month outcome in the other 3 was: 1 (streptokinase) Rankin 2; 1 (streptokinase) Rankin 3; 1 (placebo) Rankin 4 These results have not been included because the number is so small and the randomisation (because of the premature termination) so imbalanced |
| EMFATAS 1996 | Abandoned |
| EMS 1996 | rt‐PA i.a. plus i.v. versus i.v. |
| Fan 2001 | Not a thrombolytic drug; fibrinolysis‐enhancing only |
| Fisher 2003 | Not a RCT |
| Fu 2004 | No SICH, death or poor functional outcome data at all and discrepancies in dose of drug and low‐dose urokinase for 5 days starting > 24 hours |
| Fuentes 2007 | Non‐random case series |
| Geng 2004 | Confounded by heparin |
| Hartmann 2005 | Abstract only; author did not respond to multiple requests for more information between October 2007 and December 2008; no evidence that randomised comparison of rt‐PA |
| Hong Kong 1994 | This trial of i.v. streptokinase, stopped prematurely after randomisation of only a few participants because of concerns about use of streptokinase arising from termination of ASK 1996, MAST‐E 1996 and MAST‐I 1995 |
| Huang 1996 | Snake venom; not a thrombolytic agent |
| Huang 2000 | Trial of lumbrokinase; effect on blood coagulability; no neurological outcomes mentioned; no detail of control groups |
| ICTuS‐L 2006 | Interventional clot‐pulling trial |
| IMS II 2004 | Non‐random single arm pilot |
| IMS III 2008 | rt‐PA i.v. versus i.a. |
| INSTINCT 2005 | Trial of modifying physician attitudes, not of rt‐PA |
| ITAIS 2005 | Not a RCT; using multimodal MRI to decide whether to give i.a. or i.v. rt‐PA |
| Jin 2000 | Trial of effect of lumbrokinase on blood coagulability; no neurological outcome measures; further details awaited |
| Kandil 2001 | Only ever published in abstract, insufficient details, apparently RCT of i.a. urokinase plus heparin versus heparin in 36 participants; no clinical details |
| Kim 2008 | Non‐random case series |
| Konta 1996 | Comparison of 2 doses of urokinase, not clear if random |
| Lang 2013 | Not a trial of thrombolysis; is a trial of cerebrolysin in participants already treated with rt‐PA |
| Li 2003 | Urokinase versus acupuncture, not clear that it is a RCT, only recorded SICH; no information on death or dependency |
| Lindsberg 2006 | Non‐random, observational comparison of i.v. and i.a. rt‐PA in basilar artery occlusion |
| Liu 1991 | Urokinase and dextran i.v. versus i.a. and dextran |
| Liu 1994 | RCT of ahylysantifarctase once per day for 21 days plus dextran versus dextran, i.v. administration, outcome measures were all of changes in blood coagulation not neurological assessments; no mention of cerebral haemorrhages or deaths |
| Liu 2004 | No long‐term follow‐up (latest at 4 weeks), no functional outcome |
| Lu 2001 | Not a RCT, only information on blood fibrinogen levels |
| Lyden 2003a | Non‐random, dose‐escalating study of tenekteplase |
| Lyden 2003b | rt‐PA +/‐ hypothermia; not trial of rt‐PA |
| Meyer 1963 | Although randomised and controlled, this trial was conducted in the pre‐CT era and therefore there was no way of being sure that only people with ischaemic stroke were included |
| Meyer 1964 | Although randomised and controlled, this trial was conducted in the pre‐CT era and therefore there was no way of being sure that only people with ischaemic stroke were included |
| Michel 2008 | Small randomised feasibility study with PCT for participant selection ‐ stopped as unfeasible |
| MITI‐IV 2005 | Not a thrombolytic drug |
| Molina 2005 | Trial of microbubble‐enhanced sonothrombolysis in the presence of rt‐PA, i.e. not a trial of rt‐PA |
| Morris 2002 | Not relevant |
| Naito 1984 | The data are presented in 2, possibly 3, different publications; many participants were lost to follow‐up during the 4‐week trial period. Dr Naito has died and Professor Abe is unable to supply further information on those lost to follow‐up. Although there were no deaths or cerebral haemorrhages among the participants who completed the 4‐week trial period, the data are incomplete and may be badly skewed by lack of information on what happened to the participants who dropped out |
| Ohta 1997 | Non‐random case series |
| Pan 2011 | Not a randomised trial ‐ just an observational study |
| Pang 1993 | Appears to be a randomised trial (method uncertain) of lumbrokinase 2 tablets daily for 21 days versus placebo; 303 participants received lumbrokinase and 150 participants received placebo, both groups received dextran; very little known about outcome. Follow‐up was at 3 weeks only, therefore this trial was excluded. No information on deaths or intracranial haemorrhages. There is thought to have been conflict between the authors and the pharmaceutical sponsor so no further details have been published (Liu M, personal communication) |
| PRACTISE 2005 | Trial of implementation of rt‐PA, not of rt‐PA itself |
| Qiang 2001 | Not a RCT; study of DWI/PWI |
| Qureshi 2001 | Not relevant |
| Qureshi 2006 | Reteplase and abciximab dose escalation; no control |
| Ribeiro 2012 | Not a trial |
| ROSIE 2002 | Reteplase dose escalation |
| ROSIE‐2 2002 | |
| ROSIE‐CT 2002 | Non‐randomised, dose‐escalation of abciximab versus reteplase + abciximab |
| Rother 2003 | DWI/PWI comparison, not a RCT |
| Shi 2000 | Not a RCT; no relevant outcomes |
| Skoloudik 2004 | Not a RCT; ultrasound versus no ultrasound in presence of rt‐PA |
| Skoloudik 2006 | Not a RCT; ultrasound versus no ultrasound in presence of rt‐PA |
| Sobrino 2007 | Non‐random dose study of endothelial cells |
| SYNTHESIS 2007 | Comparison of i.v. versus i.a. rt‐PA within 3 hours of stroke |
| TEMPO 2013 | Trial of tenecteplase versus rt‐PA in minor stroke |
| TNK 2005 | Dose escalation study |
| TNK‐S2B 2005 | RCT of 3 doses of tenecteplase versus rt‐PA |
| TNK‐TPA 2008 | Tenecteplase versus tPA |
| Trouillas 2000 | Not a RCT; haematological study of effect of rt‐PA on clotting factors |
| TTATTS 1997 | Dose‐range‐finding study of rt‐PA on recanalisation in people with angiographic evidence of arterial occlusion; never published in full. Not a RCT |
| VASTT 2005 | Dose‐range‐finding comparison of rt‐PA variant V10153; same as Vernalis 2005 |
| Vernalis 2005 | Dose‐range‐finding comparison of rt‐PA variant V10153; same as VASTT 2005 |
| Wang 1997 | Blood results only |
| Wang 1999 | Ahylysantinfactase versus defibrase |
| Xiang 1995 | Randomised trial (method unknown) of i.a. urokinase 300,000 to 1,000,000 units for 1 hour plus heparin, plus a 'physical therapy' (nature uncertain) plus 'conventional treatment' versus 'conventional treatment' (nature uncertain) started within 6 hours of onset of acute ischaemic carotid territory stroke. 67 participants were included, 32 in the active and 35 in the placebo arms. Follow‐up was at 3 months using a neurological deficit score. The number of deaths and intracranial haemorrhages was not mentioned. The trial was excluded because of the confounding of treatment allocation |
| Xu 1997 | No details of control group and not conventional drug regimen |
| Xu 2000 | No placebo |
| Xu 2004 | Urokinase and nimodipine |
| Yuan 1995 | A randomised trial of i.v. urokinase 8000 to 10,000 u/kg (total 600,000 to 900,000 ) plus nimodipine, vitamins E and C, aspirin, mannitol, dexamethasone, nidocain and snake venom, versus 'conventional treatment' (i.e. everything except urokinase) given over 40 minutes within 2 days of onset of ischaemic stroke; urokinase 10,000 to 15,000 repeated the following day if no improvement. Follow‐up was using the Chinese Neurological Scale at only 2 weeks after treatment; no long‐term follow‐up. This scale does not appear to measure dependency. A total of 80 participants were randomised, 40 per treatment arm. There is no mention of the number who died or had SICH. The trial was excluded because of the short follow‐up period |
| Zhang 2003 | No dependency data, odd dose of urokinase |
| Zhou 1996 | Not a RCT; drug unclear; only measured endothelial function |
CT: computed tomography DWI: diffusion weighted imaging i.a.: intra‐arterial i.v.: intravenous MRI: magnetic resonance imaging MU: mega units PCT: perfusion computed tomography PWI: perfusion imaging RCT: randomised controlled trial rt‐PA: recombinant tissue plasminogen activator SICH: symptomatic intracranial haemorrhage tPA: tissue plasminogen activator
Characteristics of studies awaiting assessment [ordered by study ID]
FRALYSE.
| Methods | Data awaited |
| Participants | Data awaited |
| Interventions | Data awaited |
| Outcomes | Data awaited |
| Notes | Data awaited |
Lin 2006.
| Methods | RCT |
| Participants | 46 to 72‐year‐old participants within 6 hours of acute ischaemic stroke CT |
| Interventions | Urokinase 1,500,000 U over 40 minutes in i.v. versus control (no placebo) |
| Outcomes | SICH, death, activities of daily living scale (sounds like mRS) at 6 months |
| Notes | 95 participants; further translation required |
TESPI.
| Methods | RCT |
| Participants | Age > 80 years, < 3 hours of acute ischaemic stroke, NIHSS upper limit 17 |
| Interventions | I.v. rt‐PA as per NINDS dose versus placebo |
| Outcomes | Uncertain |
| Notes | Planned sample size 600 total |
CT: computed tomography i.v.: intravenous mRS: modified Rankin Scale NIHSS: National Institutes of Health Stroke Scale RCT: randomised controlled trial rt‐PA: recombinant tissue plasminogen activator sc: subcutaneous SICH: symptomatic intracranial haemorrhage
Characteristics of ongoing studies [ordered by study ID]
BASICS.
| Trial name or title | BASICS |
| Methods | Intra‐arterial and intravenous treatment |
| Participants | Basilar occlusion after i.v. rt‐PA within 4.5 hours of stroke |
| Interventions | Urokinase, rt‐PA, catheter devices |
| Outcomes | 12‐month mRS |
| Starting date | ‐ |
| Contact information | ‐ |
| Notes | Basically a trial of intra‐arterial plus intravenous versus intravenous ‐ probably relevant to Wardlaw 2013 |
DIAS‐3.
| Trial name or title | Desmoteplase in Acute Stroke 3 |
| Methods | Within 3 to 9 hours of onset |
| Participants | Acute ischaemic stroke selected on basis of advanced imaging |
| Interventions | Desmoteplase 90 microgrammes/kg i.v. as bolus versus placebo |
| Outcomes | 90 days |
| Starting date | ‐ |
| Contact information | ‐ |
| Notes | Nearing completion |
DIAS‐4.
| Trial name or title | Desmoteplase in Acute Stroke 4 |
| Methods | Within 3 to 9 hours of onset |
| Participants | Acute ischaemic stroke selected on basis of advanced imaging |
| Interventions | Desmoteplase 90 microgrammes/kg i.v. as bolus versus placebo |
| Outcomes | 90 days |
| Starting date | ‐ |
| Contact information | ‐ |
| Notes | Nearing completion |
DIAS‐J.
| Trial name or title | Desmoteplase in Acute Stroke Japan |
| Methods | Within 3 to 9 hours of onset |
| Participants | Acute ischaemic stroke selected on basis of advanced imaging |
| Interventions | Desmoteplase 70 or 80 microgrammes/kg i.v. as bolus versus placebo |
| Outcomes | 90 days |
| Starting date | ‐ |
| Contact information | ‐ |
| Notes | Nearing completion |
EXTEND.
| Trial name or title | EXTEND |
| Methods | RCT of rt‐PA at 3 ‐ 6 and 6 ‐ 9 hours after acute ischaemic stroke with specific lesion features |
| Participants | People with acute stroke with PWI:DWI 2:1, PWI lesion > 20 ml, Tmax 6 seconds, CTP if needed, NIHSS 4 to 26 |
| Interventions | rt‐PA i.v. dose as per NINDS 1995 |
| Outcomes | Unclear |
| Starting date | Unclear ‐ seeking funding |
| Contact information | ‐ |
| Notes | Full trial would have 15% absolute risk reduction, 80% power, 0.05 alpha, allowing for 15% drop‐out, need 200 per arm; 100‐patient Australian and New Zealand pilot study |
Ogawa ia Urokinase.
| Trial name or title | Intra‐arterial urokinase trial |
| Methods | ‐ |
| Participants | People with stroke due to MCA embolic occlusion within 6 hours; further information being sought |
| Interventions | Intra‐arterial urokinase versus placebo |
| Outcomes | Unknown |
| Starting date | April 2002 |
| Contact information | Prof Ogawa aogawa@iwate‐med.ac.jp |
| Notes | ‐ |
PROACT III.
| Trial name or title | PROACT III |
| Methods | ‐ |
| Participants | Further information awaited |
| Interventions | Unknown |
| Outcomes | Unknown |
| Starting date | Unknown |
| Contact information | Unknown |
| Notes | Believed withdrawn by pharmaceutical company (Abbott) and unlikely to start as at March 2003 |
WAKE‐UP 2011.
| Trial name or title | Wake Up stroke |
| Methods | DWI‐FLAIR mismatch to select participants for thrombolysis with acute ischaemic stroke without known time of onset |
| Participants | Acute ischaemic stroke with DWI‐FLAIR mismatch |
| Interventions | rt‐PA 9 mg/kg, 10% as bolus, rest over 1 hour |
| Outcomes | 90 day mRS |
| Starting date | 2013 |
| Contact information | ‐ |
| Notes | ‐ |
WASSABI.
| Trial name or title | WASSABI |
| Methods | Unknown |
| Participants | People who wake up with stroke |
| Interventions | Unknown |
| Outcomes | Unknown |
| Starting date | Unknown |
| Contact information | ‐ |
| Notes | Not much is known about this trial |
Yamanouchi iv rt‐PA.
| Trial name or title | Unknown |
| Methods | ‐ |
| Participants | Unknown |
| Interventions | Intravenous rt‐PA versus placebo |
| Outcomes | Unknown |
| Starting date | Unknown |
| Contact information | Unknown |
| Notes | ‐ |
CT: computed tomography CTP: computed tomography perfusion DWI: diffusion weighted imaging FLAIR: fluid attenuated inversion recovery i.a.: intra‐arterial i.v.: intravenous MCA: middle cerebral artery NIHSS: National Institutes of Health Stroke Scale PWI: perfusion imaging RCT: randomised controlled trial rt‐PA: recombinant tissue plasminogen activator SICH: symptomatic intracranial haemorrhage Tmax: a method of quantifying a cerebral perfusion abnormality
Differences between protocol and review
None in principle. The original protocol was written in 1992, since when Cochrane Reviews have become more complex, data analyses have been refined and substantially more data have become available for this review. Notwithstanding, the basic principles of this review are unchanged from the original protocol.
Contributions of authors
Four review authors contributed to the collection of data in previous versions, including non‐English language publication literature searching. Dr Eivind Berge undertook further detailed literature searching for the 2003 update. Both Dr Veronica Murray and Prof Joanna Wardlaw reviewed all new trials since 2003 and extracted, verified and entered new data for the 2009 update. Dr Veronica Murray also cross‐checked all previously extracted data. Both Prof Joanna Wardlaw and Dr Veronica Murray checked additional publications since 2003 of trials already included in the 2003 version for additional new information. All review authors also contributed to interpretation of the data. Prof Joanna Wardlaw drafted the review and the other review authors contributed to the critical revision of that review and final approval of the version to be published.
Sources of support
Internal sources
Department of Clinical Neurosciences, University of Edinburgh, Scotland, UK.
External sources
Scottish Office Chief Scientist's Office for the Cochrane Stroke Group, UK.
Previous versions of this review were supported by the Stroke Association and the UK Medical Research Council, UK.
Previous updates were supported in part by a grant from the NHS Health Technology Assessment (HTA) Programme (Grant 98/02/02), UK.
The Norwegian Research Council supported Dr Eivind Berge for a year in Edinburgh, Norway.
The Swedish Heart‐Lungfund, AFA and The Marianne and Marcus Wallenberg Foundation supported Dr Veronica Murray, Sweden.
-
The present update of the review was supported by a grant from the UK National Institutes of Health Research Cochrane Review Incentive Scheme 2013, UK.
Grant to help update the review
Declarations of interest
The Division of Clinical Neurosciences at the University of Edinburgh had a collaborative project with Boehringer Ingelheim (UK) to establish a research magnetic resonance scanner, through the UK Research Councils Joint Research Equipment Initiative in 1997. For this, the Division received a grant from Boehringer Ingelheim, manufacturers of rt‐PA in Europe, towards the purchase of the scanner. Further details of competing interests are listed on the Division's web site (www.dcn.ed.ac.uk).
The Division of Clinical Neurosciences at the University of Edinburgh are co‐ordinating the Third International Stroke Trial (IST3 2012) of intravenous tissue Plasminogen Activator within six hours of acute ischaemic stroke. Prof Joanna Wardlaw is the Imaging Principal Investigator of this trial, Dr Veronica Murray is the Swedish National Co‐ordinator and Dr Eivind Berge is the Norwegian National Co‐ordinator for IST‐3. The start‐up phase was funded by the UK Stroke Association and PPP Foundation, with a limited supply of drug and placebo for the first part of the start‐up phase from Boehringer Ingelheim; the main trial is funded by the UK Medical Research Council.
Prof Joanna Wardlaw received payment from Boehringer Ingelheim for reading scans for ECASS 3 on a cost‐per‐scan basis up to 2008. She was/is on the Steering Committees of MAST‐I 1995, IST3 2012, and contributed to the design of ECASS 3 2008 (first Steering Committee meeting and design of scan reading). Boehringer Ingelheim applied for an extension to the licence for rt‐PA from three to 4.5 hours on the basis of the ECASS 3 2008 result and supporting data, such as individual patient data analyses and the Cochrane review.
The review was assembled, analysed and reported independent of any sponsor or pharmaceutical company.
Edited (no change to conclusions), comment added to review
References
References to studies included in this review
Abe 1981 {published and unpublished data}
- Abe T. Oral urokinase: absorption, mechanisms of fibrinolytic enhancement and clinical effect on cerebral thrombosis. Folia Haematologica ‐ Leipzig 1986;113(1‐2):121‐36. [PubMed] [Google Scholar]
- Abe T. Thrombolytic therapy for cerebral infarction. International Angiology 1984;3:359‐65. [Google Scholar]
- Abe T, Kazama M, Naito I, Kinoshita T, Sasaki K. Effect of oral urokinase and its clinical application. The New Instanbul Contribution to Clinical Science 1982;13(3):161‐70. [PubMed] [Google Scholar]
- Abe T, Kazama M, Naito I, Ueda M, Tanaka T, Higuchi H, et al. Clinical evaluation for efficacy of tissue cultured urokinase (TCUK) on cerebral thrombosis by means of multi‐center double‐blind study. Blood & Vessel 1981;12:321‐41. [Google Scholar]
- Naito I. Basic and clinical studies on oral urokinase and lysyl‐plasminogen. Rinsho Ketsueki 1984;25(7):1027‐35. [PubMed] [Google Scholar]
ASK 1996 {published data only}
- Davis S, Donnan G, Mitchell P, Fitt G, Chambers B, Gates PC, et al. Australian streptokinase trial: clinical and CT outcome predictors. Cerebrovascular Diseases 1996;6(Suppl 2):128. [Google Scholar]
- Donnan G, Davis S. ASK trial: predictors of good outcome. Cerebrovascular Diseases 1996;6(3):183. [Google Scholar]
- Donnan G, Davis S. ASK trial: risk factors for poor outcome. Cerebrovascular Diseases 1996;6(3):182. [Google Scholar]
- Donnan GA, Davis SM, Chambers BR, Gates PC, Hankey GJ, McNeil JJ, et al. Streptokinase for acute ischemic stroke with relationship to time of administration. JAMA 1996;276(12):961‐6. [PubMed] [Google Scholar]
- Donnan GA, Davis SM, Chambers BR, Gates PC, Hankey GJ, McNeil JJ, et al. Trials of streptokinase in severe acute ischaemic stroke. Lancet 1995;345(8949):578‐9. [PubMed] [Google Scholar]
- Donnan GA, Davis SM, Chambers BR, Gates PC, Hankey GJ, Stewart‐Wynne EG, et al. Australian Streptokinase Trial (ASK). In: Zoppo GJ, Mori E, Hache W editor(s). Thrombolytic therapy in acute ischaemic stroke II. Springer‐Verlag, 1993:80‐5. [Google Scholar]
- Donnan GA, Hommel M, Davis SM, McNeil JJ, for the steering committees of the ASK and MAST‐E trials. Streptokinase in acute ischemic stroke. Lancet 1995;346(8966):56. [DOI] [PubMed] [Google Scholar]
- Gilligan A, Markus R, Read S, Srikanth V, Hirano T, Chambers B, et al. Early CT changes do not predict parenchymal haemorrhage following streptokinase therapy in acute stroke. Abstracts from the 6th International Symposium on Acute Stroke and Thrombolytic Therapy; 25‐29 November 2000; Hamilton Island, Australia. 2001.
- Gilligan AK, Markus R, Read SJ, Srikanth V, Fitt G, Chambers BR, et al. Early CT changes do not predict parenchymal haemorrhage following streptokinase therapy in acute stroke. Stroke 2001;32(1):370. [Google Scholar]
- Yasaka M, O'Keefe GJ, Chambers BR, Davis SM, Infield B, O'Malley H, et al. Streptokinase in acute stroke: effect on reperfusion and recanalisation. Neurology 1998;50(3):626‐32. [DOI] [PubMed] [Google Scholar]
Atarashi 1985 {published and unpublished data}
- Atarashi J, Ohtomo E, Araki G, Itoh E, Togi H, Matsuda T. Clinical utility of urokinase in the treatment of acute stage cerebral thrombosis: Multi‐center double blind study in comparison with placebo. Clinical Evaluation 1985;13:659‐709. [Google Scholar]
ATLANTIS A 2000 {published and unpublished data}
- Albers GW, Clark WM for the ATLANTIS Study Investigators. The ATLANTIS rtPA (alteplase) acute stroke trial: final results. Cerebrovascular Diseases 1999;9(Suppl 1):126. [Google Scholar]
- Clark WM, Albers GW for the ATLANTIS Stroke Study Investigators. The ATLANTIS rt‐PA (alteplase) acute stroke trial ‐ final results. Stroke 1999;30:234. [Google Scholar]
- Clark WM, Albers GW, Madden KP, Hamilton S. The rtPA (alteplase) 0‐ to 6‐ hour acute stroke trial, part A (A027g). Results of a double‐blind, placebo‐controlled, multicentre study. Thrombolytic Therapy in Acute Ischaemic Stroke Study Investigators. Stroke 2000;31(4):811‐6. [DOI] [PubMed] [Google Scholar]
- Marks M, Holmgren EB, Fox AJ, Patel S, Kummer R, Froehlich J. Evaluation of early computed tomographic findings in acute ischaemic stroke. Stroke 1999;30:389‐92. [DOI] [PubMed] [Google Scholar]
ATLANTIS B 1999 {published and unpublished data}
- Albers GW, Clark WM for the ATLANTIS Study Investigators. The ATLANTIS rtPA (alteplase) acute stroke trial: final results. Cerebrovascular Diseases 1999;9(Suppl 1):126. [Google Scholar]
- Albers GW, Clark WM, Madden KP, Hamilton SA. The ATLANTIS trial: Results for patients treated within three hours of stroke onset. Alteplase Thrombolysis for Acute Noninterventional Therapy in Ischemic Stroke.. Stroke 2002;33(2):493‐6. [DOI] [PubMed] [Google Scholar]
- Clark WM, Albers GW for the ATLANTIS Stroke Study Investigators. The ATLANTIS rt‐PA (alteplase) acute stroke trial ‐ final results. Stroke 1999;30:234. [Google Scholar]
- Clark WM, Wissman S, Albers GW, Jhamandas JH, Madden KP, Hamilton S, et al. Recombinant tissue‐type plasminogen activator (alteplase) for ischaemic stroke 3 to 5 hours after symptom onset. The ATLANTIS Study: a randomised controlled trial. JAMA 1999;282(21):2019‐26. [DOI] [PubMed] [Google Scholar]
- Kent D, Ruthazer R, Selker HP. Do some patients benefit from rt‐PA treatment for acute ischaemic stroke even when treated more than 3 hours after the onset of symptoms?. Abstracts of the 7th International Symposium on Thrombolysis and Acute Ischaemic Stroke; 2002 May 27‐28; Lyon. 2002:79.
- Kent DM, Price L, Selker HP. Are asymptomatic intracranial hemorrhages really innocuous?. Cerebrovascular Diseases 2003;16(Suppl 4):34. [Google Scholar]
- Kent DM, Ruthazer R, Selker HP. Are some patients likely to benefit from recombinant tissue‐type plasminogen activator for acute ischemic stroke even beyond 3 hours from symptom onset?. Stroke 2003;34(2):464‐7. [DOI] [PubMed] [Google Scholar]
- Marks M, Holmgren EB, Fox AJ, Patel S, Kummer R, Froehlich J. Evaluation of early computed tomographic findings in acute ischaemic stroke. Stroke 1999;30(2):389‐92. [DOI] [PubMed] [Google Scholar]
AUST 2005 {published data only}
- Davis SM, Donnan GA, Gerraty RP, Mitchell PJ, Fitt G, Bladin CF, et al. Australian Urokinase Stroke Trial. Cerebrovascular Diseases 1996;6(3):188. [Google Scholar]
- Donnan G. Australian urokinase stroke trial ‐ AUST. www.strokecenter.org/trials/list 2001.
- Donnan GA, Davis SM, Gerraty RP, Mitchell PJ, Fitt G, Bladin CF, et al. Australian urokinase stroke trial (AUST). Cerebrovascular Diseases 1996;6(Suppl 2):25. [Google Scholar]
- Gerraty RP, Mitchell PJ, Fitt G, Tress BM, Donnan GA, Davis SM. Intra‐arterial thrombolysis for basilar artery occlusion: Australian urokinase stroke study phase II results. Programme and Abstract Book of the Stroke Society of Australasia; 1995 Annual Scientific Meeting; 1995 Oct 4‐6; Melbourne. 1995.
- Macleod M. Preliminary results of the AUST/POST study. Internal Medicine Journal 2004;34(1‐2):A1. [Google Scholar]
- Macleod MR, Davis SM, Mitchell JP, Gerraty RP, Fitt G, Hankey GJ, et al. Results of a multicentre, randomised controlled trial of intra‐arterial urokinase in the treatment of acute posterior circulation ischaemic stroke. Cerebrovascular Diseases 2005;20(1):12‐7. [DOI] [PubMed] [Google Scholar]
- Mitchell PJ, Gerraty RP, Donnan GA, Fitt G, Tress BM, Thomson KR, et al. Thrombolysis in the vertebrobasilar circulation: the Australian urokinase stroke trial. Cerebrovascular Diseases 1997;7(2):94‐9. [Google Scholar]
- National Stroke Research Institute. Preliminary results of the Australian urokinase stroke study/posterior circulation thrombolysis study. Powerpoint presentation.
Chen 2000 {published data only}
- Intravenous thrombolysis with urokinase for acute cerebral infarctions. The study group of a 5 year National Project of the People's Republic of China, conducted by Qingtang Chen and Maolin He. Chinese Journal of Neurology 2002.
- Chen Q‐T. Intravenous thrombolysis with urokinase for acute cerebral infarctions. Hong Kong Medical Journal 2001;7(4):7. [Google Scholar]
- Cooperating Group for National 95's Project. Intravenous thrombolysis with urokinase for acute cerebral infarctions. Chinese Journal of Neurology 2002;35(4):210‐3. [Google Scholar]
- Wang Z‐X. Intravenous thrombolysis with urokinase for acute cerebral infarctions. Journal of Clinical Neuroscience 2004;11 Suppl 1:S7. [Google Scholar]
DEDAS 2006 {published and unpublished data}
- Conroy MM. Bat saliva drug may widen treatment window for stroke. www.medscape.com (accessed 17 February 2005).
- Fiebach JB, Al‐Rawi Y, Wintermark M, Furlan AJ, Rowley HA, Lindsten A, et al. Vascular occlusion as an imaging biomarker for selecting acute ischemic stroke patients for treatment with desmoteplase. Stroke 2012;43(2):Abst. 2595. [DOI] [PubMed] [Google Scholar]
- Fiebach JB, Al‐Rawi Y, Wintermark M, Furlan AJ, Rowley HA, Lindsten A, et al. Vascular occlusion enables selecting acute ischemic stroke patients for treatment with desmoteplase. Stroke 2012;43(6):1561‐6. [DOI] [PubMed] [Google Scholar]
- Furlan AJ. Dose Escalation of Desmoteplase in Acute Ischemic Stroke (DEDAS). www.clinicaltrials.gov/ct2/show/NCT00638248?term=Dose+Escalation+of+Desmoteplase+in+Acute+Ischemic+Stroke+%28DEDAS%29&rank=1 2003 (accessed July 6th 2014).
- Furlan AJ, Eyding D, Albers GW, Al‐Rawi Y, Lees KR, Rowley HA, et al. Dose Escalation of Desmoteplase for Acute Ischemic stroke (DEDAS): evidence of safety and efficacy 3 to 9 hours after stroke onset. Stroke 2006;37(5):1227‐31. [DOI] [PubMed] [Google Scholar]
- Hacke W, Furlan F, Jakob N. Desmoteplase in acute stroke ‐ an integrated analysis of two phase II clinical trials. International Journal of Stroke 2006;1 Suppl 1:36‐7. [Google Scholar]
- Pugsley M, Peterson K‐U, Eyding D. Recombinant desmodus salivary plasminogen activator (desmoteplase) in acute ischemic stroke. Stroke 2006;37(2):656. [Google Scholar]
- Warach S, Al‐Rawi Y, Furlan AJ, Fiebach JB, Wintermark M, Lindsten A, et al. Refinement of the magnetic resonance diffusion‐perfusion mismatch concept for thrombolytic patient selection. Stroke 2012;43(9):2313‐8. [DOI] [PubMed] [Google Scholar]
- Warach S, Al‐Rawi Y, Furlan AJ, Fiebach JB, Wintermark M, Lindstén A, et al. Refinement of the MR diffusion‐perfusion mismatch concept for thrombolytic patient selection: insights from the Desmoteplase in Acute Stroke Trials. Stroke 2012;43(2):Abst.2600. [DOI] [PubMed] [Google Scholar]
DIAS 2005 {published and unpublished data}
- Debens T. Technology evaluation: desmoteplase, PAION/Forest. Current Opinion on Molecular Therapeutics 2004;6(5):567‐74. [PubMed] [Google Scholar]
- Fiebach JB, Al‐Rawi Y, Wintermark M, Furlan AJ, Rowley HA, Lindsten A, et al. Vascular occlusion as an imaging biomarker for selecting acute ischemic stroke patients for treatment with desmoteplase. Stroke 2012;43:A2595. [DOI] [PubMed] [Google Scholar]
- Fiebach JB, Al‐Rawi Y, Wintermark M, Furlan AJ, Rowley HA, Lindstén A, et al. Vascular occlusion enables selecting acute ischemic stroke patients for treatment with desmoteplase. Stroke 2012;43(6):1561‐6. [DOI] [PubMed] [Google Scholar]
- Hacke W. Desmoteplase in Acute Ischemic Stroke (DIAS). clinicaltrials.gov/ct2/show/NCT00638781?term=Desmoteplase+in+Acute+Ischemic+Stroke+%28DIAS%29&rank=1 (accessed July 6th 2014).
- Hacke W. Results of the phase II study DIAS. Powerpoint presentation.
- Hacke W. The DIAS study: results of a phase II, MRI‐based dose finding study of desmoteplase in acute stroke. XVIIth International Congress on Fibrinolysis and Proteolysis; 2004 Mar 21‐15; Melbourne. 2004.
- Hacke W for the DIAS and DEDAS Steering Committees and Investigators. The results of the joint analysis of two phase II trials on desmoteplase in acute ischemic stroke with treatment 3 to 9 hours after stroke onset. Cerebrovascular Diseases 2005;19(Suppl 2):69. [Google Scholar]
- Hacke W, Albers G, Al‐Rawi Y, Bogousslavsky J, Davalos A, Eliasziw M, et al. The Desmoteplase In Acute ischemic Stroke trial (DIAS): A phase II MRI‐based 9‐hour window acute stroke thrombolysis trial with intravenous desmoteplase. Stroke 2005;36(1):66‐73. [DOI] [PubMed] [Google Scholar]
- Hacke, W. Desmoteplase in acute ischemic stroke (DIAS). The American Stroke Association 28th International Stroke Conference; 2003 Feb 13‐15; Phoenix. 2003:CTP14.
- Lees KR, Hacke W, Furlan AJ. Combined safety analysis of desmoteplase in acute ischemic stroke 3 to 9 hours after stroke symptom onset from two independent Phase II studies (DIAS and DEDAS). Journal of Neurological Sciences 2005;238 Suppl 1:S62. [Google Scholar]
- PAION. DIAS. www.paion.de/.
- Kummer R, Albers G, Mori E, DIAS Steering Committees. The Desmoteplase in Acute Ischemic Stroke (DIAS) clinical trial program. International Journal of Stroke 2012;7(7):589‐96. [DOI] [PubMed] [Google Scholar]
- Warach S for the DIAS Steering Committee. Early reperfusion related to clinical response in DIAS: phase II, randomized, placebo‐controlled dose finding trial of IV desmoteplase 3‐9 hours from onset in patients with diffusion‐perfusion mismatch. www.abstractsonline.com/arch/RecordPrintView.aspx?LookupKey=12345&RecordID=11015 (accessed 7 August 2007).
- Warach S for the DIAS Steering Committee. Randomized, placebo‐controlled trial of IV desmoteplase 3‐9 hours from stroke onset in patients with diffusion‐perfusion mismatch: early reperfusion related to dose and therapeutic response. Proceedings of the International Society of Magnetic Resonance Medicine 2004;11:409. [Google Scholar]
- Warach S, Al‐Rawi Y, Furlan AJ, Fiebach JB, Wintermark M, Lindstén A, et al. Refinement of the MR diffusion‐perfusion mismatch concept for thrombolytic patient selection: insights from the Desmoteplase in Acute Stroke Trials. Stroke 2012;43(9):A2600. [DOI] [PubMed] [Google Scholar]
- Warach S, Al‐Rawi Y, Furlan AJ, Fiebach JB, Wintermark M, Lindstén A, et al. Refinement of the magnetic resonance diffusion‐perfusion mismatch concept for thrombolytic patient selection: insights from the desmoteplase in acute stroke trials. Stroke 2012;43(9):2313‐8. [DOI] [PubMed] [Google Scholar]
DIAS 2 2008 {published and unpublished data}
- Eng P, Spielhofen A. DIAS‐2. Desmoteplase in acute ischemic stroke‐2. www.strokecenter.org/trials/TrialDetail.aspx?tid=515 (accessed 19 July 2007).
- Fiebach JB, Al‐Rawi Y, Wintermark M, Furlan AJ, Hacke W, Rowley HA, et al. Vascular occlusion as imaging biomarker in selecting acute ischaemic stroke patients for treatment with desmoteplase. Cerebrovascular Diseases 2011;31(Suppl 2):165. [Google Scholar]
- Fiebach JB, Al‐Rawi Y, Wintermark M, Furlan AJ, Rowley HA, Lindstén A, et al. Vascular occlusion as an imaging biomarker for selecting acute ischemic stroke patients for treatment with desmoteplase. Stroke 2012;43(6):A2595. [DOI] [PubMed] [Google Scholar]
- Fiebach JB, Al‐Rawi Y, Wintermark M, Furlan AJ, Rowley HA, Lindstén A, et al. Vascular occlusion enables selecting acute ischemic stroke patients for treatment with desmoteplase stroke. Stroke 2012;43(6):1561‐6. [DOI] [PubMed] [Google Scholar]
- Forest Laboratories. Study of Desmoteplase (International Nonproprietary Name [INN]) in Acute Ischemic Stroke (DIAS‐2). www.clinicaltrials.gov/ct2/show/NCT00111852?term=DIAS‐2&rank=1 (Accessed July 6th 2014).
- Hacke W. DIAS‐2: No benefit of desmoteplase in acute ischemic stroke. www.medscape.com/viewarticles/557663_print (accessed 23 February 2008).
- Hacke W, Furlan A. The ongoing phase III study of desmoteplase in acute ischemic stroke. International Journal of Stroke 2006;1 Suppl 1:128‐9. [Google Scholar]
- Hacke W, Furlan A. The ongoing phase III study of desmoteplase in acute ischemic stroke. International Journal of Stroke 2006;1 Suppl 2:46. [Google Scholar]
- Hacke W, Furlan A, for the DIAS‐2 Investigators. Results from the phase III study of Desmoteplase in Acute Ischemic Stroke trial 2 (DIAS‐2). Cerebrovascular Diseases 2007;23(Suppl 2):54. [Google Scholar]
- Hacke W, Furlan AJ, Al‐Rawi Y, Davalos A, Fiebach JB, Gruber F, et al. Intravenous desmoteplase in patients with acute ischaemic stroke selected by MR perfusion‐diffusion weighted imaging or perfusion CT (DIAS‐2): a prospective, randomised, double‐blind, placebo‐controlled study. Lancet Neurology 2008; Vol. 18 December 2008 online. [DOI: 10.1016/6/51474-4422(08)70267-9] [DOI] [PMC free article] [PubMed]
- Hacke W, Furlan AJ, Al‐Rawi Y, Davalos A, Fiebach JB, Gruber F, et al. Intravenous desmoteplase in patients with acute ischaemic stroke selected by MRI perfusion‐diffusion weighted imaging or perfusion CT (DIAS‐2): a prospective, randomised, double‐blind, placebo‐controlled study. Lancet Neurology 2009;8(2):141‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MD. Desmoteplase and imaging science. Lancet Neurology 2009;8(2):126‐7. [DOI] [PubMed] [Google Scholar]
- Jeffrey S. DIAS‐2 revisited: development of desmoteplase for stroke to continue. www.medscape.com/viewarticle/565145 (accessed 11 March 2008).
- Jeffrey S. DIAS‐2 revisited: development of desmoteplase for stroke to continue. www.medscape.com/viewarticle/565145_print (accessed 15 November 2007).
- Jeffrey S. DIAS‐2: No benefit of desmoteplase in acute ischemic stroke. www.medscape.com/viewarticle/557663_print (accessed 29 January 2008).
- Jeffrey S. DIAS‐2: patient enrolment resumes in trial of desmoteplase in stroke. www.medscape.com/viewarticle/547120 (accessed 25 July 2007).
- Liebeskind DS. Reversing stroke in the 2010s. Lessons from Desmoteplase In Acute ischemic Stroke‐2 (DIAS‐2). Stroke 2009;40(9):3156‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAION AG, Forest Laboratories Inc. DIAS 2. Desmoteplase (INN) in acute ischemic stroke. Assessing treatment within 3‐9 hours after onset of stroke symptoms. paiondias.01kunden.net (accessed 25 July 2007).
- Warach S. Clinical benefit of desmoteplase treatment in patients with moderate to severe stroke: further results of the DIAS‐2 trial. Proceedings of the International Stroke Conference; 2008 Feb 20‐22; New Orleans. American Stroke Association, 2008:LB P2.
- Warach S, Al‐Rawi Y, Furlan AJ, Fiebach JB, Wintermark M, Lindstén A, et al. Refinement of the MR diffusion‐perfusion mismatch concept for thrombolytic patient selection: insights from the Desmoteplase in Acute Stroke Trials. Stroke 2012;43(9):A2600. [DOI] [PubMed] [Google Scholar]
- Warach S, Al‐Rawi Y, Furlan AJ, Fiebach JB, Wintermark M, Lindstén A, et al. Refinement of the magnetic resonance diffusion‐perfusion mismatch concept for thrombolytic patient selection. Stroke 2012;43(9):2318‐8. [DOI] [PubMed] [Google Scholar]
ECASS 1995 {published and unpublished data}
- Boysen G, ECASS Study Group. European Cooperative Acute Stroke Study (ECASS): (rt‐PA ‐ thrombolysis in acute stroke) study design and progress report. European Journal of Neurology 1995;1(3):213‐9. [DOI] [PubMed] [Google Scholar]
- Ciccone A, Motta C, Aritzu E, Candelise L. Letter to the editor. JAMA 1996;275:983‐4. [DOI] [PubMed] [Google Scholar]
- Dávalos A, Toni D, Iweins F, Lesaffre E, Bastianello S, Castillo J, et al. Neurological deterioration in acute ischaemic stroke: potential predictors and associated factors in the European Cooperative Acute Stroke Study (ECASS) I. Stroke 1999;30(12):2631‐6. [DOI] [PubMed] [Google Scholar]
- ECASS Study Group. Very early identification of lacunar infarcts in the ECASS Study. Cerebrovascular Diseases 1997;7(Suppl 4):29. [Google Scholar]
- Fieschi C, Argentino C, Fiorelli M, Mau J, Roine RO, Willig V, et al. Frequency and consequences of medical complications of ischaemic stroke. The Challenge of Stroke. Lancet Conference; 1999 Oct 15‐16; Montreal. 1999:35.
- Fiorelli M, Argentino C, Falcou A, Meier D, Kaste M, Hacke W, et al. Natural history of ischaemic stroke in ECASS 1 and ECASS II placebo cohorts. Cerebrovascular Diseases 1999;9(Suppl 1):14. [Google Scholar]
- Fiorelli M, Bastianello S, Kummer R, Zoppo GJ, Larrue V, Lesaffre E, et al. Hemorrhagic transformation within 36 hours of a cerebral infarct. Relationships with early clinical deterioration and three month outcome in the European Cooperative Acute Stroke Study I (ECASS I) cohort. Stroke 1999;30(11):2280‐4. [DOI] [PubMed] [Google Scholar]
- Fiorelli M, Busse O, Mau J, Bozzao L, Bastianello S, Fieschi C, et al. Cortical extension of the infarction at 24 hours in patients with isolated hypodensity of the lentiform nucleus on 1‐6 hour CT: Incidence, predictors and outcome in the ECASS 1 cohort. Cerebrovascular Diseases 1998;8(Suppl 4):19. [Google Scholar]
- Fisher M, Pessin MS, Furlan AJ. ECASS: Lessons for future thrombolytic stroke trials. JAMA 1996;274(13):1058‐9. [PubMed] [Google Scholar]
- Hacke W, Bluhmki E, Steiner T, Tatlisumak T, Mahagne M‐H, Sacchetti M‐L, et al. Dichotomized efficacy end points and global end‐point analysis applied to the ECASS intention‐to‐treat data set. Post hoc analysis of ECASS 1. Stroke 1998;29(10):2073‐5. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Fieschi C. Reply to letter to the editor. JAMA 1996;275:984‐5. [Google Scholar]
- Hacke W, Kaste M, Fieschi C, Toni D, Lesaffre E, Kummer R, et al. Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke. The European Cooperative Acute Stroke Study (ECASS). JAMA 1995;274(13):1017‐25. [PubMed] [Google Scholar]
- Hacke W, Toni D, Steiner T, Kaste M, Kummer R, Fieschi C, et al. rt‐PA in acute ischaemic stroke ‐ results from the ECASS three hour cohort. Stroke 1997;28:272. [Google Scholar]
- Kaste M for the ECASS Study Group. Potential contributors to bad functional outcome or death in European Cooperative Acute Stroke Study (ECASS). Cerebrovascular Diseases 1996;6(3):181. [Google Scholar]
- Kaste M, Fieschi C, Hacke W, Lesaffre E, Verstraete M, Frohlich J. The European Cooperative Acute Stroke Study (ECASS). In: Zoppo G, Mori E, Hacke W editor(s). Thrombolytic therapy in acute ischaemic stroke. Berlin: Springer Verlag, 1993:66‐71. [ISBN 3‐540‐56442‐X] [Google Scholar]
- Kaste M, Mau J, Bluhmki E, Zoppo GJ, Orgogozo J‐M, Overgaard K, et al. A post‐hoc risk/benefit assessment for mortality and handicap in 615 ECASS patients randomised and treated. Upsala Journal of Medical Sciences 1997; Vol. Suppl 53.
- Kaste M, Mau J, Bluhmki E, Zoppo GJ, Orgogozo J‐M, Overgaard K, et al. Risk/benefit assessment for mortality and handicap among 615 patients randomised and treated in ECASS. European Journal of Neurology 1998;5 Suppl 1:S20. [Google Scholar]
- Kaste M, Mau J, Bluhmki E, Zoppo GJ, Orgogozo J‐M, Overgaard K, et al. Risk/benefit assessment for mortality and handicap in ECASS randomised and treated patients: a post‐hoc analysis. Stroke 1998;29:288. [Google Scholar]
- Kaste M, Overgaard K, Zoppo GJ, Kummer R, Orgogozo J‐M, Wahlgren N‐G, et al. Potential contributors for good vs bad functional outcome or death in European Cooperative Acute Stroke Study (ECASS). Cerebrovascular Diseases 1996;6(Suppl 2):129. [Google Scholar]
- Larrue V, Kummer R, Zoppo GJ, Bluhmki E. Hemorrhagic transformation in acute ischaemic stroke. Potential contributing factors in the European Cooperative Acute Stroke Study. Stroke 1997;28(5):957‐60. [DOI] [PubMed] [Google Scholar]
- Larrue V, Kummer R, Hoxter G, Bluhmki E, Manelfe G, Regesta G, et al. Predictors of hemorrhagic transformation in the ECASS trial. Cerebrovascular Diseases 1996;6(3):181. [Google Scholar]
- Manelfe C, Larrue V, Kummer R, Bozzao L, Ringleb P, Bastianello S, et al. Association of hyperdense middle cerebral artery sign with clinical outcome in patients treated with tissue plasminogen activator. Stroke 1999;30(4):769‐72. [DOI] [PubMed] [Google Scholar]
- Mau J, Jayavel S, Yong M. Would first‐day body temperature dynamics contribute independently to long‐term handicap after acute ischemic stroke?. Cerebrovascular Diseases 2006;21(Suppl 4):55. [Google Scholar]
- Mau J, Jayavel SD. First‐day body temperature dynamics ‐ a clinician's tool for monitoring penumbral tissue transformation and updating prognosis after ischemic stroke?. Cerebrovascular Diseases 2007;23(Suppl 2):96. [Google Scholar]
- Mau J, Jayavel SD, Yong M. A robust prognostic advantage with "late" rising body temperature profiles in the ECASS‐I data base. International Journal of Stroke 2006;1 Suppl 2:30. [Google Scholar]
- Mau J, Kaste M, Bluhmki E, Zoppo G, Orgogozo JM, Overgaard K, et al. A risk/benefit assessment of 1.1 mg/kg iv rt‐PA in 615 acute stroke patients of ECASS. Cerebrovascular Diseases 1997;7(Suppl 4):1. [Google Scholar]
- Mau J, Kaste M, Zoppo GJ, Kummer R, Orgogozo JM, Overgaard K, et al. Risk profile comparison in 615 patients randomised to either 1.1 mg/kg intravenous rt‐PA or placebo of ECASS 1. Stroke 1999;30:247. [Google Scholar]
- Mau J, Yong M. Beta‐blockers with thrombolysis? Cautioning evidence from the ECASS‐1 trial. Stroke 2004;35(6):e189. [Google Scholar]
- Pantano P, Caramina F, Bozzao L, Dieler C, Kummer R. Delayed increase in infarct volume after cerebral ischaemia. Correlations with thrombolytic treatment and clinical outcome. Stroke 1999;30(3):502‐7. [DOI] [PubMed] [Google Scholar]
- Steiner T, Bluhmki E, Kaste M, Toni D, Trouillas P, Kummer R, et al. The ECASS 3‐hour cohort. Secondary analysis of ECASS data by time stratification. Cerebrovascular Diseases 1998;8(4):198‐203. [DOI] [PubMed] [Google Scholar]
- The ECASS Trial. Predictors of good outcome ‐ ECASS trial. Cerebrovascular Diseases 1996;6(3):182. [Google Scholar]
- Kummer R, Allen KL, Holle R, Bozzao L, Bastianello S, Manelfe C, et al. Acute stroke ‐ usefulness of early CT findings before thrombolytic therapy. Radiology 1997;205(2):327‐33. [DOI] [PubMed] [Google Scholar]
- Kummer R, Bozzao L. Letter to the editor. JAMA 1996;275:13. [DOI] [PubMed] [Google Scholar]
- Willig V, Hoxter G, Steiner T, Fiorelli M, Kummer R, Hacke W, et al. Causes of death in ECASS. Cerebrovascular Diseases 1996;6(Suppl 2):129. [Google Scholar]
- Willig V, Pawelec D, Mau J, Hoxter G, Hacke W. Heparin effects in the European Cooperative Acute Stroke Study. Stroke 1997;28:272. [Google Scholar]
- Wood L. Letter to the editor. JAMA 1996;275:983. [Google Scholar]
- Yong M, Diener HC, Kaste M, Mau J. Characteristics of blood pressure profiles as predictors of long‐term outcome after acute ischemic stroke. Stroke 2005;36(12):2619‐25. [DOI] [PubMed] [Google Scholar]
- Yong M, Diener HC, Kaste M, Mau J. Effect of thrombolysis on relationship between blood pressure and stroke outcome. Cerebrovascular Diseases 2006;21(Suppl 4):38. [Google Scholar]
- Yong M, Diener HC, Kaste M, Mau J. Long‐term outcome as function of blood pressure in acute ischemic stroke and effects of thrombolysis. Cerebrovascular Diseases 2007;24(4):349‐54. [DOI] [PubMed] [Google Scholar]
- Yong M, Mau J. Comparison of blood pressure profiles between rt‐PA and placebo in the ECASS‐1 trial. Stroke 2004;35(6):e218. [Google Scholar]
ECASS 3 2008 {published and unpublished data}
- Bluhmki E, Chamorro A, Davalos A, Machni T, Sauce C, Wahlgren N, et al. Stroke treatment with alteplase given 3·0‐4·5 h after onset of acute ischaemic stroke (ECASS III): additional outcomes and subgroup analysis of a randomised controlled trial. Lancet Neurology 2009;8(12):1095‐102. [DOI] [PubMed] [Google Scholar]
- Boehringer Ingelheim Pharmaceuticals. Rt‐PA in the treatment of acute ischemic stroke. www.clinicaltrials.gov/ct2/show/NCT00153036?term=Rt‐PA+in+the+treatment+of+acute+ischemic+stroke&rank=1 2003 (accessed July 6th 2014).
- Cronin CA. Intravenous tissue plasminogen activator for stroke: a review of the ECASS III results in relation to prior clinical trials. Journal of Emergency Medicine 2010;38(1):99‐105. [DOI] [PubMed] [Google Scholar]
- Davis S, Donnan G. The ECASS III results and the tPA paradox. International Journal of Stroke 2009;4(1):17‐8. [DOI] [PubMed] [Google Scholar]
- Davis SM, Donnan GA. 4.5 Hours. The new time window for tissue plasminogen activator in stroke. Stroke 2009;40(6):2266‐7. [DOI] [PubMed] [Google Scholar]
- Fisher M, Hachinski V. European Cooperative Acute Stroke Study III. Support for and questions about a truly emerging therapy. Stroke 2009;40(6):2262‐3. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick AE, Noone I, O'Shea DD. Thrombolysis 3 to 4.5 hours after acute ischemic stroke (to the Editor). New England Journal of Medicine 2008;359(26):2840. [PubMed] [Google Scholar]
- Hacke W. A wider time window for regular i.v. lysis: towards the result of ECASS‐3. International Journal of Stroke 2008;3(Suppl 1):444. [Google Scholar]
- Hacke W, Baumelou B, Reess J, Bluhmki E. European cooperative acute stroke study 3 (ECASS 3): study design and progress report. European Journal of Neurology 2005;12 Suppl 2:183. [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after ischemic stroke. New England Journal of Medicine 2008;359(13):1317‐29. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Toni D. Thrombolysis 3 to 4.5 hours after acute ischemic stroke (the authors' reply). New England Journal of Medicine 2008;359(26):2841. [PubMed] [Google Scholar]
- Ingall TJ. Intravenous thrombolysis for acute ischemic stroke. Time is prime. Stroke 2009;40(6):2264‐5. [DOI] [PubMed] [Google Scholar]
- Kurth T, Tzourio C. Treating patients with ischemic stroke with tissue plasminogen activator in the 3.5‐ to 4‐hour window. Numbers support benefit but the message is to still go fast. Stroke 2009;40(7):2295‐6. [DOI] [PubMed] [Google Scholar]
- Lorenzano S, Correnti A, Toni D. Intravenous thrombolysis: results of the Italian Sits‐Most Study, ECASS III Study and SITS‐ISTR. Cerebrovascular Diseases 2008;26(Suppl 2):2‐3. [Google Scholar]
- Lyden P. Thrombolytic therapy for acute stroke ‐ not a moment to lose. New England Journal of Medicine 2008;359(13):1393‐5. [DOI] [PubMed] [Google Scholar]
- Morris DC. Thrombolysis 3 to 4.5 hours after acute ischemic stroke (to the Editor). New England Journal of Medicine 2008;359(26):2840‐1. [PubMed] [Google Scholar]
- Saver JL, Gornbein J, Grotta J, Liebeskind D, Lutsep H, Schwamm L, et al. Number needed to treat to benefit and to harm for intravenous tissue plasminogen activator therapy in the 3‐ to 4.5‐hour window. Joint outcome table analysis of the ECASS 3 Trial. Stroke 2009;40(7):2433‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamm LH, Fonarow GC. Thrombolysis 3 to 4.5 hours after acute ischemic stroke (to the Editor). New England Journal of Medicine 2008;359(26):2840. [PubMed] [Google Scholar]
- Torbey MT, Jauch E, Liebeskind DS, for the Acute Stroke Advisory Board of the National Stroke Association. Thrombolysis 3 to 4.5 hours after acute ischemic stroke. New England Journal of Medicine 2008;359(26):2839. [DOI] [PubMed] [Google Scholar]
- Uyttenbogaart M, Luijckx GJ, Kappelle LJ, Oostenbrugge RJ, Dippel DWJ, Stam J. Treatment of acute ischaemic stroke with intravenous thrombolysis: extension of the time window should not delay initiation of treatment. Nederlands Tijdschrift voor Geneeskunde 2008;152(49):2653‐5. [PubMed] [Google Scholar]
- Wahlgren N. Relation of blood pressure with outcomes after intravenous thrombolysis in the Third European Cooperative Acute Stroke Study. Stroke 2009;40(4):e107. [Google Scholar]
- Yong M, Kaste M. Association of characteristics of blood pressure profiles and stroke outcomes in the ECASS‐II trial. Stroke 2008;39(2):366‐72. [DOI] [PubMed] [Google Scholar]
ECASS II 1998 {published and unpublished data}
- Barer D. Letter to the editor. Lancet 1999;353:66‐7. [DOI] [PubMed] [Google Scholar]
- Berrouschot J, Barthel H, Hesse S, Knapp WH, Schneider D, Kummer R. Reperfusion and metabolic recovery of brain tissue and clinical outcome after ischaemic stroke and thrombolytic therapy. Stroke 2000;31(7):1545‐51. [DOI] [PubMed] [Google Scholar]
- Berrouschot J, Barthel H, Kummer R, Hesse S, Schneider D. Reperfusion after focal cerebral ischaemia does not increase the rate and severity of secondary haemorrhage. Cerebrovascular Diseases 1999;9(Suppl 1):95. [Google Scholar]
- Berrouschot J, Barthel H, Kummer R, Koester J, Knapp W, Schneider D. Thrombolysis with rt‐PA: ASPECT study. Stroke 1999;30:267. [Google Scholar]
- Brainin M, Matz K, Teuschl Y, Tatschl C, Yong M, Kaste M. The hidden burden of glucose pathway in acute stroke remains hidden. Stroke 2009;40:e3‐4. [DOI] [PubMed] [Google Scholar]
- Dzialowski I, Coutts S, Demchuk AM, Wunderlich O, Kent D, Hill MD, et al. Assessment of ischemic damage on early CT with and without the use of a quantitative score. Cerebrovascular Diseases 2004;17(5 Suppl):5. [Google Scholar]
- Dzialowski I, Hill MD, Coutts SB, Demchuk A, Kent DM, Wunderlich O, et al. Extent of ischemic edema on CT before thrombolysis: prognostic value of the Alberta Stroke Program Early CT Score in ECASS II. Stroke 2006;37(2):636. [DOI] [PubMed] [Google Scholar]
- Dzialowski I, Hill MD, Coutts SB, Demchuk AM, Kent DM, Wunderlich O, et al. Extent of early ischemic changes on computed tomography (CT) before thrombolysis: prognostic value of the Alberta Stroke Program Early CT Score in ECASS II. Stroke 2006;37(4):973‐8. [DOI] [PubMed] [Google Scholar]
- Dávalos A on behalf of the ECASS II Spanish Group. Fibrinolytic therapy for acute stroke: the Spanish experience. Cerebrovascular Diseases 1999;9(Suppl 3):9‐15. [DOI] [PubMed] [Google Scholar]
- Dávalos A, Mostacero E, Castillo J, Larracoechea J, Ruiz‐Peris M on behalf of the ECASS II Spanish Group. Type of acute stroke care, stroke outcome and safety parameters in the ECASS II trial: the Spanish experience. Cerebrovascular Diseases 1999;9(Suppl 1):33. [Google Scholar]
- Fieschi C, Hacke W, Kaste M for the ECASS II Study Group. Final results of the second European‐Australasian Acute Stroke Study of intravenous alteplase in acute ischaemic stroke (ECASS II). Cerebrovascular Diseases 1999;9(Suppl 1):128. [Google Scholar]
- Fiorelli M, Argentino C, Falcou A, Meier D, Kaste M, Hacke W, et al. Natural history of ischaemic stroke in ECASS I and ECASS II placebo cohorts. Cerebrovascular Diseases 1999;9(Suppl 1):14. [Google Scholar]
- Ford G, Freemantle N, Lees KR, Raha S, Barer D, Jenkinson D, et al. ECASS II: Intravenous alteplase in acute ischaemic stroke. Lancet 1999;353(9146):65‐8. [DOI] [PubMed] [Google Scholar]
- Hacke W. The results of the European Co‐operative Stroke Study II (ECASS II), a placebo‐controlled, randomized study of thrombolytic therapy. European Journal of Neurology 1998;5 Suppl 3:S9. [Google Scholar]
- Hacke W for the ECASS II Study Group. Thrombolysis in acute ischaemic stroke: ECASS II. The Challenge of Stroke, Lancet Conference; 1999 Oct 15‐16; Montreal. 1999.
- Hacke W, Davalos A, Kummer R, Kaste M, Larrue V for the steering committee and authors of the ECASS II Study Group. Authors' reply. Lancet 1999;353(9146):67‐8. [Google Scholar]
- Hacke W, Kaste M, Fieschi C, Kummer R, Davalos A, Meier D, et al. Randomised double‐blind placebo‐controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II). Second European‐Australasian Acute Stroke Study Investigators. Lancet 1998;352(9136):1245‐51. [DOI] [PubMed] [Google Scholar]
- Jenkinson D. Letter to the editor. Lancet 1999;353(9146):67. [DOI] [PubMed] [Google Scholar]
- Larrue V, Bluhmki E, Muller A, Kummer R. Risk factors for haemorrhagic transformation in ECASS II. Stroke 2000;31:278. [Google Scholar]
- Larrue V, Kummer R, Müller A, Bluhmki E. Risk factors for severe hemorrhagic transformation in ischemic stroke patients treated with recombinant tissue plasminogen activator: a secondary analysis of the European‐Australasian Acute Stroke Study (ECASS II). Stroke 2001;32(2):438‐41. [DOI] [PubMed] [Google Scholar]
- Lees KR. Letter to the editor. Lancet 1999;353(9146):65‐6. [DOI] [PubMed] [Google Scholar]
- Messé SR, Fonarow GC, Smith EE, Kaltenbach L, Olson DM, Kasner SE, et al. Use of tissue‐type plasminogen activator before and after publication of the European Cooperative Acute Stroke Study III in Get With The Guidelines‐Stroke. Circulation. Cardiovascular Quality and Outcomes 2012;5(3):321‐6. [DOI] [PubMed] [Google Scholar]
- Raha S. Letter to the editor. Lancet 1999;353(9146):66. [Google Scholar]
- Savitz SI, Lew R, Bluhmki E, Hacke W, Fisher M. Rankin shift analysis of the NINDS and ECASS‐II trials. Stroke 2007;38(2):576. [DOI] [PubMed] [Google Scholar]
- Savitz SI, Lew R, Bluhmki E, Hacke W, Fisher M. Shift analysis versus dichotomization of the modified Rankin scale outcome scores in the NINDS and ECASS‐II trials. Stroke 2007;38(12):3205‐12. [DOI] [PubMed] [Google Scholar]
- Schaefer E, Hacke W, Dávalos A, Donnan GA, Fieschi C, Kaste M, et al. European‐Australian cooperative acute stroke study II (ECASS II) ‐ interim demographics and baseline characteristics. Cerebrovascular Diseases 1998;8(Suppl 4):82. [Google Scholar]
- Stingele R, Bluhmki E, Hacke W. Bootstrap statistics of ECASS II data: Just another post hoc analysis of a negative stroke trial?. Cerebrovascular Diseases 2001;11(1):30‐3. [DOI] [PubMed] [Google Scholar]
- Toni D, Sacchetti ML, Chamorro A. Acute stroke trials: the problems of local investigators?. European Neurology 2003;49(2):109‐14. [DOI] [PubMed] [Google Scholar]
- Kummer R, Bastianello S, Bozzao L, Manelfe CH, Meier D, Hacke W. Baseline CT findings and the response of thrombolytic treatment in acute ischemic stroke. Radiology 2001;221 Suppl:301. [Google Scholar]
- Kummer R, Berrouschot J, Henrik B, Swen H, Deiler C, Schneider D. The effect of early and late brain tissue reperfusion on infarct volume. Stroke 2000;31:275. [Google Scholar]
- Yong M, Kaste M. Association of characteristics of blood pressure profiles and stroke outcomes in the ECASS‐II trial. Stroke 2008;39(2):366‐72. [DOI] [PubMed] [Google Scholar]
- Yong M, Kaste M. Dynamic of hyperglycemia as a predictor of stroke outcome in the ECASS‐II Trial. Stroke 2008;39(10):2749‐55. [DOI] [PubMed] [Google Scholar]
- Yong M, Mau J. Characteristics of blood pressure profiles and their prognostic value of stroke outcome. International Journal of Stroke 2006;1 Suppl 1:40. [Google Scholar]
- Yong M, Mau J. Review of characteristics of blood pressure profiles and long‐term outcome in the ECASS‐II database. International Journal of Stroke 2006;1 Suppl 2:34. [Google Scholar]
- Young M, Mau J. Prognostic value of blood pressure profiles on cerebral hemorrhages in rt‐PA treated patients. Cerebrovascular Diseases 2007;23(Suppl 2):23‐4. [Google Scholar]
EPITHET 2008 {published data only}
- Brekenfeld C, Silva DA, Christensen S, Churilov L, Parsons MW, Levi CR, et al. Dual target (mismatch and vessel obstruction) at baseline MRI does not improve stroke patient selection for thrombolysis 3‐6H. Cerebrovascular Diseases 2009;27(Suppl 6):4. [Google Scholar]
- Butcher K, Christensen S, Parsons M, Silva D, Ebinger M, Levi C, et al. Hemorrhagic transformation in the Echoplanar Imaging Thrombolysis Evaluation Trial (EPITHET) is predicted by post‐treatment blood pressure control and infarct volume. Cerebrovascular Diseases 2009;27(Suppl 6):22‐3. [Google Scholar]
- Butcher K, Christensen S, Parsons M, Silva D, Ebinger M, Levi C, et al. Post‐treatment blood pressure control predicts thrombolysis related hemorrhagic transformation. Stroke 2009;40(4):e152. [Google Scholar]
- Butcher K, Christensen S, Parsons M, Silva DA, Ebinger M, Levi C, et al. Postthrombolysis blood pressure elevation is associated with hemorrhagic transformation. Stroke 2010;41(1):72‐7. [DOI] [PubMed] [Google Scholar]
- Butcher K, Davis S. EPITHET protocol [personal communication]. email to JM Wardlaw 7 July 2002.
- Butcher K, Parsons M, Allport L, Lee SB, Barber PA, Tress B, et al. Rapid assessment of perfusion‐diffusion mismatch. Stroke 2008;39(1):75‐81. [DOI] [PubMed] [Google Scholar]
- Butcher K, Parsons M, MacGregor L, Barber PA, Levi C, Chalk J, et al. Reperfusion outweighs PWI‐DWI mismatch in determining acute stroke outcome. Internal Medicine Journal 2005;35(Suppl 2):A1. [Google Scholar]
- Butcher KS. Echoplanar imaging thrombolysis evaluation trial: EPITHET. Proceedings of the 28th International Stroke Conference. February 13‐15 2003; Phoenix, Arizona, USA. 2003.
- Butcher KS. Echoplanar imaging thrombolysis evaluation trial: EPITHET. Proceedings of the International Stroke Conference 2005. Ongoing Clinical Trials Abstracts. 2 February 2005; New Orleans, Louisiana, USA. 2005.
- Butcher KS, Macgregor L, Parsons MW, Barber PA, Levi C, Chalk J, et al. Multiple definitions of PWI‐DWI mismatch reliably predict infarct growth. Stroke 2004;35(6):e198. [Google Scholar]
- Butcher KS, Parsons M, Barber A, Levi C, Chalk J, Read S, et al. The frequency of perfusion‐diffusion mismatch decreases with objective definition. Stroke 2004;35(1):259. [Google Scholar]
- Butcher KS, Parsons M, MacGregor L, Barber PA, Chalk J, Bladin C, et al. Refining the perfusion‐diffusion mismatch hypothesis. Stroke 2005;36(6):1153‐9. [DOI] [PubMed] [Google Scholar]
- Butcher KS, Parsons MW, Allport L, MacGregor L, Lee SB, Tress B, et al. MRI ASPECTS (Alberta Stroke Program Early CT Scores) predict perfusion‐diffusion mismatch. Cerebrovascular Diseases 2005;19(Suppl 2):71. [Google Scholar]
- Butcher KS, Tress B, Davis S, MacGregor L, Parsons MW, Donnan G. Reperfusion is more important than PWI‐DWI mismatch definition in determining tissue fate. Journal of Clinical Neuroscience 2005;12(3):332. [Google Scholar]
- Campbell B, Christensen S, Butcher K, Parsons M, Desmond P, Gordon I, et al. Prediction of haemorrhagic transformation after stroke thrombolysis: very low cerebral blood volume (VLCBV) outperforms DWI/ADC lesion volume. International Journal of Stroke 2009;4(Suppl 1):7. [Google Scholar]
- Campbell B, Purushotham A, Christensen S, Desmond P, Nagakane Y, Parsons M, et al. Diffusion lesion reversal in acute ischemic stroke is uncommon in the 3‐6hr treatment window. International Journal of Stroke 2010;5(Suppl 1):19. [Google Scholar]
- Campbell BC, Christensen S, Parsons MW, Desmond PM, Barber PA, Butcher KS, et al. Regional very low cerebral blood volume with subsequent local reperfusion predicts hemorrhagic transformation in acute ischemic stroke. Stroke 2012;43(2):Abst. 95. [DOI] [PubMed] [Google Scholar]
- Campbell BC, Christensen S, Parsons MW, Desmond PM, Barber PA, Butcher KS, et al. Very low cerebral blood volume predicts hemorrhagic transformation better than diffusion lesion volume in acute ischemic stroke. Stroke 2010;41(4):e246. [DOI] [PubMed] [Google Scholar]
- Campbell BCV, Christensen S, Butcher KS, Gordon I, Parsons MW, Desmond PM, et al. Regional very low cerebral blood volume predicts hemorrhagic transformation better than diffusion‐weighted imaging volume and thresholded apparent diffusion coefficient in acute ischemic stroke. Stroke 2010;41(1):82‐8. [DOI] [PubMed] [Google Scholar]
- Campbell BCV, Christensen S, Desmond PM, Parsons MW, Barber PA, Silva DA, et al. Major infarct growth beyond 6 hours is associated with collateral circulation failure. Cerebrovascular Diseases 2010;29(Suppl 2):3‐4. [Google Scholar]
- Campbell BCV, Christensen S, Foster SJ, Desmond PM, Parsons MW, Butcher KS, et al. Visual assessment of perfusion‐diffusion mismatch is inadequate to select patients for thrombolysis. Cerebrovascular Diseases 2010;29(6):592‐6. [DOI] [PubMed] [Google Scholar]
- Campbell BCV, Christensen S, Tress BM, Desmond PM, Parsons MW, Barber PA, et al. Insights into the relationship of perfusion‐diffusion mismatch and leptomeningeal collateral quality ‐ simultaneous assessment through novel visualization of perfusion imaging. International Journal of Stroke 2012;7(Suppl 1):12. [Google Scholar]
- Campbell BCV, Costello C, Christensen S, Ebinger M, Parsons MW, Desmond PM, et al. Fluid‐attenuated inversion recovery hyperintensity in acute ischemic stroke may not predict hemorrhagic transformation. Cerebrovascular Diseases 2011;32(4):401‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BCV, Purushotham A, Christensen S, Desmond PM, Nagakane Y, Parsons MW, et al. The acute diffusion lesion reliably represents infarct core: clinically relevant reversibility is rare. Stroke 2011;42(3):e71. [Google Scholar]
- Chalk JB, Rose SE, Walsh M, Read S. Sodium MRI in acute stroke. Journal of Clinical Neuroscience 2005;12(3):351. [Google Scholar]
- Chemmanam T, Campbell BCV, Christensen S, Nagakane Y, Desmond PM, Bladin CF, et al. Ischemic diffusion lesion reversal is uncommon and rarely alters perfusion‐diffusion mismatch. Neurology 2010;75(12):1040‐7. [DOI] [PubMed] [Google Scholar]
- Christensen S, Campbell B, Parsons MW, Silva DA, Ebinger M, Butcher K. High Tmax values on perfusion MRI often reflect low CBV ‐ a pathophysiological link between the malignant perfusion profile and poor outcome?. Stroke 2011;42(3):e118. [Google Scholar]
- Christensen S, Campbell BC, Perez de la Ossa N, Lansberg M, Straka M, Silva DA, et al. Optimal perfusion thresholds for prediction of tissue destined for infarction in the combined EPITHET and DEFUSE dataset. Stroke 2010;41(4):e297. [Google Scholar]
- Christensen S, Parsons M, Silva D, Ebinger M, Butcher K, Fink J, et al. Optimal mismatch definitions for detecting treatment response in acute stroke. Cerebrovascular Diseases 2008;25(Suppl 2):33. [Google Scholar]
- Christensen S, Parsons M, Hunter J, Silva D, Ebinger M, Butcher K, et al. Optimising MR criteria for penumbral selection trials. Stroke 2009;40(4):e162‐3. [Google Scholar]
- Christensen S, Parsons MW, Silva DA, Ebinger M, Butcher K, Fink J, et al. Testing the mismatch hypothesis in the randomized EPITHET data Set: the effect of treatment, mismatch and their interaction on infarct growth. Stroke 2010;41(4):e203. [Google Scholar]
- Christensen S, Parsons MW, DeSilva DA, Ebinger M, Butcher K, Fink J, et al. Optimizing mismatch definitions in acute stroke MRI ‐ an EPITHET post hoc study. Internal Medicine Journal 2008;38(Suppl 4):A74. [Google Scholar]
- Davies S, Davis SM, Donnan GA, Parsons M, Butcher K, Tress B, et al. Thrombolysis beyond 3 hours: The EPITHET trial. Proceedings of the 7th International Symposium on Thrombolysis and Acute Stroke Therapy. 27‐28 May 2002; Lyon, France. 2002:50.
- Davis S. Clinical MRI trials: EPITHET/DEFUSE/DIAS/MR RESCUE. Stroke 2004;35(6):e177. [Google Scholar]
- Davis S. The use of MRI in the selection of thrombolytic therapy for stroke. Proceedings of the 17th International Congress on Fibrinolysis and Proteolysis. 21‐25 March 2004; Melbourne, Australia. 2004:57.
- Davis S, Donnan G. Echoplanar Imaging Thrombolysis Evaluation Trial. www.strokecenter.org (accessed 21 July 2007).
- Davis SM, Donnan G. Echoplanar imaging thrombolysis evaluation trial (EPITHET). www.clinicaltrials.gov/ct2/show/NCT00238537 (accessed July 10th 2014).
- Davis SM, Donnan GA. EchoPlanar Imaging Thrombolysis Evaluation Trial. Powerpoint presentation.
- Davis SM, Donnan GA. Echoplanar Imaging Thrombolysis Evaluation Trial: EPITHET. Proceedings of the International Stroke Conference 2006. Ongoing Clinical Trials Abstracts. 16 February 2006; Kissimmee, Florida, USA. 2006.
- Davis SM, Donnan GA, Silva DA, Butcher KS, Parsons M, Levi C, et al. Results of EPITHET: Effects of tPA beyond 3 hours on infarct growth, reperfusion and clinical outcomes. Proceedings of the International Stroke Conference 2008. 20‐22 February 2008; New Orleans, USA. 2008.
- Davis SM, Donnan GA, Parsons MW, Levi C, Butcher KS, Peeters A, et al. EPITHET investigators. Effects of alteplase beyond 3 h after stroke in the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET): a placebo‐controlled randomised trial. Lancet Neurology 2008;7(4):299‐309. [DOI] [PubMed] [Google Scholar]
- Silva D. Data query for EPITHET [personal communication]. Email to JM Wardlaw March 15 2008.
- Silva DA, Brekenfeld C, Ebinger M, Christensen S, Barber PA, Butcher KS, et al. The benefits of intravenous thrombolysis relate to the site of baseline arterial occlusion in the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET). Stroke 2010;41(2):295‐9. [DOI] [PubMed] [Google Scholar]
- Silva DA, Christensen S, Butcher KS, Parsons MW, Levi CR, Bladin CF, et al. The incidence and degree of recanalization depends on site of arterial occlusion in acute ischemic stroke. Journal of Clinical Neuroscience 2008;15:364. [Google Scholar]
- Silva DA, Christensen S, Ebinger M, Fink J, Levi C, Parsons MW, et al. Spontaneous and TPA‐induced recanalization and reperfusion in the echoplanar imaging thrombolytic evaluation trial (EPITHET). International Journal of Stroke 2008;3(Suppl 1):267‐8. [Google Scholar]
- Silva DA, Churilov L, Olivot JM, Christensen S, Lansberg MG, Mlynash M, et al. Does presence of arterial obstruction influence the treatment effect of intravenous tPA over placebo in the 3‐6 hour time window?. Stroke 2010;41(4):e207‐8. [Google Scholar]
- Silva DA, Ebinger M, Christensen S, Levi C, Parsons MW, Peeters A, et al. The impact of diabetes and admission blood glucose on outcomes in the Echoplanar Imaging Thrombolytic Evaluation Trial (Epithet). Cerebrovascular Diseases 2008;25(Suppl 2):7. [Google Scholar]
- Silva DA, Ebinger M, Christensen S, Parsons MW, Levi C, Butcher K, et al. Baseline diabetic status and admission blood glucose were poor prognostic factors in the EPITHET trial. Cerebrovascular Diseases 2010;29(1):14‐21. [DOI] [PubMed] [Google Scholar]
- Silva DA, Fink JN, Christensen S, Ebinger M, Bladin C, Levi CR, et al. Assessing reperfusion and recanalization as markers of clinical outcomes after intravenous thrombolysis in the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET). Stroke 2009;40(8):2872‐4. [DOI] [PubMed] [Google Scholar]
- Silva DA, Fink JN, Christensen S, Ebinger M, Levi C, Parsons MW, et al. Impact of baseline arterial obstruction site and degree on outcomes in the echoplanar imaging thrombolytic evaluation trial (EPITHET). International Journal of Stroke 2008;3(Suppl 1):21. [Google Scholar]
- Silva DA, Mlynash M, Lansberg MG, Lee J, Christensen S, Straka M, et al. Large and severe baseline PWI volumes predict poor response to intravenous tPA vs. placebo in the pooled DEFUSE‐EPITHET database. Stroke 2010;41(4):e208. [Google Scholar]
- EPITHET investigators. Echoplanar imaging thrombolysis evaluation trial: EPITHET. Proceedings of the 27th International Stroke Conference. Feb 7‐9 2002; San Antonio, USA. 2002:CTP330.
- Ebinger M, Christensen S, Butcher KS, Parsons MW, Levi CR, Bladin CF, et al. Expediting MRI‐based proof of concept stroke trials using an earlier primary endpoint. Journal of Clinical Neuroscience 2008;15:353. [Google Scholar]
- Ebinger M, Christensen S, Silva DA, Parsons MW, Levi CR, Butcher KS, et al. Echoplanar Imaging Thrombolytic Evaluation Trila Investigators. Expediting MRI‐based proof‐of‐concept stroke trials using an earlier imaging end point. Stroke 2009;40(4):1353‐8. [DOI] [PubMed] [Google Scholar]
- Ebinger M, Christensen S, DeSilva DA, Parsons MW, Levi CR, Peeters, et al. Expediting MRI‐based proof of concept stroke trials using an earlier primary endpoint. Cerebrovascular Diseases 2008;25(Suppl 2):34. [Google Scholar]
- Ebinger M, Iwanaga T, Prosser JF, Silva DA, Christensen S, Collins M. Clinical‐diffusion mismatch and benefit from thrombolysis 3‐6 hours after acute stroke. International Journal of Stroke 2008;3(Suppl 1):17. [Google Scholar]
- Ebinger M, Iwanaga T, Prosser JF, Silva DA, Christensen S, Collins M, et al. Clinical‐diffusion mismatch and benefit from thrombolysis 3 to 6 hours after acute stroke. Stroke 2009;40(7):2572‐4. [DOI] [PubMed] [Google Scholar]
- Finnigan SP, Rose SE, Chalk JB. Rapid EEG changes indicate reperfusion after tissue plasminogen activator injection in acute ischaemic stroke. Clinical Neurophysiology 2006;117(10):2338‐9. [DOI] [PubMed] [Google Scholar]
- Lansberg MG, Lee J, Christensen S, Straka M, Silva DA, Mlynash M, et al. DEFUSE and EPITHET: two different studies with one consistent message. Stroke 2010;41(4):e295. [Google Scholar]
- Lansberg MG, Lee J, Christensen S, Straka M, Silva DA, Mlynash M, et al. RAPID automated patient selection for reperfusion therapy: a pooled analysis of the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET) and the Diffusion and Perfusion Imaging Evaluation for Understanding Stroke Evolution (DEFUSE) Study. Stroke 2011;42(6):1608‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Lansberg MG, Mlynash M, Christensen S, Straka M, Silva DA. Effect of reperfusion in the pooled DEFUSE‐EPITHET database. International Journal of Stroke 2010;5(Suppl 2):13. [Google Scholar]
- Lee J, Lansberg MG, Mlynash M, Christensen S, Straka M, Silva DA, et al. Poor outcome in malignant profile patients is strongly associated with reperfusion and parenchymal hematoma. International Journal of Stroke 2010;5(Suppl 2):95. [Google Scholar]
- Lee J, Lansberg MG, Mlynash M, Silva DA, Christensen S, Straka M, et al. The combination of reperfusion and recanalization predicts favorable outcome better than reperfusion or recanalization alone in target mismatch patients. Stroke 2011;42(3):e66‐7. [Google Scholar]
- Lee J, Lansberg MG, Mlynash M, Silva DA, Christensen S, Straka M, et al. Validation of the malignant profile in the DEFUSE‐EPITHET pooled database. Stroke 2010;41(4):e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlynash M, Silva DA, Lansberg MG, Lee J, Christensen S, Straka M, et al. Optimal definition of the malignant profile in the DEFUSE‐EPITHET pooled database. Stroke 2010;41(4):e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlynash M, Lansberg MG, Silva DA, Lee J, Christensen S, Straka M, et al. Refining the definition of the malignant profile. Insights from the DEFUSE‐EPITHET pooled data set. Stroke 2011;42(5):1270‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagakane Y, Christensen S, Brekenfeld C, Ma H, Churilov L, Parsons MW, et al. EPITHET. Positive result after reanalysis using baseline diffusion‐weighted imaging/perfusion‐weighted imaging co‐registration. Stroke 2011;42(1):59‐64. [DOI] [PubMed] [Google Scholar]
- Nagakane Y, Christensen S, Ogata T, Churilov L, Ma H, Parsons MW, et al. Moving beyond a single perfusion threshold to define penumbra: a novel probabilistic mismatch definition. Stroke 2012;43(6):1548‐55. [DOI] [PubMed] [Google Scholar]
- Ogata T, Christensen S, Nagakane Y, Ma H, Campbell BC, Churilov L, et al. The effects of alteplase 3 to 6 hours after stroke in the EPITHET‐DEFUSE combined dataset: post hoc case‐control study. Stroke 2013;44(1):87‐93. [DOI] [PubMed] [Google Scholar]
- Ogata T, Nagakane Y, Christensen S, Ma H, Campbell B, Churilov L, et al. A topographic study of the evolution of MR mismatch patterns and clinical relevance. Journal of Stroke 2010;5(Suppl 1):36‐7. [DOI] [PubMed] [Google Scholar]
- Ogata T, Nagakane Y, Christensen S, Ma H, Campbell BCV, Churilov L, et al. EPITHET and DEFUSE Investigators. A topographic study of the evolution of the MR DWI/PWI mismatch pattern and its clinical impact: a study by the EPITHET and DEFUSE Investigators. Stroke 2011;42(6):1596‐601. [DOI] [PubMed] [Google Scholar]
- Parsons MW, Christensen S, McElduff P, Levi CR, Butcher KS, Silva DA, et al. Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET) Investigators. Pretreatment diffusion‐ and perfusion‐MR lesion volumes have a crucial influence on clinical response to stroke thrombolysis. Journal of Cerebral Blood Flow and Metabolism 2010;30(6):1214‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez de la Ossa N, Chandra RV, Campbell BCV, Christensen S, Collins M, Parsons MW, et al. Leukoaraiosis is not an independent risk factor for parenchymal hemorrhage after thrombolysis. Cerebrovascular Diseases 2010;20(Suppl 2):14. [Google Scholar]
- Ricanco MR, Campbell BC, Christensen S, Desmond PM, Churilov L, Silva DA, et al. Reperfusion beyond 4.5 hours reduces infarct growth and improves clinical outcome. Stroke 2012;43(2):Abst 61. [DOI] [PubMed] [Google Scholar]
- Sandercock P, Wardlaw J, Dennis M, Lindley R, Hankey G, Matz K, et al. EPITHET ‐‐ Where next?. Lancet Neurology 2008;7(7):570‐3. [DOI] [PubMed] [Google Scholar]
- Schellinger PD. EPITHET: failed chance or new hope?. Lancet Neurology 2008;7(4):286‐7. [DOI] [PubMed] [Google Scholar]
- Straka M, Lansberg MG, Christensen S, Mlynash M, Silva DA, Olivot JM, et al. Is reduced CBV a reliable surrogate marker for infarct core and can it be used to identify mismatch?. Stroke 2010;41(4):e295. [Google Scholar]
- Toth G, Albers GW. Use of MRI to estimate the therapeutic window in acute stroke. Is perfusion‐weighted imaging/diffusion‐weighted imaging mismatch an EPITHET for salvageable ischemic brain tissue?. Stroke 2009;40(1):333‐5. [DOI] [PubMed] [Google Scholar]
- Tu H, Campbell B, Christensen S, Butcher K, Collins M, Parsons M, et al. Atrial fibrillation is associated with increased infarct size, hemorrhagic transformation and worse outcome in patients with ischaemic stroke. International Journal of Stroke 2009;4(Suppl 1):7. [Google Scholar]
- Tu H, Campbell B, Christensen S, Churilov L, Parsons M, Silva D, et al. More severe hypoperfusion leads to greater infarct growth and worse stroke outcome in atrial fibrillation. International Journal of Stroke 2011;6(Suppl 1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu HT, Campbell BC, Christensen S, Butcher KS, Collins M, Parsons MW, et al. The effects of atrial fibrillation on infarct evolution and outcome. Stroke 2010;41(4):e337. [Google Scholar]
- Tu HT, Campbell BC, Christensen S, Collins M, Silva DA, Butcher KS, et al. Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET) Investigators. Pathophysiological determinants of worse stroke outcome in atrial fibrillation. Cerebrovascular Diseases 2010;30(4):389‐95. [DOI] [PubMed] [Google Scholar]
- Tu HT, Campbell BC, Christensen S, Silva DA, Parsons MW, Churilov L, et al. Worse stroke outcome in atrial fibrillation links to more severe hypoperfusion. Stroke 2011;42(3):e119. [Google Scholar]
- Wardlaw JM. Surrogate outcomes. A cautionary note. Stroke 2009;40(4):1029‐31. [DOI] [PubMed] [Google Scholar]
- Willats L, Connelly A, Christenson S, Donnan GA, Davis SM, Calamante F. The role of bolus delay and dispersion in predictor models for stroke. Stroke 2012;43(4):1025‐31. [DOI] [PubMed] [Google Scholar]
Haley 1993 {published data only}
- Haley EC Jr, Brott TG, Sheppard GL, Barsan W, Broderick J, Marler JR, et al. Pilot randomized trial of tissue plasminogen activator in acute ischemic stroke. Stroke 1993;24(7):1000‐4. [DOI] [PubMed] [Google Scholar]
- Haley EC, TPA Bridging Study Group. Pilot randomized trial of tissue plasminogen activator in acute ischemic stroke. Neurology 1992;42 Suppl 3:203. [DOI] [PubMed] [Google Scholar]
IST3 2012 {published and unpublished data}
- Anderson C. Thrombolysis with alteplase after stroke: extending outcomes. Lancet Neurology 2013;12(8):731‐2. [DOI] [PubMed] [Google Scholar]
- Anonymous. Department of Error (erratum). Lancet 2012;380(9843):730. [Google Scholar]
- Broom A, Broom N, Stevens C, Stoneman T, Worsfold L, Pay G. The South West Stroke Research Network PCPI Newsletter. The South West Stroke Research Network PCPI Newsletter 2012.
- Carpenter T, Wardlaw JM, Sandercock PAG, Lindley RI, Murray V, Peeters A, et al. The Third International Stroke Trial (IST‐3) perfusion and angiography study: current progress and participation. Proceedings of the 19th European Stroke Conference 2010. 25‐28 May 2010; Barcelona, Spain. 2010.
- Chang K‐C. Thrombolysis for acute ischaemic stroke: the Third International Stroke Trial (IST‐3). Proceedings of the 2003 Annual Meeting of Taiwan Stroke Society; 2003 Mar 22‐23; Taiwan. Taiwan Stroke Society, 2003:104.
- Czlonkowska A, Kobayashi A, Lewis S, Sandercock P, Lindley R, Baranska‐Gieruszczak M, et al. The Third International Stroke Trial: thrombolysis (IST‐3) in Poland: are we recruiting the right patients?. Neurologia i Neurochirurgia Polska 2009;43(3):228‐35. [PubMed] [Google Scholar]
- Diener H‐C. Third International Stroke Trial (IST‐3): Intravenous thrombolysis in ischemic insult. Arzneimitteltherapie 2012;30:400. [Google Scholar]
- Donnan GA, Davis SM. IST‐3: a major contribution to thrombolysis research. International Journal of Stroke 2012;7(7):566‐7. [DOI] [PubMed] [Google Scholar]
- Farrall A, Wardlaw J, Sandercock P, Lindley R, Cohen G, Kummer R, et al. The third international stroke trial (IST‐3) of intravenous thrombolysis with rt‐PA: Baseline imaging features among 3035 patients randomised. International Journal of Stroke 2012;7:61‐2. [Google Scholar]
- Fatovich DM. Believing is seeing: Stroke thrombolysis remains unproven after the third international stroke trial (IST‐3). Emergency Medicine Australasia 2012;24(5):477‐9. [DOI] [PubMed] [Google Scholar]
- Fatovich DM, MacDonald SP, Brown SG. Thrombolysis in acute ischaemic stroke. Lancet 2012;380(9847):1053. [DOI] [PubMed] [Google Scholar]
- Furlan AJ. IST‐3: no pragmatic answers. International Journal of Stroke 2012;7(7):568‐9. [DOI] [PubMed] [Google Scholar]
- Hankey GJ, Hand PJ, Lindley RI, Wardlaw JM, Sandercock PA, Anderson C. Thrombolysis for acute ischaemic stroke: the Third International Stroke Trial (IST‐3). Journal of Clinical Neuroscience 2002;9(4):480‐93. [Google Scholar]
- Hoffman JR, Cooper RJ. How is more negative evidence being used to support claims of benefit: The curious case of the third international stroke trial (IST‐3). Emergency Medicine Australasia 2012;24(5):473‐6. [DOI] [PubMed] [Google Scholar]
- Hughes S. IST‐3: tPA benefits sustained out to 18 months. www.medscape.com 2013:Article No. 806690.
- IST‐3 Collaborative Group. Effect of thrombolysis with alteplase within 6 h of acute ischaemic stroke on long‐term outcomes (the third International Stroke Trial [IST‐3]): 18‐month follow‐up of a randomised controlled trial. Lancet Neurology 2013;12(8):768‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IST‐3 Collaborative Group. Effect of thrombolysis with alteplase within 6 h of acute ischaemic stroke on long‐term outcomes (the third International Stroke Trial [IST‐3]): 18‐month follow‐up of a randomised controlled trial. Lancet Neurology 2013;12(8):768‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IST‐3 Collaborative Group. The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST‐3]): a randomised controlled trial. Lancet 2012;379(9834):2352‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innes K, IST‐3 Stroke Nurse Collaborative Group. Thrombolysis for acute ischaemic stroke: core nursing requirements. British Journal of Nursing 2003;12(7):416‐24. [DOI] [PubMed] [Google Scholar]
- Kane I, Lindley R, Lewis S, Sandercock P, IST‐3 Collaborative Group. Impact of stroke syndrome and stroke severity on the process of consent in the Third International Stroke Trial. Cerebrovascular Diseases 2006;21(5‐6):348‐52. [DOI] [PubMed] [Google Scholar]
- Kane I, Lindley R, Sandercock P, Lewis S, IST‐3 Collaborative Group. Consent for thrombolysis for acute ischaemic stroke. Cerebrovascular Diseases 2005;19(Suppl 2):76. [Google Scholar]
- Kane I, Sandercock P, Lindley R. The Third International Stroke Trial (IST‐3). Stroke 2004;35(6):e200. [Google Scholar]
- Kane I, Sandercock P, Lindley R on behalf of the IST‐3 Collaborative Group. The Third International Stroke Conference. www.esc‐brussels.org/esc_ongoing_trials.asp (no date given).
- Kane I, Sandercock P, Lindley R, Gubitz G, Phillips S, IST‐3 Collaborative Group. The MRC Third International Stroke Trial (IST‐3). asa.apprisor.com/snap/NET/GT/700.cfm (accessed 7 April 2006).
- Kane I, Sandercock P, Lindley R, IST‐3 Collaborative Group. Third International Stroke Trial. www.abstractsonline.com/arch/RecordPrintView.aspx?Lookupkey=12345&RecordID=10978 (accessed 9 August 2007).
- Kane I, Sandercock P, Lindley R, IST‐3 Collaborative Group. Third International Stroke Trial. www.abstractsonline.com/arch/RecordPrintView.aspx?Lookupkey=12345&RecordID=16913 (accessed 9 August 2007).
- Kane I, Sandercock P, Lindley R, Lewis S. The Third International Stroke Trial. Baseline characteristics of patients recruited in the expansion phase. Cerebrovascular Diseases 2004;17(Suppl 5):110‐1. [Google Scholar]
- Kane I, Sandercock P, Lindley R, Lewis S. The Third International Stroke Trial. Baseline characteristics of patients recruited in the expansion phase. Stroke 2004;35(6):e216. [Google Scholar]
- Kane I, Sandercock P, Lindley R, Lewis S, IST‐3 Collaborative Group. The Third International Stroke Trial (IST‐3). Thrombolysis for acute ischaemic stroke: characteristics of patients randomised in the expansion phase. Age and Ageing 2005;34 Suppl 1:i45. [Google Scholar]
- Kane I, Sandercock P, Lindley R, Lewis S, IST‐3 Collaborative Group. The Third International Stroke Trial. Baseline characteristics of patients recruited in the expansion phase. Cerebrovascular Diseases 2005;19(Suppl 2):29. [Google Scholar]
- Kane I, Sandercock P, Lindley R, Lewis S, IST‐3 Collaborative Group. The Third International Stroke Trial: baseline characteristics of patients recruited in the expansion phase. Stroke 2005;36(2):450. [Google Scholar]
- Kane I, Sandercock P, Lindley R, Lewis S, IST‐3 Collaborative Group. Third International Stroke Trial: characteristics of patients recruited in the start‐up phase. Stroke 2004;35(1):295. [Google Scholar]
- Kane I, Sandercock P, IST‐3 Collaborative Group. The Third International Stroke Trial (IST‐3). Thrombolysis for acute ischaemic stroke. Journal of Neurology, Neurosurgery & Psychiatry 2005;76(9):1314. [Google Scholar]
- Kane I, Sandercock P, IST‐3 Collaborative Group. Third International Stroke Trial (IST‐3). Thrombolysis for acute ischaemic stroke. Journal of Neurology, Neurosurgery & Psychiatry 2004;75:797. [Google Scholar]
- Kane I, Wardlaw J, Sandercock P, Lewis S, IST‐3 Collaborative Group. Baseline CT characteristics of the first 253 patients in the Third International Stroke Trial. British Association of Stroke Physicians Annual Scientific Meeting; 2005 Jan 19; Newcastle upon Tyne. 2005:4.
- Kleinig TJ, Churilov L, Parsch CS, Dewey HM, Barber PA. Stroke thrombolysis and the third international stroke trial: Examining 'the totality of the evidence'. Emergency Medicine Australasia 2013;25(2):107‐9. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Bembenek J, Sandercock P, Kane I, Skowronska M, Czlonkowska A, et al. Correlation of stroke subtype in OCSP classification and early ischaemic changes on CT: an analysis based on the first 389 patients from the IST‐3 Trial. Cerebrovascular Diseases 2006;21(Suppl 4):35. [Google Scholar]
- Kobayashi A, Sandercock P, Czlonkowska A, Lewis S, Wardlaw J. Early ischaemic changes on CT and the OCSP classification: an analysis of the first 389 patients from the IST‐3 Trial. European Journal of Neurology 2006;13 Suppl 2:26. [Google Scholar]
- Kobayashi A, Wardlaw JM, Lindley RI, Lewis SC, Sandercock PAG, Czlonkowska A, IST‐3 Collaborative Group. Oxfordshire Community Stroke Project clinical stroke syndrome and appearances of tissue and vascular lesions on pretreatment CT in hyperacute ischemic stroke among the first 510 patients in the Third International Stroke Trial (IST‐3). Stroke 2009;40(3):743‐8. [DOI] [PubMed] [Google Scholar]
- Lewis SC, Sandercock PA, Lindley RI, Wardlaw JM, Dennis MS. The MRC Third International Stroke trial (IST‐3). Proceedings of the 16th European Stroke Conference; 2007 May 29‐June 1, Glasgow. 2007.
- Leys L, Cordonnier C. rt‐PA for ischaemic stroke: what will the next question be?. Lancet 2012;379(9834):2320‐1. [DOI] [PubMed] [Google Scholar]
- Lindley R, Grimley R, Australian IST‐3 Collaborative Group. The Third International Stroke Trial (IST‐3): thrombolysis for acute ischaemic stroke. Internal Medicine Journal 2007;37 Suppl 2:A84. [Google Scholar]
- Lindley R, Kobayashi A, Wardlaw J, Sandercock P, Lewis C. Persisting hyperdense artery sign, aspects scores and risk of developing mass effect: analysis from the first 389 patients in IST‐3. Internal Medicine Journal 2008;37 Suppl 1:A7. [Google Scholar]
- Lindley RI. The Third International Stroke Trial: thrombolysis for acute ischaemic stroke. Proceedings of the American Stroke Association 28th International Stroke Conference; 2003 Feb 13‐15; Arizona. 2003:CTP22.
- Lindley RI, Sandercock PA, Wardlaw JM, Dennis MS, Cohen G, IST‐3 Collaborative Group. Third international stroke trial (IST‐3): subgroup effects of iv rt‐PA < 6 hrs in acute ischemic stroke on symptomatic intracranial haemorrhage and outcome at 6 months. Stroke 2013;44:Abst.TMP23. [Google Scholar]
- Lindley RI, Sandercock PAG, Wardlaw JM, Dennis MS, Lewis S. Hyperintense artery sign, atrial fibrillation and infarct subtype in the Third International Stroke Trial (IST‐3). International Journal of Stroke 2008;3(Suppl 1):135. [Google Scholar]
- Lindley RI, Sandercock PAG, Wardlaw JM, Dennis MS, Lewis SC, IST‐3 Collaborative Group. Hyperintense artery sign, atrial fibrillation and infarct subtype in the Third International Stroke Trial (IST‐3). Internal Medicine Journal 2008;38 Suppl 4:A96. [Google Scholar]
- Lindley RI, Sandercock PAG, Wardlaw JM, Dennis MS, Lewis SC, IST‐3 collaborative group. Thrombolysis for stroke ‐ what are the 'grey' areas of uncertainty and are they changing? Results from the Third International Stroke Trial IST‐3. Internal Medicine Journal 2008;38 Suppl 4:A96. [Google Scholar]
- Lindley RI, Sandercock PAG, Wardlaw JM, Dennis MS. The Third International Stroke Trial (IST‐3) on behalf of the IST‐3 Collaborative Group. Proceedings of the 18th European Stroke Conference 2009. 26‐29 May 2009; Stockholm, Sweden. 2009.
- Lindley RI, IST‐3 Collaborative Group. The Third International Stroke Trial. Baseline characteristics of patients recruited. Internal Medicine Journal 2005;35 Suppl:A62. [Google Scholar]
- Lueck CJ, Lindley RI. The Third International Stroke Trial (IST‐3): thrombolysis for acute ischaemic stroke. Internal Medicine Journal 2007;37(Suppl 4):A102. [Google Scholar]
- Lyden PD. In anticipation of International Stroke Trial‐3 (IST‐3). Stroke 2012;43(6):1691‐4. [DOI] [PubMed] [Google Scholar]
- Mair G, Wardlaw JM, Sandercock P, Lindley R, Kummer R. Combining CT angiography with non‐contrast CT to predict infarct on follow up CT in acute ischaemic stroke. Substudy analysis of imaging from the third International Stroke Trial (IST‐3). Cerebrovascular Diseases 2013;35(Suppl 3):237. [Google Scholar]
- Mair G, Wardlaw JM, Sandercock P, Lindley R, Kummer R, Farrall AJ. Association of non‐contrast CT and CT angiography with baseline clinical deficit and functional outcome. Substudy analysis of imaging from the third International Stroke Trial (IST‐3). Cerebrovascular Diseases 2013;35(Suppl 3):405. [Google Scholar]
- Murray V, Norrving B, Terent A, Wester P, Heijne A, Isakson E. Patient recruitment in IST‐3, Sweden so far during the years 2003 and onwards. Proceedings of the 18th European Stroke Conference 2009. 26‐29 May 2009; Stockholm, Sweden. 2009.
- Radecki RP, Chathampally YG, Press GM. Rt‐PA and stroke: Does IST‐3 make it all clear or muddy the waters?. Annals of Emergency Medicine 2012;60:666‐7. [DOI] [PubMed] [Google Scholar]
- Ricci S, Celani MG, Righetti E, Cantisani AT. [Trombolisi per l'ictus ischemico: lo studio IST 3]. Proceedings of the Conferenza Nazionale Sull'ictus Cerebrale; 2004 Mar 7‐9; Firenze. 2004:158.
- Ricci S, Celani MG, Righetti E, Cantisani TA, gruppo collaborative IST 3. [L'international Stroke Trial 3: trombolisi per l'ictus ischeico acuto]. Proceedings of the Conferenza Nazionale Sull'ictus Cerebrale; 2003 Mar 2‐4; Firenze. 2003:135.
- Ricci S, Celani MG, Righetti E, Mazzoli T. Thrombolysis for ischaemic stroke [Trombolisi per l' ictus ischemico acuto: lo studio IST 3]. www.strokeforum.org/Stroke2007/Abstracts/Freefiles/ACTER_02.htm (accessed 9 August 2007).
- Roots A, Birns J, Bhalla A, Rudd A. Nurse‐led telemedicine‐directed hyper‐acute stroke trial randomisation and management of post‐thrombolysis anaphylaxis. Cerebrovascular Diseases 2012;33(Suppl 2):477‐8. [Google Scholar]
- SCOPE (Stroke Complications and Outcomes Prediction Engine) Collaborations, IST, Lewis SC, Sandercock PA, Dennis MS. Predicting outcome in hyper‐acute stroke: validation of a prognostic model in the Third International Stroke Trial (IST3). Journal of Neurology Neurosurgery and Psychiatry 2008;79(4):397‐400. [DOI] [PubMed] [Google Scholar]
- Sakka E, Perry D, Buchanan D, Innes K, Sandercock P, Lindley RI, et al. Medical image management for multicentre trials. Experience from the Third International Stroke Trial (IST‐3) with 6576 scans. Cerebrovascular Diseases 2013;35(Suppl 3):562. [Google Scholar]
- Sandercock P. The Third International Stroke Trial (IST‐3) of thrombolysis. Main results & implications for clinical practice. Cerebrovascular Diseases 2012;34(1):6‐7. [Google Scholar]
- Sandercock P. Update on the 3rd international stroke trial; effects of thrombolysis among 3035 stroke patients within 6 h of onset. Journal of Thrombosis and Haemostasis 2012;10:e25. [Google Scholar]
- Sandercock P, Kane I, Lindley R, Lewis S, Dennis M, Wardlaw J. Baseline CT characteristics of the first 317 patients in the MRC Third International Stroke Trial (IST‐3). Journal of the Neurological Sciences 2005;238 Suppl 1:s439‐40. [Google Scholar]
- Sandercock P, Lindley R. The Third International Stroke Trial (IST3). www.eurostroke.org (no date given).
- Sandercock P, Lindley R, Wardlaw J. Protocol 06PRT/1269: Third International Stroke Trial (IST‐3). www.thelancet.com/journals/lancet/misc/protocol/06PRT‐1269 (accessed 10 August 2007).
- Sandercock P, Lindley R, Wardlaw J. The Third International Stroke Trial. Book of Abstracts of the UK Stroke Forum Conference; 2006 Dec 7‐8; Harrogate. 2006:59.
- Sandercock P, Lindley R, Wardlaw J. Third International Stroke Trial (IST‐3). A large‐scale randomised trial of thrombolytic therapy for patients with acute ischaemic stroke ISRCTN ISRCTN25765518. Proceedings of the 19th European Stroke Conference 2010. 25‐28 May 2010; Barcelona, Spain. 2010.
- Sandercock P, Lindley R, Wardlaw J. Thrombolysis for acute ischaemic stroke after ECASS 3 ‐ what are remaining uncertainties?. International Journal of Stroke 2009;4(Suppl 2):5. [DOI] [PubMed] [Google Scholar]
- Sandercock P, Lindley R, Wardlaw J, Dennis M, Innes K, Cohen G, et al. IST‐3 Collaborative Group. Update on the third international stroke trial (IST‐3) of thrombolysis for acute ischaemic stroke and baseline features of the 3035 patients recruited. Trials 2011;12:252‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandercock P, Lindley R, Wardlaw J, Dennis M, Lewis S, Venables G, et al. The third international stroke trial (IST) of thrombolysis for acute ischaemic stroke. Trials 2008;9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandercock P, Lindley R, Wardlaw J, Whiteley W, Murray G, IST‐3 collaborative group. Statistical analysis plan for the third International Stroke Trial (IST‐3); part of a `thread` of reports of the trial. International Journal of Stroke 2012;7(3):186‐7. [DOI] [PubMed] [Google Scholar]
- Sandercock P, Lindley R, IST‐3 Collaborative Group. Third International Stroke Trial (IST‐3). Proceedings of the British Association of Stroke Physicians Annual Conference; 2003 Jan 22; Stoke on Trent. 2003:59.
- Sandercock P, Lindley RI, Wardlaw J, Dennis M, Lewis S. The Third International Stroke Trial (IST‐3). International Journal of Stroke 2008;3(Suppl 1):444. [Google Scholar]
- Sandercock P, Murray V, Berge E, IST 3 Collaborative Group. Third International Stroke Trial (IST 3). Program and Abstract Book from the 12th Nordic Meeting on Cerebrovascular Diseases; 2003 Sep 17‐20; Oslo. 2003:26.
- Sandercock P, Wardlaw J, Lindley R. Age and time to randomisation in the 2017 patients recruited to date in the third International Stroke Trial (IST‐3). Cerebrovascular Diseases 2010;29(Suppl 2):161. [Google Scholar]
- Sandercock P, Wardlaw J, Lindley R. Time to randomisation following symptom onset in 2117 patients recruited to date in the third international stroke trial (IST‐3). International Journal of Stroke 2010;5(Suppl 2):194‐5. [Google Scholar]
- Sandercock P, Wardlaw JM, Lindley RI. Third International Stroke Trial (IST‐3). A large‐scale randomised trial of thrombolytic therapy for patients with acute ischaemic stroke ISRCTN25765518. Proceedings of the 20th European Stroke Conference 2011. 24‐27 May 2011; Hamburg, Germany. 2011.
- Sandercock P, Wardlaw JM, Lindley RI, Dennis MS, Cohen G. The third international stroke trial (IST‐3) main results part I: primary and secondary outcomes among 3035 patients randomised. Cerebrovascular Diseases 2012;33(Suppl 2):16. [Google Scholar]
- Sandercock PA, Lindley R, Wardlaw J, Lewis S, Dennis M. The Third International Stroke Trial (IST‐3). Proceedings of the 17th European Stroke Conference; 2008 May 13‐16; Nice. 2008:Abst 4.
- Sandercock PA, Lindley RI, Wardlaw J. Third International Stroke Trial (IST‐3). A large‐scale randomised trial of thrombolytic therapy for patients with acute ischaemic stroke ISRCTN25765518. Proceedings of the International Stroke Conference 2011. 8‐11 February 2011; Los Angeles, USA. 2011.
- Sandercock PA, Wardlaw J, Lindley R. Third International Stroke Trial (IST‐3). Proceedings of the International Stroke Conference 2010. 24‐26 February 2010; San Antonio Texas, USA. 2010.
- Sandercock PA, Wardlaw J, Lindley R, Dennis M, Lewis S. Thrombolysis for stroke ‐ what are the "grey areas" of uncertainty and are they changing? Results from the third international stroke trial (IST‐3). Cerebrovascular Diseases 2008;25(Suppl 2):192. [Google Scholar]
- Sandercock PA, Wardlaw JM, Lindley RI, Dennis MS. Third international stroke trial (IST‐3): Effect of IV RT‐PA < 6 hours in acute ischaemic stroke on living circumstances and health related quality of life at six months. Stroke 2013;44:AWMP22. [Google Scholar]
- Sandercock PAG. Imaging perfusion deficits and thrombolysis safety and efficacy in acute ischaemic stroke: the Third International Stroke Trial. www.controlled‐trials.com/mrct/trial/1988897/third+international+stroke (accessed July 10th 2014). [PubMed]
- Sandercock PAG, Lindley RI. IST3. stroke.ahajournals.org/cgi/content/full/34/2/e1 (accessed July 10th 2014).
- Sandercock PAG, Lindley RI, Wardlaw JM. Confounding factors in acute stroke trials: how do stroke severity, brain image appearances, BP and age vary with time to randomisation? IST‐3 experience. International Journal of Stroke 2010;5(Suppl 3):25. [Google Scholar]
- Sandercock PAG, Lindley RI, Wardlaw JM, Dennis MS, Cohen G. The third international stroke trial (IST‐3): benefits of iv thrombolysis on functional outcome and health‐related quality of life (HRQoL) persist to 18 months after treatment. Cerebrovascular Diseases 2013;35(Suppl 3):34. [Google Scholar]
- Sandercock PAG, Lindley RI, Wardlaw JM, Dennis MS, Lewis SC. Thrombolysis for stroke ‐ what are the 'grey areas' of uncertainty and are they changing? Results from the IST‐3. International Journal of Stroke 2008;3(Suppl 1):138. [Google Scholar]
- Sandercock PAG, Lindley RI, Wardlaw JM, Dennis MS, Lewis SC on behalf of the IST‐3 collaborative group. The MRC Third International Stroke Trial (IST‐3). Proceedings of the UK Stroke Forum Conference; 2007 Dec 4‐6; Harrogate. 2007:69.
- Sandercock PAG, Lindley RI, Wardlaw JM, Dennis MS, Lewis SC on behalf of the IST‐3 collaborative group. Thrombolysis for stroke ‐ what are the "grey areas" of uncertainty and are they changing? Results from the Third International Stroke Trial (IST‐3). Proceedings of the UK Stroke Forum Conference; 2007 Dec 4‐6; Harrogate. 2007:71‐2.
- Sandercock PAG, Wardlaw JM, Lindley RI, Cohen G. The third international stroke trial (IST‐3) of intravenous rt‐PA: Effect of age and time on treatment effect among 3035 patients randomized. International Journal of Stroke 2012;7:6. [Google Scholar]
- Skowronska M, Bembenek, J, Sandercock P, Kane I, Kobayashi A, Czlonkowska A, et al. Persisting hyperdense artery sign, ASPECTS score and risk of developing mass effect: an analysis based on the first 389 patients from the IST‐3 Trial. Cerebrovascular Diseases 2006;21(Suppl 4):33‐4. [Google Scholar]
- Slot KB, Berge E, Wardlaw J. Haemorrhagic transformation of a recent silent cerebral infarct during thrombolytic stroke treatment. BMJ Case Reports 2008;2008:bcr0620080266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables G, Kane I, Sandercock P, Wardlaw J, IST‐3 collaborative group. Baseline CT characteristics of the first 137 patients in the start‐up phase of the MRC Third International Stroke Trial (IST‐3). Journal of Neurology, Neurosurgery & Psychiatry 2006;77:136. [Google Scholar]
- Venables G, Lindley R, Sandercock P, Wardlaw J, Cohen G. The third international stroke trial (IST‐3) of intravenous RT‐PA: Effect of age and time among 3035 patients randomised. Annals of Neurology 2012;72:S14. [Google Scholar]
- Wardlaw J, Carpenter T, Kummer R, Cohen G, Lindley R, Sandercock P. Perfusion imaging in patients randomised to rt‐PA or control in the third international stroke trial (IST‐3): baseline characteristics and association with outcome. Stroke 2013;44:A10. [Google Scholar]
- Wardlaw J, Sandercock P, Carpenter T, Kummer R, Heijn A, Murray V, et al. The Third International Stroke Trial (IST‐3) a large randomised controlled trial of rt‐PA in acute ischemic stroke: perfusion and angiography study: current progress and recruitment. Proceedings of the International Stroke Conference 2011. 8‐11 February 2011; Los Angeles, USA. 2011.
- Wardlaw J, Kummer R, Farrall A, Cala L, Heijne A, Peeters A, et al. The impact of ischemic and structural brain tissue changes on response to rt‐PA: An analysis of imaging from 3035 patients in the third international stroke trial. Stroke 2013;44:A104. [Google Scholar]
- Wardlaw JM, Bath P, Sandercock P, Perry D, Palmer J, Watson G, et al. The NeuroGrid stroke exemplar clinical trial protocol. International Journal of Stroke 2007;2(1):63‐9. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Carpenter T, Cohen G, Kummer R, Lindley R, Sandercock P. Does perfusion imaging lesion size or mismatch influence six month outcomes after rt‐PA given up to six hours after acute ischaemic stroke? The third International Stroke Trial (IST‐3). Cerebrovascular Diseases 2013;35(Suppl 3):111. [Google Scholar]
- Whiteley W, Lindley R, Wardlaw J, Sandercock P, IST‐3 Collaborative Group. Third International Stroke Trial. International Journal of Stroke 2006;1(3):172‐6. [DOI] [PubMed] [Google Scholar]
- Whiteley W, Sandercock P, Lewis S, Wardlaw J, Venables G, et al. The performance of UK centres in the Third International Stroke Trial. Book of Abstracts from the UK Stroke Forum Conference; 2006 Dec 7‐8; Harrogate, UK. 2006:57‐8.
- Whiteley W, Sandercock P, Lindley R, Wardlaw J, IST‐3 Collaborative Group. CT appearance at baseline in the MRC Third International Stroke Trial: relationship with time from stroke onset to scan and stroke severity. Stroke 2007;38(2):488. [Google Scholar]
- Whiteley WN, Cohen G, Wardlaw J, Lindley R, Sandercock PAG. IST‐3 trial: impact of rt‐PA on survival to 18 months post ischaemic stroke. Cerebrovascular Diseases 2013;35(Suppl 3):19. [Google Scholar]
- Whiteley WN, Thompson D, Cohen G, Lindley R, Wardlaw JM, Sandercock PAG. Predictions of intracranial haemorrhage and the risks and benefits of rtPA in acute ischaemic stroke: an analysis of the IST‐3 trial. Cerebrovascular Diseases 2013;35(Suppl 3):169. [Google Scholar]
JTSG 1993 {published and unpublished data}
- Terashi A, Kobayashi Y, Katayama Y, Inamura K, Kazama M, Abe T. Clinical effects and basic studies of thrombolytic therapy on cerebral thrombosis. Seminars in Thrombosis and Hemostasis 1990;16(3):236‐41. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Hayakawa T, Kikuchi H, Abe T. Thrombolytic therapy in embolic and thrombotic cerebral infarction: a cooperative study. In: Hacke W, Zoppo GJ, Hirschberg M editor(s). Thrombolytic Therapy in Acute Ischemic Stroke. Berlin Heidelberg: Springer‐Verlag, 1991:168‐74. [Google Scholar]
- Yamaguchi T, Hayakawa T, Kikuchi H, Japanese Thrombolysis Study Group. Intravenous tissue plasminogen activator in acute thromboembolic stroke: A placebo‐controlled, double‐blind trial. In: Zoppo GJ, Mori E, Hacke W editor(s). Thrombolytic Therapy in Acute Ischemic Stroke II. New York: Springer Verlag, 1993:59‐65. [Google Scholar]
- Yamaguchi T, Hayakawa T, Kiuchi H, Japanese Thrombolysis Study Group. Intravenous tissue plasminogen activator ameliorates the outcome of hyperacute embolic stroke. Cerebrovascular Diseases 1993;3(4):269‐72. [Google Scholar]
- Yamaguchi T, Mori E, Minematsu K, Nakagawara J, Hashi K, Saito I, et al. Alteplase at 0.6 mg/kg for acute ischemic stroke within 3 hours of onset. Japan Alteplase Clinical Trial (J‐ACT). Stroke 2006;37(7):1810‐5. [DOI] [PubMed] [Google Scholar]
MAST‐E 1996 {published and unpublished data}
- Besson G, Moulin T, MAST Study Group. Intra observer concordance of the neuroradiologic reviewing committee in CT scan reviewing in MAST‐E. Cerebrovascular Diseases 1996;6(Suppl 2):21‐2. [Google Scholar]
- Donnan GA, Davis SM, Chambers BR, Gates PC, Hankey GJ, McNeill JJ, et al. Trials of streptokinase in severe acute ischaemic stroke. Lancet 1995;345(8949):578‐9. [PubMed] [Google Scholar]
- Hacke W, Marler J. TPA in acute stroke. European Heart Journal 1997;18(8):1360. [DOI] [PubMed] [Google Scholar]
- Hommel M, Besson G, Serradj AJ, MAST‐E group. Multicentre Acute Stroke Trial‐Europe trial: predictors of good outcome. Cerebrovascular Diseases 1996;6(3):183. [Google Scholar]
- Hommel M, Besson G, Serradj AJ, MAST‐E group. Multicentre Acute Stroke Trial‐Europe trial: risk factors for the development of oedema, haemorrhage, bad neurological, bad functional outcome and death. Cerebrovascular Diseases 1996;6(3):182. [Google Scholar]
- Hommel M, Boissel JP on behalf of the MAST Group. Trials of streptokinase in severe acute ischaemic stroke (authors' reply). Lancet 1995;345(8949):578. [PubMed] [Google Scholar]
- Hommel M, Boissel JP, Cornu C, Boutitie F, Lees KR, Besson G, et al. Termination of trial of streptokinase in severe acute ischaemic stroke. Lancet 1994;345(8941):57. [DOI] [PubMed] [Google Scholar]
- Hommel M, Lizeratti J, Serradj AJ, Besson G, MAST‐E Group. Multicentre Acute Stroke Trial‐Europe trial: predictors of good outcome. Cerebrovascular Diseases 1996;6(Suppl 2):76. [Google Scholar]
- Jaillard A, Cornu C, Durieux A, Moulin T, Boutitie F, Lees KR, et al. Haemorrhagic transformation in acute ischaemic stroke. The MAST‐E Study. Stroke 1999;30(7):1326‐32. [DOI] [PubMed] [Google Scholar]
- Jaillard AS, Hommel M. Predictive factors related to outcome at one year after IV thrombolysis/the MAST‐E study. Abstracts from the 6th International Symposium on Acute Stroke and Thrombolytic Therapy; 2000 Nov 25‐29; Hamilton Island (Australia). 2001.
- Jaillard AS, Hommel M. Predictive factors related to outcome at one year after intravenous thrombolysis: the MAST‐E study. Cerebrovascular Diseases 2000;10(Suppl 2):79. [Google Scholar]
- Moulin T, Besson G, Chavot D, Crepin‐Leblond T, Garnier P, Tatu L, et al. Predictive value of early CT signs in MCA occlusion. Cerebrovascular Diseases 1995;5(3):189. [Google Scholar]
- Moulin T, Besson G, Crepin‐Leblond T, Carnier P, Tatu L, Chavot D, et al. Predictive factors for hemorrhagic transformations in MAST‐E. Cerebrovascular Diseases 1996;6(Suppl 2):128. [Google Scholar]
- Moulin T, Besson G, Crepin‐Leblond, Garnier P, Tau L, Chavot D, et al. Haemorrhagic transformations in MAST‐E trial: predictive factors. Cerebrovascular Diseases 1996;6(3):182. [Google Scholar]
- Shahar E, McGovern PG. Trials of streptokinase in severe acute ischaemic stroke. Lancet 1995;345(8949):578. [PubMed] [Google Scholar]
- The Multicentre Acute Stroke Trial ‐ Europe Study Group. Thrombolytic therapy with streptokinase in acute ischaemic stroke. New England Journal of Medicine 1996;335(3):145‐50. [DOI] [PubMed] [Google Scholar]
MAST‐I 1995 {published and unpublished data}
- Aritzu E, Candelise L, Motto C, Ciccone A, Piana A, MAST‐I Group. Long term effect of thrombolytic treatment in acute ischaemic stroke. Cerebrovascular Diseases 1996;6(Suppl 2):129. [Google Scholar]
- Aritzu E, Motto C, Ciccone A, Candelise L, MAST‐I Study Group. Prognostic value of haemorrhagic transformations in acute ischaemic stroke. Cerebrovascular Diseases 1996;6(3):190. [Google Scholar]
- Barer D. Multicentre Acute Stroke Trial ‐ Italy. Lancet 1996;347(8998):391‐2. [PubMed] [Google Scholar]
- Candelise L. The long term effects of acute treatment with streptokinase and/or aspirin. Book of Abstracts Proceedings of the 6th International Conference on Thrombolysis in Acute Ischaemic Stroke; 2000 Nov 25‐29; Hamilton Island (Australia). 2001:3.
- Candelise L, Aritzu E, Ciccone A, Ricci S, Wardlow J. Multicentre Acute Stroke Trial ‐ Italy. Lancet 1996;347(8998):393. [PubMed] [Google Scholar]
- Candelise L, Ciccone A, Motto C. Extraparenchymal bleeding predicts an unfavorable outcome in patients with hemorrhagic transformation. Stroke 2000;31(7):1785. [DOI] [PubMed] [Google Scholar]
- Candelise L, Motto C, Aritzu E, Ciccone A, Ricci S, MAST‐I Study group. Aspirin given within 6 hours reduces long term case fatality but not stroke related disability. Cerebrovascular Diseases 1996;6(Suppl 2):23‐4. [Google Scholar]
- Candelise L, Roncaglione C, Aritzu E, Ciccone A, Maggioni AP, MAST‐I Group. Thrombolytic therapy. From myocardial to cerebral infarction. Italian Journal of Neurological Sciences 1996;17(1):5‐21. [DOI] [PubMed] [Google Scholar]
- Carolei A, Marini C, Motolese M. Multicentre Acute Stroke Trial ‐ Italy. Lancet 1996;347(8998):392. [PubMed] [Google Scholar]
- Ciccone A, Motto C, Aritzu E, Piana A, Candelise L on behalf of the MAST‐I Collaborative Group. Negative interaction of aspirin and streptokinase in acute ischaemic stroke: further analysis of the Multicentre Acute Stroke Trial‐Italy. Cerebrovascular Diseases 2000;10(1):61‐4. [DOI] [PubMed] [Google Scholar]
- Ciccone A, Motto C, Aritzu E, Piana A, Candelise L on behalf of the MAST‐I Collaborative Group. Risk of aspirin use plus thrombolysis after acute ischaemic stroke: a further MAST‐I analysis. MAST‐I Collaborative Group. Multicentre Acute Stroke Trial‐‐Italy. Lancet 1998;352(9131):880. [DOI] [PubMed] [Google Scholar]
- Fiorelli M, Kummer R, Bastianello S, Bozzoa L. Extraparenchymal bleeding predicts an unfavorable outcome in patients with hemorrhagic transformation. Stroke 2000;31(7):1785. [DOI] [PubMed] [Google Scholar]
- Horton R. MAST‐I: agreeing to disagree: Multicentre Acute Stroke Trial‐‐Italy Group. Lancet 1995;346(8989):1504. [DOI] [PubMed] [Google Scholar]
- Motto C, Aritzu E, Boccardi E, Grandi C, Piana A, Candelise SL. Reliability of hemorrhagic transformation diagnosis in acute ischemic stroke. Stroke 1997;28(2):302‐6. [DOI] [PubMed] [Google Scholar]
- Motto C, Candelise L, Aritzu E, Ciccone A, Piana A, MAST‐I Group. Aspirin in combination with thrombolysis increases the risk of early death but not of intracranial bleeding in patients with acute ischaemic stroke. Cerebrovascular Diseases 1996;6(Suppl 2):129. [Google Scholar]
- Motto C, Ciccone A, Aritzu E, Boccardi E, Grandi C, Piana A, et al. Haemorrhage after an acute ischaemic stroke. Stroke 1999;30(4):761‐4. [DOI] [PubMed] [Google Scholar]
- Muir KW. Multicentre Acute Stroke Trial‐‐Italy. Lancet 1996;347(8998):391. [PubMed] [Google Scholar]
- Multicentre Acute Stroke Trial ‐ Italy (MAST‐I) Group. Randomised controlled trial of streptokinase, aspirin, and combination of both in treatment of acute ischaemic stroke. Lancet 1995;346(8989):1509‐14. [PubMed] [Google Scholar]
- Rothwell P. Multicentre Acute Stroke Trial‐ ‐Italy. Lancet 1996;347(8998):392‐3. [PubMed] [Google Scholar]
- Sandercock PAG. Thrombolytic therapy for acute ischaemic stroke: promising, perilous or unproven?. Lancet 1996;346(8989):1504‐5. [DOI] [PubMed] [Google Scholar]
- The MAST‐I Collaborative Group. Is thrombolysis useful for acute stroke patients? The experience of the MAST‐I study. In: Yamaguchi T, Mori E, Minematsu K, Zoppo GJ editor(s). Thrombolytic Therapy in Acute Ischaemic Stroke III. Tokyo: Springer Verlag, 1995:198‐205. [Google Scholar]
- The MAST‐Italy Investigators. Predictors of good outcome in the MAST Italy trial. Cerebrovascular Diseases 1996;6(3):183. [Google Scholar]
- The MAST‐Italy Investigators. Risk factors in the MAST‐Italy trial. Cerebrovascular Diseases 1996;6(3):181. [Google Scholar]
- Tognoni G, Roncaglione MC. Dissent: an alternative interpretation of MAST‐I. Lancet 1996;346(8989):1515. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Dorman PJ. Thrombolysis in acute ischaemic stroke. Neurology Reviews International 1996;1(1):2‐6. [Google Scholar]
- Wardlaw JM, Dorman PJ, Candelise L, Signorini DF. The influence of baseline prognostic variables on outcome after thrombolysis: MAST‐Italy Collaborative Group. Journal of Neurology 1999;246(11):1059‐62. [DOI] [PubMed] [Google Scholar]
MELT 2007 {published data only}
- Ezura M, Takahashi A, Matsumoto Y, Ogawa A. MCA Embolism Local Fibrinolytic Intervention Trial (MELT) Japan. In: Kanno T, Kato Y editor(s). Minimally Invasive Neurosurgery and Multidisciplinary Neurotraumatology. Tokyo: Springer, 2006:85‐9. [Google Scholar]
- MELT. MCA Embolism Local Fibrinolytic Intervention Trial. melt.umin.ac.jp/en/ (accessed 8 May 2004).
- Mori E. CT in acute stroke: learning from MELT‐JAPAN. International Journal of Stroke 2006;1(Suppl 2):16. [Google Scholar]
- Ogawa. MCA‐Embolism Local fibrinolytic intervention Trial. www.strokecenter.org/trials/TrialDetail.aspx?tid=589&printView=true (accessed 6 November 2008).
- Ogawa A. MCA‐Embolism Local Fibrinolytic Intervention Trial. www.strokecenter.org/trials/TrialDetail.aspx?tid=589 (accessed 18 July 2007).
- Ogawa A, Inoue T. MELT Japan (MCA ‐ Embolism Local Fibrinolytic Intervention Trial Japan). Nippon Rinsho 2006;64(Suppl 7):519‐23. [PubMed] [Google Scholar]
- Ogawa A, Mori E, Minematsu K, Taki W, Takahashi A, Nemoto S, et al. Randomized trial of intraarterial infusion of urokinase within 6 hours of middle cerebral artery stroke. The Middle Cerebral Artery Embolism Local Fibrinolygic Intervention Trial (MELT) Japan. Stroke 2007;38(10):2633‐9. [DOI] [PubMed] [Google Scholar]
- Saver JL. Intra‐arterial fibrinolysis for acute ischemic stroke. The message of MELT. Stroke 2007;38(10):2627‐8. [DOI] [PubMed] [Google Scholar]
- Yong M, Mau J. Characteristics of blood pressure profiles and their prognostic value of stroke outcome. International Journal of Stroke 2006;1 Suppl:40‐1. [Google Scholar]
Mori 1992 {published and unpublished data}
- Mori E, Tabuchi M, Ohsumi Y, Yoshida T, Ohkawa S, Yoneda Y, et al. Intra‐arterial urokinase infusion therapy in acute thromboembolic stroke. Stroke 1990;21(Suppl 1):I‐74. [Google Scholar]
- Mori E, Tabuchi M, Yoshida T, Yamadori A. Intracarotid urokinase with thromboembolic occlusion of the middle cerebral artery. Stroke 1988;19(7):802‐12. [DOI] [PubMed] [Google Scholar]
- Mori E, Yoneda Y, Ohksawa S, Yoshida T, Ohsumi Y, Tabuchi M, et al. Double‐blind, placebo‐controlled trial of recombinant tissue plasminogen activator (rt‐PA) in acute carotid stroke. Neurology 1991;41(Suppl 1):347. [Google Scholar]
- Mori E, Yoneda Y, Tabuchi M, Yoshida T, Ohkawa S, Ohsumi Y, et al. Intravenous recombinant tissue plasminogen activator in acute carotid artery territory stroke. Neurology 1992;42(5):976‐82. [DOI] [PubMed] [Google Scholar]
Morris 1995 {published data only}
- Morris AD, Ritchie C, Grosset DG, Adams FG, Lees KR. A pilot study of streptokinase for acute cerebral infarction. Quarterly Journal of Medicine 1995;88(10):727‐31. [PubMed] [Google Scholar]
- Morris AD, Ritchie C, Grosset DG, Squire I, Taylor A, Bone I, et al. Streptokinase for the treatment of acute cerebral infarction: a placebo controlled pilot study. Clinical Science 1994;86(Suppl 30):4p. [Google Scholar]
NINDS 1995 {published data only}
- Anonymous. Heart treatment reduces damage from stroke. Laboratory Medicine 1996;27(5):296. [Google Scholar]
- Balucani C, Levine S, Khoury Khoury J, Khatri P, Saver JL, Broderick JP. Rapidly improving stroke symptoms: relation to stroke volumes and discharge disposition. Stroke 2012;43(2):Abst.3916. [Google Scholar]
- Barch C, Spilker J, Bratina P, Rapp K, Daley S, Donnarumma R, et al. Nursing management of acute complications following rt‐PA in acute ischemic stroke. Journal of Neuroscience Nursing 1997;29(6):367‐72. [DOI] [PubMed] [Google Scholar]
- Bonomo J, Yeatts S, Kleindorfer D, Khatri P. Dramatic early improvement in the NINDS trial: better 90 day outcomes and no increased rates of intracranial hemorrhage. Stroke 2009;40(4):e120. [Google Scholar]
- Broderick J, Lu M, Jackson C, Pancioli A, Tilley BM, Fagan S, et al. Apo E phenotype and the efficacy of intravenous t‐PA in acute ischemic stroke. Stroke 2000;31(11):2828. [Google Scholar]
- Broderick JP, Lu M, Kothari R, Levine SR, Lyden PD, Haley C, et al. Finding the most powerful measures of the effectiveness of tissue plasminogen activator in the NINDS t‐PA Stroke Trial. Stroke 2000;31(10):2335‐41. [DOI] [PubMed] [Google Scholar]
- Brott T. Thrombolysis for stroke. Archives of Neurology 1996;53(12):1305‐6. [DOI] [PubMed] [Google Scholar]
- Brott T, Kothari R, Fagan S, Frankel M, Grotta JC, Broderick J, et al. Hypertension and its treatment in the NINDS rt‐PA stroke trial. Stroke 1998;29(8):1504‐9. [DOI] [PubMed] [Google Scholar]
- Brown DL, Johnston KC, Wagner DP, Haley EC. Predicting major neurological improvement with intravenous rt‐PA treatment of stroke. Stroke 2003;34(1):320. [DOI] [PubMed] [Google Scholar]
- Brown DL, Johnston KC, Wagner DP, Haley EC Jr. Predicting major neurological improvement with intravenous recombinant tissue plasminogen activator treatment of stroke. Stroke 2004;35(1):147‐50. [DOI] [PubMed] [Google Scholar]
- Bruno A, Levine SR, Frankel MR, Brott TG, Lin Y, Tilley BC, et al. Admission glucose level and clinical outcomes in the NINDS rt‐PA stroke trial. Neurology 2002;59(5):669‐74. [DOI] [PubMed] [Google Scholar]
- Bruno A, Saha C, Williams LS. Percent change of neurological deficits in acute stroke: another useful outcome measure. Stroke 2008;39(2):693. [Google Scholar]
- Bruno A, Saha C, Williams LS. Percent change on the National Institutes of Health Stroke Scale: a useful acute stroke outcome measure. Journal of Stroke and Cerebrovascular Diseases 2009;18(1):56‐9. [DOI] [PubMed] [Google Scholar]
- Burger KM, Jordan JD, Moshier E, Godbold J, Brown DM, Levine SR. The change in National Institute of Health Stroke Scale score over time in the NINDS rtPA stroke study. Stroke 2005;36(2):494. [Google Scholar]
- Cordina SM, Vazquez G, Suri FK, Lakshminarayan K, Mohammad Y, Qureshi AI. Predictors of functional improvement after the first 3 months following ischemic stroke. Stroke 2009;40(4):e254. [Google Scholar]
- Cordina SM, Vazquez G, Suri MF, Lakhsminarayan K, Adams HP, Qureshi AI. Does Aspirin failure increase the chances of recurrent stroke and/or death among patients with ischemic stroke: a pooled analysis of two prospective trials. Stroke 2010;41(4):e214‐5. [Google Scholar]
- Dachs RJ, Burton JH, Joslin J. A user`s guide to the NINDS rt‐PA stroke trial database. PLos Medicine 2008;5(5):e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppo GJ. Tissue plasminogen activator for acute ischaemic stroke (authors' reply). New England Journal of Medicine 1996;334(21):1406. [PubMed] [Google Scholar]
- Demchuk A, Hill MD, Barber PA, Silver B, Patel S, Levine SR, et al. Importance of early ischemic CT changes using ASPECTS in NINDS rtPA stroke study. Stroke 2005;36(2):448. [DOI] [PubMed] [Google Scholar]
- Demchuk AM, Hill MD, Barber PA, Silver B, Patel SC, Levine SR, et al. Importance of early ischemic computed tomography changes using ASPECTS in NINDS rtPA stroke study. Stroke 2005;36(10):2110‐5. [DOI] [PubMed] [Google Scholar]
- Demchuk AM, Khan F, Hill MD, Barber PA, Silver B, Patel S, et al. Importance of leukoaraiosis on CT for tissue plasminogen activator decision making: evaluation of the NINDS rt‐PA Stroke Study. Cerebrovascular Diseases 2008;26(2):120‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JS, Vittinghoff E, Johnston SC. Predictors of slow recovery after acute ischemic stroke. Stroke 2007;38(2):521. [Google Scholar]
- Fagan SC, Morgenstern LB, Petitta A, Ward RE, Tilley BC, Marler JR, et al. Cost‐effectiveness of tissue plasminogen activator for acute ischemic stroke. Neurology 1998;50(4):883‐90. [DOI] [PubMed] [Google Scholar]
- Fagan SC, Morgenstern LB, Petitta A, et al. Prompt tissue plasminogen activator therapy increased quality of life and reduced costs in acute ischemic stroke. ACP Journal Club 1998;September/October:52. [Google Scholar]
- Feng W, Vasquez G, Suri MFK, Lakshminarayan K, Qureshi AI. Repeated‐measures analysis of the National Institute of Neurological Disorders and Stroke rt‐PA Stroke Trial. Journal of Stroke and Cerebrovascular Diseases 2011;20(3):241‐6. [DOI] [PubMed] [Google Scholar]
- Fiorelli M, Kummer R. Early ischemic changes on computed tomography in patients with acute stroke. JAMA 2002;287(18):2361‐2. [DOI] [PubMed] [Google Scholar]
- Flaherty ML, Karlawish J, Khoury JC, Kleindorfer D, Woo D, Broderick JP. How important is surrogate consent for stroke research?. Neurology 2008;71(20):1566‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty ML, Khoury JC, Kleindorfer D, Broderick JP. How important is surrogate consent for stroke research?. Stroke 2008;39(2):600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freidman HS. Tissue plasminogen activator for acute ischaemic stroke. New England Journal of Medicine 1996;334(21):1405. [PubMed] [Google Scholar]
- Genentech, Inc. Activase for acute ischemic stroke. Clinical Review. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm080832.pdf(accessed 25 July 2014).
- Georgiadis AL, Cordina SM, Vazquez G, Tariq N, Suri MFK, Lakshminarayan K, et al. Aspirin treatment failure and the risk of recurrent stroke and death among patients with ischemic stroke. Journal of Stroke and Cerebrovascular Diseases 2013;22(2):100‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman M, Tanne D, Shaftman SR, Tilley BC, Levine SR. Renal function, hydration status, and outcome from stroke after thrombolysis. Stroke 2010;41(4):e331. [Google Scholar]
- Grotta J for the National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group. The NINDS Stroke Study Group Response (author's response). BMJ 2002;324:723. [Google Scholar]
- Grotta J, Marler J. Intravenous rt‐PA: a tenth anniversary reflection. Surgical Neurology 2007;68(S1):12‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotta JC. Heart treatment reduces damage from stroke. Laboratory Medicine 1996;27(5):296. [Google Scholar]
- Grotta JC. The NINDS Stroke Study group responses. Journal of Stroke and Cerebrovascular Diseases 2002;11(3/4):121‐4. [DOI] [PubMed] [Google Scholar]
- Grotta JC, Chiu D, Lu M, Patel S, Levine SR, Tilley BC, et al. Agreement and variability in the interpretation of early CT changes in stroke patients qualifying for intravenous rtPA therapy. Stroke 1999;30(8):1528‐33. [DOI] [PubMed] [Google Scholar]
- Grotta JC, Welch K, Fagan SC, Lu Mei, Frankel MR, Brott T, et al. Clinical deterioration following improvement in the NINDS rt‐PA stroke trial. Stroke 2001;32(3):661‐8. [DOI] [PubMed] [Google Scholar]
- Haley EC, Lewandowski C, Tilley BC. Myths regarding the NINDS rt‐PA stroke trial: setting the record straight. Annals of Emergency Medicine 1997;30(5):676‐82. [DOI] [PubMed] [Google Scholar]
- Hemmen TM, Ernstrom K, Raman R. Two hour improvement of patients in the NINDS t‐PA Trials and prediction of final outcome. Stroke 2011;42(3):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmen TM, Ernstrom K, Raman R. Two‐hour improvement of patients in the National Institute of Neurological Disorders and Stroke trials and prediction of final outcome. Stroke 2011;42(11):3163‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmen TM, Rapp KS, Emond JA, Raman R, Lyden PD. Analysis of the National Institute of Neurological Disorders and Stroke Tissue Plasminogen Activator Studies following European Cooperative Acute Stroke Study III patient selection criteria. Journal of Stroke and Cerebrovascular Diseases 2010;19(4):290‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzberg V, Ingall T, O`Fallon W, Asplund K, Goldfrank L, Louis T, et al. Methods and processes for the reanalysis of the NINDs tissue plasminogen activator for acute ischemic stroke treatment trial. Clinical Trials: Journal of the Society for Clinical Trials 2008;5(4):308‐15. [DOI] [PubMed] [Google Scholar]
- Hoffman JR Schriger DL. A graphic reanalysis of the NINDS Trial. Annals of Emergency Medicine 2009;54(3):329‐36. [DOI] [PubMed] [Google Scholar]
- Hong KS, Saver JL. Years of disability‐adjusted life gained as a result of thrombolytic therapy for acute ischemic stroke. Stroke 2010;41(3):471‐7. [DOI] [PubMed] [Google Scholar]
- Hong KS, Saver JL. Years of disability‐adjusted life gained as a result of thrombolytic therapy for acute ischemic stroke. Stroke 2010;41(4):e389‐90. [DOI] [PubMed] [Google Scholar]
- Ingall TJ, O'Fallon WM, Asplund K, Goldfrank LR, Hertzberg VS, Louis TA, et al. Findings from the reanalysis of the NINDS tissue plasminogen activator for acute ischemic stroke treatment trial. Stroke 2004;35(10):2418‐24. [DOI] [PubMed] [Google Scholar]
- Ingall TJ, O'Fallon WM, Louis TA, Hertzberg V, Goldfrank LR, Asplund K. Initial findings of the rt‐PA acute stroke treatment review panel. Cerebrovascular Diseases 2003;16 Suppl 4:125. [Google Scholar]
- Jauch EC, Lindsell C, Broderick J, Fagan SC, Tilley BC, Levine SR, et al. Association of serial biomarkers with acute ischemic stroke: the National Institute of Neurological Disorders and stroke recombinant tissue plasminogen activator stroke study. Stroke 2006;37(10):2508‐13. [DOI] [PubMed] [Google Scholar]
- Jauch EC, Lindsell C, Broderick J, Lin Y, Fagan SC, Tilley BC, et al. Serial biochemical markers are associated with acute ischemic stroke in the NINDS rtPA stroke study. Stroke 2005;36(2):419. [Google Scholar]
- Johnston KC, Connors AF Jr, Wagner DP, Haley EC Jr. Predicting outcome in ischemic stroke: external validation of predictive risk models. Stroke 2003;34(1):200‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston KC, Connors AF Jr, Wagner DP, Haley EC Jr. Risk adjustment effect on stroke clinical trials. Stroke 2004;35(2):e43‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston KC, Wagner DP, Connors AF, Haley EC. Risk adjustment uncovers greater treatment effect in NINDS tPA trial. Stroke 2003;34(1):256. [Google Scholar]
- Johnston SC, Easton JD. Are patients with acutely recovered cerebral ischemia more unstable?. Stroke 2003;34(10):2446‐50. [DOI] [PubMed] [Google Scholar]
- Kent DM, Price L, Selker HP. Are asymptomatic intracranial hemorrhages really innocuous?. Cerebrovascular Diseases 2003;16(Suppl 4):34. [Google Scholar]
- Khatri P, Kleindorfer DO, Yeatts SD, Saver JL, Levine SR, Lyden PD, et al. Strokes with minor symptoms. An exploratory analysis of the National Institute of Neurological Disorders and Stroke Recombinant Tissue Plasminogen Activator Trials. Stroke 2010;41(11):2581‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Yeatts S, Palesch Y, Broderick JP. Modified Rankin score group distributions as a clinical trial end point: A NINDS tPA stroke study reanalysis. Stroke 2007;38(2):576‐7. [Google Scholar]
- Kirchoff KF, Shaftman SR, Zach V, Tanne D, Tilley BC, Gorman M, et al. Utility of measuring distal motor function in the NIHSS: correlation with motor arm function. Proceedings of the American Academy of Neurology 62nd Annual Meeting 2010. 10‐17 April 2010; Toronto, Canada. 2010:Abst. P03.054.
- Kirshner HS, Schneck MJ, D'Addesio JP, Caplan LR, Grotta J. Should thrombolytic therapy be the first‐line treatment for acute ischemic stroke?. New England Journal of Medicine 1998;338(11):761‐3. [DOI] [PubMed] [Google Scholar]
- Kleindorfer D, Schneider A, Kissela BM, Woo D, Khoury J, Alwell K, et al. The effect of race and gender on patterns of rt‐PA use within a population. Journal of Stroke and Cardiovascular Diseases 2003;12(5):217‐20. [DOI] [PubMed] [Google Scholar]
- Koroshetz WJ. Tissue plasminogen activator for acute ischaemic stroke. New England Journal of Medicine 1996;334(21):1405‐6. [PubMed] [Google Scholar]
- Kwiatkowski T, Libman R, Tilley BC, Lewandowski C, Grotta JC, Lyden P, et al. The impact of imbalances in baseline stroke severity on outcome in the National Institute of Neurological Disorders and stroke recombinant tissue plasminogen activator stroke study. Annals of Emergency Medicine 2005;45(4):377‐84. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TG, Libman R, Frankel M, Tilley B, Morganstern L, Lu M, et al. The NINDS rt‐PA stroke study: sustained benefit at one year. Stroke 1998;29(1):288. [Google Scholar]
- Kwiatkowski TG, Libman RB, Frankel M, Tilley BC, Morgenstern LB, Lu M, et al. Effects of tissue plasminogen activator for acute ischaemic stroke at one year. New England Journal of Medicine 1999;340(23):1781‐7. [DOI] [PubMed] [Google Scholar]
- Lenzer J. Alteplase for stroke: money and optimistic claims buttress the "brain attack" campaign. BMJ 2002;324(7339):723‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzer J. Controversial stroke trial is under review following BMJ report. BMJ 2002;325(7373):1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Questioning thrombolytic use for cerebrovascular accidents. Journal of Emergency Medicine 1998;16(5):757‐8. [DOI] [PubMed] [Google Scholar]
- Libman R, Kwiatkowski T, Lyden P, Grotta JC, Tilley BC, Fagen SC, et al. Asymptomatic hemorrhagic transformation of cerebral infarction does not worsen long‐term outcome. Journal of Stroke and Cerebrovascular Diseases 2005;14(2):50‐4. [DOI] [PubMed] [Google Scholar]
- Liebeskind DS, Levine SR, Wang HJ, Patel SC. Hyperdense ischemic infarcts in the NINDS rt‐PA trials reflect hyperglycemia, not hemorrhage. Stroke 2009;40(4):e117. [Google Scholar]
- Lou M, Selim M. Does body weight influence the response to intravenous tissue plasminogen activator in stroke patients?. Cerebrovascular Diseases 2009;27(1):84‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou M, Selim MH. Does stroke patient's weight influence the response to intravenous t‐PA?. Stroke 2008;39(2):530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyden P, Lu M, Jackson C, Marler J, Kothari R, Brott T, et al. Underlying structure of the National Institutes of Health stroke scale. Results of a factor analysis. Stroke 1999;30(11):2347‐54. [DOI] [PubMed] [Google Scholar]
- Lyden PD, Lu M, Jackson CM, Marler J, Kothari R, Brott T, et al. Underlying structure of the NIH stroke scale: results of a factor analysis. Stroke 2000;31(1):56. [DOI] [PubMed] [Google Scholar]
- Magid D, Naviaux N, Wears RL. Stroking the data: re‐analysis of the NINDS trial. Annals of Emergency Medicine 2005;45(4):385‐7. [DOI] [PubMed] [Google Scholar]
- Mandava P, Brooks ME, Krumpelman CS, Kent TA. A new more sensitive method to assess balance among stroke patient populations. Stroke 2012;43(2):Abst. 2374. [Google Scholar]
- Mandava P, Kalkonde YV, Rochat RH, Kent TA. A matching algorithm to address imbalances in study populations. Application to the National Institute of Neurological Diseases and Stroke Recombinant Tissue Plasminogen Activator Acute Stroke Trial. Stroke 2010;41(4):765‐70. [DOI] [PubMed] [Google Scholar]
- Mandava P, Kent TA. A method to generate a control population for acute ischemic stroke trials: validation with NINDS and application to a phaseI treatment series. Cerebrovascular Diseases 2009;27(Suppl 6):172. [Google Scholar]
- Mandava P, Munoz M, Dalmeida W, Sarma K, Brook MA, Anderson J, et al. Hyperglycemia worsens outcome after rt‐PA in the large artery stroke subtype. Stroke 2011;42(3):e84. [DOI] [PubMed] [Google Scholar]
- Mandava P, Munoz M, Sarma K, Brooks MA, Dalmeida W, Anderson J, et al. Gender and race interact to influence outcome of ischemic stroke after treatment with rt‐PA. Stroke 2011;42(3):e164. [Google Scholar]
- Mandava P, Murthy SB, Munoz M, McGuire D, Simon RP, Alexandrov AV, et al. Explicit consideration of baseline factors to assess recombinant tissue‐type plasminogen activator response with respect to race and sex. Stroke 2013;44(6):1525‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandava P, Murthy SB, Munoz M, McGuire D, Simon RP, Alexandrov AV, et al. Gender differences in outcome following rt‐PA are mainly due to the influence of race. Cerebrovascular Diseases 2013;35(Suppl 3):277. [Google Scholar]
- Mandava P, Sarma AK, Anderson JA, Wehner W, Kent TA. How gender and race interact to influence outcome of ischemic stroke after treatment with rt‐PA. Cerebrovascular Diseases 2011;31(Suppl 2):263. [Google Scholar]
- Mandava P, Sarma AK, Dalmeida W, Anderson JA, Kent TA. Hyperglycemia is an independent risk factor for poor outcome after rt‐PA only in the large artery stroke subtype. Cerebrovascular Diseases 2011;31(Suppl 2):255. [Google Scholar]
- Mandava P, Sarma AK, Martini SR, Kent TA. Evaluation of subject matching methods to adjust for imbalances in stroke trials. Cerebrovascular Diseases 2012;33(Suppl 2):439‐40. [Google Scholar]
- Mann J. Truths about the NINDS study: setting the record straight. Western Journal of Medicine 2002;176(3):192‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann J, Ingall TJ, O'Fallon WM, Asplund K, Goldfrank LR, Hertzberg VS, et al. NINDS Reanalysis Committee's reanalysis of the NINDS trial (response). Stroke 2005;36(2):230‐2. [DOI] [PubMed] [Google Scholar]
- Marler JR, Tilley BC, Lu M, Brott T, Lyden P, Broderick JP, et al. Earlier treatment associated with better outcome in the NINDS TPA Stroke Study. Stroke 1999;30:244. [Google Scholar]
- Marler JR, Tilley BC, Lu M, Brott TG, Lyden PC, Grotta JC, et al. Early stroke treatment associated with better outcome. The NINDS rt‐PA Stroke Study. Neurology 2000;55(11):1649‐55. [DOI] [PubMed] [Google Scholar]
- Marler JR, NINDS rt‐PA Stroke Study Group. Tissue plasminogen activator for acute ischaemic stroke. New England Journal of Medicine 1999;334(21):1406. [PubMed] [Google Scholar]
- Martini SR, Yeatts SD, Belagage SR, Khatri P. No differential effect of tPA based on final stroke etiologic subtype: A reanalysis of the NINDS tPA Trial. Stroke 2010;41(4):e331. [Google Scholar]
- Messe SR, Kasner SE, Cucchiara BL, Demchuk A, Tanne D, Ouyang B, et al. Dosing errors did not have a major impact on outcome in the NINDS t‐PA Stroke Study. Journal of Stroke and Cerebrovascular Diseases 2011;20(3):236‐40. [DOI] [PubMed] [Google Scholar]
- Mohammad YM, Nasar A, Khatri I. Congestive heart failure is associated with poor outcome at 6 months in patients with ischemic stroke. Stroke 2005;36(2):524. [Google Scholar]
- Morgenstern L. Recent results from the NINDS‐tPA trial. Acta Neurologica Scandinavica 1996;94(Suppl 167):28‐9. [Google Scholar]
- Nichols C, Khoury J, Brott T, Broderick J. IV rt‐PA improves arterial recanalization rates and reduces infarct volumes in subjects with hyperdense artery sign on baseline CT. Stroke 2007;38(2):503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols C, Khoury J, Brott T, Broderick J. Intravenous recombinant tissue plasminogen activator improves arterial recanalization rates and reduces infarct volumes in patients with hyperdense artery sign on baseline computed tomography. Journal of Stroke and Cerebrovascular Diseases 2008;17(2):64‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norby KE, Siddiq, Adil, MM, Chaudhry SA, Qureshi AI. Long‐term outcomes of post‐thrombolytic intracerebral hemorrhage in ischemic stroke patients. Neurocritical Care 2013;18(2):170‐7. [DOI] [PubMed] [Google Scholar]
- O'Fallon M, Asplund K, Goldfrank LR, Hertzberg V, Ingall, TJ, Louis TA. The NINDS trial on t‐PA in acute ischemic stroke revisited. Main results from an independent re‐analysis of primary data. Program and abstract book. 12th Nordic Meeting on Cerebrovascular Diseases; 2003 Sep 17‐20; Oslo. 2003:27.
- Ovbiagele B, Saver JL. Day‐90 acute ischemic stroke outcomes can be derived from early functional activity level. Cerebrovascular Diseases 2010;29(1):50‐6. [DOI] [PubMed] [Google Scholar]
- Ovbiagele B, Saver JL. The smoking‐thrombolysis paradox applies to stroke as well as MI: smoking has early beneficial effects following intravenous thrombolysis for acute ischemic stroke. Stroke 2005;36(2):496. [Google Scholar]
- Patel SC, Levine S, Tilley BC, Grotta JC, Lu M, Frankel M, et al. Lack of clinical significance of early ischaemic changes on computed tomography in acute stroke. JAMA 2001;286(22):2830‐8. [DOI] [PubMed] [Google Scholar]
- Pearson M. Early treatment of stroke with t‐PA provides uncertain long‐term benefits. Lancet 1999;353:2132. [Google Scholar]
- Phan TG, Demchuk A, Srikanth V, Silver B, Patel S, Barber PA, et al. Relating ASPECTS infarct location to stroke disability in the NINDS rt‐PA trial: proof of concept study using penalized logistic regression. Cerebrovascular Diseases 2013;35(Suppl 3):407. [DOI] [PubMed] [Google Scholar]
- Phan TG, Demchuk A, Srikanth V, Silver B, Patel SC, Barber PA, et al. Proof of concept study: Relating infarct location to stroke disability in the NINDS rt‐PA Trial. Cerebrovascular Diseases 2013;35(6):560‐5. [DOI] [PubMed] [Google Scholar]
- Quereshi N. Tissue plasminogen activator for acute ischaemic stroke. New England Journal of Medicine 1996;334(21):1406. [PubMed] [Google Scholar]
- Qureshi AI, Alkawi A, Georgiadis AL, Ibrahim MS. Is a maximum dose of 90 mg of recombinant tissue plasminogen activator in patients weighing greater than 90 kg justified?. Stroke 2006;37(2):658. [Google Scholar]
- Qureshi AI, Chaudhry SA, Sivagnanam K, Rodriguez GJ, Suri MFK, Lakshminarayan K, et al. Clinical‐radiological severity mismatch phenomenon: patients with severe neurological deficits without matching infarction on computed tomographic scan. Journal of Neuroimaging 2013;23(1):21‐7. [DOI] [PubMed] [Google Scholar]
- Qureshi AI, Ezzeddine MA, Nasar A, Suri MF, Kirmani JF, Janjua N, et al. Is IV tissue plasminogen activator beneficial in patients with hyperdense artery sign?. Neurology 2006;66(8):1171‐4. [DOI] [PubMed] [Google Scholar]
- Qureshi AI, Nasar A, Ahmed S, Harris‐Lane P, Divani AA. Analysis of patients with hyperdense artery sign in the National Institute of Neurological Diseases and stroke tissue plasminogen activator trial. Stroke 2005;36(2):493. [Google Scholar]
- Qureshi AI, Suri MF, Zhou J, Divani AA. African American women have poor long‐term survival following ischemic stroke. Neurology 2006;67(9):1623‐9. [DOI] [PubMed] [Google Scholar]
- Qureshi AI, Suri MFK, Ezzeddine MA. Clinical‐radiological severity mismatch phenomenon: patients with severe neurological deficits without matching infarction on computed tomographic scan. Stroke 2011;42(3):e245. [DOI] [PubMed] [Google Scholar]
- Qureshi AI, Zhou J, Suri MFK, Divani AA, Ezzeddine MA. Aspirin failure does not increase the risk of recurrent stroke or death among patients with ischemic stroke. Stroke 2007;38(2):532. [Google Scholar]
- Rao NM, Gornbein JA, Levine SR, Saver JL. The most accurate definition of clinically relevant hemorrhagic transformation after thrombolytic therapy for stroke: analysis of the NINDS‐TPA trials. Stroke 2012;43(2):Abst. 209. [Google Scholar]
- Rhoney DH, Coplin WM, Lin Y, Frankel M, Lyden PD, Levine SR. Time of day, outcome, and response to thrombolytic therapy: The National Institute of Neurological Disorders and Stroke Recombinant Tissue Plasminogen Activator Stroke Trial experience. Journal of Stroke and Cerebrovascular Diseases 2010;19(1):40‐8. [DOI] [PubMed] [Google Scholar]
- Sangha NS, Huang J, Grotta JC, Savitz S. Patients with diabetes and previous ischemic strokes in the National Institute of NINDS trial. Stroke 2012;43(2):Abst. 3797. [Google Scholar]
- Saposnik G, Demchuk A, Tu JV, Johnston CS. The iScore predicts efficacy and risk of bleeding in the NINDS t‐PA stroke trial. Stroke 2012;43(11):e116. [DOI] [PubMed] [Google Scholar]
- Saver JL. Hemorrhage after thrombolytic therapy for stroke: the clinically relevant number needed to harm. Stroke 2007;38(8):2279‐83. [DOI] [PubMed] [Google Scholar]
- Saver JL. Number needed to treat estimates incorporating effects over the entire range of clinical outcomes. Novel derivation method and application to thrombolytic therapy for acute stroke. Archives of Neurology 2004;61(7):1066‐70. [DOI] [PubMed] [Google Scholar]
- Saver JL, Altman H. Relation between neurologic deficit severity and final functional outcome shifts and strengthens during first hours after onset. Stroke 2012;43(2):Abst. 2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saver JL, Altman H. Relationship between neurologic deficit severity and final functional outcome shifts and strengthens during first hours after onset. Stroke 2012;43(6):1537‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saver JL, Gornbein J, Starkman S. Graphic reanalysis of the two NINDS‐tPA trials confirms substantial treatment benefit. Stroke 2010;41(10):2381‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saver JL, Starkman S. Impact of treatment on individual patients in the two NINDS‐TPA trials. Normalized change in NIHSS score confirms benefit graphically and statistically. Stroke 2009;40(4):e205. [Google Scholar]
- Saver JL, Yafeg B. Robust confirmation of treatment effect by baseline severity‐adjusted end point reanalysis of the NINDS‐tPA stroke trials. Stroke 2006;37(2):622. [DOI] [PubMed] [Google Scholar]
- Saver JL, Yafeh B. Confirmation of tPA treatment effect by baseline severity‐adjusted end point reanalysis of the NINDS‐tPA stroke trials. Stroke 2007;38(2):414‐6. [DOI] [PubMed] [Google Scholar]
- Savitz SI, Lew R, Bluhmki E, Hacke W, Fisher M. Rankin shift analysis of the NINDS and ECASS‐II trials. Stroke 2007;38(2):576. [DOI] [PubMed] [Google Scholar]
- Savitz SI, Lew R, Bluhmki E, Hacke W, Fisher M. Shift analysis versus dichotomization of the modified Rankin scale outcome scores in the NINDS and ECASS‐II trials. Stroke 2007;38(12):3205‐12. [DOI] [PubMed] [Google Scholar]
- Sila CA, Furlan AJ. Therapy for acute ischemic stroke: the door opens. Interpreting the NINDS rt‐PA stroke study. Cleveland Clinic Journal of Medicine 1996;63(2):77‐9. [DOI] [PubMed] [Google Scholar]
- Silver B, Lu M, Morris DC, Mitsias PD, Lewandowski C, Chopp M. Blood pressure declines and less favorable outcomes in the NINDS tPA stroke study. Journal of the Neurological Sciences 2008;271(1‐2):61‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. NINDS stroke trial data reanalysis leaves issues unresolved (response). Stroke 2005;36(3):529‐30. [DOI] [PubMed] [Google Scholar]
- Smith BJ. Thromobolysis for acute ischaemic stroke: revisiting the evidence. Medical Journal of Australia 2003;179(7):386‐9. [DOI] [PubMed] [Google Scholar]
- Swearingen CJ, Tilley BC, Adams RJ, Rumboldt Z, Nicholas JS, Bandyopadhyay D, et al. Application of regression to analyze ischemic stroke volume in NINDS rt‐PA clinical trials. Neuroepidemiology 2011;37(2):73‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanne D, Bates VE, Verro P, Kasner SE, Binder JR, Patel SC, et al. Initial clinical experience with IV tissue plasminogen activator for acute ischemic stroke: a multicenter survey. Neurology 1999;53(2):424‐7. [DOI] [PubMed] [Google Scholar]
- Tanne D, Levine SR, Brey RL, Lin H, Tilley BC, NINDS rt‐PA Stroke Study Group. Antiphospholipid antibodies and outcome of acute ischemic stroke in the NINDS rt‐PA Stroke Trial. Stroke 2003;34(1):279. [DOI] [PubMed] [Google Scholar]
- Tanne D, Levine SR, Brey RL, Lin H, Tilley BC, NINDS rt‐PA Stroke Study Group. Antiphospholipid‐protein antibodies and acute ischemic stroke in the NINDS rt‐PA stroke trial. Neurology 2003;61(8):1158‐9. [DOI] [PubMed] [Google Scholar]
- Tanne D, Macko RF, Lin H, Tilley BC, Levine SR, NINDS rt‐PA Stroke Study Group. Effect of elevated hemostatic markers on outcome after rt‐PA therapy for acute ischemic stroke: results from the NINDS rt‐PA stroke trial. Stroke 2006;37(2):704. [Google Scholar]
- Tanne D, Macko RF, Lin Y, Tilley BC, Levine SR, NINDS rtPA Stroke Study Group. Hemostatic activation and outcome after recombinant tissue plasminogen activator therapy for acute ischemic stroke. Stroke 2006;37(7):1798‐1804. [DOI] [PubMed] [Google Scholar]
- The NINDS TPA Stroke Trial. Predictors of favourable outcome. Cerebrovascular Diseases 1996;6(3):182. [Google Scholar]
- The NINDS rt‐PA Stroke Study Group. Effect of rt‐PA on CT lesion volume over time. Stroke 2000;31(11):2828. [Google Scholar]
- The NINDS rt‐PA Stroke Study Group. Effect of rt‐PA on ischaemic stroke lesion size by computed tomography: preliminary results from the NINDS rt‐PA stroke trial. Proceedings of the 23rd International Joint Conference on Stroke and Cerebral Circulation; 1998 Feb 5‐7; Orlando (FL). 1998:287.
- The NINDS rt‐PA Stroke Study Group. Tissue plasminogen activator for minor strokes: the NINDS rt‐PA stroke study experience. Stroke 2005;35(1):241. [Google Scholar]
- The NINDS rt‐PA Stroke Trial. Predictors of an unfavourable outcome. Cerebrovascular Diseases 1996;6(3):181. [Google Scholar]
- The NINDS t‐PA Stroke Study Group. Generalised efficacy of t‐PA for acute stroke: subgroup analysis of the NINDS t‐PA stroke trial. Stroke 1997;28(11):2119‐25. [DOI] [PubMed] [Google Scholar]
- The NINDS t‐PA Stroke Study Group. Intracerebral haemorrhage after intravenous t‐PA therapy for ischaemic stroke. The NINDS t‐PA Stroke Study Group. Stroke 1997;28(11):2109‐18. [DOI] [PubMed] [Google Scholar]
- The National Institute of Neurological Disorders Stroke rt‐PA Stroke Study Group. Recombinant tissue plasminogen activator for minor strokes: The National Institute of Neurological Disorders and Stroke rt‐PA stroke study experience. Annals of Emergency Medicine 2005;46(3):243‐52. [DOI] [PubMed] [Google Scholar]
- The National Institute of Neurological Disorders and Stroke (NINDS) rt‐PA Stroke Study Group. A systems approach to immediate evaluation and management of hyperacute stroke. Experience at eight centres and implications for community practice and patient care. The National Institute of Neurological Disorders and Stroke (NINDS) rt‐PA Stroke Study Group. Stroke 1997;28(8):1530‐40. [DOI] [PubMed] [Google Scholar]
- The National Institute of Neurological Disorders and Stroke (NINDS) rt‐PA Stroke Study Group. Effect of intravenous recombinant tissue plasminogen activator on ischemic stroke lesion size measured by computed tomography. The National Institute of Neurological Disorders and Stroke (NINDS) rt‐PA Stroke Study Group. Stroke 2000;31(12):2912‐9. [DOI] [PubMed] [Google Scholar]
- The National Institute of Neurological Disorders and Stroke rt‐PA stroke study group. Tissue plasminogen activator for acute ischaemic stroke. New England Journal of Medicine 1995;333(24):1581‐7. [DOI] [PubMed] [Google Scholar]
- Tilley BC, Lyden PD, Brott TG, Lu M, Levine SR, Welch KMA, et al. Total quality improvement method for reduction of delays between emergency department admission and treatment of acute ischemic stroke. The National Institute of Neurological Disorders and Stroke (NINDS) rt‐PA Stroke Study Group. Archives of Neurology 1997;54(12):1466‐74. [DOI] [PubMed] [Google Scholar]
- Trotter G. Why were the benefits of tPA exaggerated?. Western Journal of Medicine 2002;176(3):194‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer R, Hacke W. 'Self‐fulfilling prophecy' or recognition requires a concept of perception. Stroke 2000;31(1):231‐2. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Lindley RI, Lewis S. Thrombolysis for acute ischemic stroke: still a treatment for the few by the few. Western Journal of Medicine 2002;176(3):198‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wears RL, Cooper RJ, Magid DJ. Subgroups, reanalysis, and other dangerous things. Annals of Emergency Medicine 2005;46(3):253‐5. [DOI] [PubMed] [Google Scholar]
- Wein TH, Hickenbottom SL, Morgenstern LB, Demchuk AM, Grotta JC. Safety of tissue plasminogen activator for acute stroke in menstruating women. Stroke 2002;33(10):2506‐8. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Saver JL. The relation between destination on hospital discharge and final 3 month disability outcome in acute ischemic stroke. Stroke 2013;44(Abst. ATP379):1‐2. [Google Scholar]
Ohtomo 1985 {published data only}
- Ohtomo E, Araki G, Itoh E, Toghi H, Matsuda T, Atarashi J. Clinical efficacy of urokinase in patients with cerebral thrombosis: multicentre double blind study. Kiso‐to‐Rinshyo (Basic and Clinical) 1985;19:445‐78. [Google Scholar]
- Ohtomo E, Araki G, Itoh E, Tougi H, Matuda T, Atarashi J. Clinical efficacy of urokinase in the treatment of cerebral thrombosis. Multi‐center double‐blind study in comparison with placebo. Clinical Evaluation 1985;13:711‐51. [Google Scholar]
PROACT 1998 {published and unpublished data}
- Zoppo GJ, Higashida RT, Furlan AJ, Pessin MS, Rowley HA, Gent M, et al. PROACT: A phase II randomised trial of recombinant pro‐urokinase by direct arterial delivery in acute middle cerebral artery stroke. Stroke 1998;29(1):4‐11. [DOI] [PubMed] [Google Scholar]
- Zoppo GJ, Higashida RT, Furlan AJ, Pessin MS, the Pro‐urokinase Acute Stroke Investigators. Intra‐arterial middle cerebral artery pro‐urokinase acute stroke study. Cerebrovascular Diseases 1996;6(3):189. [Google Scholar]
- Furlan AJ, Abou‐Chebi A. The role of recombinant pro‐urokinase (r‐pro‐UK) and intra‐arterial thrombolysis in acute ischaemic stroke: the PROACT trials. Current Medical Research and Opinion 2002;18(Suppl 2):s44‐7. [DOI] [PubMed] [Google Scholar]
- Furlan AJ, Putney K, Barker W, Zoppo GJ, Higashida R, Pessin M, et al. The clinical trials acceleration program: experience from the Prolyse in Acute Cerebral Thromboembolism Trial (PROACT). Stroke 1996;27(1):171. [Google Scholar]
- Gutterman C, Barker W, Hodkinson G. Education, facilitation and motivation at clinical sites in an effort to increase patient enrolment in the PROACT Stroke Study. Stroke 1998;29:CT 3. [Google Scholar]
- Khatri P, Carrozzella JA, Martin RH, Yeatts SD, Hill MD, Furlan AJ, et al. Intracranial hemorrhage and its relationship to reperfusion and clinical outcome: an exploratory analysis of the PROACT II Trial. Stroke 2010;41(4):e353. [Google Scholar]
- Rahme R, Abruzzo TA, Martin RH, Tomsick TA, Ringer AJ, Furlan AJ, et al. Is intra‐arterial thrombolysis beneficial for M2 occlusions? Subgroup analysis of the PROACT‐II trial. Stroke 2013;44(1):240‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salsberg J, Tomsick T, Carrozzella J, Martin RL, Furlan A, Higashida R, et al. Futile angiographic reperfusion in the PROACT II Trial: an exploratory analysis. Stroke 2011;42(3):e64. [Google Scholar]
PROACT 2 1999 {published and unpublished data}
- Furlan A, Higashida R, Wechsler L, Schulz G, PROACT II Investigators. PROACT II: recombinant prourokinase (r‐ProUK) in acute cerebral thromboembolism. Proceedings of the 23rd Joint International Conference on Stroke and Cerebral Circulation; 1998 Feb 5‐7; Orlando (FL) USA. 1998.
- Furlan A, Higashida R, Weschler L, Gent M, Rowley H, Kase C, et al. Intraarterial pro‐urokinase for acute ischaemic stroke. The PROACT II Study: a randomised controlled trial. Prolyse in Acute Cerebral Thromboembolism. JAMA 1999;282(21):2003‐11. [DOI] [PubMed] [Google Scholar]
- Furlan AJ, Abou‐Chebi A. The role of recombinant pro‐urokinase (r‐pro‐UK) and intra‐arterial thrombolysis in acute ischaemic stroke: the PROACT trials. Prolyse in Acute Cerebral Thromboembolism. Current Medical Research and Opinion 2002;18 Suppl 2:s44‐7. [DOI] [PubMed] [Google Scholar]
- Furlan AJ, Higashida R, Wechsler L, Schulz G for the PROACT 2 Investigators: PROACT II. Recombinant prourokinase (r‐ProUK) in acute cerebral thromboembolism: initial trial results. The PROACT investigators. Stroke 1999;30:234. [Google Scholar]
- Furlan AJ, Higashida RT. Early ischemic signs should not be used as exclusion criteria in thrombolysis trials. Stroke 2004;35:e3‐4. [DOI] [PubMed] [Google Scholar]
- Hamann GF. Early ischemic signs should not be used as exclusion criteria in thrombolysis trials. Stroke 2004;35:e3. [DOI] [PubMed] [Google Scholar]
- Hill MD, Eliasziw M, Buchan AM, Rowley HA, Furlan AJ, Schulz G. Predicting functional outcome after intra‐arterial thrombolysis: aspects of ASPECTS. Stroke 2004;35:e7‐8. [DOI] [PubMed] [Google Scholar]
- Hill MD, Kent DM, Hinchey J, Rowley H, Buchan AM, Wechsler LR, et al. PROACT‐2 Investigators. Sex‐based differences in the effect of intra‐arterial treatment of stroke. Analysis of the PROACT‐2 Study. Stroke 2006;37(9):2322‐5. [DOI] [PubMed] [Google Scholar]
- Hill MD, Kent DM, Hinchey JA, Schulz G, Furlan AJ. Sex‐based differences in the effect of intra‐arterial treatment of stroke: analysis of the PROACT‐2 study. Stroke 2006;37(2):706‐7. [DOI] [PubMed] [Google Scholar]
- Hill MD, Rowley HA, Adler F, Eliasziw M, Furlan A, Higashida RT, et al. PROACT‐II Investigators. Selection of acute ischaemic stroke patients for intra‐arterial thrombolysis with pro‐urokinase by using ASPECTS. Stroke 2003;34(8):1925‐31. [DOI] [PubMed] [Google Scholar]
- Kase CS, Furlan AJ, Wechsler LR, Higashida RT, Rowley HA, Hart RG, et al. Cerebral hemorrhage after intra‐arterial thrombolysis for ischemic stroke: the PROACT II trial. Neurology 2001;57(9):1603‐10. [DOI] [PubMed] [Google Scholar]
- Kase CS, Furlan AJ, Wechsler LR, Higashida RT, Rowley HA, Hart RG, et al. Symptomatic intracranial hemorrhage after intraarterial thrombolysis with recombinant prourokinase in acute ischaemic stroke: The PROACT II Study. Neurology 2000;54 Suppl 3:A260‐1. [Google Scholar]
- Kim JJ, Fischbein NJ, Lu Y, Pham D, Dillon WP. Regional angiographic grading system for collateral flow: correlation with cerebral infarction in patients with middle cerebral artery occlusion. Stroke 2004;35(6):1340‐4. [DOI] [PubMed] [Google Scholar]
- Rahme R, Abruzzo T, Martin RH, Tomsick TA, Ringer AJ, Furlan AJ, et al. Is intra‐arterial thrombolysis beneficial for M2 occlusions? Subgroup analysis of the PROACT‐II trial. Stroke 2013;44(1):240‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts HC, Dillon WP, Furlan AJ, Rowley HA, Fischbein NJ, Higashida RT, et al. Angiographic collaterals in acute stroke ‐ relationship to clinical presentation and outcome: The PROACT II Trial. Stroke 2001;32:336‐b. [Google Scholar]
- Roberts HC, Dillon WP, Furlan AJ, Wechsler LR, Rowley HA, Fischbein NJ, et al. Computed tomographic findings in patients undergoing intra‐arterial thrombolysis for acute ischaemic stroke due to middle cerebral artery occlusion: results from the PROACT II trial. Stroke 2002;33(6):1557‐65. [DOI] [PubMed] [Google Scholar]
- Roberts HC, Rowley HA, Dillon WP, Fischbein NJ, Furlan AJ, Higashida RT, et al. Effect of intraarterial prourokinase on CT infarct volume: results of PROACT II. Stroke 2000;31(1):278. [Google Scholar]
- Sandercock P, Lewis S. Sex‐based differences in the effect of intra‐arterial treatment of stroke: a plea to stop torturing the old data and do large trials!. Stroke 2006;37(9):2198. [DOI] [PubMed] [Google Scholar]
- Saposnik G. Predicting functional outcome after intra‐arterial thrombolysis: aspects of ASPECTS. Stroke 2004;35(1):e7‐8. [DOI] [PubMed] [Google Scholar]
- Saver JL. Intra‐arterial fibrinolysis for acute ischemic stroke. The message of MELT. Stroke 2007;38(10):2627‐8. [DOI] [PubMed] [Google Scholar]
- Wechsler LR, Roberts R, Furlan AJ, Higashida R, Dillon WP, Roberts HC, et al. Factors influencing outcome and treatment effect in PROACT II. Stroke 2001;32:319. [DOI] [PubMed] [Google Scholar]
- Wechsler LR, Roberts R, Furlan AJ, Higashida RT, Dillon W, Roberts H, et al. Factors influencing outcome and treatment effects in PROACT II. Stroke 2003;34(5):1224‐9. [DOI] [PubMed] [Google Scholar]
Wang 2003 {published data only}
- Wang S, Wang X, Zheng H, Zuo Y, Hu N, Li X, et al. Early intravenous thrombolysis with recombinant tissue plasmogen activator for acute cerebral infarction. Chinese Critical Care Medicine 2003;15(9):542‐5. [PubMed] [Google Scholar]
- Wang XL, Zeng H, Fan K, Wang KY, Zuo Y, Wang SY, et al. Clinical study on early intravenous thrombolysis with rt‐PA for acute cerebral infarction. Chinese Journal of Neurology 2006;39:678‐83. [Google Scholar]
- Zeng H, Wang X, Qi X, Wang H. Thrombolytic therapy using Actilyse (rt‐PA) in patients with acute cerebral infarction. Chinese Journal of Emergency Medicine 2006;15(5):457‐9. [Google Scholar]
References to studies excluded from this review
Bao 2003 {published data only}
- Bao Z. Clinical revaluation of intravenous thrombolysis with urokinase in treatment of acute cerebral infarction. Proceedings of the 4th International Conference on Research Advances in Cerebrovascular Disease; 2003 Oct 13‐15; China. 2003:201‐3.
Boehringer Mannheim 1994 {published data only}
- Boehringer Mannheim. Double‐blind, placebo‐controlled, randomized multicenter study to evaluate the efficacy and safety of two doses of reteplase (r‐PA_) (10 MU; 10 MU + 10 MU) in acute cerebral ischemia (MF 4398). Protocol 1994.
CLEAR 2008 {published data only}
- Pancioli AM, CLEAR Trial Investigators. The combined approach to lysis utilizing eptifibatide and rt‐PA in acute ischemic stroke (the CLEAR stroke trial): final results from Tier I and II. Stroke 2008;39(2):531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Davalos 2003 {published data only}
- Dávalos A, Leira R, Pedraza S, Blanco M, Serena J, Silva Y, et al. The usefulness of clinical‐DWI mismatch in the treatment of acute ischemic stroke with reperfusion therapies. Cerebrovascular Diseases 2003;16(Suppl 4):Abst 0270. [Google Scholar]
Defuse 2001 {published data only}
- Boehringer Ingelheim. Defuse. Powerpoint presentation.
DeWinter 1999 {published data only}
- DeWinter G, Guerrero S, Mouren S, Baillard C, Bertrand M, Coriat P. Hemodynamic stability during remifentanil or sufentanil anaesthesia in patients undergoing carotid endarterectomy (CE). British Journal of Anaesthesia 1999;82 Suppl 1:46‐7. [Google Scholar]
Ding 2006 {published data only}
- Ding Y, Yin XG. Small‐dose aspirin plus lumbrukinase in improving neurological function of patients with acute cerebral infarction. Chinese Journal of Clinical Rehabilitation 2006;10(6):60‐3. [Google Scholar]
Ducrocq 2005 {published data only}
- Ducrocq C, Anxionnat R, Taillandier L, Lacour JC, Bracard S, Bollaert PE, et al. Intravenous versus intra‐arterial urokinase thrombolysis in acute ischemic stroke. Randomised study of 27 patients. Cerebrovascular Diseases 2000;10(Suppl 2):76. [Google Scholar]
- Ducrocq X, Bracard S, Taillandier L, Anxionnat R, Lacour JC, Guillemin F, et al. Comparison of intravenous and intra‐arterial urokinase thrombolysis for acute ischaemic stroke. Journal of Neuroradiology 2005;32(1):26‐32. [DOI] [PubMed] [Google Scholar]
EAST II [pers comm] 1999 {published data only}
- Mori E. Informaiton about the EAST II trial [personal communication]. Email to JM Wardlaw 18 January 1999.
Edinburgh 1991 {published data only}
- Wardlaw JM, Lindley RI, Warlow CP, Sandercock PAG. Edinburgh Stroke Trial. A pilot study of intra‐arterial thrombolysis for acute ischaemic stroke. Journal of Neurology, Neurosurgery & Psychiatry 1994;57:251. [Google Scholar]
EMFATAS 1996 {published data only}
- Fieschi C. The European multicentre four‐arm trial in acute stroke project (EMFATAS): A European Union work ‐ program on health research. Cerebrovascular Diseases 1999;9(Suppl 1):106. [Google Scholar]
- Fieschi C, Hacke W, Kaste M, EMFATAS Study Group. Early protocol of the European Multicentre Four‐arm Trial in Acute Stroke (EMFATAS) of combination therapy in acute ischaemic stroke. The Challenge of Stroke. The Lancet Confererence; 1998 Oct 15‐16; Montreal. 1998:36.
- Fieschi C, Sacchetti ML. The EMFATAS project: a four‐arm safety and efficacy European trial. Cerebrovascular Diseases 1996;6(3):192. [Google Scholar]
- Toni D, Sacchetti ML, Chamorro A. Acute stroke trials: the problems of local investigators?. European Neurology 2003;49(2):109‐14. [DOI] [PubMed] [Google Scholar]
EMS 1996 {published data only}
- Derex L. Outcome of the EMS Trial patients without angiographically demonstrated arterial occlusion. 5th Thrombolysis in Acute Stroke Symposium, 1998 May 1‐2; Bethesda (MD), USA. 1998.
- Derex L, Tomsick TA, Brott TG, Lewandowski CA, Frankel MR, Clark W, et al. Outcome of stroke patients without angiographically revealed arterial occlusion within four hours of symptom onset. American Journal of Neuroradiology 2001;22(4):685‐90. [PMC free article] [PubMed] [Google Scholar]
- Emergency Management of Stroke (EMS) Investigators. Combined intra‐arterial and intravenous tPA for stroke. Stroke 1997;28(1):273. [Google Scholar]
- Lewandowski CA, Frankel M, Tomsick TA, Broderick J, Frey J, Clark W, et al. Combined intravenous and intra‐arterial r‐TPA versus intra‐arterial therapy of acute ischemic stroke. Emergency Management of Stroke (EMS) Bridging Trial. Stroke 1999;30(12):2598‐605. [DOI] [PubMed] [Google Scholar]
- Sandercock P. Interim results of EMS trial in acute ischaemic stroke [personal communication]. Email to JM Wardlaw 1997.
- Swarnkar AS, Jungreis CA, Wechsler LR, Wehner JJ. Combined intravenous and intraarterial thrombolytic therapy for treatment of an acute ischemic stroke: a case report. Journal of Stroke and Cerebrovascular Diseases 1999;8(4):264‐7. [DOI] [PubMed] [Google Scholar]
- The EMS Bridging Trial Investigators. Combined intravenous and intra‐arterial thrombolysis versus intra‐arterial thrombolysis alone: preliminary safety and clot lysis. Cerebrovascular Diseases 1996;6(3):184. [Google Scholar]
Fan 2001 {published data only}
- Fan C, Chen Q, Chang J, et al. Antithrombosis enzyme vs dextran‐40 in treatment of acute cerebral infarction. Henan Journal of Practical Nervous Diseases 2001;4(5):9‐10. [Google Scholar]
Fisher 2003 {published data only}
- Fisher M, Brott TG. Emerging therapies for acute ischemic stroke: new therapies on trial. Stroke 2003;34(2):359‐61. [DOI] [PubMed] [Google Scholar]
Fu 2004 {published data only}
- Fu Z, Wen X, Lin T. Clinical observation of treatment through intravenous thrombolysis with low dose urokinase in 36 patients with advanced cerebral infarction. Chinese Journal of Primary Medicine and Pharmacy 2004;11(8):948‐9. [Google Scholar]
Fuentes 2007 {published data only}
- Fuentes B, Alonso de Lecinana M, Masjuan J, Egido J, Simal P, Diaz‐Otero F, et al. Is intravenous t‐PA treatment beneficial in acute ischemic stroke related to internal carotid dissection?. Cerebrovascular Diseases 2007;23(Suppl 2):108. [Google Scholar]
Geng 2004 {published data only}
- Geng S, Wang Y, Liu P. Intravenous drip of urokinase combined with low molecular weight heparin in the treatment of acute cerebral infarction. Practical Journal of Medicine and Pharmacy 2004;21(7):604‐6. [Google Scholar]
Hartmann 2005 {unpublished data only}
- Hartmann A, Fleischer N, Schlier F. Intravenous thrombolysis has a beneficial effect on clinical course but not infarct size in patients with supratentorial brain infarct. Cerebrovascular Diseases 2005;19(Suppl 2):80. [Google Scholar]
Hong Kong 1994 {unpublished data only}
- Kumana C. Trial of intravenous streptokinase in Chinese patients with acute ischaemic stroke. Unpublished.
Huang 1996 {published data only}
- Huang R, Fang Y, Su Z. 31 cases clinical report of snake venom IV on acute cerebral infarction. Chinese Journal of Nervous and Mental Diseases 1996;22(6):378. [Google Scholar]
Huang 2000 {published data only}
- Huang Z‐D, Li Z‐W, Zhang W‐X. Lumbrokinase in treating cerebral infarction. Chinese Journal of New Drugs and Clinical Remedy 2000;19(6):453‐5. [Google Scholar]
ICTuS‐L 2006 {published data only}
- Guluma KZ, Hemmen TM, Olsen SE, Rapp KS, Lyden PD. A trial of therapeutic hypothermia via endovascular approach in awake patients with acute ischemic stroke: methodology. Academic Emergency Medicine 2006;13(8):820‐7. [DOI] [PubMed] [Google Scholar]
- Hemmem TM, Cruz‐Flores S, Martin‐Schild S, Gomes JA, Wijman CA, Rapp KS, et al. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke (ICTuS‐L). asa.scientificposter.com/epsView.cfm?pid=CTP43&vr=2008 (accessed 25 February 2008).
- Hemmen TM, Cruz‐Flores S, Martin‐Schild S, Gomes JA, Wijman CA, Rapp KS, et al. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke (ICTuS‐L). Proceedings of the International Stroke Conference; 2008 Feb 20‐22; New Orleans (LA), USA. American Stroke Associate, 2008:CTP43.
- Hemmen TM, Guluma KZ, Wijman CA, Cruz‐Flores S, Meyer BC, Rapp KS, et al. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke (ICTuS‐L). Stroke 2006;37(2):706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmen TM, Raman R, Gomez JA, Cruz Florez S, Wijman CA, Rapp KS, et al. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke (ICTuS‐L): final results. Stroke 2010;41(4):e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmen TM, Raman R, Guluma KZ, Meyer BC, Gomes JA, Cruz‐Flores S, et al. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke (ICTuS‐L) final results. Stroke 2010;41(10):2265‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyden P, Hemmen T, Raman R, Gomes J, Cruz‐Flores S, Wijman C, et al. Factors that determine cooling rate in a trial of endovascular hypothermia for acute stroke‐the ICTUS‐L trial. International Journal of Stroke 2010;5(Suppl 2):12. [Google Scholar]
- Lyden PD. ICTuS‐L trial [personal communication]. Email to JM Wardlaw 30 November 2007.
- Lyden PD, Hemmen T. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke (ICTuS‐L) ‐ ongoing trial. International Journal of Stroke 2008;3(Suppl 1):439. [Google Scholar]
IMS II 2004 {published data only}
- The IMS II Trial Investigators. The interventional management of stroke (IMS) II study. Stroke 2007;38(7):2127‐35. [DOI] [PubMed] [Google Scholar]
- The IMS Study Investigators. Combined intravenous and intra‐arterial recanalization for acute ischemic stroke: the interventional management of stroke study. Stroke 2004;35(4):904‐12. [DOI] [PubMed] [Google Scholar]
IMS III 2008 {published data only}
- Broderick JP, Anderson C, Carrozzella J, Demchuk AM, Dillon C, Hill MD, et al. The Interventional Management of Stroke (IMS) III trial: an ongoing phase III trial. American Heart Association. 2008:CT P44.
INSTINCT 2005 {published data only}
- Meurer WJ, Majersik JJ, Frederiksen SM, Zhang L, Sandretto A, Scott PA. Barriers to the emergency use of thrombolysis for acute ischemic stroke: the INcreasing Stroke Treatment through INteractive Behavioural Change Tactics (INSTINCT) Trial. Stroke 2008;39(2):597. [Google Scholar]
- Scott P, Morgenstern L. INcreasing Stroke Treatment through INterventional behavior Change Tactics (the INSTINCT Trial). sitemaker.umich.edu/instinct/home (accessed 3 August 2007).
- Scott PA. INSTINCT. INcreasing Stroke Treatment through INterventional Behavior Change Tactics. www.strokecenter.org/trials/TrialDetail.aspx?tid=688 (accessed 18 July 2007).
- Scott PA, Silbergleit R, Morgenstern LB, Haan MN, Kalbfleisch JD, Cabana MD, et al. INcreasing Stroke Treatment through INterventional Behaviour Change Tactics. www.abstractsonline.com/arch/RecordPrintView.aspx?LookupKey=12345&Re... (accessed 3 August 2007).
ITAIS 2005 {published data only}
- Donnan G. Comment on ITAIS. International Journal of Stroke 2009;4:49. [DOI] [PubMed] [Google Scholar]
- Wang Y. Imaging‐based thrombolysis trial in acute ischemic stroke. www.controlled‐trials.com 2005. [DOI] [PubMed]
- Wang Y, Jiang W, Liao X, Du B, Zhao X, Dong K, et al. Comparison of intravenous and intra‐arterial tPA within 3‐6 hours guided by multi‐MRI: randomised study of 36 patients. Stroke 2008;39(2):602. [Google Scholar]
- Wang Y, Jiang W, Zhao X, Du B, Dong K, Liao X, et al. Imaging‐based thrombolysis trial in acute ischemic stroke. www.abstractonline.com/arch/RecordPrintView.aspx?LookupKey=12345&recordID=17703 (accessed 3 August 2007).
- Wang Y, Liao X, Zhao X, Wang C, Li L, Zhou Y, et al. Imaging‐based thrombolysis trial in acute ischemic stroke‐II (ITAIS‐II). International Journal of Stroke 2009;4(1):49‐53. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang Y. ITAIS. Imaging‐based thrombolysis trial in acute ischemic stroke. www.strokecenter.org/trials/TrialDetail.aspx?tid=680 (accessed 18 July 2007).
- Wang YL, Liao XL, Jiang WJ, Dong KH, Zhao XQ, Xue J, et al. Different multi‐parametric MRI patterns guide thrombolytic therapy beyond 3h of symptom onset in acute ischemic stroke patients. International Journal of Stroke 2008;3(Suppl 1):217. [Google Scholar]
- Wang, Y. The ITAIS trial [personal communication]. Email to JM Wardlaw 30 November 2007.
Jin 2000 {published data only}
- Jin L, Jin H, Zhang G, Xu G. Changes in coagulation and tissue plasminogen activator after the treatment of cerebral infarction with lumbrokinase. Clinical Hemorheology and Microcirculation 2000;23(2‐4):213‐8. [PubMed] [Google Scholar]
Kandil 2001 {published and unpublished data}
- Kandil MR, Farweez HM, Aziz A, Ahmed MA. Evaluation of intraarterial urokinase therapy in acute stroke. Journal of Neurological Science 2001;187 Suppl 1:S256. [Google Scholar]
Kim 2008 {published data only}
- Kim G‐M, Bang OY, Chung C‐S, Lee KH. Partial recanalization after intraarterial thrombolysis: angiographic consequences and clinical outcome. Stroke 2008;39(2):589‐90. [Google Scholar]
Konta 1996 {published data only}
- Konta Y, Aizu K, Fuziwara K, Matsui T. Evaluation of intravenous urokinase, ozagrel sodium and low molecular dextran therapy in acute ischemic stroke ‐ comparison of infusion methods of urokinase. Cerebrovascular Diseases 1996;6(3):187. [Google Scholar]
Lang 2013 {published data only}
- Lang W, Stadler CH, Poljakovic Z, Fleet D, Willeit J, Brainin M, et al. A prospective, randomized, placebo‐controlled, double‐blind trial about safety and efficacy of combined treatment with alteplase (rt‐PA) and Cerebrolysin in acute ischaemic hemispheric stroke. International Journal of Stroke 2013;8(2):95‐104. [DOI] [PubMed] [Google Scholar]
Li 2003 {published data only}
- Li JY, Peng YZ, Yang F, et al. Clinical observation on effect of tongnao huolo acupuncture therapy in treating acute cerebral infarction at ultra‐early or acute stage. Chinese Journal of Integrated Traditional and Western Medicine 2003;23(10):736‐9. [PubMed] [Google Scholar]
Lindsberg 2006 {published data only}
- Lindsberg PJ, Mattle HP. Therapy of basilar artery occlusion. A systematic analysis comparing intra‐arterial and intravenous thrombolysis. Stroke 2006;37(3):922‐8. [DOI] [PubMed] [Google Scholar]
Liu 1991 {published data only}
- Liu CX, Shun XY, Zhu Q. Efficacy analysis of DUM infusion interventionally on 33 cases of cerebral thrombosis. Chinese Journal of Practical Internal Medicine 1991;11(6):303‐5. [Google Scholar]
Liu 1994 {published data only}
- Yufeng L. Randomised controlled trial of Crack Ahylysantifarctase in treating cerebral infarction. Beijing Medical Journal 1994;16(5):313‐5. [Google Scholar]
Liu 2004 {published data only}
- Liu M, MA X. Clinical observation of treatment through intravenous thrombolysis with urokinase for patients with early acute cerebral infarction. Practical Journal of Medicine and Pharmacy 2004;21(6):504‐5. [Google Scholar]
Lu 2001 {published data only}
- Lu XR, Mi Z, Yuan XD, et al. Study of correlation between times of a disease and plasma fibrinogen molecular reactivity in patients with acute cerebral infarction. Hong Kong Medical Journal 2001;7(4):42. [Google Scholar]
Lyden 2003a {published data only}
- Haley EC. Pilot study of TNK‐TPA in acute ischemic stroke. commons.cit.nih.gov/crisp3...dit_numfound=53&p_keywords=stroke (accessed 12 September 2002).
- Haley EC. TNK in acute ischemic stroke. www.strokecenter.org/trials/ 2000.
- Haley EC Jr, Lyden PD, Johnston KC, Hemmen TM, TNK in Stroke Investigators. A pilot dose‐escalation safety study of tenecteplase in acute ischemic stroke. Stroke 2005;36(3):607‐12. [DOI] [PubMed] [Google Scholar]
- Lyden P, TNK for Stroke Investigators. Pilot study of tenecteplase (TNK) in acute ischemic stroke: preliminary report. Stroke 2003;34(1):246. [Google Scholar]
Lyden 2003b {published data only}
- Lyden P. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke. www.ClinicalTrials.gov (accessed ??). [DOI] [PMC free article] [PubMed]
- Lyden P. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke. www.strokecenter.org (accessed ??). [DOI] [PMC free article] [PubMed]
Meyer 1963 {published data only}
- Meyer JS, Gilroy J, Barnhart MI, Johnson JF. Therapeutic thrombolysis in cerebral thromboembolism. Neurology 1963;13:927‐37. [DOI] [PubMed] [Google Scholar]
Meyer 1964 {published data only}
- Meyer JS, Gilroy J, Barnhart MI, et al. Anticoagulants plus streptokinase therapy in progressing stroke. JAMA 1963;189:373. [DOI] [PubMed] [Google Scholar]
Michel 2008 {published data only}
- Michel P, Ntaios G, Reichhart M, Schindler C, Bogousslavsky J, Maeder P, et al. Perfusion‐CT guided intravenous thrombolysis in patients with unknown‐onset stroke: a randomized, double‐blind, placebo‐controlled, pilot feasibility trial. Neuroradiology 2012;54(6):579‐88. [DOI] [PubMed] [Google Scholar]
- Michel P, Reichhart M, Schindler C, Bogousslavsky J, Meuli R, Wintermark M. CT‐perfusion guided intravenous thrombolysis for unknown onset of stroke symptoms: clinical results of a pilot study. International Journal of Stroke 2008;3(Suppl 1):271. [Google Scholar]
- Michel P, Reichhart M, Schindler C, Bogousslavsky J, Meuli R, Wintermark M. CT‐perfusion guided intravenous thrombolysis for unknown onset stroke: clinical and radiological results of a pilot study. Cerebrovascular Diseases 2009;27(Suppl 6):77. [Google Scholar]
MITI‐IV 2005 {published and unpublished data}
- Thijs V. MITI‐IV trial [personal communication]. Email to JM Wardlaw 11 December 2007.
- Thijs V. MITI‐IV. Intravenous administration of microplasmin for treatment of acute ischemic stroke. www.strokecenter.org/trials/TrialDetail.aspx?tid=523 (accessed 19 July 2007).
- Thijs V, Vosko, Peeters A, Aichner F, Rother J, Pakola S, et al. Double blind placebo controlled IV microplasmin administration in patients with acute ischemic stroke. Powerpoint Presentation.
- Thijs VN, Vosko MR, Peeters A, Aichner F, Roether J, Pakola S. Double‐blind, placebo‐controlled, intra‐venous microplasmin administration in patients with acute ischemic stroke. International Journal of Stroke 2008;3 Suppl 1:82. [Google Scholar]
- Thijs VN, MITI‐IV Study Group. Intravenous administration of microplasmin for treatment of acute ischemic stoke (MITI‐IV). www.abstractsonline.com/arch/RecordPrintView.aspx?LookupKey=12345&RecordID=17591 (accessed 3 August 2007).
- Thijs VNS, Peeters A, Vosko M, Aichner F, Schellinger PD, Schneider D, et al. Randomized, placebo‐controlled, dose‐ranging clinical trial of intravenous microplasmin in patients with acute ischemic stroke. Stroke 2009;40(12):3789‐95. [DOI] [PubMed] [Google Scholar]
- ThromboGenics. Intravenous administration of microplasmin for treatment of acute ischemic stroke. www.clinicaltrials.gov/ct/show/NCT00123305 (accessed 18 July 2007).
Molina 2005 {published data only}
- Molina CA. Microbubble‐enhanced sonothrombolysis for acute ischemic stroke. Cerebrovascular Diseases 2008;26(Suppl 2):15‐6. [Google Scholar]
- Molina CA, Ribo M, Arenillas JF, Rubiera M, Montaner J, Santamarina E, et al. Microbubbles administration accelerates clot lysis during continuous 2 MHz ultrasound monitoring in stroke patients treated with intravenous tPA. Stroke 2005;36(2):419. [DOI] [PubMed] [Google Scholar]
- Molina CA, Ribo M, Rubiera M, Arenillas JF, Montaner J, Santamarina E, et al. Microbubbles‐enhanced sonothrombolysis for acute ischemic stroke. Cerebrovascular Diseases 2005;19(Suppl 2):30. [Google Scholar]
- Molina CA, Ribo M, Rubiera M, Montaner J, Santamarina E, Delgado‐Mederos R, et al. Microbubbles administration accelerates clot lysis during continuous 2‐MHz ultrasound monitoring in stroke patients treated with intravenous tissue plasminogen activator. Stroke 2006;37(2):425‐9. [DOI] [PubMed] [Google Scholar]
- Rubiera M, Ribo M, Delgado‐Mederos R, Santamarina E, Huertas R, Delgado P, et al. Efficacy of microbubble‐enhanced sonothrombolysis among stroke subtypes. Stroke 2006;37(2):621. [Google Scholar]
- Rubiera M, Ribo M, Delgado‐Mederos R, Santamarina E, Maisterra O, Montaner J, et al. Effects of microbubbles‐enhanced sonothrombolysis among stroke subtypes. Cerebrovascular Diseases 2006;21(Suppl 4):39. [Google Scholar]
Morris 2002 {unpublished data only}
- Morris DC. Abciximab and rt‐PA in acute ischemic stroke treatment. www.strokecenter.org/trials/TrialDetail.aspx?tid=481 (accessed 20 July 2007).
Naito 1984 {published and unpublished data}
- Naito I, Abe T. Oral urokinase: absorption, mechanisms of fibrinolytic enhancement and clinical effect on cerebral thrombosis. Folia Haematologica (Leipz) 1986;113(1‐2):122‐36. [PubMed] [Google Scholar]
Ohta 1997 {published data only}
- Ohta H, Yokogami K, Nakano S, Goya T, Wakisaka S. Acute thrombolytic therapy for middle cerebral artery occlusion. Japanese Journal of Neurosurgery 1997;6:84‐9. [DOI] [PubMed] [Google Scholar]
Pan 2011 {published data only}
- Pan S, Shen S, Dai L, Li H, Liu M. The effect of intravenous thrombolysis using recombinant tissue plasminogen activator (rt‐PA) for acute ischemic stroke. Stroke 2011;42(3):e126. [Google Scholar]
Pang 1993 {published data only}
- Pang S‐Q, et al. Clinical study of therapeutic effectiveness in treating ischaemic cerebrovascular disease with lumbrokinase. Chinese Journal of Neurology and Psychiatry 1993;26(4):229‐31. [Google Scholar]
PRACTISE 2005 {published data only}
- Dippel DWJ, Dirks M, Niessen L, Huijsman R, Minkman M, Franke C, et al. PRACTISE. Promoting acute thrombolysis for ischemic stroke ISRCTN 20405426. www.researchgate.net/publication/246838379_Promoting_acute_thrombolysis_for_ischaemic_stroke_(PRACTISE)._Protocol_for_a_cluster_randomised_controlled_trial_to_assess_the_effect_of_implementation_strategies_on_the_rate_and_effects_of_thrombolysis_for_acute_ischaemic_stroke_(ISRCTN_20405426) (accessed July 11th 2014). [ISRCTN 20405426]
- Dirks M, Niessen LW, Koudstaal PJ, Wijngaarden J, Minkman M, Franke C, et al. PRACTISE: Promoting acute thrombolysis for ischemic stroke. A cluster‐randomised trial of high intensity versus regular intensity implementation of thrombolysis. www.esc‐brussels.org/esc_ongoing_trials.asp (accessed 11th July 2014).
- Nederlands Trial Register. PRACTISE: Promoting acute thrombolysis for ischemic stroke. www.trialregister.nl/trialreg/admin/rctview.asp?TC=311 (accessed 19 July 2007).
- On behalf of the PRACTISE Investigators, Dirks M, Niessen L, Koudstaal PJ, Franke C, Oostenbrugge RJ, Huijsman R. Indications and contra‐indications for intravenous thrombolysis in acute ischemic stroke. Unpublished.
- PRACTISE. PRACTISE. Promoting acute thrombolysis for ischemic stroke. www.practise‐trial.org (accessed 6 August 2007).
- Wijngaarden JDH, Dirks M, Dippel DWJ, Minkman M, Niessen LW. Towards effective and efficient care pathways: thrombolysis in acute ischaemic stroke. Quarterly Journal of Medicine 2006;99(4):267‐72. [DOI] [PubMed] [Google Scholar]
Qiang 2001 {published data only}
- Qiang D. The dynamic study of stroke MRI in hyperacute stroke patients with thrombolysis. Hong Kong Medical Journal 2001;7(4):16. [Google Scholar]
Qureshi 2001 {unpublished data only}
- Qureshi AI. Safety and efficacy of intra‐arterial reteplase and intravenous Abciximab in patients with acute ischemic stroke. www.strokecenter.org/trials/TrialDetails.aspx?tid=201 (accessed 20 July 2007).
Qureshi 2006 {published data only}
- Qureshi AI, Harris‐Lane P, Kirmani JF, Janjua N, Divani AA, Mohammad YM, et al. Intra‐arterial reteplase and intravenous abciximab in patients with acute ischemic stroke: an open‐label, dose‐ranging, Phase 1 study. Neurosurgery 2006;59(4):789‐97. [DOI] [PubMed] [Google Scholar]
Ribeiro 2012 {published data only}
- Ribeiro PW, Cola PC, Gatto AR, Silva RG, Schelp AO, Braga GP, et al. Impact of cerebral reperfusion therapy in patients after ischemic stroke: aspects of oral intake evolution. Proceedings of the 8th World Stroke Congress. Oct 10‐13 2012; Brasilia, Brazil. 2012.
ROSIE 2002 {published data only}
- Dunn B, Davis LA, Todd JW, Chalela JA, Warach S. Safety of combined abciximab and reteplase in acute ischemic stroke: interim results of ReoPro Retavase Reperfusion of Stroke Safety Study Imaging Evaluation (ROSIE). Neurology 2005;62 Suppl 5:A462. [Google Scholar]
- Dunn B, Davis LA, Todd JW, Chalela JA, Warach S, ROSIE Investigators. ReoPro Retavase Reperfusion of Stroke Safety Study ‐ Imaging Evaluation (ROSIE). www.abstractsonline.com/arch/RecordView.aspx?LookupKey=12345&recordID=10974 (accessed 6 August 2007).
- Dunn B, Warach S. ReoPro Retavase Reperfusion of Stroke Safety Study ‐ Imaging Evaluation. www.abstractsonline.com/arch/RecordView.aspx?LookupKey=12345&recordID=16962 (accessed 6 August 2007).
- NCT00039832. ReoPro and retavase to restore brain blood flow after stroke. clinicaltrials.gov/ct/gui/show/NCT00039832 (accessed 6 August 2007).
- Thall PF, Nguyen HQ, Wstey EH. Patient‐specific dose finding based on bivariate outcomes and covariates. Biometrics 2008;64(4):1126‐36. [DOI: 10.1111/j.1541-0420.2008.01009.x] [DOI] [PubMed] [Google Scholar]
- Warach S. Personal communication 1 October 2008.
- Warach S. ROSIE. ReoPro Retavase Reperfusion of Stroke Safety Study ‐‐ Imaging Evaluation. www.strokecenter.org/trials/TrialDetail.aspx?tid=462 (accessed 20 July 2007).
- Warach S, Butman J, Davis L, Dunn B, Hsia A, Latour L, et al. ReoPro Retavase Reperfusion in Stroke Safety Study ‐‐ Imaging Evaluation (ROSIE): interim safety and efficacy results. www.abstractsonline.com/arch/RecordView.aspx?LookupKey=12345&recordID=16995 (accessed 6 August 2007).
- Warach S, Davis L, ROSIE Study Group. ReoPro Retavase Reperfusion of Stroke Safety Study ‐ Imaging Evaluation (ROSIE). The American Stroke Association 28th International Stroke Conference; 2003 Feb 13‐15; Phoenix (AZ), USA. 2003:CTP40.
ROSIE‐2 2002 {published data only}
- Davis L. ROSIE‐2. Reperfusion of stroke Safety study Imaging Evaluation ‐ 2. www.strokecenter.org.
ROSIE‐CT 2002 {published data only}
- NCT00046293. ReoPro Retavase Reperfusion of Stroke Safety Study‐Imaging Evaluation With Computed Tomography (ROSIE‐CT). www.clinicaltrials.gov/ct/gui/show/NCT00046293 (accessed 19 July 2007).
- Warach S. ROSIE‐CT study [personal communication]. Email to JM Wardlaw 1 October 2008.
- Warach S. ROSIE‐CT. ReoPro Retavase Reperfusion of Stroke Safety Study ‐ Imaging Evaluation with Computed Tomography. www.strokecenter.org/trials/TrialDetail.aspx?tid=438&printView=true (accessed 6 August 2007).
Rother 2003 {published data only}
- Rother J, Schellinger PD, Gass A, Siebler M, Villringer A, Fiebach JB, et al. Thrombolysis in patients without MRI mismatch. Stroke 2003;34(1):260‐1. [Google Scholar]
Shi 2000 {published data only}
- Shi Y, Li D, Zhao Y, et al. Clinical study on ultra‐early intravenous thrombolysis with high‐dose urokinase in treatment of acute cerebral infarction. National Medical Journal of China 2000;80(2):101‐3. [PubMed] [Google Scholar]
Skoloudik 2004 {published data only}
- Skoloudik D, Bar M, Vaclavik D, Skoda O, Hradilek P, Simickova K. Thrombotripsy study ‐ safety and efficacy of acceleration of thrombolysis by TCCS. Stroke 2004;35(6):e217. [Google Scholar]
Skoloudik 2006 {published data only}
- Skoloudik D, Bar M, Skoda O, Vaclavik D, Hradilek P, Herzig R, et al. Efficacy of sonothrombotripsy versus sonothrombolysis in recanalization of intracranial arteries. European Journal of Neurology 2006;13(Suppl 2):180. [Google Scholar]
Sobrino 2007 {published data only}
- Sobrino T, Arenillas JF, Leira R, Brea D, Castellanos M, Hurtado O, et al. T‐PA treatment increases circulating endothelial progenitor cells (SPCS) in acute ischemic stroke. Cerebrovascular Diseases 2007;23(Suppl 2):21. [Google Scholar]
SYNTHESIS 2007 {unpublished data only}
- Ciccone A, Valvassori L, Gasparotti R, Ballabio E, Sterzi R. Debunking 7 myths that hamper the realization of randomized controlled trials on intra‐arterial thrombolysis for acute ischemic stroke. Stroke 2007;38(7):2191‐5. [DOI] [PubMed] [Google Scholar]
- Ciccone A, Valvassori L, Ponzio M, Ballabio E, Gasparotti R, Sessa M, et al. Intra‐arterial or intravenous thrombolysis for acute ischemic stroke? The SYNTHESIS pilot trial. Journal of Neurointerventional Surgery 2010;2(1):74‐9. [DOI] [PubMed] [Google Scholar]
TEMPO 2013 {published data only}
- Mandzia JL. Thrombolysis for minor ischaemic stroke with proven acute symptomatic occlusion using TNK‐tPA (TEMPO‐1). Proceedings of the 22nd European Stroke Conference. 28‐31 May 2013; London, UK. 2013:Abst. OAID113.
- Mandzia JL, Coutts SB, Kenney C, Hill MD. Thrombolysis for minor ischaemic stroke with proven acute symptomatic occlusion using TNK‐tPA (TEMPO‐1). Proceedings of the International Stroke Conference 2013. 6‐8 February 2013; Honolulu, Hawaii, USA. 2013:Abst. CT P18.
TNK 2005 {published data only}
- Haley EC Jr, Lyden PD, Johnston KC, Hemmen TM, TNK Stroke Investigators. A pilot dose‐escalation safety study of tenecteplase in acute ischemic stroke. Stroke 2005;36(3):607‐12. [DOI] [PubMed] [Google Scholar]
- Wang D, Talkad A, Beck J, Mathews M, Jahnel J. TNK for ischemic lacunar syndrome ‐ the TNKilas trial. American Heart Association. 2008:CT P42.
TNK‐S2B 2005 {published data only}
- Haley EC. Study of tenecteplase (TNK) in acute ischemic stroke (TNK‐S2B). clinicaltrials.gov/ct/show/NCT00252239 (accessed 20 November 2007).
TNK‐TPA 2008 {published data only}
- Molina CA, Ribo M, Rubiera M, Santamarina E, Delgado‐Mederos R, Maisterra O, et al. TNK induces faster MCA recanalization and better short‐term outcome than native TPA. The TNK‐TPA reperfusion stroke study. Cerebrovascular Diseases 2008;25(Suppl 2):14. [Google Scholar]
- Molina CA, Ribo M, Rubiera M, Santamarina E, Delgado‐Mederos R, Maisterra O, et al. TNK induces faster MCS recanalization and leads to better short‐ and long‐term clinical outcome than native tPA. The TNK‐TPA reperfusion stroke study. Stroke 2008;39(2):563. [Google Scholar]
Trouillas 2000 {published data only}
- Troullias P, Philippeau F, Nighoghossian N, Derex L, Honnorat J, Dechavanne M. A biological syndrome predictive of cerebral bleeding in rtPA thrombolysis (data from the Lyon thrombolysis registry). Cerebrovascular Diseases 2000;10(Suppl 2):71. [Google Scholar]
TTATTS 1997 {published data only}
- Zoppo G. TTATTS Study [personal communication]. Email to JM Wardlaw 18 February 1997.
VASTT 2005 {published data only}
- Hill M, Demaerschalk B, Teal P, Pawsey S, Kwiatkowski K, Stratton G. Vastt ‐ the v10153 acute stroke thrombolysis trial. International Journal of Stroke 2008;3 Suppl 1:133. [Google Scholar]
- Hill M, Demaerschalk B, Teal P, Pawsey S, Munro S, Forton N. VASTT ‐ the V10153 Acute Stroke Thrombolysis Trial. Cerebrovascular Diseases 2007;24:abstract 26. [Google Scholar]
- Hill MD. VASTT. The V10153 acute stroke thrombolysis trial. www.strokecenter.org/trials/TrialDetail.aspx?tid=735 (accessed 19 July 2007).
- Pawsey S, Hutchinson JH, Munro S, Barsikian NA. VASTT ‐ The V10153 Acute Stroke Thrombolysis Trial. International Journal of Stroke 2006;1 Suppl 2:45. [Google Scholar]
- Vernalis Development. Safety and efficacy study in acute ischaemic stroke. www.clinicaltrials.gov (accessed 2007).
Vernalis 2005 {published data only}
- Vernalis. Thrombotic disease: V10153. www.vernalis.com/ver/rdc2/neurology/v10153 (accessed 19 July 2007).
Wang 1997 {published data only}
- Wang Y‐H, Zhao Y‐B, Chen Q‐T. A randomized, double‐blind, placebo‐controlled clinical trial of lumbrokinase capsules on the patients of blood rheology disorders with ischemic cerebrovascular diseases. Chinese Journal of Clinical Pharmacology 1997;13(2):65‐70. [Google Scholar]
Wang 1999 {published data only}
- Wang Y. Observations of effect of defibrase in treatment of 30 cases with acute cerebral thrombosis. Chinese Journal of Critical Care Medicine 1999;19(8):485. [Google Scholar]
Xiang 1995 {published data only}
- Xiang ZY, et al. Thrombolytic therapy and external counterpulsation in acute cerebral infarction. Proceedings of the 4th Chinese Stroke Conference; 1995 Oct; Chengdu. 1995:44.
Xu 1997 {published data only}
- Xu YL. Acute cerebral infarction treated by ahylsantinfarctase acupuncture: report of 60 cases. Forum on Traditional Chinese Medicine 1997;12(4):35. [Google Scholar]
Xu 2000 {published data only}
- Xu H, Bei S, Chengye Z. Comparison of the curative effects between ahylysantinfarctase and difibrinogenase on cerebral infarction. Journal of Wenzhou Medical College 2000;30(2):124‐6. [Google Scholar]
Xu 2004 {published data only}
- Xu XY, Chen XF. Clinical study on urokinase and nimodipine intravenous thrombolysis in acute cerebral infarction. Chinese Pharmacological Bulletin 2004;20(3):359‐60. [Google Scholar]
Yuan 1995 {published data only}
- Yuan D‐C, et al. High dose urokinase in the treatment of acute ischaemic stroke. Journal of Brain and Neurological Diseases 1995;3(2):111. [Google Scholar]
Zhang 2003 {published data only}
- Zhang Z, Zhou Y, Gao L. Intravenous thrombolysis with urokinase in treating acute cerebral infarctions. Sichuan Medical Journal 2003;24(8):790‐2. [Google Scholar]
Zhou 1996 {published data only}
- Zhou Y, Gao H, Ma R. Functions changes of VEC and therapeutic effect on VEC in patients with acute cerebral infarct. Zuzhong Yu Shenjing Jibing 1996;3(4):191‐3. [Google Scholar]
References to studies awaiting assessment
FRALYSE {published data only}
- Berge E. Results of the FRALYSE randomized study: effect of the duration of rt‐PA infusion on the functional outcome in patients with acute cerebral infarct [personal communication]. Email to JM Wardlaw January 24 2000.
Lin 2006 {published data only}
- Lin J, Chen DM, Wang Y. Effects of intravenous thrombolysis in treatment of acute cerebral infraction at early stage. Chinese Journal of Emergency Medicine 2006;15(10):924‐6. [Google Scholar]
TESPI {unpublished data only}
- Lorenzano S, Toni D. Intravenous thrombolysis with rt‐PA in acute stroke patients aged >80 years: the TESPI study. International Journal of Stroke 2008;3(Suppl 1):135. [DOI] [PubMed] [Google Scholar]
- Lorenzano S, Toni D. TESPI (thrombolysis in elderly stroke patients in Italy): a randomized controlled trial of alteplase (rt‐PA) versus standard treatment in acute ischaemic stroke in patients aged more than 80 years where thrombolysis is initiated within three hours after stroke onset. International Journal of Stroke 2012;7(3):250‐7. [DOI] [PubMed] [Google Scholar]
- Toni D. A randomised trial of alteplase in acute stroke [Studio clinico randomizzato controllato con alteplase (RT‐PA) in pazienti con ictus ischemio cerebrale acuto con eta superiore agli 80 anni trattati enro 3 ore dall'esordio dei sintomi]. www.agenziafarmaco.it (accessed 27 October 2006).
- Toni D. A randomized controlled trial of alteplase (rt‐PA) vs standard treatment in acute ischemic hemispheric stroke in patients aged more than 80 years, where thrombolysis is initiated within 3 hours after stroke onset [Studio clinico randomizzato controllato con alteplase (RT‐PA) in pazienti con ictus ischemico cerebrale acuto con eta superiore agli 80 anni trattati entro 3 ore dall'escorio dei sintomi]. www.agenziafarmaco.it (accessed 25 July 2007).
- Toni D. TESPI study [personal communication]. Email to JM Wardlaw 31 July 2007.
- Toni D. TESPI. Thrombolysis in Elderly Stroke Patients in Italy. www.strokecenter.org/trials/.
References to ongoing studies
BASICS {published data only}
- Hoeven EJRJ, Schonewille WJ, Vos JA. BASICS Trial. Basilar Artery International Cooperation Study Research Protocol. www.basicstrial.com/Protocol.html 2012.
DIAS‐3 {published data only}
- Albers G, Kummer R. Desmoteplase in Acute Ischemic Stroke: Phase III twin trials. Proceedings of the International Stroke Conference 2009; 2009 Feb 18‐20; San Diego (CA), USA. 2009:CT P19.
- Albers G, Kummer R. The desmoteplase DIAS‐3 and DIAS‐4 clinical trials: a status update. Neurology 2013;80(Meeting Abstracts 1):P07.249. [Google Scholar]
- Albers GW, Kummer R. Desmoteplase 3‐9 hours after Acute Ischemic Stroke: an update on the DIAS clinical trial program. Proceedings of the International Stroke Conference 2010; 2010 Feb 24‐26; San Antonio (TX) USA. 2010:CT P5.
- Albers GW, Kummer R. Desmoteplase 3‐9 hours after Acute Ischemic Stroke: the ongoing DIAS‐3 and DIAS‐4 clinical trials. Proceedings of the International Stroke Conference 2012; 2012 Feb 1‐3; New Orleans (LA), USA. 2012:CT P21.
- Albers GW, Kummer R. Desmoteplase in Acute Ischemic Stroke: status update on the DIAS clinical trial program. Proceedings of the International Stroke Conference 2011; 2011 Feb 8‐11; Los Angeles (CA), USA. 2011:CTP16.
- Albers GW, Von Kummer R: DIAS‐3 and DIAS‐4 Study Group. The desmoteplase DIAS‐3 and DIAS‐4 clinical trials: an important milestone passed. Proceedings of the 8th World Stroke Congress; 2013 Oct 10‐13; Brasilia, Brazil. 2013:Abst. 98.
- Albers GW, Kummer R, the DIAS‐3 and DIAS‐4 Study Group. The desmoteplase DIAS‐3 and DIAS‐4 clinical trials: background and status update. Proceedings of the International Stroke Conference 2013; 2013 Feb 6‐8 ; Honolulu (HI), USA. 2013:CT P3.
- Lundbeck H. DIAS‐3. Efficacy and safety study of desmoteplase to treat acute ischemic stroke. www.strokecenter.org/trials/.
- Lundbeck H. Efficacy and safety study of desmoteplase to treat acute ischemic stroke (DIAS‐3). www.ClinicalTrials.gov.
- Kummer R. The desmoteplase clinical trial programme: DIAS‐3 is nearing completion. Proceedings of the 22nd European Stroke Conference; 2013 May 28‐31; London, UK. 2013:Abst. OAID59.
- Kummer R, Albers G. Desmoteplase In Acute Stroke 3 and 4, DIAS‐3 and DIAS‐4, phase III twin trials. Proceedings of the 18th European Stroke Conference 2009; 2009 May 26‐29; Stockholm, Sweden. 2009:Abst. OAID 5.
- Kummer R, Albers GW. The Desmoteplase DIAS‐3 and DIAS‐4 clinical trials: an important milestone passed. Proceedings of the 21st European Stroke Conference; 2012 May 22‐25; Lisbon, Portugal. 2012:Abst. OAID 37.
- Kummer R, Albers GW, Mori E, DIAS Steering Committee. The desmoteplase in acute ischemic stroke (DIAS) clinical trial program. International Journal of Stroke 2012;7(7):589‐96. [DOI] [PubMed] [Google Scholar]
DIAS‐4 {published data only}
- Albers G, Kummer R. Desmoteplase in Acute Ischemic Stroke: Phase III twin trials. Proceedings of the International Stroke Conference 2009; 2009 Feb 18‐20; San Diego (CA), USA. 2009:CT P19.
- Albers G, Kummer R. The desmoteplase DIAS‐3 and DIAS‐4 clinical trials: A status update. Neurology 2013;80(Meeting Abstracts 1):P07.249. [Google Scholar]
- Albers GW, Kummer R. Desmoteplase 3‐9 hours after Acute Ischemic Stroke: an update on the DIAS clinical trial program. Proceedings of the International Stroke Conference 2010; 2010 Feb 24‐26; San Antonio (TX), USA. 2010:CT P5.
- Albers GW, Kummer R. Desmoteplase 3‐9 hours after Acute Ischemic Stroke: the ongoing DIAS‐3 and DIAS‐4 clinical trials. Proceedings of the International Stroke Conference 2012; 2012 Feb 1‐3; New Orleans (LA), USA. 2012:CT P21.
- Albers GW, Kummer R. Desmoteplase in Acute Ischemic Stroke: status update on the DIAS clinical trial program. Proceedings of the International Stroke Conference 2011; 2011 Feb 8‐11; Los Angeles (CA), USA. 2011:CTP16.
- Albers GW, Kummer R, DIAS‐3 and DIAS‐4 Study Group. The desmoteplase DIAS‐3 and DIAS‐4 clinical trials: an important milestone passed. Proceedings of the 8th World Stroke Congress; 2013 Oct 10‐13; Brasilia, Brazil. 2013:Abst. 98.
- Albers GW, Kummer R, the DIAS‐3 and DIAS‐4 Study Group. The desmoteplase DIAS‐3 and DIAS‐4 clinical trials: background and status update. Proceedings of the International Stroke Conference 2013; 2013 Feb 6‐8; Honolulu (HI), USA. 2013:CT P3.
- Gorelick PB, Albers GW, Kummer R. Desmoteplase in Acute Ischemic Stroke (DIAS) clinical trial program: regional hub‐spoke recruitment in DIAS‐4 in the United States. Proceedings of the International Stroke Conference 2011; 2011 Feb 8‐11; Los Angeles (CA), USA. 2011:CTP39.
- H Lundbeck A/S. Efficacy and Safety Study of Desmoteplase to Treat Acute Ischemic Stroke (DIAS‐4). www.ClinicalTrials.gov. [DOI] [PubMed]
- Kummer R. The desmoteplase clinical trial programme: DIAS‐3 is nearing completion. Proceedings of the 22nd European Stroke Conference. 2013 May 28‐31; London, UK. 2013:Abst. OAID59.
- Kummer R, Albers G. Desmoteplase In Acute Stroke 3 and 4, DIAS‐3 and DIAS‐4, phase III twin trials. Proceedings of the 18th European Stroke Conference 2009; 2009 May 26‐29; Stockholm, Sweden. 2009:Abst. OAID 5.
- Kummer R, Albers GW. The Desmoteplase DIAS‐3 and DIAS‐4 clinical trials: an important milestone passed. Proceedings of the 21st European Stroke Conference; 2012 May 22‐25; Lisbon, Portugal. 2012; Vol. 1:Abst. OAID 37.
- Kummer R, Albers GW, Mori E, DIAS Steering Committee. The desmoteplase in acute ischemic stroke (DIAS) clinical trial program. International Journal of Stroke 2012;7(7):589‐96. [DOI] [PubMed] [Google Scholar]
DIAS‐J {published data only}
- Lundbeck Japan KK. Clinical Study of Desmoteplase in Japanese Patients With Acute Ischemic Stroke (DIAS‐J). www.ClinicalTrials.gov.
- Mori E. Desmoteplase in Japanese patients with acute ischaemic stroke (DIAS‐J): study objectives of a randomised, double‐blind, placebo‐controlled, dose escalation trial. International Journal of Stroke 2010;5(Suppl 2):192. [Google Scholar]
- Penner R. Paion`s partner Lundbeck initiates Japanese clinical phase II trial with desmoteplase in ischaemic stroke. www.paion.de/images/stories/investoren/finanznachrichten/2010/en/pm_dias‐j_enfinal.pdf 2010.
- Kummer R, Albers GW, Mori E, DIAS Steering Committee. The desmoteplase in acute ischemic stroke (DIAS) clinical trial program. International Journal of Stroke 2012;7(7):589‐96. [DOI] [PubMed] [Google Scholar]
EXTEND {published data only}
- Campbell B, Donnan G, Davis S, Ma H, Christensen S, Connelly A, et al. EXtending the time for Thombolysis in Emergency Neurological Deficits ‐ The EXTEND Trial progress. International Journal of Stroke 2012;7:52. [DOI] [PubMed] [Google Scholar]
- Churilov L, Liu D, Ma H, Christensen S, Nagakane Y, Campbell B, et al. Multiattribute selection of acute stroke imaging software platform for extending the time for thrombolysis in emergency neurological deficits (EXTEND) clinical trial. International Journal of Stroke 2013;8(3):204‐10. [DOI] [PubMed] [Google Scholar]
- Dagonnier M, Howells DW, Donnan GA, Dewey HM. Recruitment to trials of late thrombolysis. International Journal of Stroke 2012;7(Suppl 1):61. [DOI] [PubMed] [Google Scholar]
- Donnan G. EXTEND : Extending the time for Thrombolysis in Emergency Neurological Deficits. www.anzctr.org.au/.
- Donnan G, Davis S. START‐EXTEND. STroke imAging pRevention and Treatment‐Extending the time for Thrombolysis in Emergency Neurological Deficits. www.strokecenter.org/trials/.
- Donnan G, Davis S. START‐EXTEND. Stroke Imaging Prevention And Treatment ‐ Extending The Time For Thrombolysis In Emergency Neurological Deficits. www.strokecenter.org/trials/.
- Donnan G, Davis S, Ma H, Campbell B, Christensen S, Connelly A, et al. EXtending the time for Thrombolysis in Emergency Neurological Deficits ‐ the EXTEND trial progress. International Journal of Stroke 2011;6(Suppl 1):36. [Google Scholar]
- Donnan G, Davis S, Ma H, Campbell B, Christensen S, Connelly A, et al. Extending the time for thombolysis in emergency neurological deficits‐the extend trial rationale and protocol. International Journal of Stroke 2010;5(Suppl 1):40‐1. [Google Scholar]
- Donnan GA. What's next for the ischaemic penumbra: life after EPITHET. Internal Medicine Journal 2008;38(Suppl 4):A84. [Google Scholar]
- Ma H, Parsons MW, Christensen S, Campbell BCV, Churilov L, Connelly A, et al. A multicentre, randomized, double‐blinded, placebo‐controlled Phase III study to investigate EXtending the time for Thrombolysis in Emergency Neurological Deficits (EXTEND). International Journal of Stroke 2012;7(1):74‐80. [DOI] [PubMed] [Google Scholar]
Ogawa ia Urokinase {unpublished data only}
- Intra‐arterial urokinase trial. Ongoing study April 2002.
PROACT III {unpublished data only}
- PROACT III. Ongoing study Unknown.
WAKE‐UP 2011 {published and unpublished data}
- Anonymous. The WAKE‐UP clinical trial. www.stroke.org.uk/research/wake‐clinical‐trial 2013.
- Thomalla G. A multicentre, randomized, double‐blind, placebo‐controlled trial to test efficacy and safety of MRI‐based thrombolysis in wake‐up stroke (WAKE‐UP). Proceedings of the 22nd European Stroke Conference; 2013 May 28‐31; London, UK. 2013:Abst. OAID54.
- Thomalla G. Efficacy and safety of MRI‐based thrombolysis in wake‐up stroke (WAKE‐UP). www.ClinicalTrials.gov.
- Thomalla G. Efficacy and safety of MRI‐based thrombolysis in wake‐up stroke: a randomised, double‐blind, placebo‐controlled trial. www.clinicaltrialsregister.eu. [DOI] [PubMed]
- Thomalla G, Ebinger M, Fiehler J, Fiebach JB, Endres M, Gerloff C. EU‐funded treatment study: WAKE‐UP. A randomized, placebo‐controlled MRI‐based trial of thrombolysis in wake‐up stroke. Der Nervenart 2012;83(10):1241‐51. [DOI] [PubMed] [Google Scholar]
WASSABI {published data only}
- Kass‐Hout T, Kass‐Hout O, Mokin M, Al Masry M, Nourollahzadeh E, Siddiqui A, et al. Wake up symptomatic stroke in acute brain ischemia (WASSABI) clinical trial. Journal of Neurointerventional Surgery 2012;4:A77. [Google Scholar]
Yamanouchi iv rt‐PA {unpublished data only}
- Unknown. Ongoing study Unknown.
Additional references
APT 1994
- Antiplatelet Trialists' Collaboration. Collaborative overview of randomised trials of antiplatelet therapy ‐ I: Prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. BMJ 1994;308(6923):81‐106. [PMC free article] [PubMed] [Google Scholar]
Ciccone 1998
- Ciccone A, Motto C, Aritzu E, Piana A, Candelise L. Risk of aspirin use plus thrombolysis after acute ischaemic stroke: a further MAST‐I analysis. MAST‐I Collaborative Group. Multicentre Acute Stroke Trial‐‐Italy. Lancet 1998;352(9131):880. [DOI] [PubMed] [Google Scholar]
Deeks 2001
- Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several studies in metaanalysis. In: Egger M, Davey Smith G, Altman DG editor(s). Systematic Reviews in Health Care: Metaanalysis in Context. 2nd Edition. London: BMJ Publication Group, 2001. [Google Scholar]
Dundar 2003
- Dundar Y, Hill R, Dickson R, Walley T. Comparative efficacy of thrombolytics in acute myocardial infarction: a systematic review. Quarterly Journal of Medicine 2003;96(2):103‐13. [DOI] [PubMed] [Google Scholar]
ESO Stroke Guidelines 2008
- The European Stroke Organization (ESO) Executive Committee and the ESO Writing Committee. Guidelines for management of ischaemic stroke and transient ischaemic attack 2008. Cerebrovascular Diseases 2008;25(5):457‐507. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Ingall 2003
- Ingall TJ, O'Fallon WM, Louis TA, Hertzberg V, Goldfrank LR, Asplund K. Initial findings of the rt‐PA acute stroke treatment review panel. Cerebrovascular Diseases 2003;16(Suppl 4):125. [Google Scholar]
Jin Urokinase metaanalysis 2000
- Jin R, Zheng RY. Meta analysis of thrombolytic therapy of intravenous urokinase in acute cerebral infarction. Chinese Journal of Pharmacoepidemiology 2000;9(2):57‐8. [Google Scholar]
Kent 2005a
- Kent DM, Price LL, Ringleb P, Hill MD, Selker HP. Sex‐based differences in response to recombinant tissue plasminogen activator in acute ischemic stroke. A pooled analysis of randomized clinical trials. Stroke 2005;36(1):62‐5. [DOI] [PubMed] [Google Scholar]
Kent 2005b
- Kent DM, Hill MD, Savitz SI, Selim M. Gender differences in tPA‐related arterial recanalization (response). Stroke 2005;36(12):2529‐30. [DOI] [PubMed] [Google Scholar]
Kent 2007
- Kent DM, Selker HP, Ruthazer R, Bluhmki, Hacke W. Glycemic index predicts poor prognosis and risk of thrombolytic‐related symptomatic intracerebral hemorrhage but does not appear to modify the effectiveness of rtPA therapy. Stroke 2007;38(2):455. [Google Scholar]
Mann 2005
- Mann J, Kent DM, Price LL, Selker HP, Ringleb P, Hill MD. Sex‐based differences in response to recombinant tissue plasminogen activator in acute ischemic stroke. Stroke 2005;36(5):929. [DOI] [PubMed] [Google Scholar]
NICE Stroke Guideline 2008
- National Institute for Health and Clinical Excellence (NICE). Stroke. The diagnosis and acute management of stroke and transient ischaemic attacks. National Institute of Clinical Excellence, www.dh.gov.uk/stroke 2008:1‐37. [www.dh.gov.uk/stroke]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre. The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre. The Cochrane Collaboration, 2012.
rt‐PA pooled analysis 2004
- Hacke W1, Donnan G, Fieschi C, Kaste M, Kummer R, Broderick JP, et al. ATLANTIS Trials Investigators, ECASS Trials Investigators, and NINDS rt‐PA Study Group investigators. Better outcome with early stroke treatment: a pooled analysis of the ATLANTIS, ECASS and NINDS rt‐PA stroke trials. Lancet 2004;363(9411):768‐74. [DOI] [PubMed] [Google Scholar]
rt‐PA pooled analysis 2008
- Lees KR, Bluhmki E, Kummer R, Brott TG, Toni D, Grotta JC, for the ECASS, ATLANTIS, NINDS and EPITHET rt‐PA Study Group Investigators. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS and Epithet trials. Lancet 2010;375:1965‐703. [DOI] [PubMed] [Google Scholar]
Sandercock 2002
- Sandercock PAG, Berge E, Dennis M, Hand P, Kwan J, Lewis S, et al. A systematic review of the effectiveness, cost‐effectiveness and barriers to the implementation of thrombolytic and neuroprotective treatment in the NHS. Southampton: NHS Health Technology Assessment Programme, 2002. [DOI] [PubMed] [Google Scholar]
Sandercock 2006
- Sandercock P, Lewis S. Sex‐based differences in the effect of intra‐arterial treatment of stroke: a plea to stop torturing the old data and do large trials!. Stroke 2006;37(9):2198. [DOI] [PubMed] [Google Scholar]
Sharp 1996
- Sharp SJ, Thompson SG, Altman DG. The relation between treatment benefit and underlying risk in meta‐analysis. BMJ 1996;313(7059):735‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
SITS‐MOST
- Wahlgren N, Ahmed N, Dávalos A, Ford GA, Grond M, Hacke W, et al. Thrombolysis for acute ischaemic stroke in the Safe Implementation of Thrombolysis in Stroke‐Monitoring Study (SITS‐MOST): an observational study. Lancet 2007;369(9558):275‐82. [DOI] [PubMed] [Google Scholar]
TAS‐PP 1999
- Cornu C, Boutitie F. Thrombolysis in acute stroke pooling project: a meta‐analysis on individual patient data. Journal of Clinical Neuroscience 1999;6(1):20‐3. [DOI] [PubMed] [Google Scholar]
TTAS
- Emberson J, Lees KR, Howard G, Bluhmki E, Tilley B, Albers G, et al. Details of a prospective protocol for a collaborative meta‐analysis of individual participant data from all randomized trials of intravenous rt‐PA vs. control: Statistical analysis plan for the Stroke Thrombolysis Trialists' Collaborative meta‐analysis. International Journal of Stroke 2013;8(4):278‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Von Kummer 2002
- Kummer R. Brain haemorrhage after thrombolysis: good or bad?. Stroke 2002;33(6):1446‐7. [DOI] [PubMed] [Google Scholar]
Wardlaw 1999a
- Wardlaw JM, Dorman PJ, Candelise L, Signorini DF. The influence of baseline prognostic variables on outcome after thrombolysis. MAST‐Italy Collaborative Group. Journal of Neurology 1999;246(11):1059‐62. [DOI] [PubMed] [Google Scholar]
Wardlaw 2000
- Wardlaw JM, Sandercock PAG, Warlow CP, Lindley RI. Trials of thrombolysis in acute ischaemic stroke: does the choice of primary outcome measure really matter?. Stroke 2000;31(5):1133‐5. [DOI] [PubMed] [Google Scholar]
Wardlaw 2002
- Wardlaw JM, Lindley RI, Lewis S. Thrombolysis for acute ischaemic stroke: still a treatment for the few by the few. Western Journal of Medicine 2002;176(3):198‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wardlaw 2012
- Wardlaw JM, Murray V, Berge E, Zoppo G, Sandercock P, Lindley Rl, et al. Recombinant tissue plasminogen activator for acute ischaemic stroke: an updated systematic review and meta‐analysis. Lancet 2012;379(9834):2364‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wardlaw 2013
- Wardlaw JM, Koumellis P, Liu M. Thrombolysis (different doses, routes of administration and agents) for acute ischaemic stroke. Cochrane Database of Systematic Reviews 2013, Issue 5. [DOI: 10.1002/14651858.CD000514.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Warlow 2008
- Warlow CP, Gijn J, Dennis MS, Wardlaw JM, Bamford J, Hankey G, et al. Stroke: Practical Management. 3rd Edition. Oxford, UK: Blackwell Scientific Ltd, 2008. [ISBN 978‐1‐4051‐2766‐0] [Google Scholar]
Whiteley 2012
- Whiteley WN, Slot KB, Fernandes P, Sandercock P, Wardlaw J. Risk factors for intracranial hemorrhage in acute ischemic stroke patients treated with recombinant tissue plasminogen activator: a systematic review and meta‐analysis of 55 studies. Stroke 2012;43(11):2904‐9. [DOI] [PubMed] [Google Scholar]
Zinkstok 2008
- Zinkstok SM, Roos YB, ARTIS Investigators. Early administration of aspirin in patients treated with alteplase for acute ischaemic stroke: A randomised controlled trial. Lancet 2012;380(9843):731‐7. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Wardlaw 1992
- Wardlaw JM, Warlow CP. Thrombolysis in acute ischemic stroke: does it work?. Stroke 1992;23(12):1826‐39. [DOI] [PubMed] [Google Scholar]
Wardlaw 1995
- Wardlaw JM, Yamaguchi T, Zoppo GJ, Hacke W. Thrombolysis for acute ischaemic stroke. Cochrane Database of Systematic Reviews 1995, Issue 1. [DOI: 10.1002/14651858.CD000213] [DOI] [Google Scholar]
Wardlaw 1997a
- Wardlaw JM, Yamaguchi T, Zoppo G, Hacke W. Meta‐analysis: thrombolytic therapy increases the risk for early death and intracranial hemorrhage after acute ischemic stroke. ACP Journal Club 1997;126(2):35. [Google Scholar]
Wardlaw 1997b
- Wardlaw JM, Warlow CP, Counsell C. Systematic review of evidence on thrombolytic therapy for acute ischaemic stroke. Lancet 1997;350:607‐14. [DOI] [PubMed] [Google Scholar]
Wardlaw 1999
- Wardlaw JM, Zoppo GJ, Yamaguchi T. Thrombolysis for acute ischaemic stroke. Cochrane Database of Systematic Reviews 1999, Issue 4. [DOI: 10.1002/14651858.CD000213] [DOI] [Google Scholar]
Wardlaw 2003a
- Wardlaw JM, Sandercock PAG, Berge E. Thrombolytic therapy with rt‐PA for acute ischaemic stroke: where do we go from here? A cumulative meta‐analysis. Stroke 2003;34(6):1437‐42. [DOI] [PubMed] [Google Scholar]
Wardlaw 2003b
- Wardlaw JM, Zoppo G, Yamaguchi T, Berge E. Thrombolysis for acute ischaemic stroke. Cochrane Database of Systematic Reviews 2003, Issue 3. [DOI: 10.1002/14651858.CD000213] [DOI] [PubMed] [Google Scholar]
Wardlaw 2004
- Wardlaw J, Berge E, Zoppo G, Yamaguchi T. Thrombolysis for acute ischemic stroke. Stroke 2004;35(12):2914‐5. [DOI] [PubMed] [Google Scholar]
Wardlaw 2009
- Wardlaw JM, Murray V, Sandercock PAG. Thrombolysis for acute ischaemic stroke. An update of the Cochrane thrombolysis metaanalysis. International Journal of Stroke 2008;3(Suppl 1):50. [Google Scholar]