

Abstract
An R120G mutation in αB-crystallin (CryABR120G) causes desmin-related myopathy (DRM). In mice with cardiomyocyte-specific expression of the mutation, CryABR120G-mediated DRM is characterized by CryAB and desmin accumulations within cardiac muscle, mitochondrial deficiencies, activation of apoptosis, and heart failure (HF). Excessive production of reactive oxygen species (ROS) is often a hallmark of HF and treatment with antioxidants can sometimes prevent the progression of HF in terms of contractile dysfunction and cardiomyocyte survival. It is unknown whether blockade of ROS is beneficial for protein misfolding diseases such as DRM. We addressed this question by blocking the activity of xanthine oxidase (XO), a superoxide-generating enzyme that is upregulated in our model of DRM. The XO inhibitor oxypurinol was administered to CryABR120G mice for a period of 1 or 3 months. Mitochondrial function was dramatically improved in treated animals in terms of complex I activity and conservation of mitochondrial membrane potential. Oxypurinol also largely restored normal mitochondrial morphology. Surprisingly, however, cardiac contractile function and cardiac compliance were unimproved, indicating that the contractile deficit might be independent of mitochondrial dysfunction and the initiation of apoptosis. Using magnetic bead microrheology at the single cardiomyocyte level, we demonstrated that sarcomeric disarray and accumulation of the physical aggregates resulted in significant changes in the cytoskeletal mechanical properties in the CryABR120G cardiomyocytes. Our findings indicate that oxypurinol treatment largely prevented mitochondrial deficiency in DRM but that contractility was not improved because of mechanical deficits in passive cytoskeletal stiffness.
Keywords: mitochondria, oxidative stress, antioxidants, protein misfolding, reactive oxygen species
The protein αB-crystallin (CryAB) is a small heat shock–like protein that is prevalent in cardiomyocytes and can function as a chaperone. A missense mutation, R120G, in αB-crystallin (CryABR120G) is linked to desmin-related myopathy (DRM).1 A transgenic mouse model of CryABR120G-DRM with cardiomyocyte-specific expression of the mutated protein developed hypertrophy, which gradually progressed to dilated cardiomyopathy and heart failure (HF).2 Previous data indicated that mitochondrial dysfunction presented very early in the DRM pathology before any functional cardiac deficits could be detected, and local associations between mitochondria and aggregations of misfolded CryABR120G were noted.3 Early pathological presentation included abnormalities in mitochondrial organization and localization, significant reductions in oxygen consumption, activation of the mitochondrial permeability transition pore (MPTP), and gradual loss of mitochondrial membrane potential. Complex I activity was decreased by as much as 50% by the time the animals reached sexual maturity at 6 weeks, and this decrease occurred before any functional compromises could be detected.3
Reduced complex I activity is associated with a wide spectrum of protein misfolding and neurodegenerative diseases.4 For example, inhibition of complex I in mice leads to degeneration of dopaminergic neurons in the substantia nigra, as is observed in Parkinson’s disease, through activation of apoptotic pathways.5 It is thought that complex I dysfunction and the subsequent impairment of mitochondrial respiration provoke the activation of the mitochondria-dependent apoptotic machinery by directly triggering the release of the apoptogenic molecule cytochrome c from affected mitochondria.6 A large body of evidence suggests that mitochondria and, particularly, complex I are responsible for reactive oxygen species (ROS) generation under physiological and pathological conditions.7 Besides being a major source of ROS and oxidative stress, mitochondria also appear to be highly susceptible to ROS attack. Proteins, lipids, and nucleic acids can be altered by ROS, resulting in covalent changes that affect mitochondrial structure and function, and complex I activity is decreased by oxidative stress.8 The toxic effects of ROS can be reduced by scavenging enzymes such as superoxide dismutase, glutathione peroxidase, and catalase. However, under conditions of long-term progressive disease, continuous production of mitochondrial ROS results in a pathological feed-forward loop in which considerable mitochondrial damage occurs. Irreversible damage appears to correlate more closely with ROS formation than with respiratory chain dysfunction,9 and mitochondria are at least partly responsible for the amplification, accumulation, and propagation of oxidative stress.
Within cardiac myocytes, ROS can be produced by several mechanisms, including mitochondrial electron transport, NADPH oxidase,10 and xanthine oxidase (XO), a superoxide-generating enzyme that is upregulated in animals and in humans with HF.11 In the failing heart, excessive generation of ROS and oxidative stress contributes to cardiac remodeling, mechanoenergetic uncoupling, and depressed myofilament calcium sensitivity.12 ROS can also induce myocyte hypertrophy, apoptosis, and interstitial fibrosis by activating matrix metalloproteinases.13 Inhibition of XO by allopurinol or oxypurinol can help conserve cardiac structure and function, offset alterations in fetal gene expression, and substantially improve the HF phenotype in a rat model of dilated cardiomyopathy.14 Targeted XO blockade enhances survival and improves global cardiac function after ischemic cardiomyopathy is induced in the mouse.14–18 Inhibition of XO can also effectively ameliorate or even prevent oxidation of myofilament proteins, a process that, if left unchecked, can dynamically change cross-bridge cycling and lead to HF.18
We wished to determine the efficacy of XO inhibition in attenuating DRM-induced morbidity. Oxypurinol delivered in the drinking water markedly improved mitochondrial function in CryABR120G-DRM: complex I activity and normal mitochondrial morphology were restored, and MPTP opening decreased. Despite these improvements, subsequent developing deficits in cardiac function were unaffected, suggesting that additional factors intrinsic to the contractile apparatus and/or its milieu were responsible for the contractile dysfunction observed in the CryABR120G-DRM model. Therefore, the mechanical properties of isolated adult cardiomyocytes from the mice with cardiac expression of wild type (CryABWT) and CryABR120G were determined using magnetic bead microrheology. We found that CryABR120G cardiomyocytes showed significantly increased transverse cytoskeletal stiffness and attenuated cytoskeletal viscosity compared to CryABWT cardiomyocytes, possibly caused by sarcomeric disarray or by desmin and CryAB aggregates within the cytoskeleton.
Materials and Methods
Animals and Experimental Protocols
We studied the effect of oxypurinol on CryABR120G-overexpressing transgenic mice (TGox) and compared them to controls (nontransgenic [NTG]), and an untreated transgenic (TG) group. For microrheology experiments, we used normal CryAB-overexpressing CryABWT mice, as described previously.2 Transgenic mice were identified by PCR analysis of genomic DNA isolated from tail clips. Animals were housed in an Assessment and Accreditation of Laboratory Animal Care–approved facility, and all experiments were approved by the Animal Review Board. For this study, 1-month-old CryABR120G mice were treated with oxypurinol (1 mmol/L ; n=20) in drinking water for 30 and/or 90 days. The mice treated for 30 days were used for biochemical mitochondrial analyses only.
Antibodies
Anti–glutathione reductase, anti–glutathione peroxidase, and anti-catalase antibodies were purchased from Abcam Inc (Cambridge, Mass), and antiglucose-6-phosphate dehydrogenase was from Novus Biologicals (Littleton, Colo).
Measurements of Mitochondrial Membrane Potential, Δψ, and Complex I Activity
The analyses were performed using tetra-methyl-rhodamine ester (TMRE) dye reagent (Invitrogen, Carlsbad, Calif). Isolated mitochondria were incubated with 100 nmol/L TMRE and observed under a fluorescence microscope at an excitation wavelength of 549 nm and emission wavelength of 574 nm. Oligomycin (2 μg/mL) was used for incubation with mitochondria. Mitochondrial swelling experiments were performed as described previously.3 Complex I activity was measured as described previously.19 Fifty micrograms of isolated heart mitochondria were diluted in the reaction mixture consisted of 250 mmol/L sucrose, 1.0 mmol/L EDTA, 50 mmol/L Tris-HCl (pH 7.4), 10 μmol/L decylubiquinone (Sigma, St Louis, Mo), and 2 mmol/L KCN. The reaction was initiated by adding 50 μmol/L NADH and monitored continuously at a wavelength of 272 minus 247 nm at 30°C for 1 minute. Rotenone (5 μg) was added, and any rotenone-insensitive activity was measured for 1 minute.
Magnetic Bead Microrheology and Cytoskeletal Displacement Measurements
Detailed procedures are available in the online data supplement at http://circres.ahajournal.org.
Pressure–Volume Analysis
Mice were anesthetized, the heart exposed and instrumented with a pressure–volume catheter placed retrograde though the left ventricular (LV) apex, as described.20 Transient obstruction of the inferior vena cava was used to obtain data over a loading range, and pressure–volume data from the later portion of diastole combined from multiple cycles to generate diastolic pressure–volume relations. These were fit to a monoexponential [P=Po+α(eβV−1)], with stiffness coefficient β. Additional steady-state and pressure–volume relation parameters were obtained as described.20
ROS Analyses
ROS activity was measured using 2 different methods. Peroxidase and XO activity were determined in whole-cell homogenates using Amplex Red reagent (Invitrogen) according to the protocol of the manufacturer. In addition, fresh frozen left ventricle myocardium (8-μm slices) was incubated with 4 μmol/L 2′,7′-dichlorofluorescein (DCF) diacetate for 1 hour at 37°C. Images were taken by confocal microscopy (Nikon PCM-2000). The excitation/emission spectrum for DCF was 480/535 nm.
Statistical Analysis
All data are expressed as means±SE. Comparisons between experimental were determined by 1-way or 2-way ANOVA where appropriate, followed by Student’s t test. A probability value of <0.05 was considered statistically significant.
Results
Inhibition of ROS Production in CryABR120G Hearts
There are persuasive data showing that increased oxygen radical formation participates in the development of a number of pathologies, including cardiovascular, neurodegenerative, and chronic inflammatory disease.21 Oxidative stress also can play a key role in the development of cardiac hypertrophy and dysfunction, accelerating the transition to HF.11 XO, a superoxide-generating enzyme, is upregulated in our model of DRM and elevated XO expression and activity have been demonstrated in end-stage human HF and in the canine rapid pacing-induced HF model as well.15 To define the role of oxidative stress in the progression of CryABR120G-DRM, we treated 1-month CryABR120G TG mice with oxypurinol (TGox group) for 90 days. We confirmed that ROS activity was elevated in untreated 4 month TG mice, but significantly attenuated in the TGox group (Figure 1A and 1B). The fluorescent dye DCF was used to measure the relative ROS levels in the sections from TGox mice and compared to TG and NTG. In the presence of cellular oxidants, nonfluorescent DCF is converted into the fluorescent 5-(and-6)-chloromethyl-2,7-dichlorofluorescein (CM-DCF) and can be observed by confocal microscopy. The reagent is considered to be a marker of oxidant levels rather than a direct reporter for a specific ROS species.22 Myocardial XO and peroxidase activities were increased significantly in TG hearts versus NTG controls, as assessed by Amplex Red binding, but restored to normal levels in the TGox mice (Figure 1C and 1D).
Figure 1.

Oxypurinol administration prevents ROS generation in CryABR120G TG hearts. A, DCF staining indicating total ROS accumulation in unfixed cryosections derived from the hearts of 4-month NTG and TG mice either untreated or treated with oxypurinol (TGox) mice. B, Quantification of DCF fluorescence using MetaMorph software. C, XO activity measured using an Amplex Red assay. D, Peroxide and peroxidase activities in myocardial tissue extracts measured by Amplex Red binding and normalized to the average values from NTG mice (n=5).
ROS have direct effects on cellular structure and function and may be integral signaling molecules in the processes of myocardial remodeling and failure. To understand the role of ROS in the DRM pathology, we performed a histopathologic analysis of 4-month ventricular tissue stained with Gomori’s trichrome (Figure 2A and 2B). Hearts of untreated TG mice showed extensive fibrosis and myofibril disarray. Although aggregate accumulations, disarray, and increased interstitial space were still evident in TGox hearts, fibrosis was remarkably reduced.
Figure 2.
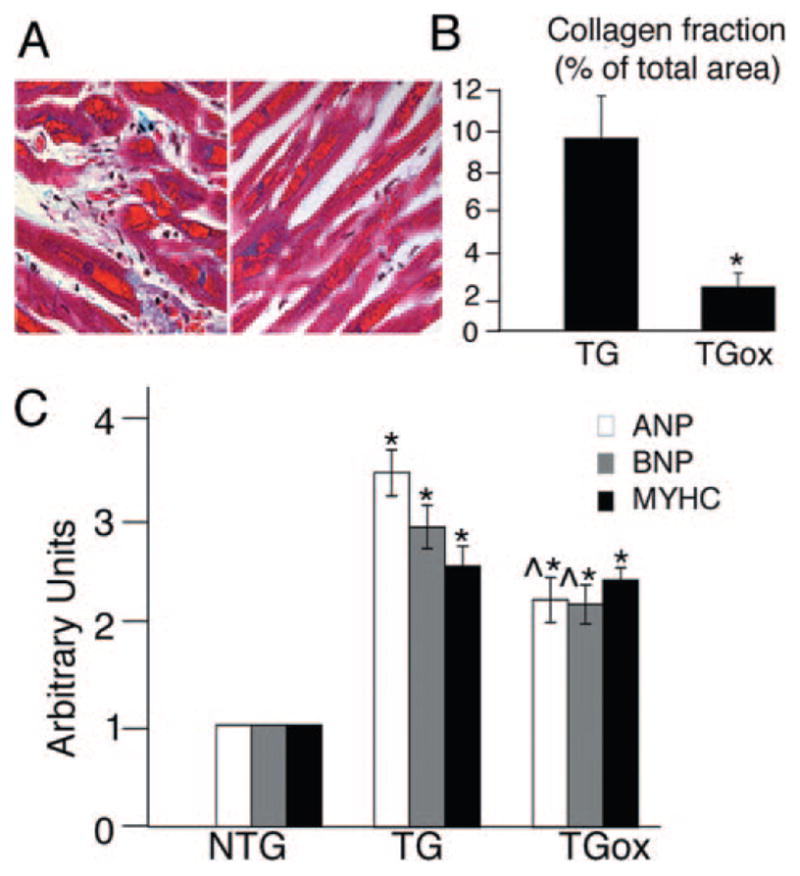
Phenotypic characterization of oxypurinol-treated CryABR120G hearts. A and B, Representative histology (A) and quantification of fibrosis (B) in heart sections from 4-month TG and TGox mice stained with Gomori’s trichrome. ANP indicates atrial natriuretic peptide; BNP, brain natriuretic peptide; MYHC, β-myosin heavy chain. Blue staining indicates collagen deposition. C, Fetal gene expression in NTG, TG, and TGox mice measured by RT-PCR (n=3). *P<0.001 vs NTG; ^P<0.001 vs TG, n=4.
Hypertrophied CryABR120G TG hearts are characterized by activation of the fetal gene program. We confirmed overexpression of atrial natriuretic peptide, brain natriuretic peptide, and β-myosin heavy chain in the left ventricles of the untreated TG group. Although oxypurinol treatment did have an effect in terms of decreasing atrial natriuretic peptide and brain natriuretic peptide (but not β-myosin heavy chain) expression, it was clearly ineffective in restoring transcription to the normal adult pattern (Figure 2C).
Having established that oxypurinol does decrease ROS levels in the CryABR120G TG hearts, we next determined the expression of markers for oxidative/reductive stress in 4-month-old NTG, TG, and TGox mice (Figure 3A and 3B). Recent data showed that overexpression of human CryABR120G in the mouse heart led to increased levels of a number of antioxidative enzymes such as catalase, glutathione peroxidase 1, and glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme in NADPH synthesis.23 In contrast to those data, we found a moderate decrease in the amount of G6PD and glutathione reductase (GSH-R), an antioxidant that catalyzes the reduction of oxidized glutathione to glutathione.24 Oxypurinol treatment restored G6PD to normal levels. In contrast, GSH-R was significantly upregulated in the oxypurinol-treated group, suggesting that oxypurinol increased the ability of mitochondria to defend against oxidative injury. The levels of catalase and glutathione peroxidase 1 were indistinguishable among all experimental groups.
Figure 3.
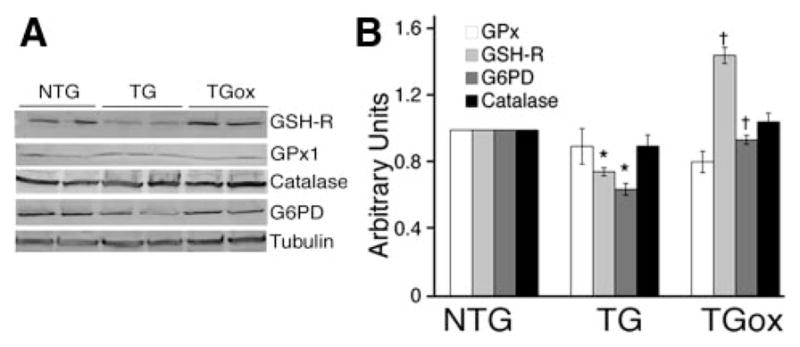
Expression of markers for oxidative/reductive stress. A, Western blots for GSH-R, glutathione peroxidase 1 (Gpx1), G6PD, and catalase. Tubulin was used as a loading control. B, Quantitation of the Western blots. *P<0.001 vs NTG, †P<0.001 vs TG (n=4).
Effects of ROS Production and Inhibition on Mitochondrial Structure and Function
We then analyzed the effect of antioxidant treatment on cardiomyocyte ultrastructure, using transmission electron microscopy to compare cardiomyocytes derived from 4-month NTG, TG, and TGox hearts. CryABR120G expression significantly perturbs sarcomere and mitochondrial organization in the myoplasm. NTG cardiomyocytes are distinguished by a regular array of sarcomeres with mitochondria juxtaposed between them and adjacent to the A and I bands (Figure 4A), but this regular architecture was lacking in CryABR120G cardiomyocytes whether or not they were treated with oxypurinol.
Figure 4.
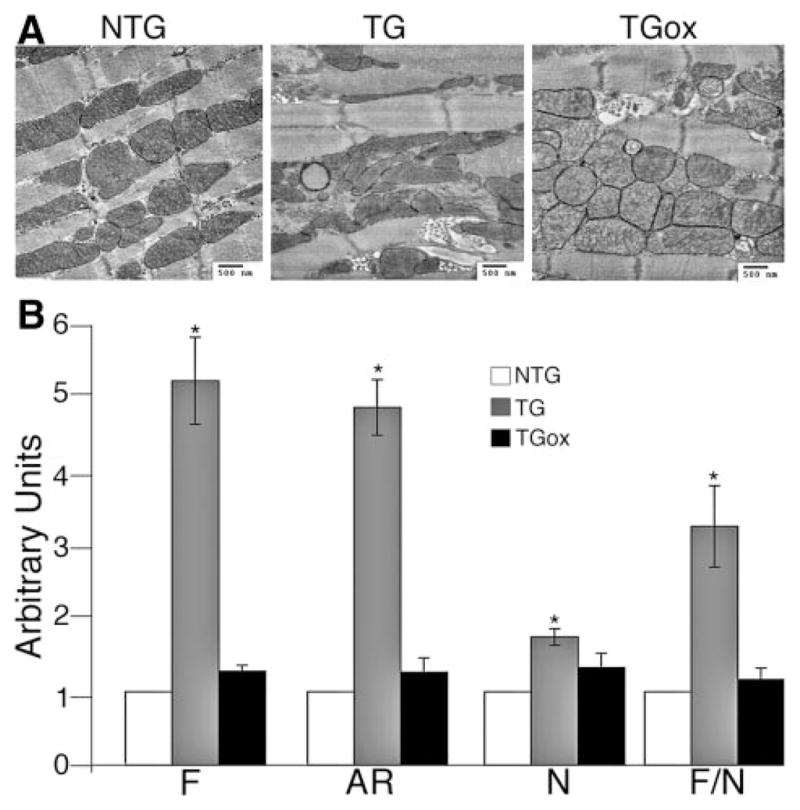
Maintenance of normal mitochondrial ultrastructure with oxypurinol treatment. A, Representative electron micrographs of the ventricles from 4-month NTG, TG, and TGox mice. B, Quantification of mitochondrial shape. *P<0.001 vs NTG.
Cells respond to respiratory deficiency and excessive production of ROS in several ways, one of which is a change in mitochondrial morphology.25 Chronic inhibition of complex I activity, for example, can significantly increase mitochondrial outgrowth and branching.26 Many of the mitochondria in the untreated TG mice are distinguished by a loss of defined cristae and changes in general shape. On average, there is also significant longitudinal elongation. Antioxidant treatment did have a significant effect on mitochondrial morphology with the cristae appearing to be largely restored to their normal shape and architecture in the oxypurinol treated mice (Figure 4A). Quantitative analysis of mitochondrial shape from the electron micrographs was used to determine the form factor, F, a measure of the mitochondrial degree of branching (perimeter2/4 π×area). Additionally, a measure of mitochondrial length, the aspect ratio (AR), which is the ratio between the major and minor axes of the ellipse equivalent to the object was also determined (Figure 4B).27 Both F and AR are independent of image magnification, and a minimum value of 1 (corresponding to a perfect circle) was routinely subtracted from each measurement. The total number of mitochondria (N) and the F/N ratio (“mitochondrial complexity” parameter)27 per each field of view was also determined. There was a slight but significant (P<0.005) increase in mitochondrial number in the TG mice but not in the TGox group. TG mice showed highly significant changes in F, AR, and F/N, with all 3 of these parameters being restored to normalcy in the TGox mice.
The relative conservation of normal cardiomyocyte mitochondrial ultrastructure in the TGox group indicated that oxypurinol had beneficial effects on the mitochondria in the CryABR120G hearts, and we wished to determine whether mitochondrial function was restored as well. One-month-old TGox mice were treated with oxypurinol for 30 days. Consistent with our previous data,3 complex I activity was significantly decreased in the untreated CryABR120G mice. Strikingly, oxypurinol treatment of the CryABR120G mice resulted in the restoration of normal levels of complex I activity (Figure 5A).
Figure 5.

Oxypurinol improves cardiac mitochondrial function in 2 month old TG mice. A, Spectrophotometric assay of complex I activity, as determined by reduction of the ubiquinone analog decylubiquinone. B, Mitochondrial swelling was induced by the addition of CaCl2. C, Changes of mitochondrial TMRE fluorescence (expressed as a percentage of the initial level) induced by addition of oligomycin, the F1F0-ATPase inhibitor. Isolated mitochondria from 2-month NTG, TG, and TGox hearts were loaded with TMRE and signal decay determined in real time using fluorescent microscopy. *P<0.001 vs NTG.
ROS Inhibition Rescues Mitochondrial Function but Fails to Rescue Cardiac Function
We had previously shown that CryABR120G expression results in MPTP activation.3 To assess the effects of oxypurinol on this process, cardiac mitochondria were isolated from NTG, TG, and TGox mice and examined for their ability to undergo Ca2+-induced swelling. As expected, mitochondria from TG hearts were significantly swollen at baseline and showed a dramatically attenuated response after addition of Ca2+ compared to NTG mice (Figure 5B). Despite these differences, mitochondria from TGox mice showed a significant increase in baseline absorption, indicating they were less swollen compared to the TG group, and displayed a similar change in absorbance compared to the NTG mitochondria when challenged with Ca2+, indicating a partial rescue of the compromised MPTP that is characteristic of CryABR120G mitochondria.
When mitochondria become dysfunctional, the F1F0 ATP synthase can hydrolyze ATP rather than synthesizing it, the net result, even in the face of respiratory deficiency, being conservation of mitochondrial membrane potential, Δψ. To test whether respiratory deficiency was masked by the ATP synthase operating in the reverse mode in the TGox mice, we loaded isolated mitochondria with TMRE to image time-dependent mitochondrial membrane potential. After loading, the mitochondria were incubated with oligomycin, an inhibitor of the mitochondrial F1F0-ATP synthase (Figure 5C). As expected, addition of oligomycin to mitochondria from TG mice quickly caused marked mitochondrial depolarization. In contrast, in NTG and TGox hearts, mitochondrial depolarization proceeded at slow rates even after extensive incubation, ruling out the possibility of a “masked” respiratory deficiency in the TGox mice.
Inhibition of XO can enhance cardiac performance at rest and under stress in several models of HF.28,29 We therefore measured cardiac hemodynamics in 4-month-old NTG, TG, and TGox animals using invasive catheterization. Surprisingly, in view of the improvements outlined above, we found significantly compromised diastolic function in both TG and TGox groups (Table). Left ventricular systolic function, as determined by dP/dtmax, was affected in both groups. Left ventricular dP/dtmin was significantly higher under baseline conditions in both the TG and TGox groups as compared with the NTG mice. The ability of TGox mice to respond to β-adrenergic stimulation was also significantly blunted versus the NTG animals. Despite the changes in the expression of hypertrophic markers, the heart weight/body weight ratios were similarly perturbed in both the TG and TGox groups (Table).
Table.
Hemodynamic Measurements
| Basal
|
Dobutamine (32 ng/min per Gram Body Weight)
|
|||||
|---|---|---|---|---|---|---|
| NTG | TG | TGox | NTG | TG | TGox | |
| Body weight, g | 26.33±1.23 | 27.22±0.85 | 27.84±0.77 | … | … | … |
| Heart weight, g | 0.12±0.01 | 0.20±0.006* | 0.192±0.007 | … | … | … |
| Heart weight/body weight | 0.005±0.0002 | 0.007±0.0003* | 0.007±0.0001 | … | … | … |
| Heart rate, bpm | 420±8.2 | 320±12* | 297±23* | 581±13.2 | 410±11.4* | 439±13.2* |
| MAP, mm Hg | 69.9±2.47 | 61.5±3.1* | 57.3±5.4* | 70.82±6 | 51.59±4.7* | 55±3.09* |
| Systolic pressure, mm Hg | 80.83±3.3 | 79.24±4.12 | 74.07±4.7 | 84.7±6.7 | 68.9±3.72 | 70.8±4.5 |
| Diastolic pressure, mm Hg | 9.3±2.36 | 11.58±2.57 | 11.45±3.14 | 2.19±0.77 | 7.15±1.23* | 4.33±1.07 |
| LVP, mm Hg | 93.4±2 | 83.6±3.17 | 83±5 | 103.92±3.1 | 83.51±2.56* | 84.5±2.5* |
| dP/dtmax, mm Hg/sec | 8828±286 | 7555±321* | 7205±547* | 20 869±672 | 9211±698* | 11 279±547*† |
| dP/dt40, mm Hg/sec | 8335±237 | 7255±392 | 6439±689 | 17 104±374 | 8792±647* | 10 855±689*† |
| dP/dtmin, mm Hg/sec | −9524±635 | −5530±544* | −5436±495* | −12 983±1707 | −6795±512* | −5820±629* |
Measurements of wet heart weight and body weight, along with the hemodynamic parameters, including heart rate, peak LV pressure (LVP), LV dP/dtmax (an index of myocardial contractility), and LV dP/dtmin (an index of myocardial relaxation), were determined at baseline (basal) and during infusion of dobutamine (32 ng/min per gram body weight). All data are presented as means±SE. MAP indicates mean arterial pressure.
P<0.001 vs NTG,
P<0.001 vs TG (n=6–7 for every group).
In addition, the hearts from NTG, TG, and TGox mice were studied by in vivo pressure–volume analysis. NTG and both groups of TG mice showed significant differences in the diastolic relations; they were shifted downward and to the right for both groups of TG mice (Figure 6). The stiffness coefficients (Figure 6B) estimated as the slope of the end-diastolic pressure–volume relations, were 0.0183±0.004 versus 0.154±0.005 and 0.147±0.01 for NTG, TG, and TGox respectively (P<0.00005). The maximal power index (PWRmx/EDV), a chamber contractility indicator, was nearly 50% lower in TG and TGox mice (not shown) indicating increased passive stiffness and reduced capacity of the heart to generate peak power.
Figure 6.
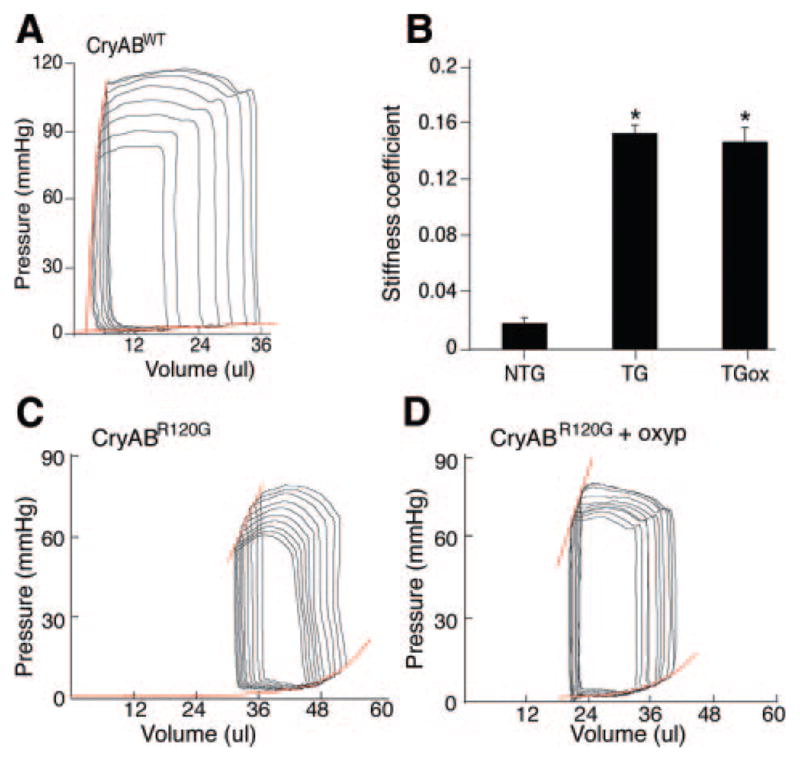
A and B, Representative pressure–volume loops and end-systolic and end-diastolic relations (in red) from 4-month-old (A) NTG, (BC, left panel) TG untreated and (BD, right panel) treated by with oxypurinol mice. CB, The stiffness coefficient estimated as the slope of the end-diastolic pressure–volume relations. *P<0.00005.
CryABR120G Expression Leads to Altered Cardiomyocyte Stiffness
Thus, despite the beneficial effects of ROS inhibition at the biochemical and mitochondrial levels, cardiac function in TGox animals was still significantly compromised. We hypothesized that the changes observed in the stiffness coefficients of the CryABR120G hearts was attributable to alterations in the overall biomechanical properties of the cardiomyocytes, as a result of the massive accumulations of aggregated proteins in the sarcoplasm and collapse of the desmin network around these granulofilamentous bodies. To address this hypothesis, we quantitated the passive mechanical properties of the cell in the longitudinal and transverse directions in isolated adult cardiomyocytes derived from TG mice and compared them to normal CryAB overexpressors (CryABWT). Magnetic bead microrheology was used to perturb laminin-coated beads bound to the cell surface, and cytoskeletal stiffness was inferred by measuring induced bead and cytoskeletal displacement.30 Because bead displacement is a combination of bulk cytoskeletal displacement and bead movement relative to the cytoskeleton through bead rolling and local membrane strain, we measured induced cytoskeletal displacement in close proximity (≈6 μm) to the bead (Figure 7A). For transverse force application, cytoskeletal displacement amplitude was significantly reduced in CryABR120G cardiomyocytes compared to CryABWT cells, indicating significantly increased cytoskeletal stiffness (Figure 7B). Furthermore, the phase lag between applied force and cytoskeletal displacement in the transverse direction was significantly reduced in the CryABR120G cardiomyocytes compared to wild-type overexpressors (Figure 7C). Force application to the longitudinal (parallel to sarcomere) direction induced only small cytoskeletal displacements, with no significant differences between CryABR120G and CryABWT cells (data not shown).
Figure 7.

Magnetic bead experiments. A, Cytoskeletal displacements were measured in 6 regions, distributed around the magnetic bead center at a distance of ≈6 μm. The magnetic trap tip is visible at the far right edge of the image, applying a forcing function in the horizontal direction. Scale bar=10 μm. B, Cytoskeletal displacement amplitude for transverse force application. C, Phase lag of cytoskeletal displacement relative to forcing function for transverse force application. P<0.001.
Discussion
The protein misfolding diseases are a heterogeneous family of disorders and are characterized by a gradually progressive, selective loss of anatomically or physiologically relevant systems.31 Despite this heterogeneity, there is strong evidence that mitochondrial dysfunction occurs early and acts causally in protein misfolding disease pathogenesis.3,31,32 The functional impairment of mitochondria is accompanied by extensive generation of ROS, but, in addition to being a major site for ROS production, mitochondria also become a target for ROS-induced damage and can be severely compromised by prolonged oxidative stress. ROS can trigger a transient burst of mitochondrial ROS production via ROS activation of the MPTP, a phenomenon termed ROS-induced ROS release.7 Treatment with oxypurinol effectively shuts down this pathogenic limb of DRM, conserving internal mitochondrial architecture and function, as measured by complex I activity. Interestingly, the key players in the ability of the cell to mount an antioxidant defense, such as catalase and glutathione peroxidase, remained unchanged in the TG hearts. Furthermore, GSH-R and G6PD levels were reduced in TG hearts, suggesting that the endogenous antioxidant defense system was unable to cope with the long-term oxidative stress induced by expression of CryABR120G. These results are in agreement with previous studies conducted on a number of cardiomyopathic models.33,34 However, Rajasekaran et al recently reported a significant upregulation of G6PD, catalase, and glutathione peroxidase in mice that overexpressed human CryABR120G in mouse cardiomyocytes.23 There are a number of possible explanations for the differences in the 2 data sets. First, our TG animals are made using the homologous mouse sequences, whereas Rajasekaran et al used human DNA. Second, the experiments were done in different strains of mice, which can often lead to significant differences in phenotype presentation and/or severity.35
Increased myocardial oxidative stress by enhanced ROS generation can have potent effects on the extracellular matrix, stimulating cardiac fibroblast proliferation, which, in turn, can trigger fibrosis and matrix remodeling. In vivo, mutant models lacking antioxidant proteins such as thioredoxin36 display myocyte hypertrophy, apoptosis, and interstitial fibrosis, whereas overexpression of antioxidants such as glutathione peroxidase37 or peroxiredoxin-338 can prevent these processes. Our data are consistent with but do not prove a causal link between ROS production and fibrosis because significant decreases in interstitial fibrosis occurred in the oxypurinol treated CryABR120G mice.
Changes in contractility can be caused by altered sarcomeric function, by modifications in passive cytoskeletal stiffness, or by structural changes in the sarcomere organization. In contrast to other studies, in which inhibition of XO improved both in vivo and ex vivo contractile function of failing hearts,14–17,28 oxypurinol treatment failed to materially rescue cardiac function in the CryABR120G mice. The results of the present study provide several new insights into the mechanisms that contribute to the origin of contractile dysfunction in CryAB-DRM. In our experiments, laminin-coated magnetic beads bound to surface of isolated cardiac myocytes from CryABR120G mice displaced significantly less under transverse force application and induced cytoskeletal displacement was markedly reduced compared to wild-type littermates, indicating significantly increased transverse cytoskeletal stiffness. CryABWT myocytes have uniformly oriented myofibrils aligned at the Z-band through desmin filaments, resulting in anisotropic cell mechanics with a significantly increased stiffness in the longitudinal direction compared to the transverse direction. In contrast, CryABR120G mutant cells show less obvious anisotropic mechanics and the transverse stiffness approaches the level of the longitudinal stiffness. We speculate that altered sarcomeric alignment and cytoplasmic desmin and CryAB aggregates function as lateral reinforcements or cross-linkers in the cardiomyocytes from CryABR120G mice, comparable to the function of diagonal struts in stabilizing a scaffold, resulting in increased transverse stiffness. In addition to CryAB being able to stabilize the cytoskeleton through its interactions with desmin under conditions of stress, recruitment of CryAB to cardiac myofibrils and a role of the protein in preventing spontaneous unfolding of titin has been described.39
In addition to the increased transverse cytoskeletal stiffness, we found that CryABR120G mutant cells had decreased cytoskeletal viscosity, characterized by a significant reduction in the phase lag between force application and induced displacement. The reason for the loss of transverse cytoskeletal viscosity is not clear, but most likely reflects the same ultrastructural changes described above, ie, increased lateral reinforcement and cross-linking of myofibrils. Interestingly, similar measurements on the cardiomyocytes from another, less severe DRM model (mice overexpressing a 7-aa deletion [R173 through E179] in desmin), did not exhibit any difference compared to wild type littermates (data not shown). The correlation between the amount of sarcomeric disarray and desmin/CryAB aggregates and the increase in transverse cytoskeletal stiffness when comparing the CryABR120G and D7-des mutants suggests that these ultrastructural changes are at least in part responsible for the observed changes in cytoskeletal stiffness and the more severe phenotype caused by CryABR120G mutation. Although these ultrastructural changes do not, by themselves, lead to cardiomyocyte cell death,40 our data point to the potential importance of their physical impediment to continuous and normal cardiomyocyte contractility, with increased cytoskeletal stiffness and altered viscoelastic mechanical properties on the level of the single cardiomyocyte and a concomitant increase in chamber stiffness.
The results of this study, although correlative, suggest that the biological processes driving the transition of CryABR120G hearts to failure in vivo are complex and dependent on multiple toxic stimuli that accumulate as a result of mutant protein expression. First and foremost is the accumulation of toxic preamyloid oligomers, which, via inducible transgenic means, we have shown are causative for cardiomyocyte cell death.40 The present study further suggests that accumulation of misfolded CryABR120G protein itself into the perinuclear aggresomes could play an important role in chronic LV dysfunction in CryABR120G TG mice. Recently, we have been able to reduce aggregate levels by cardiac restricted overexpression of Bcl2 in these mice and noted significantly improved cardiac function. These data, although also correlative, emphasize the multifactorial nature of the pathology and indicate that combinatorial therapies will be needed to develop an effective therapy for preexisting disease.
Acknowledgments
Sources of Funding
This work was supported by NIH grants P01HL69799, P50HL074728, P50HL077101, P01HL059408, and R01HL087862 and an International Collaboration Research Project with the Japanese Health Science Ministry (to J.R.).
Footnotes
This manuscript was sent to Hans Michael Piper, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
Disclosures
None.
References
- 1.Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, Chateau D, Chapon F, Tome F, Dupret JM, Paulin D, Fardeau M. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 2.Wang XOH, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 3.Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-B-crystallin desmin-related cardiomyopathy. Circulation. 2005;112:3451–3461. doi: 10.1161/CIRCULATIONAHA.105.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schon EA, Manfredi G. Neuronal degeneration and mitochondrial dysfunction. J Clin Invest. 2003;111:303–312. doi: 10.1172/JCI17741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 6.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 7.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–363. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- 9.Barrientos A, Moraes CT. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem. 1999;274:16188–16197. doi: 10.1074/jbc.274.23.16188. [DOI] [PubMed] [Google Scholar]
- 10.Tsutsui H. Mitochondrial oxidative stress and heart failure. Intern Med. 2006;45:809–813. doi: 10.2169/internalmedicine.45.1765. [DOI] [PubMed] [Google Scholar]
- 11.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49:241–248. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 12.Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol. 2002;34:379–388. doi: 10.1006/jmcc.2002.1526. [DOI] [PubMed] [Google Scholar]
- 13.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. 2001;280:C53–C60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 14.Minhas KM, Saraiva RM, Schuleri KH, Lehrke S, Zheng M, Saliaris AP, Berry CE, Barouch LA, Vandegaer KM, Li D, Hare JM. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res. 2006;98:271–279. doi: 10.1161/01.RES.0000200181.59551.71. [DOI] [PubMed] [Google Scholar]
- 15.Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, Marban E, Hare JM. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation. 2001;104:2407–2411. doi: 10.1161/hc4501.098928. [DOI] [PubMed] [Google Scholar]
- 16.Engberding N, Spiekermann S, Schaefer A, Heineke A, Wiencke A, Muller M, Fuchs M, Hilfiker-Kleiner D, Hornig B, Drexler H, Landmesser U. Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation. 2004;110:2175–2179. doi: 10.1161/01.CIR.0000144303.24894.1C. [DOI] [PubMed] [Google Scholar]
- 17.Naumova AV, Chacko VP, Ouwerkerk R, Stull L, Marban E, Weiss RG. Xanthine oxidase inhibitors improve energetics and function after infarction in failing mouse hearts. Am J Physiol Heart Circ Physiol. 2006;290:H837–H843. doi: 10.1152/ajpheart.00831.2005. [DOI] [PubMed] [Google Scholar]
- 18.Stull LB, Leppo MK, Szweda L, Gao WD, Marban E. Chronic treatment with allopurinol boosts survival and cardiac contractility in murine post-ischemic cardiomyopathy. Circ Res. 2004;95:1005–1011. doi: 10.1161/01.RES.0000148635.73331.c5. [DOI] [PubMed] [Google Scholar]
- 19.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 20.Segers P, Georgakopoulos D, Afanasyeva M, Champion HC, Judge DP, Millar HD, Verdonck P, Kass DA, Stergiopulos N, Westerhof N. Conductance catheter-based assessment of arterial input impedance, arterial function, and ventricular-vascular interaction in mice. Am J Physiol Heart Circ Physiol. 2005;288:H1157–H1164. doi: 10.1152/ajpheart.00414.2004. [DOI] [PubMed] [Google Scholar]
- 21.Gutteridge JM, Halliwell B. Free radicals and antioxidants in the year 2000. A historical look to the future. Ann N Y Acad Sci. 2000;899:136–147. doi: 10.1111/j.1749-6632.2000.tb06182.x. [DOI] [PubMed] [Google Scholar]
- 22.Koopman WJ, Verkaart S, van Emst-de Vries SE, Grefte S, Smeitink JA, Willems PH. Simultaneous quantification of oxidative stress and cell spreading using 5-(and-6)-chloromethyl-2′,7′-dichlorofluorescein. Cytometry A. 2006;69:1184–1192. doi: 10.1002/cyto.a.20348. [DOI] [PubMed] [Google Scholar]
- 23.Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, Barry WH, Loscalzo J, Odelberg SJ, Benjamin IJ. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kehrer JP, Lund LG. Cellular reducing equivalents and oxidative stress. Free Radic Biol Med. 1994;17:65–75. doi: 10.1016/0891-5849(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 25.Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 26.Koopman WJ, Verkaart S, Visch HJ, van der Westhuizen FH, Murphy MP, van den Heuvel LW, Smeitink JA, Willems PH. Inhibition of complex I of the electron transport chain causes O2-mediated mitochondrial outgrowth. Am J Physiol Cell Physiol. 2005;288:C1440–C1450. doi: 10.1152/ajpcell.00607.2004. [DOI] [PubMed] [Google Scholar]
- 27.Koopman WJ, Verkaart S, Visch HJ, van Emst-de Vries S, Nijtmans LG, Smeitink JA, Willems PH. Human NADH:ubiquinone oxidoreductase deficiency: radical changes in mitochondrial morphology? Am J Physiol Cell Physiol. 2007;293:C22–C29. doi: 10.1152/ajpcell.00194.2006. [DOI] [PubMed] [Google Scholar]
- 28.Ekelund UE, Harrison RW, Shokek O, Thakkar RN, Tunin RS, Senzaki H, Kass DA, Marban E, Hare JM. Intravenous allopurinol decreases myocardial oxygen consumption and increases mechanical efficiency in dogs with pacing-induced heart failure. Circ Res. 1999;85:437–445. doi: 10.1161/01.res.85.5.437. [DOI] [PubMed] [Google Scholar]
- 29.Perez NG, Gao WD, Marban E. Novel myofilament Ca2+-sensitizing property of xanthine oxidase inhibitors. Circ Res. 1998;83:423–430. doi: 10.1161/01.res.83.4.423. [DOI] [PubMed] [Google Scholar]
- 30.Lammerding J, Huang H, So PT, Kamm RD, Lee RT. Quantitative measurements of active and passive mechanical properties of adult cardiac myocytes. IEEE Eng Med Biol Mag. 2003;22:124–127. doi: 10.1109/memb.2003.1256282. [DOI] [PubMed] [Google Scholar]
- 31.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 32.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 33.Dhalla AK, Singal PK. Antioxidant changes in hypertrophied and failing guinea pig hearts. Am J Physiol. 1994;266:H1280–H1285. doi: 10.1152/ajpheart.1994.266.4.H1280. [DOI] [PubMed] [Google Scholar]
- 34.Singh N, Dhalla AK, Seneviratne C, Singal PK. Oxidative stress and heart failure. Mol Cell Biochem. 1995;147:77–81. doi: 10.1007/BF00944786. [DOI] [PubMed] [Google Scholar]
- 35.Kokkotou E, Jeon JY, Wang X, Marino FE, Carlson M, Trombly DJ, Maratos-Flier E. Mice with MCH ablation resist diet-induced obesity through strain-specific mechanisms. Am J Physiol Regul Integr Comp Physiol. 2005;289:R117–R124. doi: 10.1152/ajpregu.00861.2004. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, Wagner T, Vatner SF, Sadoshima J. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest. 2003;112:1395–1406. doi: 10.1172/JCI17700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiomi T, Tsutsui H, Matsusaka H, Murakami K, Hayashidani S, Ikeuchi M, Wen J, Kubota T, Utsumi H, Takeshita A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2004;109:544–549. doi: 10.1161/01.CIR.0000109701.77059.E9. [DOI] [PubMed] [Google Scholar]
- 38.Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara T, Oikawa S, Kinugawa S, Tsutsui H. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2006;113:1779–1786. doi: 10.1161/CIRCULATIONAHA.105.582239. [DOI] [PubMed] [Google Scholar]
- 39.Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, Ding L, Labeit S, Horwitz J, Leonard KR, Linke WA. Association of the chaperone alphaB-crystallin with titin in heart muscle. J Biol Chem. 2004;279:7917–7924. doi: 10.1074/jbc.M307473200. [DOI] [PubMed] [Google Scholar]
- 40.Sanbe A, Osinska H, Villa C, Gulick J, Klevitsky R, Glabe CG, Kayed R, Robbins J. Reversal of amyloid-induced heart disease in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2005;102:13592–13597. doi: 10.1073/pnas.0503324102. [DOI] [PMC free article] [PubMed] [Google Scholar]