

Abstract
The mechanical properties of the interphase nucleus have important implications for cellular function and can reflect changes in nuclear envelope structure and/or chromatin organization. Mutations in the nuclear envelope proteins lamin A and C cause several human diseases, such as Emery–Dreifuss muscular dystrophy, and dramatic changes in nuclear stiffness have been reported in cells from lamin A/C–deficient mice. We have developed a cellular strain technique to measure nuclear stiffness in intact, adherent cells and have applied this experimental method to fibroblasts from mouse models of Emery–Dreifuss muscular dystrophy and to skin fibroblasts from laminopathy patients and healthy control subjects. The experimental protocol is based on measuring induced nuclear deformations in cells plated on a flexible silicone substrate; the nuclear stiffness can subsequently be inferred from the ratio of induced nuclear strain to the applied membrane strain. These experiments reveal that lamins A and C are important determinants of nuclear stiffness and that lamin mutations associated with muscular dystrophies and other laminopathies often result in disturbed nuclear stiffness that could contribute to the tissue-specific disease phenotypes.
Keywords: Nucleus, Nuclear stiffness, Lamin, Muscular dystrophy, Cell mechanics, Strain
1 Introduction
Mutations in the nuclear envelope proteins lamin A and C cause a variety of human diseases that include Emery-Dreifuss muscular dystrophy and limb-girdle muscular dystrophy (reviewed in refs. (1, 2)). Although the mechanisms leading to these tissue-specific phenotypes remain unclear, it is thought that decreased nuclear stability could contribute to the muscular phenotypes in these diseases (1, 3). Because the nucleus is surrounded by the cytoskeleton, direct measurements of its physical properties are inherently difficult. Micropipette aspiration of single nuclei allows direct measurements of nuclear mechanics (see Chap. 1 by Rowat in this volume), but is limited by the risks that the nuclear isolation procedure might damage nuclei (especially in mutant cells) and/or alter nuclear or chromatin structure depending on the buffer conditions. Here, we describe an alternative method in which cells plated on transparent silicone membranes are subjected to uniform strain. The applied substrate strain is transmitted to the cytoskeleton through focal adhesion complexes, resulting in cytoskeletal strain that closely matches the applied membrane strain (4), while the stiffer nucleus deforms less. Applied membrane strain and induced nuclear strain are quantified based on phase contrast and fluorescence images of fluorescently labeled nuclei taken before, during, and after strain application. An advantage of this technique is that it probes nuclear mechanics in the normal cellular environment without having to isolate the nucleus, thus preserving the normal nuclear and cytoskeletal architecture; furthermore, the techniques resemble physiological load application as found in many tissues such as muscle or blood vessel walls. However, the experiments only provide information on the nuclear stiffness relative to the surrounding cytoskeleton, and the induced nuclear deformation depends on cell adhesion to a substrate and intracellular force transmission through the cytoskeleton. We have adapted this technique to measure the contribution of specific nuclear envelope proteins to nuclear stiffness and fragility, and are currently studying the effect of specific lamin mutations (3, 5, 6).
2 Materials
2.1 Preparation of Silicone Membrane Dishes
Components for nine strain dishes, each consisting of a bottomless dish and a plastic O-ring to hold the silicone membrane (see Note 1 and Fig. 2.1). The strain dishes are autoclavable for sterile cell culture conditions. The cell culture dishes are equipped with threads and a wide collar on the outside to fit securely into the dish-holder plate of the strain device in a predetermined position.
Silicone membrane: 0.005-inch-thick silicone sheeting in 12×12-inch sheets (Gloss/Gloss non-reinforced silicone sheeting; Specialty Manufacturing Inc., Saginaw, MI, USA).
Scalpel or razor blade and 12-inch ruler to cut silicone sheets.
Human plasma fibronectin (Invitrogen, Carlsbad, CA, USA) is dissolved in sterile water at 1 mg/mL and stored in aliquots of 500 μL at −20°C. For frequent use, keep one aliquot at 4°C and use it within ~4 weeks.
Phosphate-buffered saline solution (PBS) without calcium and magnesium. Final 1× buffer is prepared from 10× concentrate (Invitrogen) adjusted to pH 7.1, autoclaved, and stored at 4°C.
Fig. 2.1.

Overview of the strain device. a Schematic cross-section. The strain dish (1) contains cells plated on a transparent silicone membrane (2) held in place by a plastic O-ring (3) wedged into a groove of the strain dish. The strain dish is mounted onto the dish-holder plate (4) and carefully placed on the base plate (5) that fits firmly onto the microscope stage (6). Nylon spacers (7) limit the vertical displacement of the dish-holder plate and thus the applied membrane strain. Polytetrafluoroethylene bearings in the dish-holder plate and vertical pins (8) on the base plate provide alignment and stabilization of the movable plate relative to the stationary platen (9). A central bore in the base plate enables access of the microscope objective (10) mounted on an extension tube. Pushing down the dish/dish-holder plate with a weight plate (11) results in uniform membrane strain. b Select components of the strain device, using the same labels as above. c Photos of strain device in the resting condition (left) and during strain application (right). The weight to push down the dish-holder plate is visible on top of the strain dish
2.2 Cell Culture and Plating Cells
Appropriate cell culture medium for the desired cell type, e.g., Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum and antibiotics for mouse embryo fibroblasts.
Hanks buffered saline solution (HBSS) without calcium and magnesium. Final 1× buffer is prepared from 10× concentrate (Invitrogen), adjusted to pH 7.1, sterile filtered or autoclaved, and stored at 4°C. HBSS+ with calcium and magnesium is also required in Section 3.3.1.
Solution of 0.05% (w/v) trypsin and ethylenediamine tetraacetic acid (Trypsin-EDTA; Invitrogen).
Phosphate-buffered saline solution (PBS) without calcium and magnesium: see Section 2.1.5.
10-cm style untreated polystyrene dishes (Corning, Acton, MA, USA). These dishes are used to carry the silicone membrane dishes and to keep their contents sterile when taken outside the hood.
2.3 Strain Experiments
Inverted microscope (e.g., IX-70, Olympus, Center Valley, PA, USA) with a digital camera suited for fluorescence microscopy (e.g., CoolSNAP HQ, Roper Scientific, Tucson, AZ, USA) and appropriate image acquisition software (e.g., IPLab, ImagePro, etc.).
High-power objective (at least ×40 magnification) appropriate for the inverted microscope used in the experiments. We use a ×60 phase contrast objective (Olympus LCPlanFl ×60, 0.70NA Ph2).
Microscope objective extension tube (ThorLabs, Newton, NJ, USA) to compensate for the raised sample plane of the strain device (~20 mm above the normal sample position).
Strain device consisting of microscope platform, spacers, and dish-holder plate.
2.5-pound rubberized weight plate with 2-inch central bore (Olympia Sports, Westbrook, ME, USA).
Chemically inert, silicone-impermeant grease (Braycote 804; Castrol, Irvine, CA, USA).
HBSS without calcium and magnesium (see Section 2.2.2).
Cell-permeable Hoechst 33342 nucleic acid stain (Invitrogen/Molecular Probes): 10 mg/mL solution in water, stored at 4°C.
2.4 Analysis
Image analysis software. We use MATLAB (Mathworks, Natick, MA, USA) to analyze nuclear and membrane strain based on TIFF images taken during the experiments, but other image analysis software should be equally suitable.
Spreadsheet program. We use Microsoft Excel for the final data analysis to exclude contracted and damaged cells and for the statistical analysis of the results.
3 Methods
The experiments are carried out on a custom-made strain system mounted on an inverted epifluorescence microscope. Cells are plated on custom-made plastic strain dishes with a silicone membrane serving as the cell culture substrate that are described in Fig. 2.1 and elsewhere (7). The strain dish is then mounted on a strain device consisting of a base plate that fits onto the microscope, a movable dish holder plate that can slide up and down on four guidance pins, and a separate “weight plate” to apply a load (see 11 in Fig. 2.1). The base is made of an aluminum plate that securely fits onto the microscope stage. A low-friction poly-tetrafluoroethylene-impregnated Delrin platen is located in the center of the base and serves to apply biaxial strain to a silicone membrane when the membrane is pressed down over the platen. The platen has a large central bore to accommodate the microscope objective mounted on an objective extension tube. Vertical steel pins positioned at each corner of the plate are used to align the dish-holder plate with the base plate. The dish-holder plate contains four polytetrafluoroethylene bearings, one in each corner, to allow precise alignment with the vertical guidance pins from the base plate and to keep the dish/plate assembly in a horizontal position during strain application. In the resting position, the strain dish rests with the silicone membrane on the central platen. To apply biaxial strain, weights are used to press down the strain dish mounted on the dish-holder plate, resulting in a homogeneous and uniform biaxial strain field in the central section of the silicone membrane. Friction between the platen and the membrane is minimized by application of chemically inert, silicone-impermeant grease. The maximal membrane strain is limited by nylon spacers placed on the vertical alignment pins, effectively limiting the vertical displacement of the dish.
Images of cells and fluorescently labeled nuclei are acquired before, during, and after strain application and subsequently analyzed to compute the normalized nuclear strain, defined as the ratio of induced nuclear strain to applied membrane strain. The analysis is carried out using custom-written MATLAB software and is based on calculating the scaling factor of the linear affine image transformation (i.e., image transformation that allows for translation, rotation, and scaling) that best fits manually selected control points from the pre-strain image to the corresponding positions in the full-strain image (3). Measuring nuclear strain based on the displacements of distinct markers is more reliable than measurements that rely on changes in feature length or cross-sectional area, because the apparent dimensions can vary with the fluorescence intensity, whereas the centroid position of small distinct features is not affected as long as the corresponding features can be identified.
3.1 Preparation of Silicone Membrane Dishes
3.1.1 Assembly of Strain Dishes
Thoroughly clean the plastic components of the strain dishes with 70% alcohol and subsequently soak in hot water for at least 30 min; rinse with deionized water and then air-dry. Prepare nine sets of dishes for each 12×12-inch silicone membrane (see Note 2 on how to reuse dishes).
Cut out nine 4×4-inch pieces from a single silicone sheet using a ruler and a razor blade.
Carefully place a 4×4-inch silicone membrane piece on top of one of the plastic rings (rounded side facing up), making sure not to have any creases or folds in the membrane.
Concentrically place the plastic strain dish on top of the ring and push down over the membrane and plastic ring, thereby fixing the silicone membrane in the groove between the ring and the strain dish.
Cut the access membrane with scissors (see Note 3).
Rinse the assembled strain dish under hot water, followed by a quick rinse with deionized water.
Place the assembled strain dishes in an autoclave bag and autoclave with a dry cycle for 30 min.
3.1.2 Fibronectin-Coating Silicone Membranes
Remove the sterilized strain dishes from the autoclave bag inside a sterile cell culture hood. For better image quality during the strain experiments, wipe down the outside of the silicone membrane with a 70% alcohol sterilization pad. Place the strain dish inside an inverted, 10-cm cell culture dish, which allows easy movement of the dishes outside the cell culture hood while maintaining a sterile interior.
Mark a small dot on the center of the bottom of the silicone membrane (outside) with a fine-tip marker. This ink spot will serve as a reference frame during the strain experiments and will help in locating the cells throughout the experiments as the membrane moves during the strain application.
Prepare fibronectin solution (see Note 4). Fill a 50-mL tube with PBS for 1–4 dishes (each dish requires 11 mL of PBS), then add the appropriate amount of fibronectin stock solution for a final concentration of 2 μg/mL. Briefly invert or vortex to mix, then transfer 11 mL of fibronectin solution into each dish.
Incubate the dishes with fibronectin solution overnight at 4°C.
Before plating cells in strain dishes, aspirate off the fibronectin solution, rinse dishes once with PBS (~5 mL per dish), then fill each dish with ~10 mL of growth media.
3.2 Plating Cells
Once the cells cultured in a T25 flask reach confluence, rinse them once with HBSS and detach them with trypsin/EDTA (see Note 5).
Inactivate trypsin by addition of growth medium with FCS (use 1–2× the amount of trypsin), collect in a 50-mL tube, and spin down the cells for 5 min at 380×g.
Resuspend the cell pellet in 3–5 mL of growth media and transfer an appropriate amount of cell suspension into a strain dish containing 10 mL of growth media, then gently pipette up and down to mix (see Note 6).
Transfer the dishes into the cell culture incubator and incubate for 48 h before use for nuclear strain experiments (see Note 7).
3.3 Strain Experiments
3.3.1 Strain Experiments
Mount the high-power objective on the objective extension tube on the inverted fluorescence microscope; bring the objective into the lowest possible position to avoid contact with the silicone membrane when mounting the strain dish.
Place the base of the strain device on the microscope stage.
Apply the desired amount of spacers on the strain device.
Set up the CCD camera, image acquisition software, and microscope.
Incubate the cells in the strain dish with growth media containing 1 μg/mL of Hoechst 33342 nuclear stain for 15 min at 37°C.
Aspirate off the medium and replace with 20 mL of HBSS+ (see Note 8).
Carefully apply grease to the perimeter of the bottom of the silicone membrane, making sure to leave the center of the membrane free to allow for unobstructed imaging. In addition, apply a small amount of grease to the strain device platen.
Tightly screw the strain dish into the dish holder plate and carefully mount the dish-holder plate with the strain dish onto the strain device on the microscope.
Adjust the microscope focus and stage position to locate the dot marked in the center of the membrane through the eyepiece; the dot will serve as the starting point for all image acquisitions and aids in locating the same cells as the membrane initially moves during strain application.
Starting from the dot, move the microscope stage to locate a field of view containing well-spread cell(s) with centrally located nuclei.
Acquire a phase contrast image and a fluorescence image (blue channel) of the first section (see Note 9).
Move the microscope stage to a new field with well-spread cells (see Note 10). Acquire phase contrast and fluorescence images of this field of view and repeat the procedure for three to six additional sections. These images are saved as the pre-strain images. The pre-strain image acquisition process (and all subsequent stages) should be completed in less then 10 min to avoid remodeling of the cells when strain is applied later on.
Move the stage back to locate the starting point (ink dot). If using a water or oil-immersion objective, retract the objective to avoid pushing the membrane over the objective. Slowly place the weight plate on the top of the dish-holder plate until it rests firmly on the spacers, resulting in reproducible membrane strain. 14. Adjust the microscope focus and locate the ink dot on the silicone membrane, then find the initial starting section based on the phase contrast images taken at the pre-strain stage.
Acquire phase contrast and fluorescence images of the strained cells as described above, carefully adjusting the microscope stage and focus to precisely match the nuclear cross-section imaged at the pre-strain stage. Once again, this process should not exceed 10 min to avoid active remodeling and adaptation of the cell to the strained substrate.
After all the corresponding images have been acquired, move the microscope stage back to the starting point. Carefully remove the weight from the dish-holder plate to allow the silicone membrane to relax (see Note 11).
Adjust the microscope focus and locate the ink dot again. Subsequently, acquire phase contrast and fluorescence images of the post-strain cells, carefully adjusting the microscope stage and focus to match the images taken at pre-strain and full-strain.
Carefully remove the dish from the strain device and repeat the entire procedure with additional dishes if necessary. Add bleach to the strain dishes to disinfect them and see Notes for Section 3.1.1 for reusing the strain dishes for new experiments.
3.4 Analysis of Results
3.4.1 Membrane Strain Measurements
-
1
Select pairs of control points in the corresponding image sections taken before and during strain application, as shown in Fig. 2.2. For accurate measurements of membrane strain, select three to eight image features that are located directly on the silicone membrane and that are distributed throughout the field of view (see Note 12).
Compute the linear conformal spatial transformation matrix based on the pairs of control points selected above (see Note 13). Each control point pi in the pre-strain image can be represented in its spatial coordinates (xi, yi) and written as one row of the control point matrix u as:(Eq. 2.1) The corresponding positions of control points in the matching image taken during strain application (full-strain image), represented by the matrix u′, can be computed by applying the transformation matrix T to the control point matrix u of the pre-strain image:
where T is the spatial transformation matrix of the form:(Eq. 2.2) (Eq. 2.3) The angle of rotation α and the scaling factor s can then be determined from the least-square fit solution of Eq. 2.2 to the control point matrix u* of the actual coordinates of the control points in the full-strain image, while taking advantage of the restriction that a11 = a22 and a12 = −a21 (see Note 14):(Eq. 2.4) In the case of biaxial strain application, rotations are generally negligible, i.e., α → 0 and thus cosα ≈ 1, reducing the expression for the scaling factor s to:(Eq. 2.5) See Note 14 for details. The scaling factor s is related to the membrane stain in percent strain by:(Eq. 2.6) -
3
To validate the solution to the transformation matrix, apply the transformation matrix to the pre-strain image (see Eq. 2.2) and overlay the resulting image with the actual full-strain image. This procedure will also reveal any nonuniform deformations and partial cell detachment.
-
4
Subsequently, repeat this procedure for each pair of image sections taken during the strain application. Also use the procedure for the combination of pre-strain and post-strain images to make sure that the membrane has returned to baseline after the experiments.
-
5
Compute the induced nuclear strain for each cell following the same procedure, using bright chromatin spots within a single nucleus in the pre-strain and full-strain images as control points (see Note 12).
-
6
For each nucleus, determine the residual strain after strain application by selecting control points based on images taken before and after the strain application. The residual strain yields important information on potential cellular damage during the strain application.
-
7
Compute the normalized nuclear strain for each nucleus as the ratio of induced nuclear strain to applied membrane strain. Normalized nuclear strain data are pooled from at least three independent experiments (each containing measurements on ~5–10 nuclei) and compared with other cell or treatment groups by statistical analysis (see Note 15). Increased normalized nuclear strain indicates reduced nuclear stiffness.
-
8
Because the normalized nuclear strain is a function of the ratio of nuclear stiffness to cytoskeletal stiffness, it is recommended that cytoskeletal structure and function be compared between different experimental groups by, for example, magnetic bead microrheology (3, 8) or at least by immunofluorescence labeling of cytoskeletal components.
Fig. 2.2.
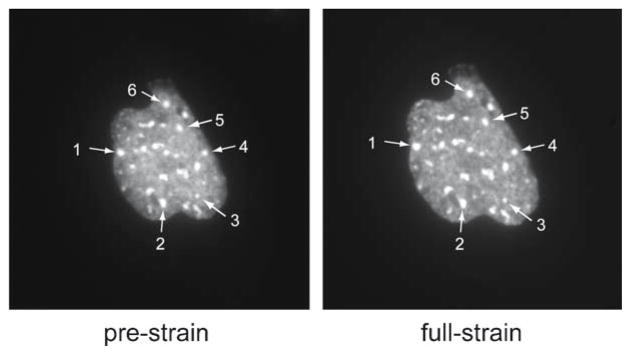
Example of control points selected for nuclear strain analysis. Shown is a pair of images of a lamin A/C-deficient mouse embryo fibroblast nucleus with fluorescently labeled chromatin before (left) and during (right) strain application. The nucleus is visibly enlarged as a result of the strain application. Distinct spots of high chromatin density are apparent in the fluorescence images, and six of these speckles are used as control points for the strain calculations (#1–6). These pairs of control points of matching image features are subsequently used to compute the best-fit affine linear conformal transformation matrix
Footnotes
We use a custom-made strain device in our experiments. A similar setup can be reproduced based on the design described here. Similar systems might also be available commercially, but one should make sure that they apply uniform strain across the image field. Our particular strain device can apply up to 35% membrane strain, but we routinely perform nuclear strain experiments with membrane strains of only 5%. Larger strain magnitudes improve the precision of the nuclear strain measurements because they generally result in larger induced nuclear deformation, but excessive membrane strain can cause cell detachment and structural damage. Because cellular sensitivity to strain varies with cell type, we recommend performing initial experiments with increasing strain magnitudes while carefully monitoring cell appearance during and after the strain application.
Assembled strain dishes can be reused after the experiments. Disinfect the interior of the cell dish with 10% bleach, then rinse repeatedly under hot water. Fill the inside with deionized water and incubate at room temperature for at least 24 h, occasionally changing the water. Subsequently, rinse the inside of the dishes again with hot water, and remove the grease on the outside (bottom side) of the strain dish with Kim-wipes and 70% alcohol, because residual grease might impair the optical transparency in subsequent experiments. Also, clean the inside of the silicone membrane with 70% ethanol to remove residual cell and extracellular matrix debris. Finally, rinse the dish with deionized water and autoclave with a dry cycle for 30 min.
Excess membrane material can interfere with screwing the strain dish into the strain device. Cutting away excess membrane works best by keeping one of the scissor blades in contact with the strain dish during the cutting process and slowly rotating the dish around.
Depending on the cell type used, other extracellular matrix proteins such as laminin, collagen, or gelatin can be substituted. It is recommended that a suitable concentration be established for each protein and cell type combination when beginning work with new cell types.
The experimental technique is limited to adherent cells that spread well on the appropriately coated substrate without detaching during strain application. Nonadherent or weakly adherent cells are not suitable for these experiments. A confluent T25 flask of human or mouse fibroblasts is generally sufficient for one to three strain dishes.
The number of cells per dish will depend on the specific cell type. In general, it is best to plate cells at an intermediate cell density, because experiments should be performed in subconfluent (~30–60% confluence) cell layers to avoid cell-to-cell interactions, while too low cell densities can negatively affect cell viability. For human and mouse fibroblasts, we found that ~1,000–2,000 cells/cm2 is a good starting cell density. The optimal cell density is best determined by initially plating cells at a range of densities. Once the optimal density has been determined, it is often not necessary to count cells for each experiment but instead to use the appropriate fraction of the cell suspension, e.g., 0.5 mL of 4-mL cell suspension harvested from a confluent T25 flask.
Cells should become sufficiently attached for experiments after overnight incubation and can be used once they reach the appropriate cell density. For fast-growing cells, we recommend incubating cells for 48 h with serum-free media supplemented with insulin, transferrin, and selenium to minimize the number of mitotic cells because the experiment is designed to measure physical properties of interphase nuclei.
Generally, experiments are performed at room temperature, and the HBSS should be equilibrated to room temperature before use. However, temperature-sensitive cells might require working at 37°C, which can be achieved by using a microscope stage incubation chamber or a perfusion system. In this case, HBSS should be prewarmed to 37°C.
In the phase contrast images it is best to focus on the membrane and the cell outline, because this can be subsequently used to compute the membrane strain and to detect potential cell detachment; in the fluorescence images of the nucleus, focus on the center cross-section of the nucleus; optionally, acquire 3D image stacks of the entire nucleus, but this can lead to photobleaching. Optionally, acquire images in other color channels if additional fluorescent markers are used.
It is easiest to locate the imaged cells again by moving in only one direction from the initial starting point, or, alternatively, moving along the perimeter of the ink dot. In order to avoid excessive photobleaching, avoid overlap of subsequently imaged sections.
In order to make sure that the silicone membrane completely returns to the pre-strain state, it can help to gently lift up the dish-holder plate a little and then gently lower it onto the platen.
The silicone membrane generally contains sufficient intrinsic distinct marks (e.g., impurities or debris) visible in the phase contrast images that can be used as control points to calculate the applied membrane strain. Alternatively, additional membrane markers consisting of markings with a felt-tip pen or fluorescently labeled polystyrene beads absorbed to the silicone membrane can be used. For the nuclear strain determination, bright fluorescent chromatin speckles serve as excellent markers in mouse embryo fibroblasts. If measurements of cytoskeletal strain are required, these can be obtained by using endocytosed small, fluorescently labeled polystyrene beads (Molecular Probes-Invitrogen) as control points. The 0.2-μm beads are small enough to be internalized by the cell, yet not so small that they diffuse freely through the cytoskeletal network (4).
Determining the four independent transformation matrix entries (a11= a22; a12 = −a21, a31, a32) requires a minimum of two pairs of control points. In our experiments, we generally use 4–15 pairs of control points to reduce the influence of a single control point on the overall results, and we improve the localization of the manually selected control points with a normalized cross-correlation algorithm (function cpcorr in the Matlab Image Processing toolbox). In this case, the transformation matrix is computed based on the least-square fit solution for all control points.
| (Eq. 2.7) |
| (Eq. 2.8) |
| (Eq. 2.9) |
| (Eq. 2.10) |
Because cytoskeletal damage and partial cell detachment during strain application generally result in collapse and apparent shrinkage of nuclei, we exclude any nuclei from the analysis that have a nuclear scaling factor of less than 0.99 (corresponding to a 1% size reduction for the transformation between pre- and post-strain images).
References
- 1.Burke B, Stewart CL. The laminopathies: the functional architecture of the nucleus and its contribution to disease. Annu Rev Genomics Hum Genet. 2006;7:369–405. doi: 10.1146/annurev.genom.7.080505.115732. [DOI] [PubMed] [Google Scholar]
- 2.Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7:940–952. doi: 10.1038/nrg1906. [DOI] [PubMed] [Google Scholar]
- 3.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caille N, Tardy Y, Meister JJ. Assessment of strain field in endothelial cells subjected to uniaxial deformation of their substrate. Ann Biomed Eng. 1998;26:409–416. doi: 10.1114/1.132. [DOI] [PubMed] [Google Scholar]
- 5.Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem. 2006;281:25768–25780. doi: 10.1074/jbc.M513511200. [DOI] [PubMed] [Google Scholar]
- 6.Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol. 2005;170:781–791. doi: 10.1083/jcb.200502148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng GC, Briggs WH, Gerson DS, Libby P, Grodzinsky AJ, Gray ML, Lee RT. Mechanical strain tightly controls fibroblast growth factor-2 release from cultured human vascular smooth muscle cells. Circ Res. 1997;80:28–36. doi: 10.1161/01.res.80.1.28. [DOI] [PubMed] [Google Scholar]
- 8.Bausch AR, Ziemann F, Boulbitch AA, Jacobson K, Sackmann E. Local measurements of viscoelastic parameters of adherent cell surfaces by magnetic bead microrheometry. Biophys J. 1998;75:2038–2049. doi: 10.1016/S0006-3495(98)77646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]