

Abstract
Evidence suggests that the tumor necrosis factor receptor (TNFR)-signaling pathway contributes to the pathogenesis of Alzheimer’s disease (AD). TNF-α converting enzyme (TACE/ADAM-17) can cleave both pro-TNF-α and TNF receptors. Recently, we have shown that TACE activity in the cerebrospinal fluid (CSF) of subjects with mild cognitive impairment (MCI) and AD patients is significantly higher than that of cognitively healthy controls (HC). To date, it is not clear whether TACE activity could be detected in the human plasma and whether TACE activity in MCI and AD patients is different from that in HC. We analyze TACE expression and activity in a large clinical sample of 64 patients with AD, 88 subjects with MCI, and 50 age-matched HC recruited from two distinct academic centers. Plasma TACE protein levels did not differ significantly in the three study groups (AD, MCI, and HC). However, plasma TACE activity in subjects with MCI and AD patients was significantly higher than that in HC. Moreover, in MCI and AD groups, we found a significant correlation between plasma TACE activity and CSF t-tau and Aβ42 levels and CSF Aβ42/tau ratios. In AD patients, the levels of plasma TACE activity correlated significantly and negatively with cognition. These findings further support the role of the TNF-α receptor complex in AD-related neuroinflammation and propose TACE plasma activity as a promising hypothesis-driven biomarker candidate for detection, diagnosis, and prognosis of prodromal and clinical AD.
Keywords: Alzheimer’s disease, biomarker, mild cognitive impairment, plasma, tumor necrosis factor converting enzyme
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia worldwide, characterized by the presence of amyloid plaques composed of amyloid-β (Aβ) peptide aggregates and neurofibrillary tangles consisting of hyperphosphorylated and aggregated tau protein in the gray matter structures of the brain [1]. Mild cognitive impairment (MCI) is a heterogeneous clinical condition with several underlying causes. However, the large proportion of MCI represents a transitional state between healthy aging and very mild AD [2–4]. Accordingly, studies suggest that MCI subjects tend to progress to clinically probable AD at a rate of approximately 10%–15% per year [4, 5].
The development of effective treatments of AD would be facilitated by easily accessible biomarkers. Although most biomarkers of AD are currently measured in the cerebrospinal fluid (CSF), the invasive nature of lumbar puncture has limited more widespread applicability [6, 7]. A major challenge therefore lies in the development of blood-based biomarkers which can reliably detect disease presence and predict disease risk. Blood-based biomarkers of AD could provide a cost- and time-effective way to enhance the utility of CSF and imaging biomarkers, such as the first step in a multi-stage screening and diagnostic process, which is common in medical practice [8–10]. This multistep screening process also has cost-savings potential for recruitment into clinical trials. Since sporadic AD is a genetically and etiologically complex and multifaceted disease characterized by various molecular mechanisms and signaling pathways pathophysiologically altered during chronic disease progression, we chose to explore hypothesis-driven candidate systems and derived biomarkers in blood (plasma and serum) to test their clinical utility and performance.
Neuroinflammation has been demonstrated in AD-affected brains by the presence of activated microglia, reactive astrocytes, and the complement system [11, 12], as well as increased cytokine, altered cytokine receptor complex expression [13], and elevated acute-phase proteins [14]. Extensive evidence supports the concept that neuroinflammation is an “on-off” phenomenon that contributes to neurodegeneration in AD [15]. Recently published genetic evidence provides further substantial support toward a significant role of neuroinflammation and involvement of the complement system in AD pathophysiology [16, 17].
Tumor necrosis factor α (TNF-α) is one of the major inflammatory cytokines produced by many cell types, including macrophages, monocytes, lymphocytes, keratinocytes, and fibroblasts, in response to inflammation, infection, injury, and other environmental challenges [18]. In the brain, TNF-α is expressed by neurons and glia and promotes inflammatory responses by recruiting microglia or astrocytes to lesion sites, leading to glial cell activation [18]. Interestingly, TNF-α may elicit the production of Aβ in vitro [19]. Using double transgenic mice, we have previously shown that the inhibition of TNF-receptor (TNFR)-signaling in transgenic APP23 mice results in a significant reduction of brain amyloid plaques and Aβ [20]. Furthermore, a recent clinical study has suggested that the inhibition of TNF-α expression may improve cognitive function in AD patients [21].
TNF-α exerts its proinflammatory effects by binding to two transmembrane receptor subtypes termed TNFR1 and TNFR2 [22]. Soluble forms of TNF-α receptors 1 and 2 (sTNFR1 and sTNFR2) represent the circulating isoforms of the corresponding membrane-bound receptors [22]. These stable receptors render soluble TNF-α receptors more reliable markers of TNF-α-dependent inflammatory activity. Interestingly, TACE (tumor necrosis factor-α-converting enzyme) can cleave both TNFR1 and TNFR2 to the corresponding soluble forms [23]. TACE, also known as ADAM17, is a transmembrane disintegrin metalloprotease that cleaves precursor TNF-α to generate soluble, secreted TNF-α in macrophages and monocytes [24, 25]. Both the cell-associated and the released forms of TNF are biologically active, but full inflammatory responses require the soluble form in at least some situations [26].
Previous studies have shown that CSF levels of TNF-α are increased in individuals with AD and MCI [27]. Accordingly, elevated plasma TNF-α levels have been associated with incident AD in subjects with MCI [28]. Subjects with MCI who subsequently progressed to AD dementia were also characterized by higher plasma and CSF levels of sTNFR1 and sTNFR2 compared to MCI subjects who did not convert to AD dementia [29]. Recently, we have shown that TACE activity is significantly increased in the CSF of MCI subjects and AD patients [30]. In addition, the concentrations of both sTNFR1 and sTNFR2 were found to be significantly correlated with TACE activity [30]. Therefore, the TNF-α receptor complex is a mechanistically-linked promising candidate biomarker for both MCI and AD.
To date, however, data on TACE expression and activity in plasma during the different stages of dementia remain scarce. To answer these questions, the aim of this study was to analyze TACE expression and activity in a large clinical sample of MCI and AD subjects recruited from two distinct academic centers.
MATERIALS AND METHODS
Subjects
A total of 202 individuals were recruited from two independent international academic research centers specialized in AD. The clinical material included 64 patients with AD, 88 subjects with MCI, and 50 age-matched healthy controls (HC) recruited from the Alzheimer Memorial Center, Department of Psychiatry, Ludwig-Maximilian University, in Germany and the Department of Clinical Neuroscience, University of Goteborg, Sahlgren’s University Hospital, in Sweden. As we have previously described [31, 32], the diagnosis of AD was performed according to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria [33, 34], which included the Mini-Mental State Examination (MMSE). The diagnosis of MCI was performed according to the Petersen criteria [35]. MCI subjects performed 1.5 SD below the age-adjusted reference average in memory scales, as assessed using the CERAD cognitive battery [36] which included verbal learning, recognition, and recall tests. The global cognitive function and activities of daily living were unimpaired. Patients with other causes of cognitive impairment, including brain tumors, sub-dural hematomas, CNS infections, major depressive episodes, schizophrenias, and current alcohol abuse were excluded.
Controls were cognitively healthy individuals whose psychiatric comorbidity was excluded by means of medical history, clinical examination, and Composite International Diagnostic Interview [37]. The baseline demographic variables of these AD and MCI patients and controls are given in Table 1.
Table 1.
General demographics of subject populations in plasma TACE study
| Variables | HC | MCI | AD |
|---|---|---|---|
| Group size | 50 | 88 | 64 |
| Age (years) | 68.9 ± 6.8 | 69.6 ± 7.6 | 70.1 ± 8.9 |
| Gender (M/F) | 23/27 | 33/55 | 18/46 |
| APOE ε4 carrier (%) | 28.0 | 42.0 | 59.4a,b |
| MMSE at baseline | 29.6 ± 0.6 | 27.2 ± 2.5a | 22.6 ± 3.6a,c |
| Plasma TACE activity (mFU/min/μg) | 6.52 ± 2.14 | 8.94 ± 2.74a | 10.64 ± 2.93a,b |
Values are means ± S.D., except as noted otherwise. HC, healthy controls, with unimpaired cognition after at least 3 years follow-up; MCI, mild cognitive impairment patients; AD, Alzheimer’s disease patients; MMSE, Mini-Mental State Examination; APOE ε4 carrier, at least one apolipoprotein E ε4 allele; TACE, tumor necrosis factor-α-converting enzyme.
p < 0.001 versus Controls;
p < 0.05 versus MCI;
p < 0.001 versus MCI.
At baseline, non-fasting plasma samples were collected in the morning by venipuncture in EDTA-containing tubes. After centrifugation, plasma samples were aliquoted in polypropylene tubes and stored at −80°C until biochemical analyses, without being thawed and re-frozen. All samples were processed and evaluated in a blinded fashion. Informed consent was obtained either by direct subject consent (controls) or by consent of both patient and caregiver (subjects with dementia).
TACE enzymatic activity assay
TACE activity assays were performed by using synthetic peptide substrates containing the TACE cleavage site (Mca-Pro-Leu-Ala-Gln-Ala-Val-Dpa-Arg-Ser-Ser-Ser-Arg-NH2) (R&D System, Inc., Minneapolis, MN, USA) at a 10 mM concentration in reaction buffer (50 mM acetic acid, pH 4.5, 100 mM sodium chloride). The study has shown that this peptide is the main substrate for TACE [38]. Ten microliters of each sample was used to examine TACE activity. The fluorescence was measured using a fluorescent microplate reader with an excitation wave length at 320 nm and an emission wavelength at 405 nm. TACE activity results were normalized to total protein concentration of plasma samples.
TACE western blot assay
The plasma samples from each group were mixed with an equal volume of sodium dodecyl sulfate (SDS) sample buffer and separated using 10% SDS-PAGE gel. A total of 50 μg of protein was loaded. Protein was then transferred to a nitrocellulose membrane (Bio-Rad, USA) by wet transfer according to the standard procedure. The membrane was stained with 1% Ponceau S as loading control and a brain lysate from an AD patient was used as positive control. The membrane was probed using an anti-TACE polyclonal antibody (Sigma, USA).
Assays of levels of Aβ42 and tau in CSF
CSF-Aβ1–42 ELISA concentration was determined by INNOTEST β-amyloid (1–42) (Innogenetics, Ghent, Belgium) as previously described [31]. CSF-tau concentration was quantified with Luminex xMAP technique as previously described [29].
Statistical analysis
Separate one-way analyses of covariance were performed for the comparisons between diagnostic groups for continuous data and multiple comparisons adjusted for Bonferrone tests were used. The Pearson’s χ2-test was employed for dichotomous variables. Spearman’s correlation coefficient was used for correlation analyses. Multiple linear regression analysis was performed to analyze the associations of TACE activity with diagnosis, age, gender, and recruiting centers. The statistical analyses were accomplished with SPSS for Windows, version 17.0.
RESULTS
TACE protein expression in plasma samples
To investigate whether TACE protein could be identified in human plasma, western blot analysis was used. The membrane was stained with 1% Ponceau S as loading control and a brain lysate from an AD patient was used as positive control. Western blots revealed a prominent immunoreactive band at the expected molecular weight of the ~80-kD TACE protein in all of plasma samples and in AD brain lysate. No statistically significant differences were found in plasma TACE protein levels among the three study groups (AD patients, MCI subjects, and controls) (Fig. 1).
Fig. 1.
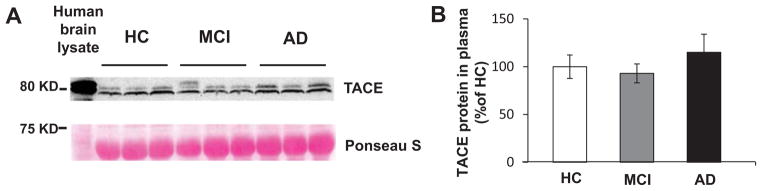
TACE protein expression in human plasma samples. A) Western blot analysis of TACE protein expression in human plasma samples from AD, MCI, and HC groups (n = 10, separately). The membrane was stained with 1% Ponceau S as loading control and a brain lysate from an AD patient was used as positive control. B) represents the quantitative data of TACE protein expression in plasma samples. No statistically significant differences were found among the three study groups.
Plasma TACE enzymatic activity is increased in samples of MCI subjects and AD patients
Plasma TACE activity assays were performed by using synthetic peptide substrates containing the TACE cleavage site (Mca-Pro-Leu-Ala-Gln-Ala-Val-Dpa-Arg-Ser-Ser-Ser-Arg-NH2). We found that plasma TACE activity of HC, MCI, and AD subjects is 6.52 ± 2.14, 8.94 ± 2.74, and 10.64 ± 2.93 (mFU/min/ug), respectively. When comparing TACE activity in plasma to the HC group, TACE activity was found to be increased by 37.1% in MCI subjects (p < 0.001) and by 63.2% in the AD subjects (p < 0.001) (Fig. 2). In addition, TACE activity was significantly higher in AD patients than in MCI subjects (p < 0.05) (Fig. 2).
Fig. 2.

TACE enzymatic activity in human plasma samples. TACE enzymatic activity analysis was performed by using synthetic peptide substrates containing the TACE cleavage site in human plasma samples from AD (64 cases), MCI (88 cases), and HC (50 cases). TACE activity was found to be increased by 37.1% in MCI subjects (p < 0.001) and by 63.2% in the AD subjects (p < 0.001). In addition, TACE activity was significantly higher in AD patients than in MCI subjects (p < 0.05). Error bars indicate SEM. *p < 0.05, **p < 0.01.
Multiple linear regression analysis for TACE activity
To study the association of plasma TACE enzymatic activity with diagnostic subgroups, age, gender, and recruiting centers, multiple linear regression analysis was performed. Plasma TACE enzymatic activity was significantly and positively associated with the presence of MCI and AD. No other associations were observed between TACE activity and age, gender, or recruiting centers (Table 2).
Table 2.
Multiple linear regression model for TACE enzymatic activity
| Model | β | p |
|---|---|---|
| TACE enzymatic activity | ||
| Subgroups | ||
| MCIa | 0.340 | 0.001* |
| ADa | 0.591 | 0.001* |
| Age | −0.027 | 0.678 |
| Gender | 0.037 | 0.563 |
| Center | 0.079 | 0.215 |
HC was set as the control subgroup.
p < 0.05.
Correlations of the baseline levels of plasma TACE activity with CSF tau and Aβ42 levels, and CSF Aβ42/tau ratios
CSF Aβ42 and tau concentrations in the same cases were determined as previously described [29, 31]. To explore whether the measured plasma TACE activities were related to AD neuropathology, the correlation between the baseline levels of plasma TACE activity with CSF tau and Aβ42 levels and CSF Aβ42/tau ratios in AD and MCI groups was performed. In MCI and AD subjects, the levels of plasma TACE activity showed a moderate positive correlation with CSF tau levels (rs = 0.347, 0.469, p < 0.05) (Fig. 3A, D). In both MCI and AD groups, there were statistically negative correlations between the levels of plasma TACE activity and CSF Aβ42 levels (rs = −0.383, −0.351, p < 0.05) (Fig. 3B, E). Furthermore, we found that in MCI and AD subjects, the levels of plasma TACE activity showed a negative correlation with CSF Aβ42/tau ratios (rs = −0.575, −0.476, p < 0.05) (Fig. 3C, F).
Fig. 3.

Correlations of the baseline levels of plasma TACE activity with CSF tau (A, D), Aβ42 levels (B, E), and CSF Aβ42/tau ratios (C, F) in AD and MCI groups. A, D) The levels of plasma TACE activity showed a moderate positive correlation with CSF tau levels (rs = 0.347, 0.469, p < 0.05) in MCI and AD subjects. B, E) There were statistically negative correlations between the levels of plasma TACE activity and CSF Aβ42 levels (rs = −0.383, −0.351, p < 0.05) in MCI and AD groups. The levels of plasma TACE activity showed a negative correlation with CSF Aβ42/tau ratios (rs = −0.575, −0.476, p < 0.05) in MCI and AD groups.
Correlations between baseline plasma TACE activity and MMSE scores
To study whether the measured plasma TACE activities were related to cognitive decline in patients, the correlation between the baseline levels of plasma TACE activity with MMSE scores in AD and MCI subjects was performed. In the AD subgroup, the levels of plasma TACE activity correlated significantly and negatively with clinical MMSE scores (rs = −0.393, p < 0.05) (Fig. 4A). However, we found no significant association between plasma TACE activity and MMSE scores in the MCI subgroup, possibly because these subjects all have high MMSE scores (Fig. 4B).
Fig. 4.
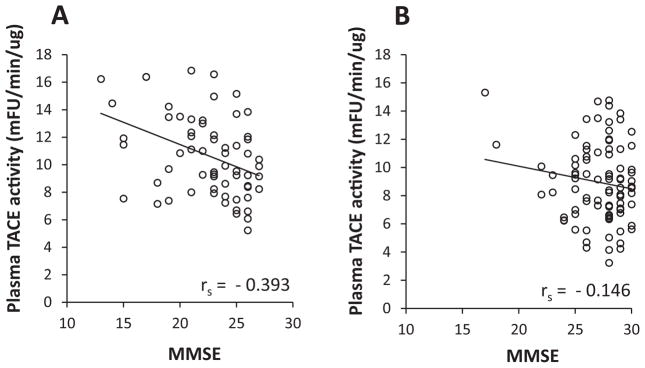
Correlations of the baseline levels of plasma TACE activity and MMSE scores in AD and MCI groups. A) The levels of plasma TACE activity correlated significantly and negatively with clinical MMSE scores (rs = −0.393, p < 0.05) in the AD subgroup. B) No significant association between plasma TACE activity and MMSE scores in the MCI subgroup.
DISCUSSION
Recently, we have shown that TACE activity is significantly increased in the CSF of both MCI and AD subjects as compared with healthy controls [30]. To date, there have been few reports about TACE expression and activity in peripheral plasma. The aim of the present study was to investigate TACE protein expression and enzymatic activity in a large clinical sample of patients with different stages of dementia recruited from two independent academic centers.
We found no significant statistical differences in plasma TACE protein levels between the three study groups. However, plasma TACE activity was significantly higher in both MCI and AD subjects compared with healthy controls. Moreover, in MCI and AD subgroups, the levels of plasma TACE activity showed significant associations with CSF T-tau and Aβ42 levels and CSF Aβ42/tau ratios. We also identified a significant negative association between plasma TACE activity and MMSE scores in AD patients. Given the increase in TACE activity, with no change in TACE protein levels, one possible explanation is that there may be increased levels of endogenous TACE activators or decreased levels of TACE inhibitors in MCI and AD. The synthetic peptide we used for TACE enzymatic activity assay contains the TACE cleavage site (Mca-Pro-Leu-Ala-Gln-Ala-Val-Dpa-Arg-Ser-Ser-Ser-Arg-NH2) is in widespread use for TACE enzymatic activity assay as the substrate is favorable for TACE (ADAM17) compared to ADAM10 [38, 39]. TACE activity and protein expression have been found in both blood cells and plasma and ADAM10 is so far only found in blood cells. Nonetheless, we cannot exclude the possibility that other enzymes may cleave the peptide in human plasma.
TNF-α functions through binding to its two transmembrane receptor subtypes, TNFR1 and TNFR2 [22]. Soluble forms of TNF-α receptors 1 and 2 (sTNFR1 and sTNFR2) represent the circulating isoforms of the membrane-bound receptors [22]. Interestingly, evidence suggests that both TNFR1 and TNFR2 can be shed as soluble isoforms from the cell surface through the activity of TACE [23]. TACE, also known as ADAM17, is a transmembrane disintegrin metalloprotease that cleaves the TNF-α precursor to generate soluble, secreted TNF-α in macrophages and monocytes [24, 25]. Both the cell-associated and the released forms of TNF are biologically active, but the full inflammatory responses require the soluble form [26].
Growing evidence suggests that neuroinflammation is common in AD-affected brains as revealed by the presence of activated glial cells, complementary proteins, pro-inflammatory cytokines expression, and acute-phase proteins [11–15]. In addition to inflammation in the CNS, peripheral immune system activation has been reported in AD patients. This has been demonstrated by cross-sectional studies reporting elevated plasma levels of the inflammatory proteins α-1-antichymotrypsin [40, 41], C-reactive protein [42], interleukin-1, and interleukin-6 [43] in AD patients compared with control subjects. TNF-α is one of the main pro-inflammatory cytokines and its role in the pathogenesis of AD has been proposed in a number of studies [18–21]. Plasma and CSF levels of TNF-α and soluble TNF receptors tend to increase in both MCI and AD subjects [27–29]. Since TACE is being shed from cell membranes after the cleavage of TNF-α and the two TNF receptors, TACE activity can be detected both in the plasma and CSF throughout different stages of AD.
In our study, plasma TACE activity was significantly higher in both MCI subjects and AD patients than in healthy controls. These results suggest that plasma TACE activity may increase progressively over the clinical course of AD, thus suggesting a substantial and dynamic (deviation from normal) inflammatory response during the progression from the prodromal to the mild-to-moderate dementia stages of AD. Another interesting finding of this study is that plasma TACE was significantly associated with the established CSF markers (CSF tau and CSF Aβ42) in both the AD and MCI subgroups. These results suggest that plasma TACE is not only associated with the presence of either the MCI or the AD groups but may reflect the pathophysiology of the disease as emphasized by the activity and dynamic of established core CSF biomarkers during the progression from prodromal to the mild-to-moderate dementia stages of AD.
TACE has been shown to be responsible for the α-secretase cleavage of the amyloid-β protein precursor (AβPP) [44, 45]. In cultured cells, TACE and β-secretase (BACE) compete for the cleavage of AβPP. Importantly, the increased cleavage of AβPP by TACE results in decreased generation of Aβ [46]. In addition, the release of sAβPPα precludes the formation of amyloidogenic peptides by BACE. The increase of TACE in the CSF of AD patients suggests that the high TACE activity may be paralleled by increased sAβPPα and reduced Aβ42 levels. However, the most abundant form of Aβ, i.e., Aβ40, is not decreased in AD [7], which argues against a role of TACE activity in the modulation of CSF Aβ levels. Indeed, several independent studies demonstrate that modifying TACE activity does not affect AβPP cleavage by BACE and Aβ production [47–50]. This would explain why increased TACE activity in AD does not alter the Aβ production from AβPP. Inhibition or knockdown of TACE also does not affect sAβPP formation and Aβ secretion under normal conditions [44, 51]. The most frequently named α-secretases are three members of the ADAM (a disintegrin and metalloproteasedomain) family: ADAM9, 10, and 17 [45, 46]. Two very recent studies have reported that ADAM10, but not ADAM9 or ADAM17, is the physiologically relevant, constitutive α-secretase in primary neurons [52, 53]. Therefore, our analyses of TACE in CSF and in plasma suggest that TACE may predominantly cleave TNFRs rather than AβPP, and contribute to upregulation of BACE and consecutive increase of Aβ production through the TNFR signaling pathway as discussed above.
In conclusion, our results demonstrated that plasma TACE activity is significantly increased in MCI and AD subjects compared with age-matched cognitively healthy controls. Notably, we found that TACE activity may increase progressively over the clinical course of AD. Taken together, these positive findings further support the role of the TNF-α receptor complex in the neuroinflammatory pathogenesis of AD. We conclude that plasma TACE activity may serve as a promising hypothesis-driven biomarker candidate for the detection, diagnosis, and prognosis of prodromal and clinical AD.
Acknowledgments
This study is supported by grants from the National Institute on Aging (NIH R01AG032441-01 to RL and R01AG025888 to YS; P50 AG025688 and P30 NS055077 to AL); Alzheimer’s Association (Zenith Award and IIRG-07-59510 to YS); the American Health Assistance Foundation (G2006-118 to RL), and the Katharina-Hardt-Foundation, Bad Homburg vor der Höhe, Germany (to HH). QS is the recipient from China Scholarship Council of Natural Science, No.2011637095.
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=2183).
References
- 1.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 2.Grundman M, Petersen RC, Ferris SH, Thomas RG, Aisen PS, Bennett DA, Foster NL, Jack CR, Jr, Galasko DR, Doody R, Kaye J, Sano M, Mohs R, Gauthier S, Kim HT, Jin S, Schultz AN, Schafer K, Mulnard R, van Dyck CH, Mintzer J, Zamrini EY, Cahn-Weiner D, Thal LJ Alzheimer’s Disease Cooperative S. Mild cognitive impairment can be distinguished from Alzheimer disease and normal aging for clinical trials. Arch Neurol. 2004;61:59–66. doi: 10.1001/archneur.61.1.59. [DOI] [PubMed] [Google Scholar]
- 3.DeCarli C. Mild cognitive impairment: Prevalence, prognosis, aetiology, and treatment. Lancet Neurol. 2003;2:15–21. doi: 10.1016/s1474-4422(03)00262-x. [DOI] [PubMed] [Google Scholar]
- 4.Markesbery WR. Neuropathologic alterations in mild cognitive impairment: A review. J Alzheimers Dis. 2010;19:221–228. doi: 10.3233/JAD-2010-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersen RC, Doody R, Kurz A, Mohs RC, Morris JC, Rabins PV, Ritchie K, Rossor M, Thal L, Winblad B. Current concepts in mild cognitive impairment. Arch Neurol. 2001;58:1985–1992. doi: 10.1001/archneur.58.12.1985. [DOI] [PubMed] [Google Scholar]
- 6.Hampel H, Frank R, Broich K, Teipel SJ, Katz RG, Hardy J, Herholz K, Bokde AL, Jessen F, Hoessler YC, Sanhai WR, Zetterberg H, Woodcock J, Blennow K. Biomarkers for Alzheimer’s disease: Academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9:560–574. doi: 10.1038/nrd3115. [DOI] [PubMed] [Google Scholar]
- 7.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 8.Hampel H, Burger K, Teipel SJ, Bokde AL, Zetterberg H, Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimers Dement. 2008;4:38–48. doi: 10.1016/j.jalz.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Schneider P, Hampel H, Buerger K. Biological marker candidates of Alzheimer’s disease in blood, plasma, and serum. CNS Neurosci Ther. 2009;15:358–374. doi: 10.1111/j.1755-5949.2009.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hampel H, Lista S, Khachaturian ZS. Development of biomarkers to chart all Alzheimer’s disease stages: The royal road to cutting the therapeutic Gordian Knot. Alzheimers Dement. 2012;8:312–336. doi: 10.1016/j.jalz.2012.05.2116. [DOI] [PubMed] [Google Scholar]
- 11.Eikelenboom P, Stam FC. Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol. 1982;57:239–242. doi: 10.1007/BF00685397. [DOI] [PubMed] [Google Scholar]
- 12.Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am J Pathol. 1999;154:927–936. doi: 10.1016/S0002-9440(10)65340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffin WS, Sheng JG, Roberts GW, Mrak RE. Interleukin-1 expression in different plaque types in Alzheimer’s disease: Significance in plaque evolution. J Neuropathol Exp Neurol. 1995;54:276–281. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 14.Abraham CR, Shirahama T, Potter H. Alpha 1-antichymotrypsin is associated solely with amyloid deposits containing the beta-protein. Amyloid and cell localization of alpha 1-antichymotrypsin. Neurobiol Aging. 1990;11:123–129. doi: 10.1016/0197-4580(90)90045-2. [DOI] [PubMed] [Google Scholar]
- 15.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tobinick EL, Gross H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J Neuroinflammation. 2008;5:2. doi: 10.1186/1742-2094-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 23.Ermert M, Pantazis C, Duncker HR, Grimminger F, Seeger W, Ermert L. In situ localization of TNFalpha/beta, TACE and TNF receptors TNF-R1 and TNF-R2 in control and LPS-treated lung tissue. Cytokine. 2003;22:89–100. doi: 10.1016/s1043-4666(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 24.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 25.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 26.Ruuls SR, Hoek RM, Ngo VN, McNeil T, Lucian LA, Janatpour MJ, Korner H, Scheerens H, Hessel EM, Cyster JG, McEvoy LM, Sedgwick JD. Membrane-bound TNF supports secondary lymphoid organ structure but is subservient to secreted TNF in driving autoimmune inflammation. Immunity. 2001;15:533–543. doi: 10.1016/s1074-7613(01)00215-1. [DOI] [PubMed] [Google Scholar]
- 27.Tarkowski E, Andreasen N, Tarkowski A, Blennow K. Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:1200–1205. doi: 10.1136/jnnp.74.9.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA, Harris TB, Benjamin EJ, Au R, Kiel DP, Wolf PA, Seshadri S. Inflammatory markers and the risk of Alzheimer disease: The Framingham Study. Neurology. 2007;68:1902–1908. doi: 10.1212/01.wnl.0000263217.36439.da. [DOI] [PubMed] [Google Scholar]
- 29.Buchhave P, Zetterberg H, Blennow K, Minthon L, Janciauskiene S, Hansson O. Soluble TNF receptors are associated with Abeta metabolism and conversion to dementia in subjects with mild cognitive impairment. Neurobiol Aging. 2010;31:1877–1884. doi: 10.1016/j.neurobiolaging.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 30.Jiang H, Hampel H, Prvulovic D, Wallin A, Blennow K, Li R, Shen Y. Elevated CSF levels of TACE activity and soluble TNF receptors in subjects with mild cognitive impairment and patients with Alzheimer’s disease. Mol Neurodegene. 2011;6:69. doi: 10.1186/1750-1326-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ewers M, Zhong Z, Burger K, Wallin A, Blennow K, Teipel SJ, Shen Y, Hampel H. Increased CSF-BACE 1 activity is associated with ApoE-epsilon 4 genotype in subjects with mild cognitive impairment and Alzheimer’s disease. Brain. 2008;131:1252–1258. doi: 10.1093/brain/awn034. [DOI] [PubMed] [Google Scholar]
- 32.Zhong Z, Ewers M, Teipel S, Burger K, Wallin A, Blennow K, He P, McAllister C, Hampel H, Shen Y. Levels of beta-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch Gen Psychiatry. 2007;64:718–726. doi: 10.1001/archpsyc.64.6.718. [DOI] [PubMed] [Google Scholar]
- 33.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 34.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen RC. Clinical practice. Mild cognitive impairment. N Engl J Med. 2011;364:2227–2234. doi: 10.1056/NEJMcp0910237. [DOI] [PubMed] [Google Scholar]
- 36.Morris JC, Heyman A, Mohs RC, Hughes JP, van Belle G, Fillenbaum G, Mellits ED, Clark C. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology. 1989;39:1159–1165. doi: 10.1212/wnl.39.9.1159. [DOI] [PubMed] [Google Scholar]
- 37.Robins LN, Wing J, Wittchen HU, Helzer JE, Babor TF, Burke J, Farmer A, Jablenski A, Pickens R, Regier DA, Sartorius N, Towle LH. The Composite International Diagnostic Interview. An epidemiologic Instrument suitable for use in conjunction with different diagnostic systems and in different cultures. Arch Gen Psychiatry. 1988;45:1069–1077. doi: 10.1001/archpsyc.1988.01800360017003. [DOI] [PubMed] [Google Scholar]
- 38.Alvarez-Iglesias M, Wayne G, O’Dea KP, Amour A, Takata M. Continuous real-time measurement of tumor necrosis factor-alpha converting enzyme activity on live cells. Lab Invest. 2005;85:1440–1448. doi: 10.1038/labinvest.3700340. [DOI] [PubMed] [Google Scholar]
- 39.Scott AJ, O’Dea KP, O’Callaghan D, Williams L, Dokpesi JO, Tatton L, Handy JM, Hogg PJ, Takata M. Reactive oxygen species and p38 mitogen-activated protein kinase mediate tumor necrosis factor alpha-converting enzyme (TACE/ADAM-17) activation in primary human monocytes. J Biol Chem. 2011;286:35466–35476. doi: 10.1074/jbc.M111.277434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsubara E, Hirai S, Amari M, Shoji M, Yamaguchi H, Okamoto K, Ishiguro K, Harigaya Y, Wakabayashi K. Alpha 1-antichymotrypsin as a possible biochemical marker for Alzheimer-type dementia. Ann Neurol. 1990;28:561–567. doi: 10.1002/ana.410280414. [DOI] [PubMed] [Google Scholar]
- 41.DeKosky ST, Ikonomovic MD, Wang X, Farlow M, Wisniewski S, Lopez OL, Becker JT, Saxton J, Klunk WE, Sweet R, Kaufer DI, Kamboh MI. Plasma and cerebrospinal fluid alpha1-antichymotrypsin levels in Alzheimer’s disease: Correlation with cognitive impairment. Ann Neurol. 2003;53:81–90. doi: 10.1002/ana.10414. [DOI] [PubMed] [Google Scholar]
- 42.Strandberg TE, Tilvis RS. C-reactive protein, cardiovascular risk factors, and mortality in a prospective study in the elderly. Arterioscler Thromb Vasc Biol. 2000;20:1057–1060. doi: 10.1161/01.atv.20.4.1057. [DOI] [PubMed] [Google Scholar]
- 43.Licastro F, Pedrini S, Caputo L, Annoni G, Davis LJ, Ferri C, Casadei V, Grimaldi LM. Increased plasma levels of interleukin-1, interleukin-6 and alpha-1-antichymotrypsin in patients with Alzheimer’s disease: Peripheral inflammation or signals from the brain? J Neuroimmunol. 2000;103:97–102. doi: 10.1016/s0165-5728(99)00226-x. [DOI] [PubMed] [Google Scholar]
- 44.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 45.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113(Pt 11):1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- 47.Blacker M, Noe MC, Carty TJ, Goodyer CG, LeBlanc AC. Effect of tumor necrosis factor-alpha converting enzyme (TACE) and metalloprotease inhibitor on amyloid precursor protein metabolism in human neurons. J Neurochem. 2002;83:1349–1357. doi: 10.1046/j.1471-4159.2002.01228.x. [DOI] [PubMed] [Google Scholar]
- 48.Dyrks T, Monning U, Beyreuther K, Turner J. Amyloid precursor protein secretion and beta A4 amyloid generation are not mutually exclusive. FEBS Lett. 1994;349:210–214. doi: 10.1016/0014-5793(94)00671-7. [DOI] [PubMed] [Google Scholar]
- 49.Gandhi S, Refolo LM, Sambamurti K. Amyloid precursor protein compartmentalization restricts beta-amyloid production: Therapeutic targets based on BACE compartmentalization. J Mol Neurosci. 2004;24:137–143. doi: 10.1385/JMN:24:1:137. [DOI] [PubMed] [Google Scholar]
- 50.LeBlanc AC, Koutroumanis M, Goodyer CG. Protein kinase C activation increases release of secreted amyloid precursor protein without decreasing Abeta production in human primary neuron cultures. J Neurosci. 1998;18:2907–2913. doi: 10.1523/JNEUROSCI.18-08-02907.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim ML, Zhang B, Mills IP, Milla ME, Brunden KR, Lee VM. Effects of TNFalpha-converting enzyme inhibition on amyloid beta production and APP processing in vitro and in vivo. J Neurosci. 2008;28:12052–12061. doi: 10.1523/JNEUROSCI.2913-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29:3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30:4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]