

Abstract
Objective:
We present a series of unrelated patients with isolated hypomyelination, with or without mild cerebellar atrophy, and de novo TUBB4A mutations.
Methods:
Patients in 2 large institutional review board–approved leukodystrophy bioregistries at Children's National Medical Center and Montreal Children's Hospital with similar MRI features had whole-exome sequencing performed. MRIs and clinical information were reviewed.
Results:
Five patients who presented with hypomyelination without the classic basal ganglia abnormalities were found to have novel TUBB4A mutations through whole-exome sequencing. Clinical and imaging characteristics were reviewed suggesting a spectrum of clinical manifestations.
Conclusion:
Hypomyelinating leukodystrophies remain a diagnostic challenge with a large percentage of unresolved cases. This finding expands the phenotype of TUBB4A-related hypomyelinating conditions beyond hypomyelination with atrophy of the basal ganglia and cerebellum. TUBB4A mutation screening should be considered in cases of isolated hypomyelination or hypomyelination with nonspecific cerebellar atrophy.
Hypomyelinating leukodystrophies remain a diagnostic challenge with a large percentage of unresolved cases.1 Herein, we report on a series of unrelated patients with isolated hypomyelination, with or without mild cerebellar atrophy, and de novo TUBB4A mutations.
Mutations in TUBB4A (MIM 602662) are known to cause either dystonia type 4 (DYT4 [MIM 128101]) or hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC [MIM 612438]).2,3 In DYT4, an autosomal dominant mutation (c.4C>G [p.Arg2Gly]) in TUBB4A (NM_006087.2) was identified in patients presenting with a “whispering” dysphonia, generalized dystonia, and gait ataxia, but normal MRI features.2 In H-ABC, a cohort of 11 individuals were found to have a common de novo mutation at c.745G>A (p.Asp249Asn) in TUBB4A.3
H-ABC is a rare leukodystrophy diagnosed on the basis of distinctive MRI findings including hypomyelination, cerebellar atrophy, and absence or disappearance of the putamen at an early age.4,5 Individuals with H-ABC present with developmental delay, extrapyramidal movement disorders (dystonia, choreoathetosis, rigidity, opisthotonos, and oculogyric crises), ataxia, and spastic tetraplegia with variable onset and in some cases seizures.4,5
Herein, we describe novel de novo mutations in TUBB4A in 5 patients belonging to 4 families with hypomyelinating leukodystrophy, and who lack the full complement of features associated with H-ABC. This finding expands the phenotype of TUBB4A-related hypomyelinating conditions beyond H-ABC and suggests that TUBB4A should be considered in cases of isolated hypomyelination.
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was performed under the approval of institutional review boards at Children's National Medical Center or Montreal Children's Hospital. Blood was collected and DNA extracted with informed consent from all subjects and their parents. The DNA was analyzed using exome sequencing as previously described.3 Of note, these cases were tested as part of a larger cohort analysis in patients with unsolved leukodystrophies, and not due to any prior suspicion of TUBB4A-related disorders. Analysis was performed on trio or great family groups in all cases. Sanger sequencing was used to validate and perform segregation analysis of all candidate mutations.
RESULTS
In all 5 cases, we identified novel de novo mutations in TUBB4A (table). All of the patients have similar MRI features including hypomyelination but did not present with severe basal ganglia involvement characteristic of H-ABC (figure 1). These 5 patients presented with a diverse clinical spectrum (see supplemental data on the Neurology® Web site at Neurology.org) as well as a broad range of neuroradiologic features, some of which are also seen in H-ABC (table). Patient 4 presented with cerebellar atrophy from a young age while patients 3 and 5 have isolated hypomyelination. Patients 1 and 2 have global atrophy, as often seen in patients with long-standing hypomyelinating disorders.
Table.
Clinical characteristics and de novo TUBB4A mutations of the patients
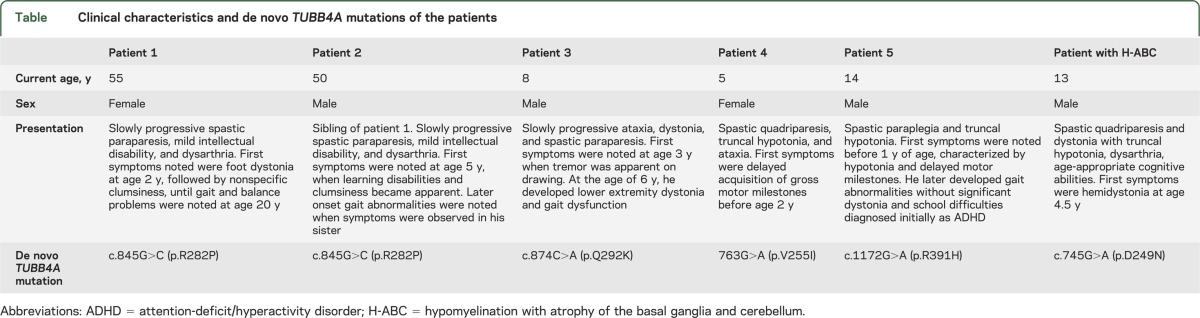
Figure 1. Brain sagittal T1, axial T2, and axial T1 MRIs of participants.

(A) Patient 1 at age 45 years. Note the generalized atrophy with preserved putamen (white arrow) and hypomyelination (T1- and T2-hyperintensity of the cerebral white matter). (B) Patient 2 at age 42 years. Note the generalized atrophy with preserved putamen (white arrow) and hypomyelination (T1- and T2-hyperintensity of the cerebral white matter). (C) Patient 3 at age 4 years. Note the hypomyelination (T1- and T2-hyperintensity of the white matter) with preserved putamen (white arrow) and cerebellum. Only postcontrast T1-weighted image available. (D) Patient 4 at age 5 years. Note the preserved putamen (white arrow) and hypomyelination (T1- and T2-hyperintensity of the white matter). Mild atrophy of the cerebellar vermis is seen (thick white arrow). Only postcontrast T1-weighted image available. (E) Patient 5 at age 10 years notable for preserved putamen (white arrow) and mild hypomyelination (T1- and T2-hyperintensity of the white matter). No axial T1-weighted images were available. A repeat study at 14 years showed no putamen atrophy. (F) Typical H-ABC MRI of the brain at age 9 years with absences of putamen (white arrow) and hypomyelination (T1- and T2-hyperintensity of the white matter), as well as cerebellar atrophy (thick white arrow). H-ABC = hypomyelination with atrophy of the basal ganglia and cerebellum.
DISCUSSION
These patients were ascertained as cases of unsolved hypomyelination without any radiologic or clinical features permitting a more specific diagnosis. These cases unexpectedly all had de novo TUBB4A mutations distinct from the original H-ABC–related mutation, at c.745G>A (p.Asp249Asn). The c.845G>C variant shared by patients 1 and 2 was not identified in either parent when tested by exome or Sanger sequencing. We hypothesize that there is likely low-level parental mosaicism, as was identified in the sibling group in the original description of TUBB4A mutations in H-ABC.3 Unfortunately, because of the older age of these patients (in their fifth decade), it was no longer possible to obtain new parental samples to test this hypothesis. In the other families, parental testing by exome and standard sequencing did not identify these variants in the parents.
Missense mutations in genes that code for α- and β-tubulin proteins, essential for assembly of neuronal microtubules, have been shown to cause a group of neurologic disorders characterized by abnormal neuronal migration, differentiation, axon guidance, and maintenance.6 Mutations in TUBA1A (MIM 602529), TUBA8 (MIM 605742), TUBB2B (MIM 612850), TUBB3 (MIM 602661), and TUBB4A have all contributed to this spectrum of disorders.
The clinical variability between DYT4 and H-ABC (late-childhood or juvenile-onset dystonia vs severe pediatric-onset dystonia and quadriparesis) suggests that the TUBB4A gene is associated with a spectrum of clinical manifestations. Other genes associated with hypomyelinating leukodystrophies are also known to cause a disease spectrum, varying from severe spastic quadriparesis with limited functional ability to mild spastic paraplegia. For example, PLP1 (MIM 300401) mutations cause Pelizaeus-Merzbacher disease (MIM 312080) as well as spastic paraplegia 2 (MIM 312920).7 Similar examples exist for Pol III–related leukodystrophies (4H or Hypomyelination with Hypogonadotropic Hypogonadism and Hypodontia)8 and Pelizaeus-Merzbacher–like disease caused by GJC2 mutations.9 It is therefore reasonable to predict that TUBB4A mutations could present similar variability in the context of hypomyelinating leukodystrophy.
TUBB4A is a neuronally expressed member of the β-tubulin protein family, and forms heterodimers with α-tubulins. The αβ heterodimers polymerize to form microtubules, an essential component of the cytoskeleton. The previously described residues associated with H-ABC (p.Asp249) and DYT4 (p.Arg2) both sit near the intradimer interface between α- and β-tubulin (figure 2). The c.763G>A de novo mutation found in patient 4 results in the amino acid change p.Val255Ile; this residue is located in the same α-helix as p.Asp249 (figure 2). Val255 is exposed to the intradimer interface. Mutations at this position may affect heterodimer formation or stability.
Figure 2. Structure of the αβ-tubulin heterodimer.

α-Tubulin is shown in gray, β-tubulin is olive, and the GTP (guanosine triphosphate) at the intradimer interface is shown in green. Each mutant residue is shown as spheres; R2 (blue), V255 (green), D249 (red), R282 (cyan), Q292 (brown), and R391 (pink). The M-loop region of β-tubulin is highlighted in red.
The c.845G>C variant shared by patients 1 and 2 results in the amino acid change p.Arg282Pro and is located in the structure known as the M-loop (figure 2). The M-loop extends from the side of tubulin monomers and is largely responsible for stabilizing the lateral contact between adjacent tubulin protofilaments.10 The conformational change resulting from the introduction of a proline into the middle of the M-loop is likely to destabilize this interaction. The p.Q292K change caused by the c.874C>A mutation in patient 3 is located in the α-helix H9; this helix is also believed to participate in the lateral contacts between adjacent protofilaments.10 Finally, the c.1172G>A mutation in patient 5 results in the amino acid change p.Arg391His. This residue is located near the interdimer interface (figure 2), thus this mutation may affect the polymerization of heterodimers.
Overall, these mutations are hypothesized to have a similar impact of tubulin polymerization and stability as the previously described c.745G>A (p.Asp249Asn) in TUBB4A identified in H-ABC. Although all patients have hypomyelination with or without cerebellar atrophy, as seen in H-ABC, no patients in the cohort have putamen atrophy, despite nearly 5 decades of disease progression in patients 1 and 2. This suggests that mutations in TUBB4A other than p.Asp249Asn can result in a phenotype of isolated hypomyelination, albeit with or without nonspecific cerebellar atrophy.
Further monitoring is required to determine the full clinical spectrum of these mutations because it is difficult to ascertain what may develop over time, in particular for the younger 2 patients. However, even if it is possible that these patients could develop additional imaging features consistent with H-ABC over time, it remains important, based on these findings, to consider TUBB4A mutation screening in cases of isolated hypomyelination or hypomyelination with nonspecific cerebellar atrophy.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and families; the Genome Quebec Innovation Center, McGill University, and Perkin Elmer for their services; members from the Center for Applied Genomics (CAG) at the Children's Hospital of Philadelphia (CHOP), especially Fengxiang Wang, James Snyder, and Haijun Qiu for sample preparation and processing, as well as Lifeng Tian for bioinformatics support; members from BGI-Shenzhen, particularly Jianguo Zhang and Jinlong Liang, for providing whole-exome sequencing and raw data processing; and Bruce Nmezi for technical assistance.
GLOSSARY
- DYT4
dystonia type 4
- H-ABC
hypomyelination with atrophy of the basal ganglia and cerebellum
- TUBB4A
tubulin, beta 4A class IVa
Footnotes
Supplemental data at Neurology.org
AUTHOR AFFILIATIONS
From the Department of Neurology (A.P., G.H., J.L.P.M., A.V.), and Center for Genetic Medicine Research (A.V.), Children's National Medical Center, Washington, DC; Departments of Pediatrics (T.M.P., S.S.) and Neurology (T.M.P., T.F.), Regenerative Medicine Institute (T.M.P., T.F.), and Medical Genetics Institute (S.S., T.F.), Cedars-Sinai Medical Center, Los Angeles, CA; Center for Applied Genomics (Y.G., H.H.), and Division of Human Genetics (H.H.), The Children's Hospital of Philadelphia, PA; Department of Pediatric Neurology (S.F.), Hospital Nacional de Pediatria Juan P. Garrahan, Buenos Aires, Argentina; Departments of Pediatrics, Neurology, and Neurosurgery (K.G., G.B.), Division of Pediatric Neurology, Montreal Children's Hospital, McGill University Heath Center, Montreal, Canada; Department of Human Genetics (Q.P.), Graduate School of Public Health, University of Pittsburgh, PA; Guangdong Enterprise Key Laboratory of Human Disease Genomics (Y.X., X.X.), BGI-Shenzhen (Y.X., X.X.), China; Department of Pediatrics (H.H.), Perelman School of Medicine, University of Pennsylvania, Philadelphia; Department of Pediatrics (M.F., S.S.), University of California, Los Angeles; Institute for Molecular Bioscience (R.J.T., C.S.), University of Queensland, St. Lucia, Australia; Department of Child Neurology (M.S.v.d.K.), VU University Medical Center, Amsterdam, the Netherlands; and Institute of Metabolic Disease (R.S.), Baylor Research Institute, Dallas, TX.
AUTHOR CONTRIBUTIONS
A.V., G.H., A.P., C.S., and M.S.v.d.K. managed the project. T.M.P., S.F., S.S., J.L.P.M., R.S., and A.V. performed clinical examination. A.P., T.F., and M.F. provided genetic counseling. T.M.P., Y.G., K.G., Q.P., Y.X., H.H., X.X., T.F., R.J.T., G.B., and C.S. performed sequencing. A.V. and C.S. designed the analyses. C.S., G.B., Y.H., Y.G., and K.G. performed the data analyses of sequencing results. A.P., T.M.P., G.H., S.F., J.L.P.M., M.S.K., C.S., and A.V. wrote the manuscript.
STUDY FUNDING
The study was funded by NHMRC APP1068278. The study was also supported by Institutional Development Funds to CAGCHOP and Kubert Estate Foundation Funds. This research was also supported by the research grant from the Shenzhen Municipal Government of China (CXZZ20130517144604091).
DISCLOSURE
A. Pizzino reports no disclosures relevant to the manuscript. T. Pierson was supported by the Diana and Steve Marienhoff Fashion Industries Guild Endowed Fellowship in Pediatric Neuromuscular Diseases. Y. Guo reports no disclosures relevant to the manuscript. G. Helman receives support from the Delman fund. S. Fortini, K. Guerrero, S. Saitta, J. Murphy, Q. Padiath, Y. Xie, H. Hakonarson, X. Xu, T. Funari, M. Fox, R. Taft, and M. van der Knaap report no disclosures relevant to the manuscript. G. Bernard has received a Research Scholar Junior 1 of the Fonds de Recherche du Québec en Santé and operating grant from the Canadian Institutes of Health Research. G.B. has received compensation from Actelion Pharmaceuticals, Genzyme, Shire, and Santhera Pharmaceuticals (speakers honoraria and for serving on scientific advisory boards). R. Schiffmann and C. Simons report no disclosures relevant to the manuscript. A. Vanderver receives support from the National Institute of Neurological Disorders NIH1K08NS060695. A.V. provides unpaid consulting to StemCells Inc. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010;133:2971–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hersheson J, Mencacci NE, Davis M, et al. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann Neurol 2013;73:546–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013;92:767–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Knaap MS, Linnankivi T, Paetau A, et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: follow-up and pathology. Neurology 2007;69:166–171 [DOI] [PubMed] [Google Scholar]
- 5.van der Knaap MS, Naidu S, Pouwels PJW, et al. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Neuroradiol 2002;23:1466–1474 [PMC free article] [PubMed] [Google Scholar]
- 6.Tischfield MA, Cederquist GY, Gupta ML, Jr, Engle EC. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr Opin Genet Dev 2011;21:286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saugier-Veber P, Munnich A, Bonneau D, et al. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat Genet 1994;6:257–262 [DOI] [PubMed] [Google Scholar]
- 8.Daoud H, Tétreault M, Gibson W, et al. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J Med Genet 2013;50:194–197 [DOI] [PubMed] [Google Scholar]
- 9.Hobson GM, Garbern JY. Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin Neurol 2012;32:62–67 [DOI] [PubMed] [Google Scholar]
- 10.Löwe J, Li H, Downing KH, Nogales E. Refined structure of αβ-tubulin at 3.5 Å resolution. J Mol Biol 2001;313:1045–1057 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.