

Abstract
Treatment resistant depression (TRD) is a global health concern affecting a large proportion of depressed patients who then require novel therapeutic options. One such treatment option that has received some attention in the past several years is vagal nerve stimulation (VNS). The present review briefly describes the relevance of this treatment in the light of other existing pharmacological and non-pharmacological options. It then summarizes clinical findings with respect to the efficacy of VNS. The anatomical rationale for its efficacy and other potential mechanisms of its antidepressant effects as compared to those employed by classical antidepressant drugs are discussed. VNS has been approved in some countries and has been used for patients with TRD for quite some time. A newer, fast-acting, non-invasive pharmacological option called ketamine is currently in the limelight with reference to TRD. This drug is currently in the investigational phase but shows promise. The clinical and preclinical findings related to ketamine have also been summarized and compared with those for VNS. The role of neurotrophin factors, specifically brain derived neurotrophic factor and its receptor, in the beneficial effects of both VNS and ketamine have been highlighted. It can be concluded that both these therapeutic modalities, while effective, need further research that can reveal specific targets for intervention by novel drugs and address concerns related to side-effects, especially those seen with ketamine.
Keywords: Depression, Vagal nerve stimulation, Ketamine, TrkB
INTRODUCTION
Major depressive disorder (MDD) is a serious worldwide public health concern. The point prevalence of MDD is 4.7% (4.4-5.0%) at the global level and 4.0% (3.4-4.6%) within East/Southeast Asia.1) MDD also imposes a huge economic burden. A recent study in South Korea estimates the total cost of the disorder at about 4 billion with USD 153 million being contributed by direct healthcare costs.2) Treatment options for MDD primarily involve antidepressant (AD) drugs. One of the first agents with AD properties, iproniazid, was discovered in 1952. This drug acts by slowing down the enzymatic breakdown of monoamines such as norepinephrine (NE) or serotonin (5-HT) and belongs to a class of ADs called monoamine oxidase inhibitors (MAOIs) due to their mechanism of action. Another class of drugs called tricyclic ADs (TCAs), named based on their three-ring chemical structure, was discovered around the same time.3) Based on mechanism of action, AD drugs can be classified as selective 5-HT reuptake inhibitors (SSRIs), selective NE reuptake inhibitors (selective NRIs) and dual 5-HT/NE reuptake inhibitors (SNRIs), 5-HT modulators and NE/5-HT modulators. All of these classes mainly target central serotonergic and noradrenergic systems whereas MAOIs and dopamine (DA)-NE reuptake inhibitors also affect the dopaminergic system.4,5,6)
MAIN SUBJECTS
Response, Remission, Recovery, Relapse and Recurrence with Reference to Treatment of Major Depressive Disorder
'Response' is defined as a clinically meaningful reduction in symptoms of depression usually seen along with better mood, reduction in pain/distress and improved daily function. It has been defined as a ≥50% reduction in the severity of symptoms that were seen pretreatment, based on standardized rating scales for the severity of depression such as the Hamilton or Montgomery-Asberg Rating Scales for Depression.7,8) 'Remission' is used when the signs and symptoms of depression are either absent or almost absent. Based on the Hamilton Rating Scale for Depression, remission would require a score of ≤5 or 7. 'Recovery' suggests an extended period of remission implying that a major depressive episode (MDE) is not probable in the near future. There is no clear consensus on the duration of remission required to define recovery. The American College of Neuropsychopharmacology Task Force recommends 4 months whereas previously, others have used 6 months of remission to define recovery. Both 'remission' and 'recovery' may be used irrespective of whether the patients are undergoing treatment. 'Relapse' and 'recurrence' both imply the reappearance of an MDE. The term 'relapse' is used if the MDE occurs during remission; however, 'recurrence' occurs only after the onset of recovery.9,10)
The classical AD drugs mentioned earlier are frequently only moderately effective and most studies have found no difference in efficacy between different classes of ADs or even between individual drugs.11) In fact, up to two-thirds of the patient population does not respond to the first prescribed AD. Further, about 15% of the population does not respond to multiple treatment options including psychotherapy.12) These patients are diagnosed with treatment resistant depression (TRD).
Treatment Resistant Depression
The impairment, morbidity and economic burden associated with MDD are further augmented in TRD.13) Over the years, the definition of TRD has been subject to debate; however, the general consensus is that for a diagnosis of TRD, treatment with ADs from different pharmacological classes is needed.14) TRD has been classified by several staging methods that incorporate factors such as treatment trials, severity and chronicity of the disorder. An example of one such model is the Thase and Rush Staging Model (Table 115)), developed as a guideline for psychiatrists and based on the number and classes of ADs that have not produced a response.
Table 1.
Thase and Rush Staging Model
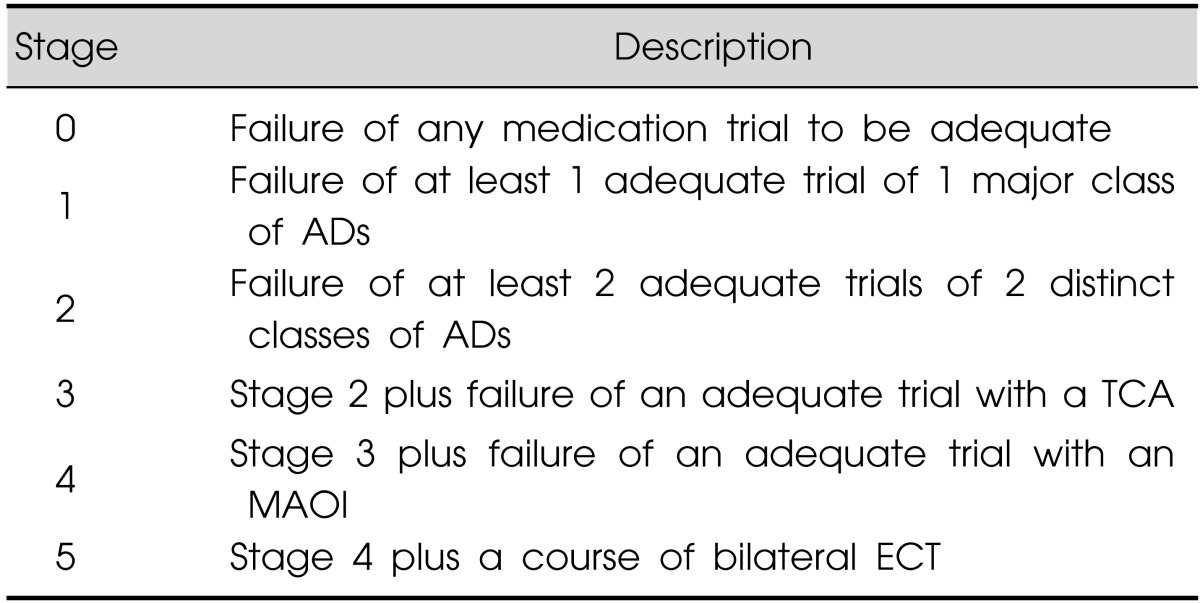
Adapted from the article of Thase and Rush15) (J Clin Psychiatry 1997;58(Suppl)13:23-29).
AD, antidepressant; TCA, tricyclic AD; MAOI, monoamine oxidase inhibitor; ECT, electroconvulsive therapy.
Other staging methods or models include the Antidepressant Treatment History Form, the European Staging Model, the Massachusetts General Hospital Staging Model and the Maudsley Staging Method. These models have different advantages and disadvantages leading to varying predictive utility and reliability.16) However, TRD must not be confused with chronic depression.17)
Treatment Options for Treatment Resistant Depression
Pharmacological treatment
Switching strategies
This involves switching treatment from the initially prescribed drug to another AD. The newer prescription may be for an AD from the same class (within-class switching) or from another class with a different mechanism of action (between-class switching). There is a lack of evidence suggesting that one of these two strategies is better than the other to achieve response or remission.10,18,19,20) The first line of treatment is often an SSRI. If this is the case, the second prescribed drug for the between-class switching strategy belongs to one of the following groups: TCAs, mianserin, dual acting agents (such as venlafaxine), MAOIs or agents acting on the dopaminergic and/or noradrenergic systems (such as bupropion and reboxetine).19)
Combination/augmentation strategies
This strategy involves combining one AD with another or with some drug with a different primary indication. Drugs commonly used for augmentation include lithium, bupropion, mirtazapine, atypical antipsychotics, anticonvulsants, DA agonists, stimulants, buspirone, modafinil, pindolol, thyroid hormone, estrogen or testosterone, herbal agents etc. Although these drugs can be beneficial, there are a variety of side-effects associated with most of their use. Upon scanning through data for the response and remission rates, it appears that lithium and liothyronine (a form of triiodothyronine) are most effective as drugs for this strategy.5)
Psychotherapy
Psychotherapy in the form of cognitive behavioral therapy (CBT) is based on the premise that MDD is associated with negative cognitive biases.21) Several studies have shown its effectiveness in reducing vulnerability and in preventing relapse of MDD when used alone,22) in combination with23,24) or sequentially after pharmacotherapy.25) The use of CBT in treating TRD also seems promising.26,27,28,29) Efficacy of psychotherapy may depend on mechanisms underlying the etiology of depression in each patient. Some studies30,31) suggest regional differences in brain activity in response to psychotherapy versus pharmacotherapy.
Somatic therapies
Other non-pharmacological options include electroconvulsive therapy (ECT), repetitive transcranial magnetic stimulation (rTMS), magnetic seizure therapy, deep brain stimulation (DBS) and vagal nerve stimulation (VNS).5,32) The latter two options require surgical intervention. VNS and rTMS were approved by the US Food and Drug Administration (FDA) for treatment of TRD in 2005 and 2008 respectively. The use of DBS for treating TRD is still not FDA-approved and remains in the experimental stage.
Vagal Nerve Stimulation
VNS therapy consists of implantation of a generator connected to bipolar electrodes (Cyberonics Inc., Houston, TX, USA) that deliver chronic intermittent electrical signals at a low frequency to the vagus nerve. The pulse generator is surgically implanted subcutaneously in the anterior chest wall and the electrodes wrapped around the left vagus nerve through an incision in the neck (Fig. 1). The parameters of the stimulation provided by the generator via the electrodes can be programmed and controlled noninvasively using a telemetric wand that is linked to a hand-held computer.
Fig. 1.
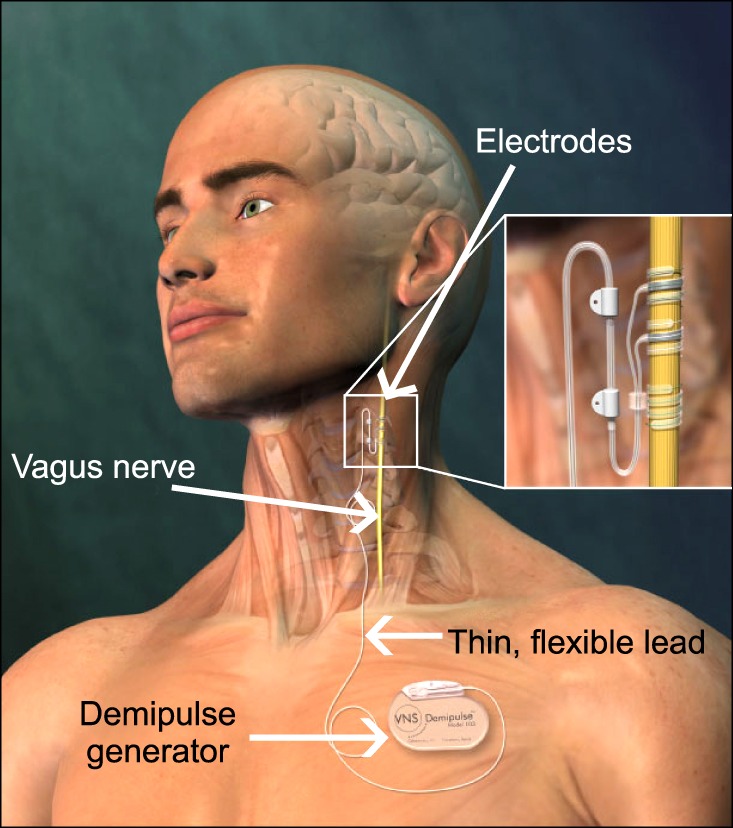
Illustration depicting the electrodes (negative electrode, positive electrode and anchor tether) wrapped around the left vagus nerve and connected to a Demipulse generator via a thin flexible lead. The Demipulse generator is subcutaneously implanted in the chest region. Figure kindly supplied by Cyberonics, Inc. (Houston, TX, USA) and used with their permission.
The left nerve is preferred to the right for electrode placement as there are more cardiac efferent fibers from the right and stimulation of this side would cause more frequent adverse cardiac effects.33) Further, pre-clinically, we have evaluated the stimulation parameters that we use in our studies, which are the same ones as those used initially in clinical studies, and have observed no significant effects on heart rate and blood pressure.34) Chronic VNS in patients does produce a few side-effects such as voice alteration, cough, pharyngitis, throat discomfort and dyspnea. Other common complications include headache, nausea, vomiting and dyspepsia.33)
Approval of VNS for TRD and clinical studies
VNS therapy is now approved for the treatment of TRD in the United States, Europe, Canada, Mexico, Brazil, Australia and New Zealand (personal communication from Cyberonics Inc.). Clinical studies show promising results with long-term treatment with VNS for patients with TRD. An early 10-week randomized controlled trial of TRD patients with VNS in combination with treatment as usual showed no significant efficacy of VNS over just treatment as usual (TAU).35) However, subsequent treatment of these patients for 9 months or 12 months (for those that initially received sham treatment) produced an increase in the efficacy of VNS over time with response and remission rates of 27.2% and 15.8% respectively.36) In another long-term observational study, George et al.37) compared response and remission rates for patients receiving TAU to rates for patients from the previous study36) who received TAU plus VNS for 1 year. Among the VNS+TAU group, 27% were responders whereas only 13% of the patients in the TAU group responded. Further, the number of patients achieving remission in the VNS+TAU group was twice that in the TAU group. A study conducted in Europe by Schlaepfer et al.38) also showed a reduction in the severity of depression with VNS treatment. This study reported response and remission rates of 37% and 13%, respectively, after 3 months of treatment and 53% and 33%, respectively, after one year of treatment. A more recent re-analysis of this study and additional clinical data confirmed the beneficial effects following VNS in at least one out of three patients suffering from chronic and highly medication-refractory unipolar depression.39) Studies looking at the two-year outcome of VNS treatment show response rates of 42-53% and remission rates of 22-39%.40,41) Several single cases with positive effects of VNS have also been reported.42) Overall, these clinical studies show an increase in efficacy of VNS over time with the effect being sustained in most patients who respond.36,43) Further, another recent clinical study showed that VNS significantly lowered relative risk of suicidal ideation and reduced all-cause mortality and suicide rates by about half as compared to those for patients with just TAU.44)
VNS and dosage
For VNS, 'dosage' refers to a combination of various stimulation parameters, which include current (milliamperes, mA), pulse width (microsec, µs), frequency (hertz, Hz) and the duty cycle (amount of time the stimulation is ON [in seconds] and OFF [in minutes]). These parameters determine the electrical output characteristics. Not many studies have accounted for the effects of different doses of VNS. Aaronson et al.45) addressed this issue by conducting a dose-response study for VNS in TRD patients. They studied the effects of three different doses defined as 'low', 'medium' and 'high' based on the magnitude of the output current (0.25, 0.5-1.0 and 1.25-1.5 mA, respectively) and pulse width (150 µs for 'low' and 250 µs for 'medium' and 'high'). The frequency and duty cycle were identical for all three doses. Patients were assessed at the end of 50 weeks of treatment. There were more frequent reports of suicide attempts among the 'low' dose group than among the 'medium' or 'high' dose groups. Also, patients in the 'low' dose group were significantly more likely to relapse at 50 weeks than patients in the 'medium' and 'high' dose groups combined suggesting a more sustained AD effect of VNS at the higher doses.
Anatomical rationale for use of VNS
The vagus nerve is a mixed nerve with efferent fibers having an autonomic function and afferent fibers carrying sensory information from the periphery to the dorsal medullary complex to the nucleus tractus solitarius (NTS).46) The left vagus nerve bifurcates in the medulla and innervates the NTS bilaterally. The NTS projects to the parabrachial nucleus, cerebellum, dorsal raphae nucleus (DRN), locus coeruleus (LC) and the periaqueductal gray.47,48) There are several projections from these primary areas to limbic, paralimbic and cortical regions. Brain areas activated in response to acute VNS have been studied in rats by immunohistochemical staining of brain slices for c-fos, an immediate early gene product.34,49) These studies were followed by immunohistochemical staining for ΔFosB, an indicator of long-term neuroadaptations, in brain slices from rats treated chronically (2-3 weeks) with VNS.34,50) Collectively, these pre-clinical studies indicate that stimulation of the vagus nerve activates areas of the brain that are involved in mood regulation and implicated in MDD. Using immunohistochemical analysis, we have also compared ΔFosB staining in different brain areas with VNS to that with classical ADs, desipramine and sertraline, and found a higher number of ΔFosB-positive cells in the dorsal raphae, bed nucleus of the stria terminalis, nucleus accumbens and peripeduncular nucleus in the VNS-treated rats.50)
Consistent with these observations, clinical neuroimaging studies (fMRI, PET, SPECT) also show changes in medial temporal regions such as the hippocampus, parahippocampus and amygdala as well as the orbito- and pre-frontal cortices.51,52,53,54,55,56) However, a thorough review of these reports reveals that the changes were bilateral in some studies whereas they were limited to one side in others. The direction of change for some brain regions, such as the amygdala,51,56) was also inconsistent. These few contradictory observations may be due to small sample sizes and differences in the duration and parameters of the VNS treatment used for the studies. In addition, there may be an influence of the varied pharmacological treatments that the participants were undergoing along with VNS therapy.
Theories for depression
There are several hypotheses regarding the cause of depression as well as the mechanisms of action of AD treatments such as the monoaminergic neurotransmission theory, the neurotrophic hypothesis and the neurogenesis theory. Here, we briefly review the current status of research associated with these theories specifically with reference to VNS.
As mentioned earlier, most classical ADs target either the serotonergic and/or noradrenergic systems4,6) and result in an enhancement of neurotransmission in these systems, forming the basis of the monoaminergic neurotransmission theory of depression.57,58,59)
VNS increases 5-HT and NE neuronal firing rates in the DRN and LC, respectively, in a time-dependent manner. Spontaneous firing activity increases in the LC first, after an hour to 3 days of stimulation whereas it increases after 14 days in 5-HT neurons in the DRN. SSRIs cause a time dependent subsensitivity of serotonergic autoreceptors but this does not occur with VNS.60) Reanalysis of these data revealed that VNS enhances the firing rate of 5-HT neurons and the burst firing activity of NE neurons, with a greater effect on NE firing activity.61) Additionally, lesion and pharmacological studies revealed that VNS increases tonic activity of 5-HT neurons in the DRN via noradrenergic neurons in the LC that target α1-adrenergic receptors on the 5-HT neurons.61)
Consistent with the increase in firing activity in NE neurons in the LC, acute VNS treatment also increases NE release in the cortex, medial prefrontal cortex (PFC), amygdala and hippocampus as measured using microdialysis.62,63,64) Chronic administration of VNS for 14 days has been reported to cause an increase in extracellular NE in the PFC and hippocampus. Extracellular levels of 5-HT were only increased in the DRN but not in the PFC or hippocampus.65) In line with these observations, we have shown that the anxiolytic-like and AD-like effects of chronic administration of VNS are abolished by lesioning either serotonergic or noradrenergic systems.66) These results were obtained using the novelty suppressed feeding test (NSFT) and the forced swimming test (FST), widely used to study anxiolytic and AD effects of drugs, respectively.67,68) In fact, the AD-like effect of VNS in the FST had been shown previously by Krahl et al.69) with 30 min of continuous VNS, administered daily for 4 days.
Interestingly, in spite of DA cells in the ventral tegmental area (VTA) decreasing their firing rate in response to VNS, extracellular DA levels in the PFC and nucleus accumbens were increased.65) Thus, in contrast to traditional ADs, VNS does seem to have effects on dopaminergic neurons.
The neurogenesis theory suggests that there is a stress-induced decrease in adult neurogenesis in the hippocampus, leading to depression and this decrease is reversed by AD treatment.70) Biggio et al.71) reported that acute VNS (3 h) increased neurogenesis in the dentate gyrus (of the hippocampus) as seen by an increase in BrdU-positive cells 24 h and 3 weeks post-cessation of VNS. Revesz et al.72) showed that acute (48 h) VNS increased proliferation but did not affect progenitor cell survival within the dentate gyrus. Chronic VNS (4 weeks), however, did not have this effect 3 weeks post-cessation of the treatment,71) so the effect on neurogenesis may be relatively short-lived. A recent study looked at the effects of chronic VNS treatment (8 weeks) on hippocampal neurogenesis in an animal model of depression (bulbectomized rats). The decrease in neuronally differentiated BrdU-positive cells within the dentate gyrus, seen due to bulbectomy, was prevented by VNS. However, there was no increase in BrdU-positive cells upon chronic stimulation in the sham-bulbectomized rats.73)
Unlike positive results with acute VNS, acute treatment with classical ADs does not increase neurogenesis; however, acute ECT does.74) ADs usually require at least 2 weeks to produce an increase in proliferation although this effect has not been consistently reproduced over various studies.75,76,77,78) The neurogenesis theory of depression has been expanded to suggest that plasticity and synaptic remodeling (independent of neurogenesis) in the hippocampus and PFC may be more critical for therapeutic efficacy of ADs.79) Both, acute and chronic VNS increased dendritic arborization of doublecortin-positive (DCX+) neurons. DCX or doublecortin is a microtubule associated protein expressed by neuronal precursor cells and marker for adult neurogenesis80) in the dentate gyrus. This effect was detected 3 weeks post-cessation of treatment and was similar to the effect of chronic treatment with fluoxetine.71,81) Ketamine, a rapidly acting drug with AD-like effects (discussed later), also induces synaptogenesis and synaptic protein synthesis approximately 2 h after treatment.82)
The neurotrophic theory of depression is based on studies that show opposite effects of stress and ADs on expression of certain neurotrophic factors in brain areas related to mood regulation. The most widely studied and perhaps the most relevant neurotrophic factor in this context is brain derived neurotrophic factor (BDNF). Along with BDNF, TrkB, the tyrosine kinase receptor for BDNF (and neurotrophin-4 [NT-4]) has also been studied (Fig. 2). From a functional perspective, the neurotrophic hypothesis is linked to the neurogenesis hypothesis as the increase in expression of neurotrophins with AD treatment may block or reverse the neuronal loss associated with depression.83)
Fig. 2.
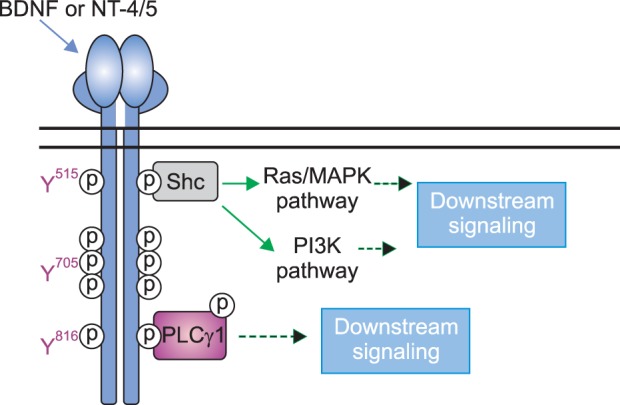
TrkB signaling. Upon ligand-binding (BDNF or NT-4/5), the receptor (which itself is a tyrosine kinase) undergoes transactivation by phosphorylation at tyrosine residues within the catalytic domain (Y705, is one such residue) and at other tyrosine residues such as Y515 and Y816. Phosphorylation at these latter two residues triggers distinct downstream signaling cascades which can eventually lead to gene transcription, cell proliferation and survival. BDNF, brain derived neurotrophic factor; NT, neurotrophin.
Chronic treatment (21 days) with classical ADs belonging to different classes increases expression of mRNAs for BDNF and TrkB in the hippocampus (CA1, CA3 and dentate gyrus).84,85) Others have reported similar results as well.83) Acute as well as chronic (10 days) treatment with ECT increased expression of mRNA for BDNF and the truncated form of TrkB in the frontal cortex and hippocampus.84) Similarly, chronic (11 weeks) repeated TMS also increased mRNA and immunoreactivity for BDNF in the hippocampus (CA3 and dentate gyrus) and in the parietal and piriform cortices.86) Acute VNS treatment (3 h) increased mRNA for BDNF in the hippocampus as well as in the cortex.62)
Drawing conclusions based on changes in mRNA for BDNF or protein levels can be complicated due to factors such as translation, proteolytic cleavage and release that are not accounted for. Hence, analyzing activation of the receptor, TrkB, may provide additional clues about the effect of AD treatment on BDNF function. We have shown that both acute (2 h) and chronic (14 days) VNS activate the TrkB receptor as evident from its phosphorylation at 3 tyrosine residues (Y705, Y816 and Y515). In contrast, acute and chronic AD treatment caused phosphorylation at Y705 and Y816 but not at Y515.87) Similar results have been shown with AD drugs in mice.88,89) Y705 is the autophosphorylation site whereas Y816 and Y515 are linked with the PLCγ1 and MAPK/PI3K signaling pathways, respectively. Consistent with the phosphorylation of the tyrosine residues, acute VNS as well as ADs caused phosphorylation of PLCγ1, however, this was not maintained with chronic treatments. However, only acute and chronic VNS caused phosphorylation of ERK1/2 and Akt that are downstream of Y515.87,90)
There is an abundance of literature showing that serum levels of BDNF are decreased in patients with MDD and that ADs normalize these levels.91) The source of this BDNF in the periphery, however, is still not clear. There are reports suggesting that BDNF in the periphery may derive from the brain via active transport through the blood brain barrier92) and from cells such as leukocytes and vascular endothelial cells in the periphery.93,94,95) To the authors' knowledge, there have not been many studies looking at serum BDNF levels in VNS treated patients. Lang et al.96) report no change in serum BDNF levels in patients treated for 4 weeks with VNS or rTMS. Considering that the patients had been previously treated with AD drugs, one explanation could be that the serum BDNF levels before initiating VNS/rTMS treatments were already normalized. Also, although patients showed clinical improvement, one could also speculate that a longer duration of treatment may lead to different results.
Ketamine
Both pre-clinical and clinical research have been carried out focusing on other pharmacological agents for TRD rather than the classical ones targeting serotonergic and/or noradrenergic systems. These so called "novel" or "alternative" targets for TRD might include ones aimed at dopaminergic or glutamatergic systems, but are not necessarily limited to them.97,98)
Regarding the glutamatergic system, N-methyl-D-aspartate receptor (NMDA-R) antagonists in particular have long been linked to the pathophysiology of MDD and the mechanism of action of AD drugs.97) The first pre-clinical work testing the hypothesis that various NMDA-R antagonists have AD-like effects was carried out in mice, where the authors showed that such agents caused a significant reduction of immobility in the FST and the tail suspension test,99) two tests with high predictive validity for AD treatments.68,100)
Among NMDA-R antagonists, the anesthetic ketamine in particular is clinically relevant for treatment refractory depression. The first randomized controlled trial investigating the AD properties of a single intravenous low dose of ketamine (0.5 mg/kg) in patients diagnosed with MDD showed a significant improvement in depressive symptoms within 72 hours after ketamine administration, and not with placebo.101) Since this first trial, others have corroborated the rapid AD effects of ketamine administration.101,102,103,104) When given to TRD patients, single subanesthetic dose of ketamine typically improves mood within hours following ketamine administration and can persist, for the most part, for about two weeks.105,106,107,108,109,110,111) It also produces a rapid decrease in suicidal ideation in both bipolar depression and MDD.104,106,112) Rapid-acting pharmacotherapy could readily reduce hospitalization time and allow disabled people to resume their daily routine including being able to work.
It is important to note that aside from the promising and consistent results gained in the clinic, ketamine poses some serious problems with regard to its acute psychotomimetic and physiological (increased blood pressure and heart rate) side effects. In addition, chronic use of ketamine has been associated with dependence.113,114,115)
Sub-anesthetic low dose ketamine can also produce rapid and sustained AD-like responses in rodents. Studies carried out in mice reported a decrease in immobility in the FST shortly after a single systemic injection, with a persistent response for up to one week,116,117,118) analogous to some of its effects previously reported in humans as discussed above.108,109,111,116,117,118)
Pre-clinical studies have focused on the mechanisms of action of ketamine. The AD-like effects of ketamine are blocked by pretreatment with 2, 3-dihydroxy-6-nitro-7-sulfamoyl-benzol[f]quinoxaline-2,3-dione (NBQX), an a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist.117) An early study revealed decreased spontaneous activity of GABAergic interneurons and an increased firing rate of glutamatergic pyramidal neurons in the PFC of rats given ketamine,119) suggesting that NMDA-R antagonism by ketamine blocked spontaneous GABAergic activity resulting in enhanced glutamatergic transmission. Stimulation of AMPA receptors leads to activity-dependent BDNF release.120) The use of conditional knockout mice has suggested that BDNF is required for the AD-like effect of ketamine.116) Similar to VNS, ketamine also shows an activation of neurotrophin signaling. In particular studies have shown a rather transient increase in the phosphorylation of the BDNF/NT-4 receptor, namely TrkB, following a single sub-anesthetic injection of ketamine in rodents.116,121) In a randomized control trial with ketamine, plasma BDNF levels were found to be significantly higher in responders compared to that in non-responders.122) However, as mentioned, VNS therapy was not associated with increased plasma BDNF levels.96)
Moreover, both VNS (as mentioned earlier) and ketamine seem to modulate signaling pathways downstream of the TrkB receptor, for example the mammalian target of rapamycin (mTor) within the hippocampus.90,121) Interestingly whereas ketamine cause a rapid and transient activation of the mTor pathway, VNS seems to require more chronic treatment for the same effect.90,121) Pre-clinical studies reviewed by Dwyer and Duman123) report that activation of the mTor pathway following single low-dose of ketamine administration is required to rescue the maladaptative responses to chronic stress.
Some studies have shown a positive correlation between plasma levels of mTor124,125) and its downstream effectors, glycogen synthase kinase-3beta (GSK-3β) and dephosphorylation of eukaryotic elongation factor 2 (e-EF2)125) in patients who responded to ketamine. This has not yet been tested with respect to the VNS-induced mTor signaling activation in humans. Since phosphorylated e-EF2 is associated with inhibition of translation, its dephosphorylation, via mTor signaling activation, leads to increased protein synthesis, including BDNF synthesis.123) It is worth noting that patients carrying a single nucleotide polymorphism, namely Val66Met in the gene encoding for BDNF, show a reduced response to ketamine.126)
CONCLUSION
In conclusion, TRD, is a serious public health concern given the morbidity, and mortality due to suicide, associated with it. With improved and more standardized criteria for its definition, it has become more amenable to proper scientific investigation. This review covered in some detail two treatments for it. One, VNS has been approved for use in many countries but not yet in Korea. The other, ketamine is still in the investigational stage but the clinical data are very encouraging, especially in light of the rapidity of its clinical response. These two treatments do share some effects in common that differ from those produced by traditional AD drugs, e.g., mTor, p70S6 kinase, etc. downstream of the TrkB receptor. The role that such effects play in their effectiveness in TRD, which by definition does not respond to traditional ADs, is an important subject for future research.
Acknowledgments
Dr. Frazer has served on advisory boards for Lundbeck and for Takeda Pharmaceuticals International, Inc. and Eli Lilly and Co. Previously, Dr. Frazer had received financial compensation as a consultant for Cyberonics Inc. and had also obtained grant support from them for a preclinical study.
We would like to acknowledge Dr. J. Hensler, from our department, for generously providing the illustration used for TrkB signaling. In addition, aspects of this review were part of a thesis for a doctorate degree (for AS).
References
- 1.Ferrari AJ, Somerville AJ, Baxter AJ, Norman R, Patten SB, Vos T, et al. Global variation in the prevalence and incidence of major depressive disorder: a systematic review of the epidemiological literature. Psychol Med. 2013;43:471–481. doi: 10.1017/S0033291712001511. [DOI] [PubMed] [Google Scholar]
- 2.Chang SM, Hong JP, Cho MJ. Economic burden of depression in South Korea. Soc Psychiatry Psychiatr Epidemiol. 2012;47:683–689. doi: 10.1007/s00127-011-0382-8. [DOI] [PubMed] [Google Scholar]
- 3.Lieberman JA. History of the use of antidepressants in primary care. J Clin Psychiatry. 2003;5(Suppl 7):6–10. [Google Scholar]
- 4.Morilak DA, Frazer A. Antidepressants and brain systems: a dimensional approach to understanding their behavioural effects in depression and anxiety disorders. Int J Neuropsychopharmacol. 2004;7:193–218. doi: 10.1017/S1461145704004080. [DOI] [PubMed] [Google Scholar]
- 5.Shelton RC, Osuntokun O, Heinloth AN, Corya SA. Therapeutic options for treatment-resistant depression. CNS Drugs. 2010;24:131–161. doi: 10.2165/11530280-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 6.Lenox RH, Frazer A. Mechanism of action of antidepressants and mood stabilizers. In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: The fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins; 2002. pp. 1139–1163. [Google Scholar]
- 7.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- 9.Frank E, Prien RF, Jarrett RB, Keller MB, Kupfer DJ, Lavori PW, et al. Conceptualization and rationale for consensus definitions of terms in major depressive disorder. Remission, recovery, relapse, and recurrence. Arch Gen Psychiatry. 1991;48:851–855. doi: 10.1001/archpsyc.1991.01810330075011. [DOI] [PubMed] [Google Scholar]
- 10.Rush AJ, Kraemer HC, Sackeim HA, Fava M, Trivedi MH, Frank E, et al. ACNP Task Force. Report by the ACNP Task Force on response and remission in major depressive disorder. Neuropsychopharmacology. 2006;31:1841–1853. doi: 10.1038/sj.npp.1301131. [DOI] [PubMed] [Google Scholar]
- 11.Murrough JW, Charney DS. Is there anything really novel on the antidepressant horizon? Curr Psychiatry Rep. 2012;14:643–649. doi: 10.1007/s11920-012-0321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burrows GD, Norman TR. Treatment-resistant unipolar depression. In: Lader M, Naber D, editors. Difficult clinical problems in psychiatry. London (UK): Martin Dunitz Ltd.; 1999. pp. 57–73. [Google Scholar]
- 13.Olchanski N, McInnis Myers M, Halseth M, Cyr PL, Bockstedt L, Goss TF, et al. The economic burden of treatment-resistant depression. Clin Ther. 2013;35:512–522. doi: 10.1016/j.clinthera.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Berlim MT, Turecki G. Definition, assessment, and staging of treatment-resistant refractory major depression: a review of current concepts and methods. Can J Psychiatry. 2007;52:46–54. doi: 10.1177/070674370705200108. [DOI] [PubMed] [Google Scholar]
- 15.Thase ME, Rush AJ. When at first you don't succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry. 1997;58(Suppl 13):23–29. [PubMed] [Google Scholar]
- 16.Ruhé HG, van Rooijen G, Spijker J, Peeters FP, Schene AH. Staging methods for treatment resistant depression. A systematic review. J Affect Disord. 2012;137:35–45. doi: 10.1016/j.jad.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 17.Birkenhäger TK, Ruhé HG. The pharmacological frontiers in treatment resistant major depression. In: Schoepf D, editor. Psychiatric disorders - New frontiers in affective disorders. Morn Hill (UK): InTech; 2013. [Google Scholar]
- 18.Bschor T, Baethge C. No evidence for switching the antidepressant: systematic review and meta-analysis of RCTs of a common therapeutic strategy. Acta Psychiatr Scand. 2010;121:174–179. doi: 10.1111/j.1600-0447.2009.01458.x. [DOI] [PubMed] [Google Scholar]
- 19.Ruhé HG, Huyser J, Swinkels JA, Schene AH. Switching antidepressants after a first selective serotonin reuptake inhibitor in major depressive disorder: a systematic review. J Clin Psychiatry. 2006;67:1836–1855. doi: 10.4088/jcp.v67n1203. [DOI] [PubMed] [Google Scholar]
- 20.Souery D, Serretti A, Calati R, Oswald P, Massat I, Konstantinidis A, et al. Switching antidepressant class does not improve response or remission in treatment-resistant depression. J Clin Psychopharmacol. 2011;31:512–516. doi: 10.1097/JCP.0b013e3182228619. [DOI] [PubMed] [Google Scholar]
- 21.Beck AT. The current state of cognitive therapy: a 40-year retrospective. Arch Gen Psychiatry. 2005;62:953–959. doi: 10.1001/archpsyc.62.9.953. [DOI] [PubMed] [Google Scholar]
- 22.Driessen E, Hollon SD. Cognitive behavioral therapy for mood disorders: efficacy, moderators and mediators. Psychiatr Clin North Am. 2010;33:537–555. doi: 10.1016/j.psc.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keller MB, McCullough JP, Klein DN, Arnow B, Dunner DL, Gelenberg AJ, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med. 2000;342:1462–1470. doi: 10.1056/NEJM200005183422001. [DOI] [PubMed] [Google Scholar]
- 24.Thase ME, Friedman ES, Biggs MM, Wisniewski SR, Trivedi MH, Luther JF, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry. 2007;164:739–752. doi: 10.1176/ajp.2007.164.5.739. [DOI] [PubMed] [Google Scholar]
- 25.Guidi J, Fava GA, Fava M, Papakostas GI. Efficacy of the sequential integration of psychotherapy and pharmacotherapy in major depressive disorder: a preliminary meta-analysis. Psychol Med. 2011;41:321–331. doi: 10.1017/S0033291710000826. [DOI] [PubMed] [Google Scholar]
- 26.Chaput Y, Magnan A, Gendron A. The co-administration of quetiapine or placebo to cognitive-behavior therapy in treatment refractory depression: a preliminary trial. BMC Psychiatry. 2008;8:73. doi: 10.1186/1471-244X-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thase ME, Friedman ES, Howland RH. Management of treatment-resistant depression: psychotherapeutic perspectives. J Clin Psychiatry. 2001;62(Suppl 18):18–24. [PubMed] [Google Scholar]
- 28.Trivedi RB, Nieuwsma JA, Williams JW., Jr Examination of the utility of psychotherapy for patients with treatment resistant depression: a systematic review. J Gen Intern Med. 2011;26:643–650. doi: 10.1007/s11606-010-1608-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiles N, Thomas L, Abel A, Ridgway N, Turner N, Campbell J, et al. Cognitive behavioural therapy as an adjunct to pharmacotherapy for primary care based patients with treatment resistant depression: results of the CoBalT randomised controlled trial. Lancet. 2013;381:375–384. doi: 10.1016/S0140-6736(12)61552-9. [DOI] [PubMed] [Google Scholar]
- 30.Goldapple K, Segal Z, Garson C, Lau M, Bieling P, Kennedy S, et al. Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 2004;61:34–41. doi: 10.1001/archpsyc.61.1.34. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy SH, Konarski JZ, Segal ZV, Lau MA, Bieling PJ, McIntyre RS, et al. Differences in brain glucose metabolism between responders to CBT and venlafaxine in a 16-week randomized controlled trial. Am J Psychiatry. 2007;164:778–788. doi: 10.1176/ajp.2007.164.5.778. [DOI] [PubMed] [Google Scholar]
- 32.Cusin C, Dougherty DD. Somatic therapies for treatment-resistant depression: ECT, TMS, VNS, DBS. Biol Mood Anxiety Disord. 2012;2:14. doi: 10.1186/2045-5380-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatton KW, McLarney JT, Pittman T, Fahy BG. Vagal nerve stimulation: overview and implications for anesthesiologists. Anesth Analg. 2006;103:1241–1249. doi: 10.1213/01.ane.0000244532.71743.c6. [DOI] [PubMed] [Google Scholar]
- 34.Cunningham JT, Mifflin SW, Gould GG, Frazer A. Induction of c-Fos and DeltaFosB immunoreactivity in rat brain by Vagal nerve stimulation. Neuropsychopharmacology. 2008;33:1884–1895. doi: 10.1038/sj.npp.1301570. [DOI] [PubMed] [Google Scholar]
- 35.Rush AJ, Marangell LB, Sackeim HA, George MS, Brannan SK, Davis SM, et al. Vagus nerve stimulation for treatment-resistant depression: a randomized, controlled acute phase trial. Biol Psychiatry. 2005;58:347–354. doi: 10.1016/j.biopsych.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 36.Rush AJ, Sackeim HA, Marangell LB, George MS, Brannan SK, Davis SM, et al. Effects of 12 months of vagus nerve stimulation in treatment-resistant depression: a naturalistic study. Biol Psychiatry. 2005;58:355–363. doi: 10.1016/j.biopsych.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 37.George MS, Rush AJ, Marangell LB, Sackeim HA, Brannan SK, Davis SM, et al. A one-year comparison of vagus nerve stimulation with treatment as usual for treatment-resistant depression. Biol Psychiatry. 2005;58:364–373. doi: 10.1016/j.biopsych.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 38.Schlaepfer TE, Frick C, Zobel A, Maier W, Heuser I, Bajbouj M, et al. Vagus nerve stimulation for depression: efficacy and safety in a European study. Psychol Med. 2008;38:651–661. doi: 10.1017/S0033291707001924. [DOI] [PubMed] [Google Scholar]
- 39.Christmas D, Steele JD, Tolomeo S, Eljamel MS, Matthews K. Vagus nerve stimulation for chronic major depressive disorder: 12-month outcomes in highly treatment-refractory patients. J Affect Disord. 2013;150:1221–1225. doi: 10.1016/j.jad.2013.05.080. [DOI] [PubMed] [Google Scholar]
- 40.Bajbouj M, Merkl A, Schlaepfer TE, Frick C, Zobel A, Maier W, et al. Two-year outcome of vagus nerve stimulation in treatment-resistant depression. J Clin Psychopharmacol. 2010;30:273–281. doi: 10.1097/JCP.0b013e3181db8831. [DOI] [PubMed] [Google Scholar]
- 41.Nahas Z, Marangell LB, Husain MM, Rush AJ, Sackeim HA, Lisanby SH, et al. Two-year outcome of vagus nerve stimulation (VNS) for treatment of major depressive episodes. J Clin Psychiatry. 2005;66:1097–1104. doi: 10.4088/jcp.v66n0902. [DOI] [PubMed] [Google Scholar]
- 42.Daban C, Martinez-Aran A, Cruz N, Vieta E. Safety and efficacy of Vagus Nerve Stimulation in treatment-resistant depression. A systematic review. J Affect Disord. 2008;110:1–15. doi: 10.1016/j.jad.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 43.Sackeim HA, Brannan SK, Rush AJ, George MS, Marangell LB, Allen J. Durability of antidepressant response to vagus nerve stimulation (VNS) Int J Neuropsychopharmacol. 2007;10:817–826. doi: 10.1017/S1461145706007425. [DOI] [PubMed] [Google Scholar]
- 44.Olin B, Jayewardene AK, Bunker M, Moreno F. Mortality and suicide risk in treatment-resistant depression: an observational study of the long-term impact of intervention. PLoS One. 2012;7:e48002. doi: 10.1371/journal.pone.0048002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aaronson ST, Carpenter LL, Conway CR, Reimherr FW, Lisanby SH, Schwartz TL, et al. Vagus nerve stimulation therapy randomized to different amounts of electrical charge for treatment-resistant depression: acute and chronic effects. Brain Stimul. 2013;6:631–640. doi: 10.1016/j.brs.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 46.Kalia M, Sullivan JM. Brainstem projections of sensory and motor components of the vagus nerve in the rat. J Comp Neurol. 1982;211:248–265. doi: 10.1002/cne.902110304. [DOI] [PubMed] [Google Scholar]
- 47.Groves DA, Bowman EM, Brown VJ. Recordings from the rat locus coeruleus during acute vagal nerve stimulation in the anaesthetised rat. Neurosci Lett. 2005;379:174–179. doi: 10.1016/j.neulet.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 48.Saper CB. The spinoparabrachial pathway: shedding new light on an old path. J Comp Neurol. 1995;353:477–479. doi: 10.1002/cne.903530402. [DOI] [PubMed] [Google Scholar]
- 49.Naritoku DK, Terry WJ, Helfert RH. Regional induction of fos immunoreactivity in the brain by anticonvulsant stimulation of the vagus nerve. Epilepsy Res. 1995;22:53–62. doi: 10.1016/0920-1211(95)00035-9. [DOI] [PubMed] [Google Scholar]
- 50.Furmaga H, Sadhu M, Frazer A. Comparison of DeltaFosB immunoreactivity induced by vagal nerve stimulation with that caused by pharmacologically diverse antidepressants. J Pharmacol Exp Ther. 2012;341:317–325. doi: 10.1124/jpet.111.188953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chae JH, Nahas Z, Lomarev M, Denslow S, Lorberbaum JP, Bohning DE, et al. A review of functional neuroimaging studies of vagus nerve stimulation (VNS) J Psychiatr Res. 2003;37:443–455. doi: 10.1016/s0022-3956(03)00074-8. [DOI] [PubMed] [Google Scholar]
- 52.Kosel M, Brockmann H, Frick C, Zobel A, Schlaepfer TE. Chronic vagus nerve stimulation for treatment-resistant depression increases regional cerebral blood flow in the dorsolateral prefrontal cortex. Psychiatry Res. 2011;191:153–159. doi: 10.1016/j.pscychresns.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Nahas Z, Teneback C, Chae JH, Mu Q, Molnar C, Kozel FA, et al. Serial vagus nerve stimulation functional MRI in treatment-resistant depression. Neuropsychopharmacology. 2007;32:1649–1660. doi: 10.1038/sj.npp.1301288. [DOI] [PubMed] [Google Scholar]
- 54.Nemeroff CB, Mayberg HS, Krahl SE, McNamara J, Frazer A, Henry TR, et al. VNS therapy in treatment-resistant depression: clinical evidence and putative neurobiological mechanisms. Neuropsychopharmacology. 2006;31:1345–1355. doi: 10.1038/sj.npp.1301082. [DOI] [PubMed] [Google Scholar]
- 55.Pardo JV, Sheikh SA, Schwindt GC, Lee JT, Kuskowski MA, Surerus C, et al. Chronic vagus nerve stimulation for treatment-resistant depression decreases resting ventromedial prefrontal glucose metabolism. Neuroimage. 2008;42:879–889. doi: 10.1016/j.neuroimage.2008.04.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zobel A, Joe A, Freymann N, Clusmann H, Schramm J, Reinhardt M, et al. Changes in regional cerebral blood flow by therapeutic vagus nerve stimulation in depression: an exploratory approach. Psychiatry Res. 2005;139:165–179. doi: 10.1016/j.pscychresns.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 57.Gyermek L. The pharmacology of imipramine and related antidepressants. Int Rev Neurobiol. 1966;9:95–143. doi: 10.1016/s0074-7742(08)60137-3. [DOI] [PubMed] [Google Scholar]
- 58.Prange AJ., Jr The pharmacology and biochemistry of depression. Dis Nerv Syst. 1964;25:217–221. [PubMed] [Google Scholar]
- 59.Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122:509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- 60.Dorr AE, Debonnel G. Effect of vagus nerve stimulation on serotonergic and noradrenergic transmission. J Pharmacol Exp Ther. 2006;318:890–898. doi: 10.1124/jpet.106.104166. [DOI] [PubMed] [Google Scholar]
- 61.Manta S, Dong J, Debonnel G, Blier P. Enhancement of the function of rat serotonin and norepinephrine neurons by sustained vagus nerve stimulation. J Psychiatry Neurosci. 2009;34:272–280. [PMC free article] [PubMed] [Google Scholar]
- 62.Follesa P, Biggio F, Gorini G, Caria S, Talani G, Dazzi L, et al. Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res. 2007;1179:28–34. doi: 10.1016/j.brainres.2007.08.045. [DOI] [PubMed] [Google Scholar]
- 63.Hassert DL, Miyashita T, Williams CL. The effects of peripheral vagal nerve stimulation at a memory-modulating intensity on norepinephrine output in the basolateral amygdala. Behav Neurosci. 2004;118:79–88. doi: 10.1037/0735-7044.118.1.79. [DOI] [PubMed] [Google Scholar]
- 64.Roosevelt RW, Smith DC, Clough RW, Jensen RA, Browning RA. Increased extracellular concentrations of norepinephrine in cortex and hippocampus following vagus nerve stimulation in the rat. Brain Res. 2006;1119:124–132. doi: 10.1016/j.brainres.2006.08.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Manta S, El Mansari M, Debonnel G, Blier P. Electrophysiological and neurochemical effects of long-term vagus nerve stimulation on the rat monoaminergic systems. Int J Neuropsychopharmacol. 2013;16:459–470. doi: 10.1017/S1461145712000387. [DOI] [PubMed] [Google Scholar]
- 66.Furmaga H, Shah A, Frazer A. Serotonergic and noradrenergic pathways are required for the anxiolytic-like and antidepressant-like behavioral effects of repeated vagal nerve stimulation in rats. Biol Psychiatry. 2011;70:937–945. doi: 10.1016/j.biopsych.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 67.Bodnoff SR, Suranyi-Cadotte B, Aitken DH, Quirion R, Meaney MJ. The effects of chronic antidepressant treatment in an animal model of anxiety. Psychopharmacology (Berl) 1988;95(3):298–302. doi: 10.1007/BF00181937. [DOI] [PubMed] [Google Scholar]
- 68.Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–336. [PubMed] [Google Scholar]
- 69.Krahl SE, Senanayake SS, Pekary AE, Sattin A. Vagus nerve stimulation (VNS) is effective in a rat model of antidepressant action. J Psychiatr Res. 2004;38:237–240. doi: 10.1016/j.jpsychires.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 70.Jacobs BL, van Praag H, Gage FH. Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol Psychiatry. 2000;5:262–269. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- 71.Biggio F, Gorini G, Utzeri C, Olla P, Marrosu F, Mocchetti I, et al. Chronic vagus nerve stimulation induces neuronal plasticity in the rat hippocampus. Int J Neuropsychopharmacol. 2009;12:1209–1221. doi: 10.1017/S1461145709000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Revesz D, Tjernstrom M, Ben-Menachem E, Thorlin T. Effects of vagus nerve stimulation on rat hippocampal progenitor proliferation. Exp Neurol. 2008;214:259–265. doi: 10.1016/j.expneurol.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 73.Gebhardt N, Bär KJ, Boettger MK, Grecksch G, Keilhoff G, Reichart R, et al. Vagus nerve stimulation ameliorated deficits in one-way active avoidance learning and stimulated hippocampal neurogenesis in bulbectomized rats. Brain Stimul. 2013;6:78–83. doi: 10.1016/j.brs.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 74.Madsen TM, Treschow A, Bengzon J, Bolwig TG, Lindvall O, Tingström A. Increased neurogenesis in a model of electroconvulsive therapy. Biol Psychiatry. 2000;47:1043–1049. doi: 10.1016/s0006-3223(00)00228-6. [DOI] [PubMed] [Google Scholar]
- 75.Hanson ND, Owens MJ, Nemeroff CB. Depression, antidepressants, and neurogenesis: a critical reappraisal. Neuropsychopharmacology. 2011;36:2589–2602. doi: 10.1038/npp.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 77.Wainwright SR, Galea LA. The neural plasticity theory of depression: assessing the roles of adult neurogenesis and PSA-NCAM within the hippocampus. Neural Plast. 2013;2013:805497. doi: 10.1155/2013/805497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Warner-Schmidt JL, Duman RS. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci U S A. 2007;104:4647–4652. doi: 10.1073/pnas.0610282104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, et al. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry. 2009;14:764–773. 739. doi: 10.1038/mp.2008.119. [DOI] [PubMed] [Google Scholar]
- 80.Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, et al. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- 81.Wang JW, David DJ, Monckton JE, Battaglia F, Hen R. Chronic fluoxetine stimulates maturation and synaptic plasticity of adult-born hippocampal granule cells. J Neurosci. 2008;28:1374–1384. doi: 10.1523/JNEUROSCI.3632-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Duman RS, Li N. A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. Philos Trans R Soc Lond B Biol Sci. 2012;367:2475–2484. doi: 10.1098/rstb.2011.0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 84.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16:2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Müller MB, Toschi N, Kresse AE, Post A, Keck ME. Long-term repetitive transcranial magnetic stimulation increases the expression of brain-derived neurotrophic factor and cholecystokinin mRNA, but not neuropeptide tyrosine mRNA in specific areas of rat brain. Neuropsychopharmacology. 2000;23:205–215. doi: 10.1016/S0893-133X(00)00099-3. [DOI] [PubMed] [Google Scholar]
- 87.Furmaga H, Carreno FR, Frazer A. Vagal nerve stimulation rapidly activates brain-derived neurotrophic factor receptor TrkB in rat brain. PLoS One. 2012;7:e34844. doi: 10.1371/journal.pone.0034844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rantamäki T, Hendolin P, Kankaanpää A, Mijatovic J, Piepponen P, Domenici E, et al. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology. 2007;32:2152–2162. doi: 10.1038/sj.npp.1301345. [DOI] [PubMed] [Google Scholar]
- 89.Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci. 2003;23:349–357. doi: 10.1523/JNEUROSCI.23-01-00349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carreno FR, Frazer A. Activation of signaling pathways downstream of the brain-derived neurotrophic factor receptor, TrkB, in the rat brain by vagal nerve stimulation and antidepressant drugs. Int J Neuropsychopharmacol. 2014;17:247–258. doi: 10.1017/S1461145713000977. [DOI] [PubMed] [Google Scholar]
- 91.Sen S, Duman R, Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol Psychiatry. 2008;64:527–532. doi: 10.1016/j.biopsych.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology. 1998;37:1553–1561. doi: 10.1016/s0028-3908(98)00141-5. [DOI] [PubMed] [Google Scholar]
- 93.Cattaneo A, Bocchio-Chiavetto L, Zanardini R, Milanesi E, Placentino A, Gennarelli M. Reduced peripheral brain-derived neurotrophic factor mRNA levels are normalized by antidepressant treatment. Int J Neuropsychopharmacol. 2010;13:103–108. doi: 10.1017/S1461145709990812. [DOI] [PubMed] [Google Scholar]
- 94.Gielen A, Khademi M, Muhallab S, Olsson T, Piehl F. Increased brain-derived neurotrophic factor expression in white blood cells of relapsing-remitting multiple sclerosis patients. Scand J Immunol. 2003;57:493–497. doi: 10.1046/j.1365-3083.2003.01260.x. [DOI] [PubMed] [Google Scholar]
- 95.Nakahashi T, Fujimura H, Altar CA, Li J, Kambayashi J, Tandon NN, et al. Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett. 2000;470:113–117. doi: 10.1016/s0014-5793(00)01302-8. [DOI] [PubMed] [Google Scholar]
- 96.Lang UE, Bajbouj M, Gallinat J, Hellweg R. Brain-derived neurotrophic factor serum concentrations in depressive patients during vagus nerve stimulation and repetitive transcranial magnetic stimulation. Psychopharmacology (Berl) 2006;187:56–59. doi: 10.1007/s00213-006-0399-y. [DOI] [PubMed] [Google Scholar]
- 97.Pilc A, Wierońska JM, Skolnick P. Glutamate-based antidepressants: preclinical psychopharmacology. Biol Psychiatry. 2013;73:1125–1132. doi: 10.1016/j.biopsych.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 98.Willner P. The mesolimbic dopamine system as a target for rapid antidepressant action. Int Clin Psychopharmacol. 1997;12(Suppl 3):S7–S14. doi: 10.1097/00004850-199707003-00002. [DOI] [PubMed] [Google Scholar]
- 99.Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol. 1990;185:1–10. doi: 10.1016/0014-2999(90)90204-j. [DOI] [PubMed] [Google Scholar]
- 100.Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 1985;85:367–370. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- 101.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 102.Zarate C, Jr, Machado-Vieira R, Henter I, Ibrahim L, Diazgranados N, Salvadore G. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18:293–303. doi: 10.3109/10673229.2010.511059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ibrahim L, Diazgranados N, Luckenbaugh DA, Machado-Vieira R, Baumann J, Mallinger AG, et al. Rapid decrease in depressive symptoms with an N-methyl-d-aspartate antagonist in ECT-resistant major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1155–1159. doi: 10.1016/j.pnpbp.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67:793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 106.Zarate CA, Jr, Brutsche N, Laje G, Luckenbaugh DA, Venkata SL, Ramamoorthy A, et al. Relationship of ketamine's plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry. 2012;72:331–338. doi: 10.1016/j.biopsych.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Price RB, Nock MK, Charney DS, Mathew SJ. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry. 2009;66:522–526. doi: 10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, aan het Rot M, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry. 2013;74:250–256. doi: 10.1016/j.biopsych.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Murrough JW. Ketamine as a novel antidepressant: from synapse to behavior. Clin Pharmacol Ther. 2012;91:303–309. doi: 10.1038/clpt.2011.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Larkin GL, Beautrais AL. A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int J Neuropsychopharmacol. 2011;14:1127–1131. doi: 10.1017/S1461145711000629. [DOI] [PubMed] [Google Scholar]
- 111.Aan Het Rot M, Zarate CA, Jr, Charney DS, Mathew SJ. Ketamine for depression: where do we go from here? Biol Psychiatry. 2012;72:537–547. doi: 10.1016/j.biopsych.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.DiazGranados N, Ibrahim LA, Brutsche NE, Ameli R, Henter ID, Luckenbaugh DA, et al. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. J Clin Psychiatry. 2010;71:1605–1611. doi: 10.4088/JCP.09m05327blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bowdle TA, Radant AD, Cowley DS, Kharasch ED, Strassman RJ, Roy-Byrne PP. Psychedelic effects of ketamine in healthy volunteers: relationship to steady-state plasma concentrations. Anesthesiology. 1998;88:82–88. doi: 10.1097/00000542-199801000-00015. [DOI] [PubMed] [Google Scholar]
- 114.Ghoneim MM, Hinrichs JV, Mewaldt SP, Petersen RC. Ketamine: behavioral effects of subanesthetic doses. J Clin Psychopharmacol. 1985;5:70–77. [PubMed] [Google Scholar]
- 115.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 116.Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 118.Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jourdi H, Hsu YT, Zhou M, Qin Q, Bi X, Baudry M. Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci. 2009;29:8688–8697. doi: 10.1523/JNEUROSCI.6078-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Duman RS, Li N, Liu RJ, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62:35–41. doi: 10.1016/j.neuropharm.2011.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Haile CN, Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Foulkes A, et al. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int J Neuropsychopharmacol. 2014;17:331–336. doi: 10.1017/S1461145713001119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dwyer JM, Duman RS. Activation of mammalian target of rapamycin and synaptogenesis: role in the actions of rapid-acting antidepressants. Biol Psychiatry. 2013;73:1189–1198. doi: 10.1016/j.biopsych.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Denk MC, Rewerts C, Holsboer F, Erhardt-Lehmann A, Turck CW. Monitoring ketamine treatment response in a depressed patient via peripheral mammalian target of rapamycin activation. Am J Psychiatry. 2011;168:751–752. doi: 10.1176/appi.ajp.2011.11010128. [DOI] [PubMed] [Google Scholar]
- 125.Yang C, Zhou ZQ, Gao ZQ, Shi JY, Yang JJ. Acute increases in plasma mammalian target of rapamycin, glycogen synthase kinase-3β, and eukaryotic elongation factor 2 phosphorylation after ketamine treatment in three depressed patients. Biol Psychiatry. 2013;73:e35–e36. doi: 10.1016/j.biopsych.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 126.Laje G, Lally N, Mathews D, Brutsche N, Chemerinski A, Akula N, et al. Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol Psychiatry. 2012;72:e27–e28. doi: 10.1016/j.biopsych.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]