

Abstract
Psychiatric disorders are among the most debilitating of all medical illnesses. Whilst there are drugs that can be used to treat these disorders, they give sub-optimal recovery in many people and a significant number of individuals do not respond to any treatments and remain treatment resistant. Surprisingly, the mechanism by which psychotropic drugs cause their therapeutic benefits remain unknown but likely involves the underlying molecular pathways affected by the drugs. Hence, in this review, we have focused on recent findings on the molecular mechanism affected by antipsychotic, mood stabilizing and antidepressant drugs at the levels of epigenetics, intracellular signalling cascades and microRNAs. We posit that understanding these important interactions will result in a better understanding of how these drugs act which in turn may aid in considering how to develop drugs with better efficacy or increased therapeutic reach.
Keywords: Antipsychotics, Mood stabilizers, Antidepressants, Epigenetics, Intracellular signalling, MicroRNA
INTRODUCTION
Schizophrenia and mood disorders are chronic and severe psychiatric disorders that affect the lives and functioning of many people worldwide.1,2) There have been some successes in developing pharmacological treatments to treat such disorders that have been generally divided into categories based on their primary clinical response and are thus termed antipsychotics, mood stabilizers and antidepressant drugs. However, these generalised divisions are being challenged by, for example, the use of second generation antipsychotic as effective treatments for bipolar disorder.3) The clinical use of psychotropic drugs in the treatment of psychiatric disease has been occurring for decades, mechanisms of action by which the drugs induce their therapeutic benefits are far from being fully defined. This lack of understanding is inhibiting efforts to identify why current drugs are only partially effective, having high rates of relapse (which may be due to non-compliance due to side-effects), poor impact on cognitive and functional impairment all of which result in ongoing psychosocial disability and diminished well-being.4,5,6) Important components of the mechanism of action of available psychotropic drugs are their ability to modulate a number of intra-cellular pathways and in this regard, the understanding of the molecular and cellular pathways that are modulated by current drug is necessary to aid in consideration as to how to develop new drugs with few side-effects, improved efficacy and broader therapeutic reach. Thus, in this review, we discuss the direct and indirect molecular mechanisms of antipsychotics, mood stabilizers and antidepressant drugs on the critical markers of gene expression. These markers include DNA methylation, histone modification (epigenetics), intracellular signalling pathways and post-transcription (microRNA) processes.
MAIN SUBJECTS
Epigenetics
Epidemiological research suggests that the aetiology of psychiatry disorders is underpinned by interaction between genes and the environment.7,8) Epigenetics is the study of potentially heritable changes in levels of gene expression that occur without a change in the DNA sequence and involve mitotically-heritable factors, such as DNA methylation and histone modification.9) DNA methylation changes the appearance and structure of DNA and interferes with the transcriptional machinery by cytosine methylation, specifically on cytosine-phosphate-guanine (CpG) island.10,11) This is because CpG methylation inhibits transcription factors requiring cytosine/guanine (CG)-rich sites to bind to the DNA or facilitates the assembly of transcription repressor complexes that contain histone deacetylases and methylases that mediate chromatin remodelling-activates.12) At the most basic level, a cytosine that is followed by guanine can be methylated on the CpG dinucleotide sequences.10) The mechanism involves the methylation of cytosines which in turn recruit methyl-DNA binding proteins such as methyl CpG binding protein 2 (MeCP2). Methyl-DNA binding proteins then recruit macromolecular complexes of chromatin proteins which compact chromatin structure, limiting accessibility of transcriptional machinery, and thus suppress gene transcription (Fig. 1).10)
Fig. 1.

Schematic of transcriptional suppression by DNA methylation. HADCs, histone deacetylase; MeCP2, methyl CpG binding protein 2; CpG, cytosine-phosphate-guanine; CH3, a methyl group.
Histones are the primary protein components of eukaryotic chromatin and play a role in gene regulation. There are 5 histone proteins: histone 1 (H1), histone 2A (H2A), histone 2B (H2B), histone 3 (H3), and histone 4 (H4). The histone tail amino acid residues are subject to covalent modifications, such as acetylation, phosphorylation and methylation.13) In the biochemical mechanism, the acetylation is catalyzed by histone acetyl transferase (HAT) and deacetylated by histone deacetylase (HDAC). Phosphorylation is catalyzed by protein kinases (PK) and reversed by protein phosphatase (PP). Methylation is catalyzed by histone methyltrasferase (HMT) and the reverse reaction is catalyzed by histone demethylases (HDM). Depending on the amino acid residue involved, there can be different effects on gene transcription. Acetylation or phosphorylation are linked to gene activation14) while methylation is more complicated; having various roles in the regulation of gene expression,15) most likely because effector proteins, such as transcriptional co-activators, recognize mono-, di-, and tri-methylated epitopes on the chromatin structure with different affinities.16) Therefore, such particular changes within histone methylation are crucial for determining its effect on gene transcription.
Epigenetic characteristics of antipsychotics
Epigenetics has been shown to be involved in the pharmacological effects of antipsychotics as well as in the pathophysiology of psychiatric illness. The evidence pertaining to the specific effects of antipsychotics on epigenetic mechanism come from both DNA methylation and histone modification studies. At the level of DNA methylation, it has been shown that haloperidol has different effects on DNA methylation in various rat tissues. A study examining the methyl cytosine content of DNA in global methylation from the leukocytes, liver and brain of rats has demonstrated haloperidol treatment induced a decrease in the methyl cytosine content of leukocytes in male rats, but unexpectedly, an increase in females compared to vehicle-treated animals.17) In addition to leukocytes of rats, haloperidol increased global DNA methylation in the liver in males, but resulted in a significant decrease in the brain in females.17) These data suggest drugs with a pharmacological profile similar to haloperidol will differentially affect DNA methylation in various tissues and could indicate that sex-specific hormones may play a role in modifying the DNA methylation state by haloperidol. Antipsychotic drugs have also been shown to have an effect on specific epigenetic mechanisms involving the regulation of specific gene regulations. This notion initially came from findings that there are low levels of reelin and glutamic acid decarboxylase 67 (GAD67) are in the brains of people with schizophrenia and bipolar disorder.18,19,20) These data suggested that the down regulation of these transcripts were due to hyper-methylation of their gene promoters.21,22,23) Subsequently, it has been shown that clozapine and sulpiride, but not haloperidol or olanzapine, induced de-methylation of the hyper-methylated of the reelin and GAD67 promoters in mice pre-treated with L-methionine to hyper-methylate the promoters of these genes in the cortex and striatum of mice brain.24) Together these data would support the argument that one mechanism by which antipsychotic drugs could facilitate improvements in symptomatology is by chromatin remodelling, which at least in the GABAergic system, appears to normalize changes that have occurred in the people with schizophrenia and bipolar disorder. The hypothesis that chromatin remodelling may be a widespread effect of antipsychotic drugs comes from recent data that haloperidol can induced demethylation and expression of MKP3 gene which in turn inhibited cell proliferation of MIA PaCa-2, human pancreatic cancer cells.25) In addition, a clinical study has revealed that haloperidol treatment is associated with higher levels of global DNA methylation in leukocytes of patients with schizophrenia.26) Therefore, existing data suggests further studies examining the effects of antipsychotic drugs in patterns or levels of DNA methylation may lead to a better understanding of their effects on gene expression and subsequent efficaciousness.
Antipsychotic drugs have also shown to regulate histone modification by modifying histone structure which is achieved by changing protein methylation, phosphorylation and acetylation.13) For example, it has been reported that haloperidol impact on phosphorylation and acetylation of histone H3. A study demonstrated that haloperidol induces both phosphorylation and acetylation of histone H3, defined phospho-serine 10 in conjunction with acetyl-lysine 14 (H3pS10-acK14), genome wide, in the mouse striatum by blockading dopamine D2-like receptor signalling.27) By contrast, another study has suggested haloperidol increases levels of Ser10 phosphorylated histone H3 in the striato-pallidal medium spiny neurons (MSNs) of the dorsal striatum without causing a change of acetylation at lysine14 histone H3.28) More recently, it has been reported that chronic clozapine and risperidone, but not haloperidol, treatment strongly decreased histone H3 acetylation at the metabotropic glutamate receptor 2 (mGlu2) promoter in mouse frontal cortex.29) In this study, HDAC2 mRNA levels were reported to significantly increase by clozapine treatment but there was no change in the methylation pattern of CpG sites at the mGlu2 promoter; data suggest DNA methylation does not seem to contribute to the regulation of the mGlu2 gene. Potentially consistent with these finding in mice, are the findings of decreased histone H3 acetylation at the mGlu2 promoter and increased expression of HDAC2 in the frontal cortex of people with schizophrenia who had been treated with atypical antipsychotics. Together, these findings suggest that atypical antipsychotic drugs can down-regulate mGlu2 expression through an epigenetic mechanism that involves decreased histone acetylation at its promoter and interaction of HDAC2 to the mGlu2 promoter.29)
In conclusion, available data suggest antipsychotic drug treatment appears to have complex and region-dependent effects on epigenetic mechanisms. This is supported by the finding that olanzapine, clozapine, clomipramine resulted in significant increases in acetylation of histone H3 in accumbens, in levels of HDAC2 in striatum and in HDAC3 in both striatum and cingulate cortex from mice brain whereas olanzapine alone induces a significant increase in HDAC5 in amygdala.30) Understanding these complex mechanisms will be necessary to fully comprehend the effects of antipsychotic drug treatment on DNA methylation and histone modification.
Epigenetic characteristics of mood stabilizers
Valproic acid (VPA) is a drug that can act as a mood stabiliser and has one mechanism of action by which it acts as a HDAC inhibitor to cause hyper-acetylation of histones and enhanced gene expression.31) As an example, VPA can inhibit methionine-induced reelin promoter hyper-methylation leading to a down-regulation of reelin expression as well as inducing an increase in GAD67 and enhanced total acetylated histone H3 levels.32) VPA also causes hyper-acetylation of histone H3 in the α-synuclein promoter region in neuroblastoma SH-SY5Y cells.33) Furthermore, VPA was shown to induce hyper-acetylation of both histones H3 and H4 in genome-wide DNA methylation in primary rat astrocytes, but did not affect the levels of DNA methyltransferases1 (DNMT1), which catalyses the methylation of cytosines in CpG dinucleotides.10) It showed no interaction of the histone modification and DNA methylation.34) Importantly, VPA and lithium are both widely used mood stabilizers and therefore it is significant that both drugs have been shown to reduced DNA methylation of brain derived neurotrophic factor (BDNF) promoter in peripheral blood mononuclear cells (PBMCs) of patients with bipolar disorder.35) This finding is balance by another study that found that the levels of DNA methylation at BDNF promoter were not significantly affected by mood stabilizers, including VPA, lithium and carbamazepine,36) whereas these mood stabilizers were associated with hypo-methylation in SLC6A4 CpG3 and 4 sites. Significantly these sites have been reported to be hyper-methylated in patients with bipolar disorder.37) Another study to examine regional specific changes in mice demonstrated VPA, lamotrigine and lithium increased levels of acetylated histone H3 in nucleus accumbens while carbamazepine, lamotrigine and levetiracetam induced a significant increase in HDAC3.30) In addition, the treatment with VPA, lamotrigine and levetiracetam results in significant increase in HDAC3 in cingulate cortex and HDAC5 in amygdala in mice brain,30) providing the information regarding differences in gene expression profiles in particular regions of brain. Finally, a clinical study has revealed that lithium treatment showed a decrease in the levels of global DNA methylation in lymphoblast of patient with bipolar disorder.38) Resolving the clinically significant effects of mood stabilisers on epigenetic mechanisms may be worthwhile as more common effects of these drugs are separated from effect which are limited to specific drugs and are therefore less likely to associate with their common effects on mood stability.
Epigenetic characteristics of antidepressant drugs
Antidepressant drugs have also been shown to affect epigenetic mechanisms, for example, chronic treatment of imipramine produces a selective hyper-acetylation of histone H3 at the BDNF III and BDFN IV promoters as well as de-methylation in histone H3 tri-methylation at lysine 4 (H3K4) at the BDNF III promoter in mice to chronic social defeat stress.39) These changes were associated with an enhancement of BDNF transcription in these mice, and this hyper-acetylation by chronic imipramine was associated with a selective down-regulation of HDAC5 whilst overexpression of HDAC5 inhibited the effect of imipramine in the social defeat paradigm. Thus, the observed histone and HDAC modifications with imipramine treatment support the therapeutic potential for histone methylation and deacetylation inhibitors in depression.39) Another study has reported that fluoxetine increased levels of MeCP2, which is thought to silence transcription of downstream gene by recruiting a HDAC, enhanced HDAC2 and reduced the acetylated forms of histone H3 in rat brain.40) Another potential of antidepressant drugs that included amitriptyline, venlafaxine and citalopram to affect epigenetic parameters in astrocytes showed amitriptyline reduced levels of genome-wide DNA methylation at CCpGG sites and inhibited enzymatic DNMT activity in primary astrocytes.34) Furthermore, the further study demonstrated this reduction of DNMT activity was neither due to reduced DNMT1 expression nor direct drug interference but decrease in levels of histone methyltransferase G9a, known modulator of DNMT1, indicating a functional impact of G9a on DNMT1.41) However, the fact that this effect was limited to one of a number of antidepressant drugs suggests it is not a "class" effect and therefore may not be linked to the therapeutic efficacy of the amitriptyline. It has also been reported that treatment with escitalopram reduced the levels of methylation in P11 gene and levels of DNMT1 and DNMT3a mRNA in the prefrontal cortex of the flinders sensitive line (FSL) rat; a genetic rodent model of depression.42) The P11 gene is associated with functional expression of 5-HT (1B) in both the human and rodent43,44) and that receptor has been linked to the symptom of depression.42) Therefore, these findings suggest P11 could be one gene that could be affected by changes in epigenetic mechanisms during antidepressant treatment.
Epigenetic effects of poly-pharmacological treatments
Over the last several years, a variety of adjunctive treatments have been used to enhance the response to antipsychotics in preclinical and clinical studies45) that includes drugs such as VPA. As previously stated, VPA is a nonspecific HDCA inhibitor. The observation that VPA is efficacious when given in combination with antipsychotic drugs may suggest that the combined impact of both drug types on epigenetic mechanisms may give improved therapeutic benefit. Mechanistically, it has been shown that combined treatment with clozapine or sulpiride and VPA results in dose-related increases in de-methylation of hyper-methylated of reelin and GAD67 promoters in cortical and striatal mice pre-treated with L-methionine compared to normal brain.24) Another study also revealed, when co-administered with VPA, clozapine and sulpiride reverse the hyper-methylation of the reelin or GAD67 DNA that can be induced in mouse frontal cortex.46) In addition, the treatment with clozapine and VPA has been shown to increase growth arrest and DNA damage 45-β (GADD45-β) mRNA and protein expression in the frontal cortex of mice.47) It is therefore possible that GADD45-β is involved in the regulation of DNA-methylation after treatment with a combination of psychotropic drugs.
Intracellular Signalling Pathways
The regulation of gene expression involves intracellular signalling pathways and they are fundamental for the regulation of transcription of gene. A comprehensive understanding of neuronal functioning and the various mechanisms of drug action is a key factor for developing the future psychiatric pharmacotherapy. This is especially the case as psychotropic drugs target multiple sites that include neurotransmitter receptors, neurotransmitter transporters and specific molecules in signalling pathways. In this review, we specifically focused on intracellular signalling cascades, cyclic adenosine monophosphate (cAMP), glycogen synthase kinase-3 (GSK-3) and mitogen-activated protein kinase (MAPK) pathways which have been explored as common important mediators that may critically involve in the actions of psychiatry drugs in the central nervous system (CNS).
Antipsychotics
The common pharmacological characteristic of all antipsychotic drugs, including typical and atypical is to antagonise dopamine D2-like receptors.48) In addition, whilst first-generation antipsychotic drugs are antagonist of dopamine D2 receptors the second generation drugs target a number of other receptors with high affinity.49,50) Finally, clozapine is considered a unique antipsychotics drug possessing superior therapeutic efficacy as demonstrated in particular with respect to patients not responding to first-generation typical antipsychotics such as haloperidol.51,52) A unique action of clozapine seems to be that it has some receptor agonist activity and has a high affinity for four (M1-M4) of the five muscarinic receptors.53,54) However, the particular mechanism underpinning the superior efficacy action of clozapine and its adverse side effects are poorly understood.52,55) Accordingly, it may allow a more precise characterization of available drugs to establish differences not only between typical and atypical antipsychotics, but also within the groups of atypical antipsychotics.
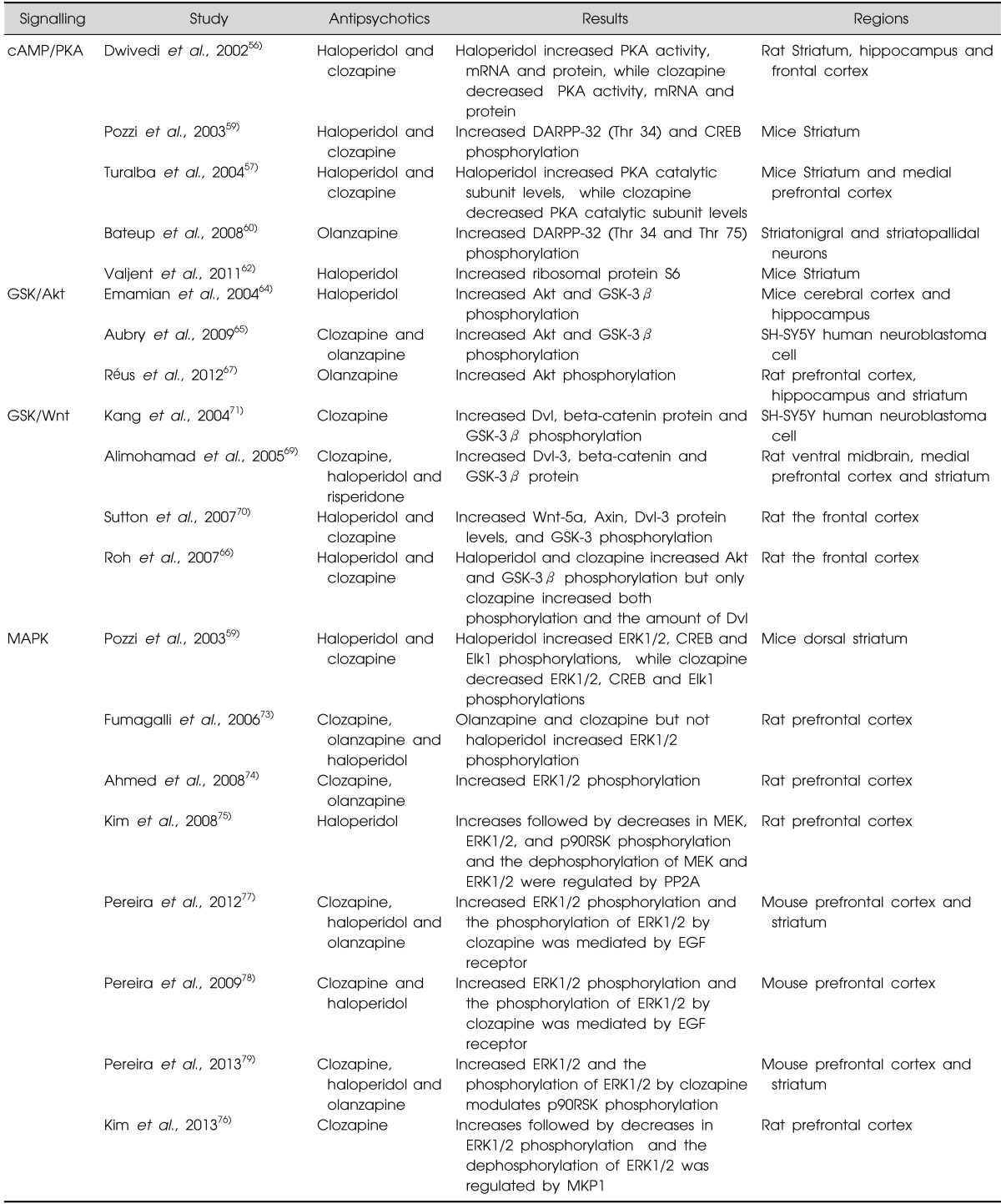
In regard with D2, 5-HT2A and muscarinic receptors, these receptors are 7-transmembrane domain protein coupled to G proteins. The various functions of G protein coupled receptors have been associated with the regulation of cAMP and protein kinase A (PKA) via G protein dependent signalling. There is growing evidence that different way antipsychotics modulate the cAMP and PKA pathway may contribute to their differing clinical and/or side-effect profiles. For example, treatment with haloperidol has been shown to increase levels of PKA activity, mRNA and protein in the rat striatum whereas clozapine reduces these parameters in the striatum, hippocampus and frontal cortex of rats.56) In another study haloperidol was shown to increase levels of the PKA catalytic subunit in the mouse dorsal striatum whereas clozapine had the opposite effect in the medial prefrontal cortex.57) Thus, current data also suggests the different effects of typical and atypical antipsychotics on cAMP and PKA appeared to be complex and region-dependent, possibly due to their affinities for different G-protein coupled receptors that stimulate adenylyl cyclase signalling.56,57) Dopamine- and cAMP-regulated phosphoprotein with molecular weight 32 (DARPP-32), a downstream to PKA, is another important mediator of PKA pathway, particularly in dopaminergic transmission.58) DARPP-32 phosphorylation catalysed by PKA at Thr 34 converts it into an inhibitor of proteinphosphates 1 (PP1), while phosphorylation at Thr 75 by cyclin-dependent kinase 5 (CDK5) converted into an inhibitor of PKA (Fig. 2). Importantly, DARPP-32 phosphorylation at Thr 34 has been shown to be increased in the mouse dorsal striatum after acute administration of haloperidol and clozapine,59) suggesting that both typical and atypical antipsychotics may modulate PKA downstream mechanisms. Another study has shown that both haloperidol and clozapine increase PKA dependent phosphorylation at Thr 32.60) However, another atypical antipsychotic drug, olanzapine, up-regulated the phosphorylation at Thr 75, which represented the specific site for the CDK5.61) In addition, haloperidol has been shown to induce activation of PKA, and DARPP-32, which has been proposed to affect gene transcription by acting on nuclear targets, including the cAMP response element-binding (CREB) protein in mouse dorsal striatum.59) This has implications for the finding that haloperidol increases the phosphorylation of the ribosomal protein S6 (rpS6), a component of the small 40S ribosomal subunit, an effect exerted by promoting the c-AMP-DARPP-32 signalling.62) These findings suggest that regulation of protein synthesis through rpS6 may be a potential target of antipsychotic drugs (Table 1).
Fig. 2.

Schematic of mechanism of DARPP32. GPCR, G protein coupled receptors; AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; DARPP-32, dopamineand cAMP-regulated phosphoprotein with molecular weight 32; CDK5, cyclin-dependent kinase 5.
Table 1.
Summary of studies of intracellular signalling in antipsychotics
PKA, protein kinase A; DARPP-32, dopamine-and cAMP-regulated phosphoprotein with molecular weight 32; CREB, cAMP response element-binding; GSK-3, glycogen synthase kinase-3; Dvl, dishevelled; ERK1/2, extralcellular signal-regulated kinases 1/2; MEK, mitogen-activated protein kinase kinase; p90RSK, p90 ribosomal S6 kinase; PP2A, protein phosphatase 2A; EGF, epidermal growth factor; MKP1, mitogen-activated protein kinase phosphatases 1.
Another intracellular mechanism potentially relevant to antipsychotics is GSK-3 signalling. GSK-3 is a serine-threonine protein kinase that controls several cellular functions, negatively regulated by the phosphorylation of N-terminal serine residues Ser 21 of GSK-3α and Ser 9 of GSK-3β.63) It has been shown that the antipsychotic drugs haloperidol, risperidone, olanzapine, clozapine, quetiapine or ziprasidone act to increase the phosphorylation of GSK-3β in different brain regions regardless of their class.64,65,66) Based on these data, Akt and GSK-3 signalling may represent a common pathway in these actions of antipsychotics. One study has revealed that treatment of mice with haloperidol induced the phosphorylation of Akt and leaded to an increased phosphorylation of GSK-3β64) whilst it has also been shown that clozapine and olanzapine produced phosphorylation of Akt and GSK-3β in SH-SY5Y human neuroblastoma cell and stimulate the expression of Akt-1 mRNA and protein.65) Another study has shown that acute administration of olanzapine increased Akt levels in the prefrontal cortex, hippocampus and striatum of rats.67) Hence, typical and atypical antipsychotics have similar effects regardless their class (Table 1).
GSK-3 is also mediated with the Wnt pathway (Fig. 3), a complex network of proteins with roles in embryogenesis and cancer as well as in adult physiological processes.68) It has been shown that significant increases in the levels of dishevelled 3 (Dvl-3), beta-catenin and GSK-3β protein occur after chronic treatment of clozapine, haloperidol or risperidone in rat ventral midbrain, medial prefrontal cortex and striatum.69) Chronic treatment with clozapine or haloperidol also increased levels of Wnt-5a, Axin, Dvl-3, total and phosphorylation of GSK-3β, and β-catenin in rat the frontal cortex.70) In addition, both haloperidol and clozapine induced phosphorylation of Akt and GSK-3β but, significantly, only clozapine increased both phosphorylation and the amount of Dvl in the rat frontal cortex.66) Furthermore, clozapine has been shown to enhance GSK-3β signalling via the activation of Wnt pathway but not via the PI3K/Akt pathway in SH-SY5Y cells.71) It is therefore possible that both first and second generation antipsychotics drugs differentially regulate PI3K/Akt and Wnt pathways depending on their relative D2- or 5HT2-receptor antagonism and that clozapine has unique effects that may be related to its increased therapeutic reach or its differential side-effects (Fig. 3, Table 1).
Fig. 3.

Schematic showing the pathways of Wnt and Akt in antipsychotics. Fz, frizzled; GPCR, G protein coupled receptors; Dvl, dishevelled; APC, adenomatous polyposis coli.
One intracellular mechanism by which antipsychotic drugs have been shown to act is the MAPK pathway. This pathway is important in a number of neuronal functions, including the regulation of gene expression, protein synthesis, and receptor modulation, which contribute to synaptic plasticity and adaptive behaviours such as learning and memory.72) Several studies have reported different impacts of typical and atypical antipsychotics on extralcellular signal-regulated kinases 1/2 (ERK1/2) phosphorylation in the brain. In particular, while a single injection of haloperidol increased phosphorylations of ERK1/2 and Elk1 in mice dorsal striatum, these phosphorylations were decreased following clozapine administration.59) Chronic administration of the olanzapine and clozapine enhanced ERK1/2 activation in rat prefrontal cortex,73,74) whereas haloperidol does not produce any significant in ERK1/2 phosphorylation.73) Antipsychotic drugs may modulate the MAPK pathway in different ways. For example, a single injection of haloperidol has been to induce an acute increase followed by a decrease in mitogen-activated protein kinase kinas (MEK), ERK1/2, and p90 ribosomal S6 kinase (p90RSK) phosphorylation in the rat frontal cortex; this occurs without changes in Raf-1 phosphorylation, and these dephosphorylation of MEK and ERK1/2 were regulated by protein phosphatase 2A (PP2A).75) Clozapine also induces an initial increase followed by a decrease ERK1/2 phosphorylation.76) Interestingly, the dephosphorylation of ERK1/2 was shown to be mediated by mitogen-activated protein kinase phosphatases 1 (MKP1), which regulates ERK1/2 through direct binding.76) These results provided critical information on the dephosphorylation mechanism of MAPK in the typical and atypical antipsychotics. In addition, a study examining a novel mechanism of action of clozapine demonstrated clozapine but not olanzapine or haloperidol induced initial inhibition and subsequent activation of cortical and striatal ERK1/277,78) and p90RSK79) phosphorylations in mice, and uniquely, the clozapine-induced ERK1/2 activation was mediated by the epidermal growth factor (EGF) receptor (Table 1).77,78,79) It showed a unique action of clozapine which differently modulates the phosphorylation of ERK1/2 compared to other antipsychotics.
Mood stabilizers
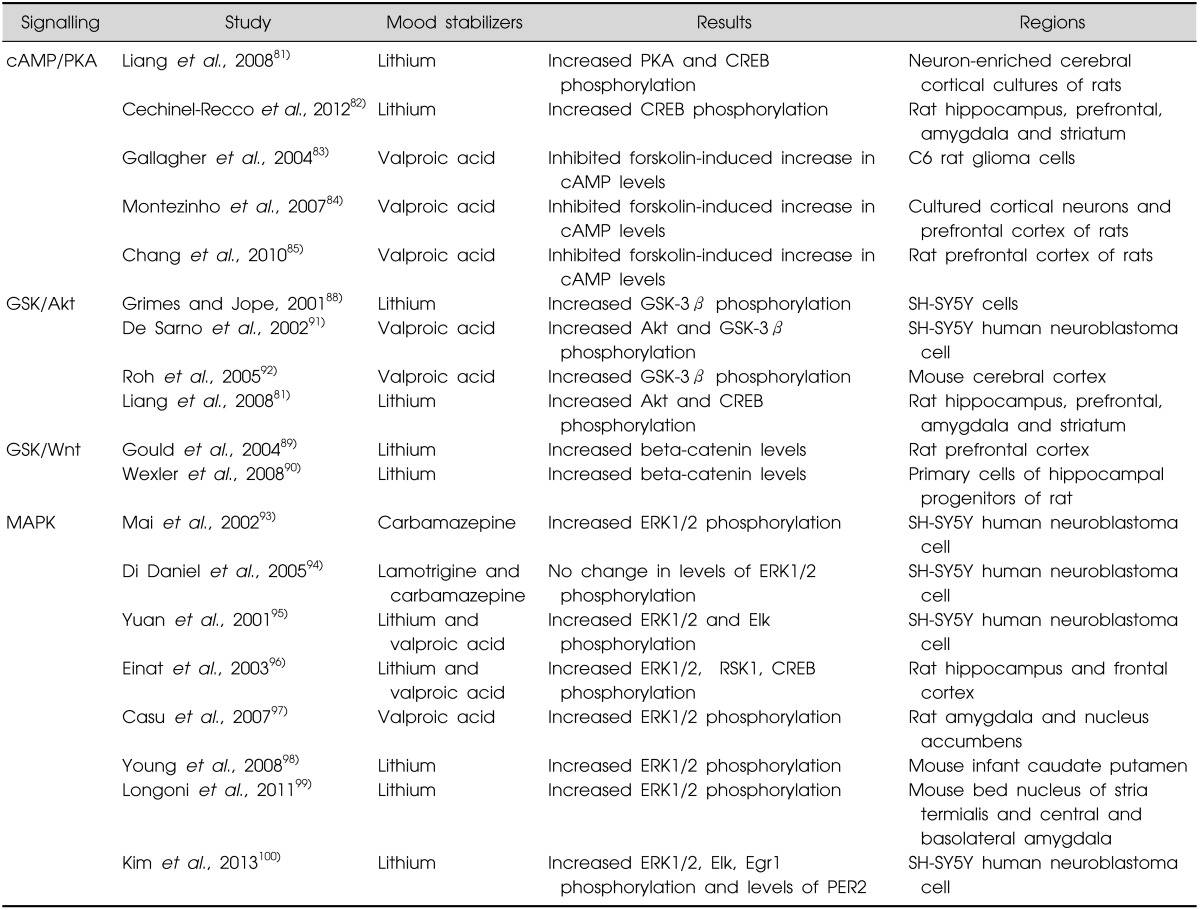
Mood stabilizers also modulate the cAMP-PKA signalling pathway. It has been shown lithium increased activation of PKA leading to enhance the phosphorylation of GSK-3β80) and CREB in neuron-enriched cerebral cortical cultures from rats81) and in the intact rat brain.82) Conversely, VPA has been suggested to reduce forskolin-induced increase in cAMP levels in C6 rat glioma cells83) in the cultured cortical neurons84) and in the prefrontal cortex of rats.85) It remains unclear whether these mood stabilizers affect cAMP directly or indirectly by inhibiting the formation of cAMP. Furthermore, the roles of the mood stabilizers in cAMP and PKA have yet to be explored (Table 2).
Table 2.
Summary of studies of intracellular signalling in mood stabilizers
PKA, protein kinase A; CREB, cAMP response element-binding; cAMP, cycline adenosine monophosphate; GSK-3, glycogen synthase kinase-3; ERK1/2, extralcellular signal-regulated kinases 1/2; RSK1, ribosomal S6 kinase; PER2, clock gene period 2.
The archetypal mood stabilizer, lithium, has been shown to directly inhibit GSK-3β activity, possibly by competing with magnesium which constituently required for GSK-3β to function optimally. Alternatively, it is possible that lithium acts indirectly by increasing Aktmediated inhibition of GSK-3β;86,87) this mechanism is supported by data showing that lithium activates Akt signalling with a subsequent inhibition of GSK-3β.81,88) Lithium acts on multiple signalling pathways, hence it acutely affects the Wnt signalling to increase levels of β-catenin in the rat brain89) and in rat hippocampal progenitor cells.90) By affecting this pathway it can also down-regulate GSK-3β activity, a key translator of the activation of Wnt pathway. Considering the notion that drugs mechanism of therapeutic importance should constitute a drug class effect it is significant that there is some data to suggest that VPA inhibits GSK-3β.91,92) Thus, in vitro studies revealed that VPA elevated levels of phosphorylation in Akt and GSK-3β in SH-SY5Y cells,91) and a single VPA treatment to mice prevented hypoxia-induced reduction in phosphorylation of GSK-3β,92) suggesting VPA has a role to inhibit the activity of GSK-3 (Table 2).
In addition, carbamazepine has been reported to increase the phosphorylation of ERK1/2 in SH-SY5Y cells93); however, this finding was not replicated using lamotrigine or carbamazepine in another study.94) In vivo studies have shown that lithium and VPA increases the phosphorylation of ERK1/2, Elk, and RSK195,96) whilst it has also been shown that these two drugs cause widespread ERK1/2 phosphorylations across rat amygdala, nucleus accumbens,97) and caudate putamen of infant mouse brains,98) bed nucleus of stria termialis and central and basolateral amygdala of mouse.99) One interesting study now links an action of lithium to circadian rhythms by showing the drug enhances the ERK1/2-Elk-Egr1 cascade which in turn increases levels of the clock gene period 2 (PER2) in SH-SY5Y cells and the mouse frontal cortex. Furthermore, lithium-induced PER2 expression was inhibited by depletion of Egr1 by siRNA in SH-SY5Y cells and Egr1 knockout mice, and ERK1/2-Elk pathway also regulated lithium-induced Egr1 and PER2 expression, thus it indicated the PER2 expression was regulated by ERK-Elk-Egr1 pathway (Table 2).100) This mechanism may prove to be significant in understanding how treating with lithium can modulate sleep patterns in people with bipolar disorder.101)
Antidepressant drugs
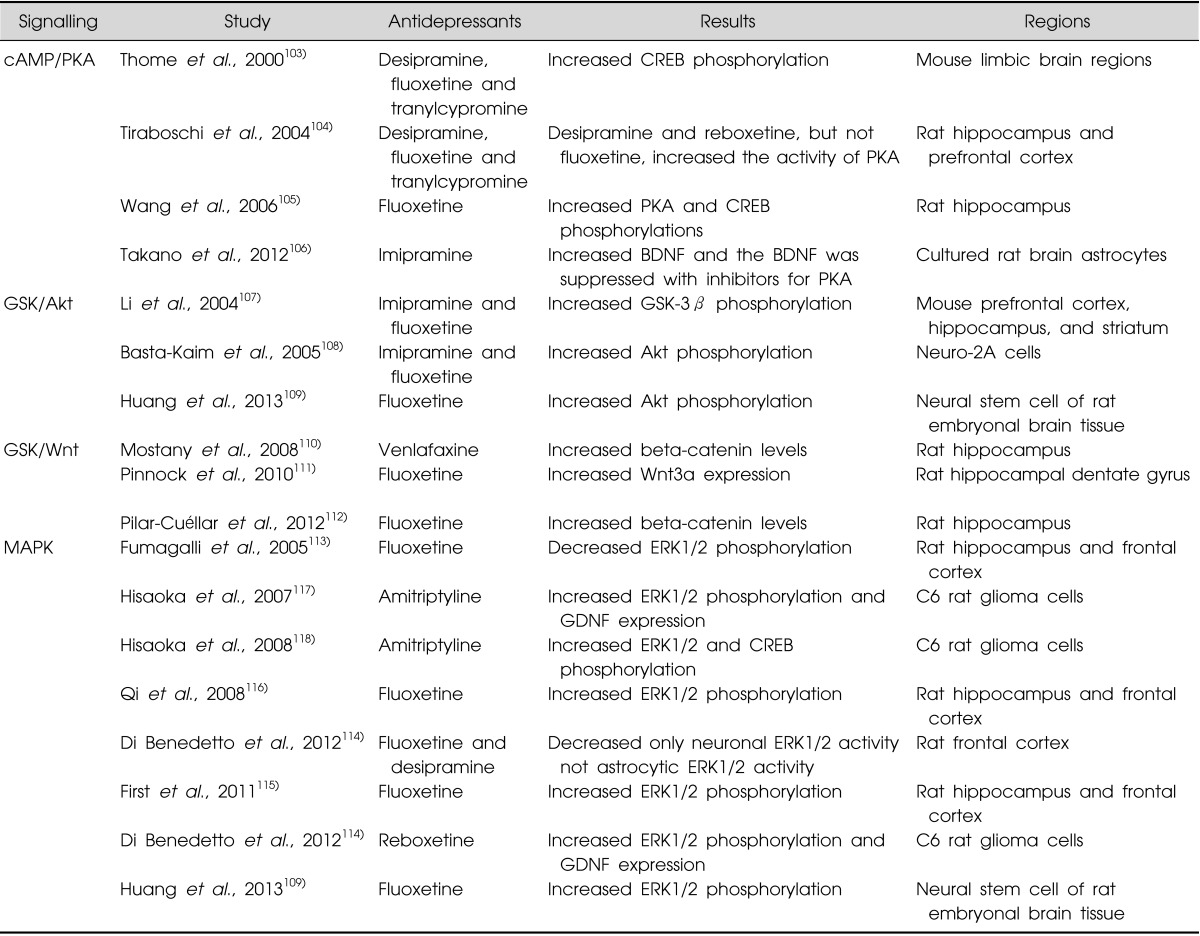
A number of studies reported the cAMP pathway is up-regulated by antidepressant treatment, for example, long-term treatment with citalopram increased the adenylyl cyclase (AC) type 1 mRNA in the hippocampus increasing cAMP signalling.102) Unfortunately, antidepressant drugs do not seem to have consistent effects on the cAMP pathway. Thus, desipramine, fluoxetine and tranylcypromine significantly increase the phosphorylation of CREB in several mouse limbic brain regions, including the cerebral cortex, hippocampus, amygdala, and hypothalamus regardless their class.103) However, desipramine and reboxetine, but not fluoxetine, increase the activity of PKA in rat hippocampus and prefrontal frontal cortex;104) these latter data suggest that PKA does not seem to account for increase of CREB induced by fluoxetine, a selective serotonin reuptake inhibitors (SSRI).104) This conclusion must be tempered by the findings from another study which show that fluoxetine activates both PKA and CREB phosphorylations in the rat hippocampus.105) In addition to cAMP-CREB pathway, a recent study demonstrated that imipramine increased BDNF mRNA expression in cultured rat brain astrocytes and the imipramine-induced BDNF increase was suppressed with inhibitors for PKA (PKI 14-22 amide), suggesting imipramine induced BDNF expression through PKA (Table 3).106)
Table 3.
Summary of studies of intracellular signalling in antidepressants
CREB, cAMP response element-binding; PKA, protein kinase A; BDNF, brain derived neurotrophic factor; GSK-3, glycogen synthase kinase-3; ERK1/2, extralcellular signal-regulated kinases 1/2; GDNF, glial cell-derived neurotrophic factor.
Much like antipsychotics and mood stabilisers, GSK-3 pathway is also affected by antidepressant drugs. A single treatment with imipramine and fluoxetine increases GSK-3β phosphorylation in mouse brain.107) Both imipramine and fluoxetine also enhanced the phosphorylation of Akt but, did not affect total Akt levels in the differentiated neuro-2A cells108) or neural stem cells (NSCs) from rat embryonal brain tissue.109) In addition, on Wnt pathways, it has been reported that venlafaxine elevated nuclear translocation of β-catenin protein in the rat hippocampus110) and fluoxetine induced Wnt3a expression improving neurogenesis in the hippocampal dentate gyrus111) and increased levels of β-catenin protein in the hippocampus of rat (Table 3).112)
Altered MAPK activity has been also observed in the intracellular mechanism of antidepressant drugs. Interestingly, the various intracellular effects of antidepressant drugs on MAPK are reported. Chronic treatment with fluoxetine inhibited ERK1/2 phosphorylation in hippocampus and frontal cortex of rat brain,113) and a recent study also revealed that acute treatments with fluoxetine and desipramine decreased neuronal, but not astrocytic, ERK1/2 activity in the frontal cortex.114) These data highlight a complexity in understanding drug action in the CNS because of the potential for cell-type specific effects. Another study has reported chronic treatment with fluoxetine reversed the reduced ERK1/2 phosphorylation caused by chronic mild stress (CMS) in hippocampus and frontal cortex of rats in an animal model of depression, suggesting ERK1/2 may have a role in mediating the neural stress response and the mode of action of fluoxetine.115),116) In addition, a further study to investigate upstream of ERK1/2 reported upregulated ERK1/2 phosphorylation by fluoxetine in neural stem cells were blocked by both PI3-K inhibitor (LY294002) and MEK inhibitor (PD98059), showing a crosstalk mechanism between Akt and ERK1/2 in the action of antidepressant drugs.109) More interestingly, it has been also demonstrate a relationship between ERK1/2 and glial cell line-derived neurotrophic factor (GDNF) by antidepressants. Treatment with amitriptyline increased both ERK1/2 phosphorylation and levels of GDNF mRNA in rat C6 glioma cells (C6 cells), and this ERK1/2 activation was correlated with antidepressant-induced GDNF production.117) In addition, it has revealed that CREB was also activated by amitriptyline in ERK1/2 dependent manners, and it indicated ERK1/2-CREB activations might contribute to gene expression in GDNF in glial cells.118) In the similar manner, recent data have shown in vitro that reboxetine can activate the ERK1/2 and GDNF expression in C6 cells and pre-treatment with a ERK inhibitor reversed GDNF expression.114) It suggests that ERK1/2 is a critical role in GDNF expression by antidepressant drugs (Table 3).
MicroRNA
MicroRNAs (miRNAs) are a class of small non-coding RNAs with relatively few nucleotides (an average of 22) that recognise binding sites located in the three prime untranslated region (3'UTR) of an mRNA target.119) In miRNA biogenesis, most of the miRNAs have the same transcription machinery as mRNA. The miRNA genes are transcribed as primary miRNAs (pri-miRNAs), with 5'Cap and 3'poly (A) tails, in the nucleus by RNA polymerase II. The pri-miRNAs are further processed into stem loop precursor miRNAs (pre-miRNAs), about 70 nucleotides long, by the microprocessor complex containing RNase III-type enzyme Drosha and its partner protein DiGeorge syndrome critical region gene 8 (Dgcr8) in human. These pre-miRNAs are then exported to the cytoplasm via Ecportin-5. In the cytoplasm, pre-miRNAs are further cleaved into the ~22 nucleotides mature miRNA duplexes by another RNase III-type enzyme, Dicer1. Only one stand of miRNA duplex, referred to as the mature miRNA, assembles into the RNA-induced silencing complex (RISC) while the opposite strand is generally not active and is degraded. The miRNA-associated RISC binds to the target mRNA to inhibit its translation or cause the degradation of the target mRNA (Fig. 4). Nowadays, the sequences of 1872 pre-miRNAs and 2578 mature miRNAs have been identified (miRBase 20, released in June 2013). The particular miRNAs expression and regulation have been found to be important for a wide range of biological processes and have subsequently been reported to be involved in the pathophysiology of psychiatric disorders.120,121)
Fig. 4.

Schematic showing biogenesis of microRNAs (miRNAs). Pri-miRNAs, primary miRNAs; Pre-miRNAs, precursor miRNAs; RISC, RNA-induced silencing complex; mRNA, messenger RNA.
There are still only a few studies investigating the effects of the current psychotropic drugs on miRNA expression and subsequent gene expression.
Antipsychotics and miRNA
Whilst there are still few studies on the ability of antipsychotic drugs to modulate miRNA these affects may be profound as one study has shown that treatment with haloperidol affected levels of 179 miRNA including miR-199a, miR-128a and miR-128b which were expressed at higher levels in the frontal cortex of treated rats.122) It has also been shown that pre-treatment with antipsychotic drugs haloperidol or clozapine can prevented the reduction of miR-219 caused by the phencyclidine-like NMDA-R antagonist (MK-801), dizocilpine, in prefrontal cortex of rat.123) This suggests antipsychotic drugs may interfere with neurotransmitter mediated changes in gene expression. However, a recent study using a miRNA microarray followed with quantitative real-time PCR (qPCR) found that only 3 miRNA were down-regulated following treatment with olanzapine (n=1; mmu-miR-193) or haloperidol (n=2; mmu-miR-434-5p and mmu-miR-22) in whole brain of mice.124) These data would suggest that antipsychotic drug treatment minimally, but differentially, affect levels of miRNAs. Another study has used microarray technology to investigated the effects of chlorpromazine, haloperidol and clozapine on miRNA levels in human T-lymphocyte cells.125) By contrast to earlier studies, this study reported that both haloperidol and clozapine appeared to regulate the expression of a large number of miRNA which are thought to be related to oxidative stress and metabolism.125) Significantly, the data suggested that 73 haloperidol-modulated mRNA and miRNA pairs are involved in a wide variety of metabolic pathways including 'carbohydrate metabolism', 'lipid metabolism' and 'small molecule biochemistry'.125) In addition, it was argued that, overall, the antipsychotics driven changes associated with oxidative stress and metabolic pathways were more relevant to the drug side-effect profiles that include weight gain, metabolic syndrome, dyslipidemia and insulin resistance.125,126,127)
Mood stabilizers and miRNA
A few studies have investigated miRNA expressions in response to mood stabilizers, such as lithium or VPA. Thus, a study in Wister rats showed treatment with lithium changed levels of 37 miRNAs whereas treatment with VPA caused changes in 31 miRNAs.128) Again, looking for drug-class effects lithium and VPA significantly decreased levels of let-7b, let-7c, miR-128a, miR-24a, miR-30c, miR-34a and miR-221 and up-regulated expression of miR-144. These changes will have functional consequences because it has been shown that a VPA associated down-regulated of miR-34a leads to an increase in expression of one of its target gene, metabotropic glutamate receptor 7 (mGlu7).128,129) These findings are the first to demonstrate that miRNAs and their predicted effectors are targets for the action of psychotherapeutic drugs. Expanding this notion, it has been shown that chronic treatment with lithium increased levels of muscarinic M1 receptor expression and this occurs at least in part through by a down-regulation of lithium-sensitive miRNA, let-7b.130)
Returning to more generalised effects of mood stabilizers, studies using a human lymphoblastoid cell lines (LCLs) showed that levels of 7 miRNAs were altered in response to lithium at treatment day 4 with 4 miRNAs being up-regulated (miR-34a, miR-152, miR-155, and miR221) at a longer treatment day 16.131) Using a microarray technique and qPCR confirmation, lithium and VPA treatment was reported to down-regulation of miR-34a, miR-495 and miR-690 and up-regulation of miR-147b, miR-182, and miR-222 after lithium and VPA treatment in neuron cultured from rat cerebellar granule cells.132) It is therefore clear that mood stabilisers have a varied effect on miRNA and further work is required to better understand these effects and their likely consequences.
Antidepressant drugs and miRNA
MiRNA may play a role in modulating depressive neurophysiology via the regulation of serotonergic signalling.133) It is shown that miR-16 is involved in fate determination of serotonergic and noradrenergic cells in 1C11 neuroectodermal cells, which can differentiate into either serotonergic or noradrenergic neuronal cells by the adaptation of serotonin transporter (SERT). In 1C11 cells, the basal levels of miR-16 expressed at higher levels in the noradrenergic locus coeruleus than in serotonergic raphe nuclei that suppresses translation of SERT mRNA within the locus coeruleus. However, after fluoxetine treatment, cells within the locus coeruleus showed decreased miR-16.134) Further, the exposure of raphe to fluoxetine released the neurotropic factor, S100β, which acts on down-regulation of miR-16, and S100β promoted the reduction in miR-16 and turned on the expression of serotonergic functions in locus coeruleus.134) In the similar line, recent data has revealed that treatment with fluoxetine also induced a decrease in the levels of miR-16 in hippocampus.135) This study investigated whether the fluoxetine-induced secretion of S100β protein by the raphe acts on the hippocampus and found that the fluoxetine-induced changes in hippocampal miR-16 and SERT were partly reversed (40-50%) upon si-RNA-mediated knockout of S100β mice in the raphe.135) Given the data that the action of fluoxetine on the locus coeruleus is mediated by the secretion of S100β by the raphe,134) it may suggest the down-regulation of miR-16 in the hippocampus is relayed, in part, by S100β via the noradrenergic neurons of the locus coeruleus.135) Another study indicated treatment with paroxetine increased BDNF expression and these effects were potentially limited by up-regulation of miR-30e-5p in the human glioblastoma-astrocytoma cells (U87).136) Finally, a clinical study investigated the levels of miRNA in the blood of patients with major depression before and after chronic treatment with escitalopram.137) It has been found 28 miRNAs up-regulated and 2 miRNAs (miR-34c-5p and miR-770-5p) down-regulated after escitalopram treatment. Taken together, these studies suggest there are several pathways mediating the differential expression of certain miRNAs in action of psychiatric drugs and it could contribute to the pathophysiology of clinical depression.
CONCLUDING REMARKS
This review clearly highlights the fact that psychotropic drugs affect many pathways that modulate cell signalling and gene expression. The challenge is that these affects are extremely diverse and complex. Based on the hypothesis that "class actions" by different drug types may be an important guide to which changes may result in therapeutic outcomes. It would appear the effects of antipsychotic drugs on schizophrenia, mood stabilisers on bipolar disorder and antidepressant drugs on major depressive disorder may be of particular interest. However, the growing use of drug types outside of their classically defined areas, for example second generation antipsychotic drugs in mood disorders, may suggest that using drug "class actions" as a primary divider does not reflect their usefulness in clinical practice. Therefore there is a clear need to better understand the molecular mechanisms that are target by psychotropic drugs and to try and relate these findings to how the drugs improve symptom severity. Unfortunately, current data suggest psychotropic drugs have both widespread and localised effects on different molecular mechanisms. Hence, it will be necessary to dissect out what are CNS wide, region specific or cell type-specific effects of different drugs to gain a comprehensive understanding of their potential mechanisms of action. This is a challenging goal for the field moving forward but could produce great rewards in underpinning the development of drugs that may have a broader therapeutic reach with lesser unwanted side-effects.
Acknowledgments
Brian Dean is an NHMRC Senior Research Fellow (APP1002240) and Elizabeth Scarr is an ARC Future Fellow (FT100100689). This work was supported in part by NHMRC project grants (APP628699) and (APP1045619), the Victorian Government's Operational Infrastructure Support and the Rebecca Cooper Medical Research Foundation. The authors declare no conflict of interest.
References
- 1.American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-IV-TR. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- 2.Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Surja AA, Tamas RL, El-Mallakh RS. Antipsychotic medications in the treatment of bipolar disorder. Curr Drug Targets. 2006;7:1217–1224. doi: 10.2174/138945006778226598. [DOI] [PubMed] [Google Scholar]
- 4.Belmaker RH, Bersudsky Y. Bipolar disorder: Treatment. Discov Med. 2004;4:415–420. [PubMed] [Google Scholar]
- 5.Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. New York: Oxford University Press; 2007. [Google Scholar]
- 6.Tandon R, Belmaker RH, Gattaz WF, Lopez-Ibor JJ, Jr, Okasha A, Singh B, et al. Section of Pharmacopsychiatry, World Psychiatric Association. World Psychiatric Association Pharmacopsychiatry Section statement on comparative effectiveness of antipsychotics in the treatment of schizophrenia. Schizophr Res. 2008;100:20–38. doi: 10.1016/j.schres.2007.11.033. [DOI] [PubMed] [Google Scholar]
- 7.Roth TL, Lubin FD, Sodhi M, Kleinman JE. Epigenetic mechanisms in schizophrenia. Biochim Biophys Acta. 2009;1790:869–877. doi: 10.1016/j.bbagen.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oh G, Petronis A. Environmental studies of schizophrenia through the prism of epigenetics. Schizophr Bull. 2008;34:1122–1129. doi: 10.1093/schbul/sbn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavin DP, Sharma RP. Histone modifications, DNA methylation, and schizophrenia. Neurosci Biobehav Rev. 2010;34:882–888. doi: 10.1016/j.neubiorev.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 11.Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213:384–390. doi: 10.1002/jcp.21224. [DOI] [PubMed] [Google Scholar]
- 12.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ. Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol. 1997;272:301–311. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- 14.Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, et al. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 15.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 16.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8:307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 17.Shimabukuro M, Jinno Y, Fuke C, Okazaki Y. Haloperidol treatment induces tissue- and sex-specific changes in DNA methylation: a control study using rats. Behav Brain Funct. 2006;2:37. doi: 10.1186/1744-9081-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- 19.Benes FM, Lim B, Matzilevich D, Walsh JP, Subburaju S, Minns M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc Natl Acad Sci U S A. 2007;104:10164–10169. doi: 10.1073/pnas.0703806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fatemi SH, Earle JA, McMenomy T. Reduction in Reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol Psychiatry. 2000;5:654–663. 571. doi: 10.1038/sj.mp.4000783. [DOI] [PubMed] [Google Scholar]
- 21.Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134B:60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- 22.Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Costa E, Chen Y, Dong E, Grayson DR, Kundakovic M, Maloku E, et al. GABAergic promoter hypermethylation as a model to study the neurochemistry of schizophrenia vulnerability. Expert Rev Neurother. 2009;9:87–98. doi: 10.1586/14737175.9.1.87. [DOI] [PubMed] [Google Scholar]
- 24.Dong E, Nelson M, Grayson DR, Costa E, Guidotti A. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci U S A. 2008;105:13614–13619. doi: 10.1073/pnas.0805493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim SH, Lee HY, Yi H, Ahn YM, Kim YS. Haloperidol induces demethylation and expression of the dual specificity phosphatase 6 gene in MIA PaCa-2 human pancreatic cancer cells. Life Sci. 2012;91:1317–1322. doi: 10.1016/j.lfs.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Melas PA, Rogdaki M, Ösby U, Schalling M, Lavebratt C, Ekström TJ. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J. 2012;26:2712–2718. doi: 10.1096/fj.11-202069. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Guo Y, Schroeder FA, Youngs RM, Schmidt TW, Ferris C, et al. Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling. J Neurochem. 2004;90:1117–1131. doi: 10.1111/j.1471-4159.2004.02569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bertran-Gonzalez J, Håkansson K, Borgkvist A, Irinopoulou T, Brami-Cherrier K, Usiello A, et al. Histone H3 phosphorylation is under the opposite tonic control of dopamine D2 and adenosine A2A receptors in striatopallidal neurons. Neuropsychopharmacology. 2009;34:1710–1720. doi: 10.1038/npp.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurita M, Holloway T, García-Bea A, Kozlenkov A, Friedman AK, Moreno JL, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ookubo M, Kanai H, Aoki H, Yamada N. Antidepressants and mood stabilizers effects on histone deacetylase expression in C57BL/6 mice: Brain region specific changes. J Psychiatr Res. 2013;47:1204–1214. doi: 10.1016/j.jpsychires.2013.05.028. [DOI] [PubMed] [Google Scholar]
- 31.Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91:137–168. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- 32.Tremolizzo L, Doueiri MS, Dong E, Grayson DR, Davis J, Pinna G, et al. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57:500–509. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 33.Leng Y, Chuang DM. Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci. 2006;26:7502–7512. doi: 10.1523/JNEUROSCI.0096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perisic T, Zimmermann N, Kirmeier T, Asmus M, Tuorto F, Uhr M, et al. Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology. 2010;35:792–805. doi: 10.1038/npp.2009.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D'Addario C, Dell'Osso B, Palazzo MC, Benatti B, Lietti L, Cattaneo E, et al. Selective DNA methylation of BDNF promoter in bipolar disorder: differences among patients with BDI and BDII. Neuropsychopharmacology. 2012;37:1647–1655. doi: 10.1038/npp.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asai T, Bundo M, Sugawara H, Sunaga F, Ueda J, Tanaka G, et al. Effect of mood stabilizers on DNA methylation in human neuroblastoma cells. Int J Neuropsychopharmacol. 2013;16:2285–2294. doi: 10.1017/S1461145713000710. [DOI] [PubMed] [Google Scholar]
- 37.Sugawara H, Iwamoto K, Bundo M, Ueda J, Miyauchi T, Komori A, et al. Hypermethylation of serotonin transporter gene in bipolar disorder detected by epigenome analysis of discordant monozygotic twins. Transl Psychiatry. 2011;1:e24. doi: 10.1038/tp.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huzayyin AA, Andreazza AC, Turecki G, Cruceanu C, Rouleau GA, Alda M, et al. Decreased global methylation in patients with bipolar disorder who respond to lithium. Int J Neuropsychopharmacol. 2014;17:561–569. doi: 10.1017/S1461145713001569. [DOI] [PubMed] [Google Scholar]
- 39.Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 40.Cassel S, Carouge D, Gensburger C, Anglard P, Burgun C, Dietrich JB, et al. Fluoxetine and cocaine induce the epigenetic factors MeCP2 and MBD1 in adult rat brain. Mol Pharmacol. 2006;70:487–492. doi: 10.1124/mol.106.022301. [DOI] [PubMed] [Google Scholar]
- 41.Zimmermann N, Zschocke J, Perisic T, Yu S, Holsboer F, Rein T. Antidepressants inhibit DNA methyltransferase 1 through reducing G9a levels. Biochem J. 2012;448:93–102. doi: 10.1042/BJ20120674. [DOI] [PubMed] [Google Scholar]
- 42.Melas PA, Rogdaki M, Lennartsson A, Björk K, Qi H, Witasp A, et al. Antidepressant treatment is associated with epigenetic alterations in the promoter of P11 in a genetic model of depression. Int J Neuropsychopharmacol. 2012;15:669–679. doi: 10.1017/S1461145711000940. [DOI] [PubMed] [Google Scholar]
- 43.Svenningsson P, Chergui K, Rachleff I, Flajolet M, Zhang X, El Yacoubi M, et al. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science. 2006;311:77–80. doi: 10.1126/science.1117571. [DOI] [PubMed] [Google Scholar]
- 44.Anisman H, Du L, Palkovits M, Faludi G, Kovacs GG, Szontagh-Kishazi P, et al. Serotonin receptor subtype and p11 mRNA expression in stress-relevant brain regions of suicide and control subjects. J Psychiatry Neurosci. 2008;33:131–141. [PMC free article] [PubMed] [Google Scholar]
- 45.Van Sant SP, Buckley PF. Pharmacotherapy for treatment-refractory schizophrenia. Expert Opin Pharmacother. 2011;12:411–434. doi: 10.1517/14656566.2011.528200. [DOI] [PubMed] [Google Scholar]
- 46.Guidotti A, Dong E, Kundakovic M, Satta R, Grayson DR, Costa E. Characterization of the action of antipsychotic subtypes on valproate-induced chromatin remodeling. Trends Pharmacol Sci. 2009;30:55–60. doi: 10.1016/j.tips.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 47.Matrisciano F, Dong E, Gavin DP, Nicoletti F, Guidotti A. Activation of group II metabotropic glutamate receptors promotes DNA demethylation in the mouse brain. Mol Pharmacol. 2011;80:174–182. doi: 10.1124/mol.110.070896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- 49.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 50.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 51.Conley RR, Kelly DL. Treatment of the special patient with schizophrenia. Dialogues Clin Neurosci. 2001;3:123–135. doi: 10.31887/DCNS.2001.3.2/rrconley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joober R, Boksa P. Clozapine: a distinct, poorly understood and under-used molecule. J Psychiatry Neurosci. 2010;35:147–149. doi: 10.1503/jpn.100055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zorn SH, Jones SB, Ward KM, Liston DR. Clozapine is a potent and selective muscarinic M4 receptor agonist. Eur J Pharmacol. 1994;269:R1–R2. doi: 10.1016/0922-4106(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 54.Olianas MC, Maullu C, Onali P. Effects of clozapine on rat striatal muscarinic receptors coupled to inhibition of adenylyl cyclase activity and on the human cloned m4 receptor. Br J Pharmacol. 1997;122:401–408. doi: 10.1038/sj.bjp.0701357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mailman RB, Murthy V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16:488–501. doi: 10.2174/138161210790361461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dwivedi Y, Rizavi HS, Pandey GN. Differential effects of haloperidol and clozapine on [(3)H]cAMP binding, protein kinase A (PKA) activity, and mRNA and protein expression of selective regulatory and catalytic subunit isoforms of PKA in rat brain. J Pharmacol Exp Ther. 2002;301:197–209. doi: 10.1124/jpet.301.1.197. [DOI] [PubMed] [Google Scholar]
- 57.Turalba AV, Leite-Morris KA, Kaplan GB. Antipsychotics regulate cyclic AMP-dependent protein kinase and phosphorylated cyclic AMP response element-binding protein in striatal and cortical brain regions in mice. Neurosci Lett. 2004;357:53–57. doi: 10.1016/j.neulet.2003.11.059. [DOI] [PubMed] [Google Scholar]
- 58.Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- 59.Pozzi L, Håkansson K, Usiello A, Borgkvist A, Lindskog M, Greengard P, et al. Opposite regulation by typical and atypical anti-psychotics of ERK1/2, CREB and Elk-1 phosphorylation in mouse dorsal striatum. J Neurochem. 2003;86:451–459. doi: 10.1046/j.1471-4159.2003.01851.x. [DOI] [PubMed] [Google Scholar]
- 60.Bateup HS, Svenningsson P, Kuroiwa M, Gong S, Nishi A, Heintz N, et al. Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat Neurosci. 2008;11:932–939. doi: 10.1038/nn.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, et al. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- 62.Valjent E, Bertran-Gonzalez J, Bowling H, Lopez S, Santini E, Matamales M, et al. Haloperidol regulates the state of phosphorylation of ribosomal protein S6 via activation of PKA and phosphorylation of DARPP-32. Neuropsychopharmacology. 2011;36:2561–2570. doi: 10.1038/npp.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296:15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 65.Aubry JM, Schwald M, Ballmann E, Karege F. Early effects of mood stabilizers on the Akt/GSK-3beta signaling pathway and on cell survival and proliferation. Psychopharmacology (Berl) 2009;205:419–429. doi: 10.1007/s00213-009-1551-2. [DOI] [PubMed] [Google Scholar]
- 66.Roh MS, Seo MS, Kim Y, Kim SH, Jeon WJ, Ahn YM, et al. Haloperidol and clozapine differentially regulate signals upstream of glycogen synthase kinase 3 in the rat frontal cortex. Exp Mol Med. 2007;39:353–360. doi: 10.1038/emm.2007.39. [DOI] [PubMed] [Google Scholar]
- 67.Réus GZ, Abelaira HM, Agostinho FR, Ribeiro KF, Vitto MF, Luciano TF, et al. The administration of olanzapine and fluoxetine has synergistic effects on intracellular survival pathways in the rat brain. J Psychiatr Res. 2012;46:1029–1035. doi: 10.1016/j.jpsychires.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 68.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 69.Alimohamad H, Sutton L, Mouyal J, Rajakumar N, Rushlow WJ. The effects of antipsychotics on beta-catenin, glycogen synthase kinase-3 and dishevelled in the ventral midbrain of rats. J Neurochem. 2005;95:513–525. doi: 10.1111/j.1471-4159.2005.03388.x. [DOI] [PubMed] [Google Scholar]
- 70.Sutton LP, Honardoust D, Mouyal J, Rajakumar N, Rushlow WJ. Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled-3. J Neurochem. 2007;102:153–169. doi: 10.1111/j.1471-4159.2007.04527.x. [DOI] [PubMed] [Google Scholar]
- 71.Kang UG, Seo MS, Roh MS, Kim Y, Yoon SC, Kim YS. The effects of clozapine on the GSK-3-mediated signaling pathway. FEBS Lett. 2004;560:115–119. doi: 10.1016/S0014-5793(04)00082-1. [DOI] [PubMed] [Google Scholar]
- 72.Sweatt JD, Weeber EJ, Lombroso PJ. Genetics of childhood disorders: LI. Learning and memory, Part 4: Human cognitive disorders and the ras/ERK/CREB pathway. J Am Acad Child Adolesc Psychiatry. 2003;42:741–744. doi: 10.1097/01.CHI.0000046859.56865.A8. [DOI] [PubMed] [Google Scholar]
- 73.Fumagalli F, Frasca A, Spartà M, Drago F, Racagni G, Riva MA. Long-term exposure to the atypical antipsychotic olanzapine differently up-regulates extracellular signal-regulated kinases 1 and 2 phosphorylation in subcellular compartments of rat prefrontal cortex. Mol Pharmacol. 2006;69:1366–1372. doi: 10.1124/mol.105.019828. [DOI] [PubMed] [Google Scholar]
- 74.Ahmed MR, Gurevich VV, Dalby KN, Benovic JL, Gurevich EV. Haloperidol and clozapine differentially affect the expression of arrestins, receptor kinases, and extracellular signal-regulated kinase activation. J Pharmacol Exp Ther. 2008;325:276–283. doi: 10.1124/jpet.107.131987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim SH, Seo MS, Jeon WJ, Yu HS, Park HG, Jung GA, et al. Haloperidol regulates the phosphorylation level of the MEK-ERK-p90RSK signal pathway via protein phosphatase 2A in the rat frontal cortex. Int J Neuropsychopharmacol. 2008;11:509–517. doi: 10.1017/S1461145707008292. [DOI] [PubMed] [Google Scholar]
- 76.Kim SH, Yu HS, Park HG, Park S, Seo MS, Jeon WJ, et al. Role of MKP-1 (DUSP1) in clozapine-induced effects on the ERK1/2 signaling pathway in the rat frontal cortex. Psychopharmacology (Berl) 2013;230:425–437. doi: 10.1007/s00213-013-3165-y. [DOI] [PubMed] [Google Scholar]
- 77.Pereira A, Sugiharto-Winarno A, Zhang B, Malcolm P, Fink G, Sundram S. Clozapine induction of ERK1/2 cell signalling via the EGF receptor in mouse prefrontal cortex and striatum is distinct from other antipsychotic drugs. Int J Neuropsychopharmacol. 2012;15:1149–1160. doi: 10.1017/S1461145711001404. [DOI] [PubMed] [Google Scholar]
- 78.Pereira A, Fink G, Sundram S. Clozapine-induced ERK1 and ERK2 signaling in prefrontal cortex is mediated by the EGF receptor. J Mol Neurosci. 2009;39:185–198. doi: 10.1007/s12031-009-9188-5. [DOI] [PubMed] [Google Scholar]
- 79.Pereira A, Zhang B, Malcolm P, Sundram S. Clozapine regulation of p90RSK and c-Fos signaling via the ErbB1-ERK pathway is distinct from olanzapine and haloperidol in mouse cortex and striatum. Prog Neuropsychopharmacol Biol Psychiatry. 2013;40:353–363. doi: 10.1016/j.pnpbp.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 80.Jope RS. Anti-bipolar therapy: mechanism of action of lithium. Mol Psychiatry. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- 81.Liang MH, Wendland JR, Chuang DM. Lithium inhibits Smad3/4 transactivation via increased CREB activity induced by enhanced PKA and AKT signaling. Mol Cell Neurosci. 2008;37:440–453. doi: 10.1016/j.mcn.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cechinel-Recco K, Valvassori SS, Varela RB, Resende WR, Arent CO, Vitto MF, et al. Lithium and tamoxifen modulate cellular plasticity cascades in animal model of mania. J Psychopharmacol. 2012;26:1594–1604. doi: 10.1177/0269881112463124. [DOI] [PubMed] [Google Scholar]
- 83.Gallagher HC, Bacon CL, Odumeru OA, Gallagher KF, Fitzpatrick T, Regan CM. Valproate activates phosphodiesterase-mediated cAMP degradation: relevance to C6 glioma G1 phase progression. Neurotoxicol Teratol. 2004;26:73–81. doi: 10.1016/j.ntt.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 84.Montezinho LP, Mørk A, Duarte CB, Penschuck S, Geraldes CF, Castro MM. Effects of mood stabilizers on the inhibition of adenylate cyclase via dopamine D(2)-like receptors. Bipolar Disord. 2007;9:290–297. doi: 10.1111/j.1399-5618.2007.00354.x. [DOI] [PubMed] [Google Scholar]
- 85.Chang P, Chandler KE, Williams RS, Walker MC. Inhibition of long-term potentiation by valproic acid through modulation of cyclic AMP. Epilepsia. 2010;51:1533–1542. doi: 10.1111/j.1528-1167.2009.02412.x. [DOI] [PubMed] [Google Scholar]
- 86.Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun. 2001;280:720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- 88.Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem. 2001;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gould TD, Chen G, Manji HK. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology. 2004;29:32–38. doi: 10.1038/sj.npp.1300283. [DOI] [PubMed] [Google Scholar]
- 90.Wexler EM, Geschwind DH, Palmer TD. Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol Psychiatry. 2008;13:285–292. doi: 10.1038/sj.mp.4002093. [DOI] [PubMed] [Google Scholar]
- 91.De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 92.Roh MS, Eom TY, Zmijewska AA, De Sarno P, Roth KA, Jope RS. Hypoxia activates glycogen synthase kinase-3 in mouse brain in vivo: protection by mood stabilizers and imipramine. Biol Psychiatry. 2005;57:278–286. doi: 10.1016/j.biopsych.2004.10.039. [DOI] [PubMed] [Google Scholar]
- 93.Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- 94.Di Daniel E, Mudge AW, Maycox PR. Comparative analysis of the effects of four mood stabilizers in SH-SY5Y cells and in primary neurons. Bipolar Disord. 2005;7:33–41. doi: 10.1111/j.1399-5618.2004.00164.x. [DOI] [PubMed] [Google Scholar]
- 95.Yuan PX, Huang LD, Jiang YM, Gutkind JS, Manji HK, Chen G. The mood stabilizer valproic acid activates mitogen-activated protein kinases and promotes neurite growth. J Biol Chem. 2001;276:31674–31683. doi: 10.1074/jbc.M104309200. [DOI] [PubMed] [Google Scholar]
- 96.Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, et al. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci. 2003;23:7311–7316. doi: 10.1523/JNEUROSCI.23-19-07311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Casu MA, Sanna A, Spada GP, Falzoi M, Mongeau R, Pani L. Effects of acute and chronic valproate treatments on p-CREB levels in the rat amygdala and nucleus accumbens. Brain Res. 2007;1141:15–24. doi: 10.1016/j.brainres.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 98.Young C, Straiko MM, Johnson SA, Creeley C, Olney JW. Ethanol causes and lithium prevents neuroapoptosis and suppression of pERK in the infant mouse brain. Neurobiol Dis. 2008;31:355–360. doi: 10.1016/j.nbd.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Longoni R, Spina L, Vinci S, Acquas E. The MEK inhibitor SL327 blocks acquisition but not expression of lithium-induced conditioned place aversion: a behavioral and immunohistochemical study. Psychopharmacology (Berl) 2011;216:63–73. doi: 10.1007/s00213-011-2192-9. [DOI] [PubMed] [Google Scholar]
- 100.Kim SH, Yu HS, Park HG, Ahn YM, Kim YS, Lee YH, et al. Egr1 regulates lithium-induced transcription of the Period 2 (PER2) gene. Biochim Biophys Acta. 2013;1832:1969–1979. doi: 10.1016/j.bbadis.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 101.Plante DT, Winkelman JW. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008;165:830–843. doi: 10.1176/appi.ajp.2008.08010077. [DOI] [PubMed] [Google Scholar]
- 102.Jensen JB, Mikkelsen JD, Mørk A. Increased adenylyl cyclase type 1 mRNA, but not adenylyl cyclase type 2 in the rat hippocampus following antidepressant treatment. Eur Neuropsychopharmacol. 2000;10:105–111. doi: 10.1016/s0924-977x(99)00064-4. [DOI] [PubMed] [Google Scholar]
- 103.Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S, et al. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci. 2000;20:4030–4036. doi: 10.1523/JNEUROSCI.20-11-04030.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tiraboschi E, Tardito D, Kasahara J, Moraschi S, Pruneri P, Gennarelli M, et al. Selective phosphorylation of nuclear CREB by fluoxetine is linked to activation of CaM kinase IV and MAP kinase cascades. Neuropsychopharmacology. 2004;29:1831–1840. doi: 10.1038/sj.npp.1300488. [DOI] [PubMed] [Google Scholar]
- 105.Wang Z, Hu SY, Lei DL, Song WX. Effect of chronic stress on PKA and P-CREB expression in hippocampus of rats and the antagonism of antidepressors. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2006;31:767–771. [PubMed] [Google Scholar]
- 106.Takano K, Yamasaki H, Kawabe K, Moriyama M, Nakamura Y. Imipramine induces brain-derived neurotrophic factor mRNA expression in cultured astrocytes. J Pharmacol Sci. 2012;120:176–186. doi: 10.1254/jphs.12039fp. [DOI] [PubMed] [Google Scholar]
- 107.Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS. In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology. 2004;29:1426–1431. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Basta-Kaim A, Budziszewska B, Jaworska-Feil L, Tetich M, Kubera M, Leśkiewicz M, et al. Inhibitory effect of imipramine on the human corticotropin-releasing-hormone gene promoter activity operates through a PI3-K/AKT mediated pathway. Neuropharmacology. 2005;49:156–164. doi: 10.1016/j.neuropharm.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 109.Huang W, Zhao Y, Zhu X, Cai Z, Wang S, Yao S, et al. Fluoxetine upregulates phosphorylated-AKT and phosphorylated-ERK1/2 proteins in neural stem cells: evidence for a crosstalk between AKT and ERK1/2 pathways. J Mol Neurosci. 2013;49:244–249. doi: 10.1007/s12031-012-9822-5. [DOI] [PubMed] [Google Scholar]
- 110.Mostany R, Valdizán EM, Pazos A. A role for nuclear beta-catenin in SNRI antidepressant-induced hippocampal cell proliferation. Neuropharmacology. 2008;55:18–26. doi: 10.1016/j.neuropharm.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 111.Pinnock SB, Blake AM, Platt NJ, Herbert J. The roles of BDNF, pCREB and Wnt3a in the latent period preceding activation of progenitor cell mitosis in the adult dentate gyrus by fluoxetine. PLoS One. 2010;5:e13652. doi: 10.1371/journal.pone.0013652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pilar-Cuéllar F, Vidal R, Pazos A. Subchronic treatment with fluoxetine and ketanserin increases hippocampal brain-derived neurotrophic factor, β-catenin and antidepressant-like effects. Br J Pharmacol. 2012;165:1046–1057. doi: 10.1111/j.1476-5381.2011.01516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fumagalli F, Molteni R, Calabrese F, Frasca A, Racagni G, Riva MA. Chronic fluoxetine administration inhibits extracellular signal-regulated kinase 1/2 phosphorylation in rat brain. J Neurochem. 2005;93:1551–1560. doi: 10.1111/j.1471-4159.2005.03149.x. [DOI] [PubMed] [Google Scholar]
- 114.Di Benedetto B, Radecke J, Schmidt MV, Rupprecht R. Acute antidepressant treatment differently modulates ERK/MAPK activation in neurons and astrocytes of the adult mouse prefrontal cortex. Neuroscience. 2012:pii: S0306-4522(12)01179-7. doi: 10.1016/j.neuroscience.2012.11.061. [DOI] [PubMed] [Google Scholar]
- 115.First M, Gil-Ad I, Taler M, Tarasenko I, Novak N, Weizman A. The effects of fluoxetine treatment in a chronic mild stress rat model on depression-related behavior, brain neurotrophins and ERK expression. J Mol Neurosci. 2011;45:246–255. doi: 10.1007/s12031-011-9515-5. [DOI] [PubMed] [Google Scholar]
- 116.Qi X, Lin W, Li J, Li H, Wang W, Wang D, et al. Fluoxetine increases the activity of the ERK-CREB signal system and alleviates the depressive-like behavior in rats exposed to chronic forced swim stress. Neurobiol Dis. 2008;31:278–285. doi: 10.1016/j.nbd.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 117.Hisaoka K, Takebayashi M, Tsuchioka M, Maeda N, Nakata Y, Yamawaki S. Antidepressants increase glial cell line-derived neurotrophic factor production through monoamine-independent activation of protein tyrosine kinase and extracellular signal-regulated kinase in glial cells. J Pharmacol Exp Ther. 2007;321:148–157. doi: 10.1124/jpet.106.116558. [DOI] [PubMed] [Google Scholar]
- 118.Hisaoka K, Maeda N, Tsuchioka M, Takebayashi M. Antidepressants induce acute CREB phosphorylation and CRE-mediated gene expression in glial cells: a possible contribution to GDNF production. Brain Res. 2008;1196:53–58. doi: 10.1016/j.brainres.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 119.Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol. 2009;21:452–460. doi: 10.1016/j.ceb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 120.Soifer HS, Rossi JJ, Saetrom P. MicroRNAs in disease and potential therapeutic applications. Mol Ther. 2007;15:2070–2079. doi: 10.1038/sj.mt.6300311. [DOI] [PubMed] [Google Scholar]
- 121.Xu B, Karayiorgou M, Gogos JA. MicroRNAs in psychiatric and neurodevelopmental disorders. Brain Res. 2010;1338:78–88. doi: 10.1016/j.brainres.2010.03.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Perkins DO, Jeffries CD, Jarskog LF, Thomson JM, Woods K, Newman MA, et al. microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol. 2007;8:R27. doi: 10.1186/gb-2007-8-2-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kocerha J, Faghihi MA, Lopez-Toledano MA, Huang J, Ramsey AJ, Caron MG, et al. MicroRNA-219 modulates NMDA receptor-mediated neurobehavioral dysfunction. Proc Natl Acad Sci U S A. 2009;106:3507–3512. doi: 10.1073/pnas.0805854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Santarelli DM, Liu B, Duncan CE, Beveridge NJ, Tooney PA, Schofield PR, et al. Gene-microRNA interactions associated with antipsychotic mechanisms and the metabolic side effects of olanzapine. Psychopharmacology (Berl) 2013;227:67–78. doi: 10.1007/s00213-012-2939-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gardiner E, Carroll A, Tooney PA, Cairns MJ. Antipsychotic drug-associated gene-miRNA interaction in T-lymphocytes. Int J Neuropsychopharmacol. 2014;17:929–943. doi: 10.1017/S1461145713001752. [DOI] [PubMed] [Google Scholar]
- 126.Newcomer JW. Antipsychotic medications: metabolic and cardiovascular risk. J Clin Psychiatry. 2007;68(Suppl 4):8–13. [PubMed] [Google Scholar]
- 127.Miljevic C, Nikolic M, Nikolic-Kokic A, Jones DR, Niketic V, Lecic-Tosevski D, et al. Lipid status, anti-oxidant enzyme defence and haemoglobin content in the blood of long-term clozapine-treated schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:303–307. doi: 10.1016/j.pnpbp.2009.11.024. [DOI] [PubMed] [Google Scholar]
- 128.Zhou R, Yuan P, Wang Y, Hunsberger JG, Elkahloun A, Wei Y, et al. Evidence for selective microRNAs and their effectors as common long-term targets for the actions of mood stabilizers. Neuropsychopharmacology. 2009;34:1395–1405. doi: 10.1038/npp.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shibata H, Tani A, Chikuhara T, Kikuta R, Sakai M, Ninomiya H, et al. Association study of polymorphisms in the group III metabotropic glutamate receptor genes, GRM4 and GRM7, with schizophrenia. Psychiatry Res. 2009;167:88–96. doi: 10.1016/j.psychres.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 130.Creson TK, Austin DR, Shaltiel G, McCammon J, Wess J, Manji HK, et al. Lithium treatment attenuates muscarinic M(1) receptor dysfunction. Bipolar Disord. 2011;13:238–249. doi: 10.1111/j.1399-5618.2011.00915.x. [DOI] [PubMed] [Google Scholar]
- 131.Chen H, Wang N, Burmeister M, McInnis MG. MicroRNA expression changes in lymphoblastoid cell lines in response to lithium treatment. Int J Neuropsychopharmacol. 2009;12:975–981. doi: 10.1017/S1461145709000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hunsberger JG, Fessler EB, Chibane FL, Leng Y, Maric D, Elkahloun AG, et al. Mood stabilizer-regulated miRNAs in neuropsychiatric and neurodegenerative diseases: identifying associations and functions. Am J Transl Res. 2013;5:450–464. [PMC free article] [PubMed] [Google Scholar]
- 133.Millan MJ. MicroRNA in the regulation and expression of serotonergic transmission in the brain and other tissues. Curr Opin Pharmacol. 2011;11:11–22. doi: 10.1016/j.coph.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 134.Baudry A, Mouillet-Richard S, Schneider B, Launay JM, Kellermann O. miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science. 2010;329:1537–1541. doi: 10.1126/science.1193692. [DOI] [PubMed] [Google Scholar]
- 135.Launay JM, Mouillet-Richard S, Baudry A, Pietri M, Kellermann O. Raphe-mediated signals control the hippocampal response to SRI antidepressants via miR-16. Transl Psychiatry. 2011;1:e56. doi: 10.1038/tp.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Angelucci F, Croce N, Spalletta G, Dinallo V, Gravina P, Bossù P, et al. Paroxetine rapidly modulates the expression of brain-derived neurotrophic factor mRNA and protein in a human glioblastoma-astrocytoma cell line. Pharmacology. 2011;87:5–10. doi: 10.1159/000322528. [DOI] [PubMed] [Google Scholar]
- 137.Bocchio-Chiavetto L, Maffioletti E, Bettinsoli P, Giovannini C, Bignotti S, Tardito D, et al. Blood microRNA changes in depressed patients during antidepressant treatment. Eur Neuropsychopharmacol. 2013;23:602–611. doi: 10.1016/j.euroneuro.2012.06.013. [DOI] [PubMed] [Google Scholar]