

Abstract
Restoration of p53 function in tumor cells would be an attractive strategy for lung cancer therapy because p53 mutations are found in more than 50% of lung cancers. The small molecule PRIMA-1 has been shown to restore the tumor suppression function of p53 and to induce apoptosis in human tumor cells. The mechanism of apoptosis induced by PRIMA-1 remains unclear. We investigated the effects of PRIMA-1 in apoptosis with Western immunoblot analysis, TaqMan microRNA real-time PCR, cell viability analysis and flow cytometry using human lung cancer cell lines containing mutant (H211 and H1155), wild-type (A549) or null (H1299) p53. PRIMA-1 induced massive apoptosis in the H211 and H1155 cells, but was less toxic to the A549 and H1299 cells. Western immunoblot analysis showed cleavage of PARP in H211 and H1155 cells but not in A549 and H1299 cells following treatment with PRIMA-1. In addition, p53 protein was also phosphorylated in H211 and H1155 cells. TaqMan microRNA assay showed that the expression of microRNA-34a was increased in the H211 and H1155 cells posttreatment. Knockdown microRNA-34a decreased the rate of apoptosis caused by PRIMA-1. The above results suggest that microRNA-34a is one of the important components of PRIMA-1-induced apoptotic network in the cancer cells harboring mutant p53.
Keywords: microRNA-34, PRIMA-1, apoptosis, lung cancer
The tumor suppressor p53 mediates cell growth by activating the transcription of multiple genes specifically involved in cell cycle regulation, apoptosis and genomic stability.1-3 P53 is also involved in response to various stress signals including anticancer therapies.2 Mutation of the p53 tumor suppressor gene is the most frequently reported genetic defect in human cancer, with mutations observed in greater than 50% of lung tumors.4,5 Although thousands of mutations have been reported, the vast majority of mutations occurring in tumors are missense mutations, which result in full length, but functionally aberrant proteins, and occur within the sequence-specific DNA-binding domain of the protein (amino acid residues 100–300). As a result, mutant p53 proteins are unable to bind to and transactivate the target genes that mediate tumor suppression.6,7 By providing a key difference between normal cells and tumor cells, the p53 protein represents an important target for drug development.8,9
It has been reported that restoration of wild-type p53 function leads to regression of cancers in mice.10,11 These results support efforts to treat cancers through pharmacological reactivation of p53 with small molecules such as CP-31398 or PRIMA-1 (p53-dependent reactivation and induction of massive apoptosis).12-14
The small molecule PRIMA-1 has been shown to induce apoptosis in human tumor cells containing mutant p53 by restoration of the tumor suppressor function of p53.13 This is believed to be mediated by a change in the conformation of mutated p53 protein, restoring DNA binding and activation of p53 target genes.13 It has been reported that Bax, PUMA and other p53 downstream genes are involved in PRIMA-1-mediated apoptosis.15,16 However, it has also been reported that PRIMA-1-induced apoptosis involves the c-Jun N-terminal kinase (JNK) pathway but not the Bcl-2 pathway.17 The mechanism of apoptosis induced by PRIMA-1 remained under investigation.
MicroRNAs are small, 18–24 nucleotide noncoding RNAs. MicroRNAs regulate target genes by regulation of mRNA level and direct regulation of protein synthesis of target genes.18 Increasing evidence has suggested that microRNAs can function as either oncogenes or tumor suppressor genes.19 Recently, microRNA-34 family members (34a, 34b and 34c) were identified as targets of the p53 gene.20-22 The microRNA-34 family members are regulated directly by interaction of p53 with these consensus sites and were reported to induce apoptosis in cancer cells.20-28 In many tumor types, the promoters of the miR-34a and the miR-34b/c genes are subject to inactivation by CpG methylation.29 However, the involvement of PRIMA-1 in regulating expression of microRNA to induce apoptosis in cancer cells has not yet been shown. We investigated the effects of PRIMA-1 in regulation of microRNA-34 family members as a potential mechanism for the mutant p53-depedent apoptosis induced by this agent.
Material and Methods
Cell culture and reagent
Human lung cancer cell lines A549 (wild-type p53); H211 (mutant p53, residue 248, from R to Q); H1155 (mutant p53, residue 273 from R to H) and H1299 (p53 null) were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and were grown in RPMI 1640 medium with 10% fetal bovine serum and 1% penicillin/streptomycin in a humidified incubator (37°C, 5% of CO2). PRIMA-1 (p53-dependent reactivation and induction of massive apoptosis) was obtained from Calbiochem (San Diego, CA). PRIMA-1 diluted in water was used for cellular treatments.
Cell viability analysis
Five thousand cells were seeded in each well of a 96-well plate 24 hr before treatment. Cells were treated with PRIMA-1 (100 μM). Dimethylthiazolyl-2-5-diphenyltetrazolium bromide (MTT) dye solution (Sigma, St. Louis, MO) was added into the 96-well plate 20 hr posttreatment. The plate was incubated at 37°C for 4 hr, and then the treatment was terminated by adding stop solution (isopropanol with 0.04 N HCl). MTT was cleaved by live cells to a colored formazan product. Absorbance at 560 nm wavelength was recorded using a Bio-Rad microplate reader 680 (Bio-Rad Laboratories, Hercules, CA). Each treatment was repeated in quadruplicate. An averaged absorbance of blank values (containing all reagents except cells) was subtracted from all absorbance to yield corrected absorbance. The relative absorbance of each sample was calculated by comparing the average of corrected absorbance with an average of corrected untreated control.
Quantitation of microRNAs by real-time PCR
Total RNA was isolated from the cell lines using Trizol RNA isolation following the protocol supplied by the manufacturer (GIBCO BRL, Rockville, MD). RNA samples were treated with DNase (Ambion, Austin, TX) to remove contaminating DNA and stored in −70°C freezer. Quantitation of micro-RNAs was carried out using TaqMan microRNA assays (Applied Biosystems, Foster City, CA). Reverse transcription was performed by using TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Briefly, 500 ng of template total RNA was reverse transcribed in a 15 μl reaction. MicroRNA-specific stem-loop primers for hsa-miR-34a, hsa-miR-34b, hsa-miR-34c and RUN48 (Applied Biosystems) were used for the reverse transcription. Primers and probes used for the real-time PCR analysis of microRNA were also obtained from Applied Biosystems (Applied Biosystems, Foster City, CA). The PCR amplification was conducted in 25 μl reaction using the TaqMan Universal PCR Master Mixture, No AmpErase UNG kit (Applied Biosystems, Foster City, CA) according to the protocol supplied by the manufacturer. Real-time PCR was carried out in a 96-well plate using an Applied Biosystems 7900HT Sequence Detection System at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Each sample was done in triplicate, and each reaction was repeated at least once to ensure reproducibility. The PCR cycle number at threshold was used for the comparison. The relative quantitative method was used for the quantitative analysis. Calibrator was the averaged ΔCt from the untreated cells. Endogenous control was RUN48.
Inhibition of microRNA-34a, p53 and apoptosis analysis
To determine the effect of microRNA-34a on PRIMA-1-induced apoptosis in lung cancer cells H211 and H1155, 4 × 105 cells were transfected with either 40 nM of miRIDIAN hairpin inhibitor of microRNA-34a or 40 nM of p53 ON-TARGET plus smart pool siRNA or 40 nM of miRIDIAN microRNA hairpin inhibitor negative control (Dharmacon, Chicago, IL) using Lipofectamine 2000 transfection system (Invitrogen, CA) according to the manufacturer’s protocol. One day after incubation in medium containing Lipofect-amine 2000 agent, these transfected cells were transferred to a dish that contains normal growth medium. H211 and H1155 cells were transfected with miRIDIAN hairpin inhibitor to inhibit has-miR-34a or siRNA antip53 or negative control. The transfected cells were treated with PRIMA-1 (25 μM for H211 and 100 μM for H1155) 24 hr posttransfection. After an additional incubation of 24 (H211) or 48 hr (H1155), the cells were harvested, stained with Annexin V-FITC apoptosis kit (BD Pharmingen, CA) and analyzed by fluorescence activated cell sorting (FACS). In addition, PARP cleavage in these cells was analyzed with Western blot.
Western immunoblot analysis and antibodies
Western immunoblot analysis was performed as described previously.30,31 Briefly, cells were digested with lysis buffer, which contained 250 mM NaCl, 5 mM EDTA, 1% Igepal, 5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride and 1% protease inhibitor cocktail (Sigma, Saint Louis, MO). Protein concentrations were evaluated using the Bradford reagent (Bio-Rad, Hercules, CA). One hundred micrograms of total protein was loaded onto NuPAGE™ 4–12% Bis-Tris Gel (Invitrogen, Carlsbad, CA). Protein on the gels was electro-transferred onto nitrocellulose membranes and blocked with blocking buffer (5% of nonfat milk, 500 mM of NaCl, 20 mM Tris and 0.1% Tween-20). The membranes were incubated with primary antibodies at 4°C overnight. After washing with TBS-T (blocking buffer without milk) 5 times, 10 min each, the membranes were incubated with anti-mouse Ig or anti-rabbit Ig horseradish peroxidase linked to whole secondary antibodies (Amersham Pharmacia Biotech, Piscataway, NJ) at room temperature for 1 hr. After washing 5 times, 10 min each, a chemiluminescent detection system (ECL Western blotting detection reagents, Amersham Pharmacia Biotech, Piscataway, NJ) was used to detect the secondary antibody. Finally, the membranes were exposed to X-ray films.
Antibodies used in current studies were as follows: DO-7, anti-human p53 specific monoclonal antibody (BD PharMingen, San Diego, CA); AC-15, anti-β-Actin monoclonal (Sigma, St. Louis, MO); C2-10, anti-PARP [poly (ADP-ribose) polymerase] monoclonal (BD PharMingen, San Diego, CA); anti-PARP polyclonal (Cell Signaling Technology, Danvers, MA); anti phosphorylated p53 (Ser15) rabbit polyclonal (Santa Cruz, Santa Cruz, CA).
Results
PRIMA-1 induces significant reduction of cell viability in H211 and H1155 cells
To investigate the influence of PRIMA-1 in apoptotic cell death in the lung cancer cells, we treated the lung cancer cell lines H1299 (null), A549 (wild-type), H211 (mutant 248Q) and H1155 (mutant 273H) with PRIMA-1 at a dose of 100 μM. MTT assay was used for the cell viability analysis, and an averaged absorbance was recorded 24 hr posttreatment. Cell viability analysis showed that the PRIMA-1 was cytotoxic to these cells, and the H211 and H1155 cells had much lower percentage of viable cells when compared with untreated controls. There were only 31% viable cells in the H211 and 54% in the H1155 cells posttreatment. In contrast, 82 and 89% viable cells were detected in the A549 and H1299 cells, respectively (Fig. 1). These results demonstrate that the PRIMA-1 is more toxic to mutant p53-containing cells H211 and H1155 when compared with the other 2 cell lines tested.
Figure 1.
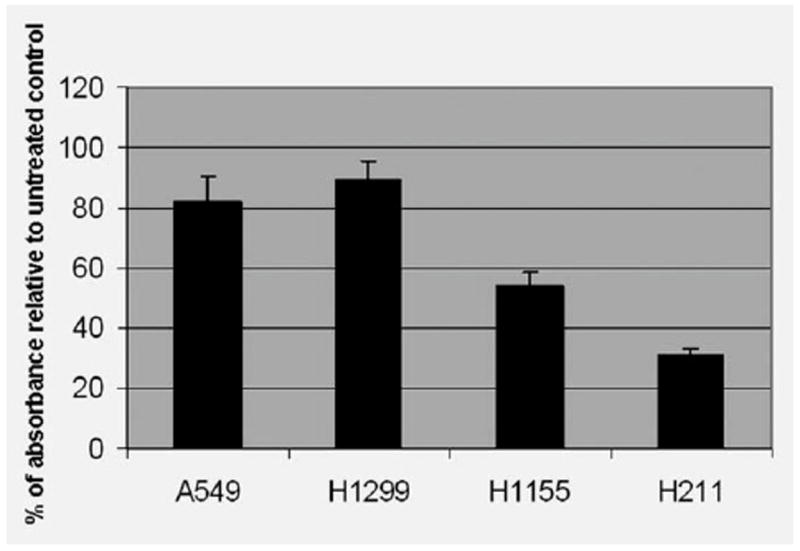
Analysis of cell viability post PRIMA-1 treatment. Cells were treated with the PRIMA-1 (100 μM), and the MTT assay was performed to determine cell viability 24 hr posttreatment. Each treatment was repeated in quadruplicate. An averaged absorbance of blank values (with no cells) was subtracted from all absorbance to yield corrected absorbance. The relative absorbance of each sample was calculated by comparing the average of corrected absorbance with an average of corrected untreated control. Each value presented in this figure was an average value obtained from 4 measurements.
PRIMA-1 induces apoptosis in cells with p53 mutations
To investigate the relationship between the PRIMA-1 and p53 status in apoptosis, we used human lung cancer cell lines as described in the MTT experiment. First, we treated these lung cancer cells with 50 and 100 μM of PRIMA-1 for 24 hr. Western immunoblot analysis with anti-PARP antibody was used to detect apoptosis. As shown in Figure 2a, PARP cleavage was found in H211 and H1155 cells post PRIMA-1 treatment, indicating the existence of an apoptotic process in these cells. In comparison, no PARP cleavage was found in the A549 and H1299 cells.
Figure 2.
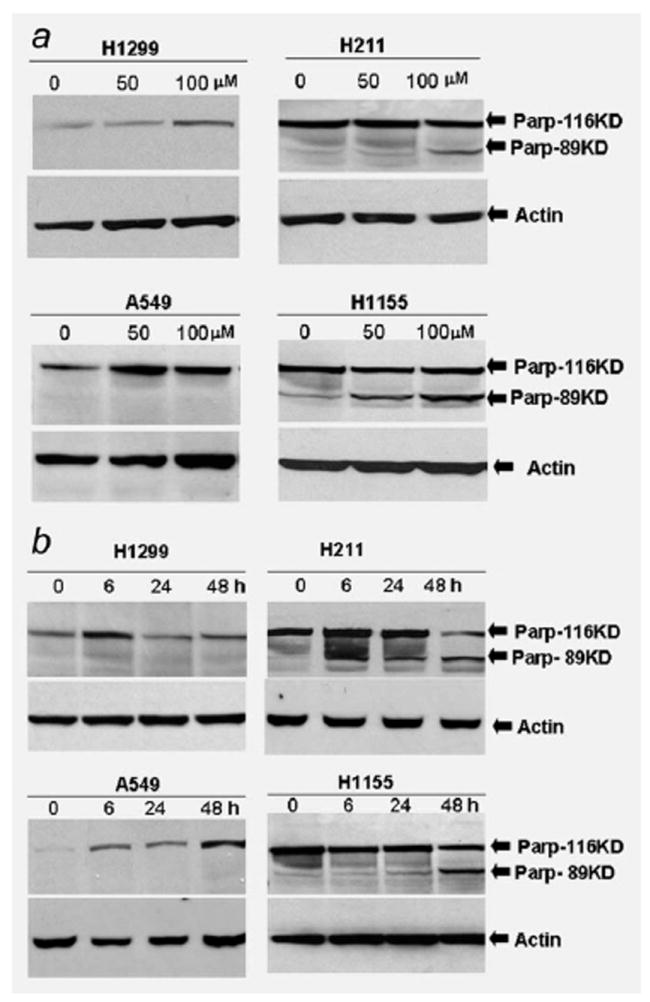
Western immunoblot analysis of PARP cleavage induced by PRIMA-1 in lung cancer cell lines. (a) Human lung cancer cells A549 (wild-type p53), H1299 (p53 null), H211 (mutant p53, 248Q) and H1155 (mutant p53, 273H) were treated with PRIMA-1 at a dose of 50 or 100 μM. Cells were harvested 24 hr posttreatment. A total of 100 μg cell lysate each sample was subjected for Western immunoblot analysis. Western immunoblot membrane was probed with antibodies against full length and cleaved PARP. (b) Human lung cancer cells were treated with PRIMA-1 (100 μM). Cells were harvested and lysed at 6, 24 and 48 hr posttreatment. Western immunoblot analysis was used to analyze the cleavage of PARP protein in the lung cancer cells at indicated time point. Membrane was probed with antibodies against full length and cleaved PARP. Loading control was β-actin. The cleaved PARP was detected in the H211 and H1155 cells after treatment with PRIMA-1.
To further evaluate the role of PRIMA-1 in induction of apoptosis, these cell lines were exposed to PRIMA-1 at a dose of 100 μM, and then cells were harvested at 6, 24 and 48 hr posttreatment. Western immunoblot analysis showed that PARP cleavage was observed 6 hr posttreatment, and the cleaved PARP (89 KD) was increased 24 and 48 hr posttreatment in the H211 and H1155 cells, respectively (Fig. 2b). In contrast, the PARP cleavage was absent from the A549 and H1299 cells at all time points. These results suggest that PRIMA-1 is a highly selective small molecule toxic to cells containing mutant p53.
PRIMA-1 induces apoptosis involving phosphorylation of p53
Phosphorylation of p53 is a critical step for p53 activation. We investigated the status of p53 phosphorylation in those PRIMA-1-treated lung cancer cells in a time course of samples. A549, H211, H1155 and H1299 cells were treated with PRIMA-1 for 6, 24 and 48 hr at a dose of 100 μM. Cell lysates were subjected to Western immunoblot analysis probed with antiphosphorylated p53 antibody. Western immunoblot results showed that PRIMA-1 exposure led to increase phosphorylation of p53 at Ser15 (Fig. 3). As shown in Figure 3, phosphorylated p53 protein was increased in the H211 and H1155 cells post PRIMA-1 exposure. Increased phosphorylation of Ser15 appeared as early as 6 hr posttreatment in the H1155 cells and 24 hr in the H211 cells. The increased phosphorylated p53 protein was maintained through 48 hr posttreatment in the H211 and H1155 cells. In contrast, no phosphorylation of p53 at Ser15 was detected in the A549 and H1299 cells at all time points.
Figure 3.
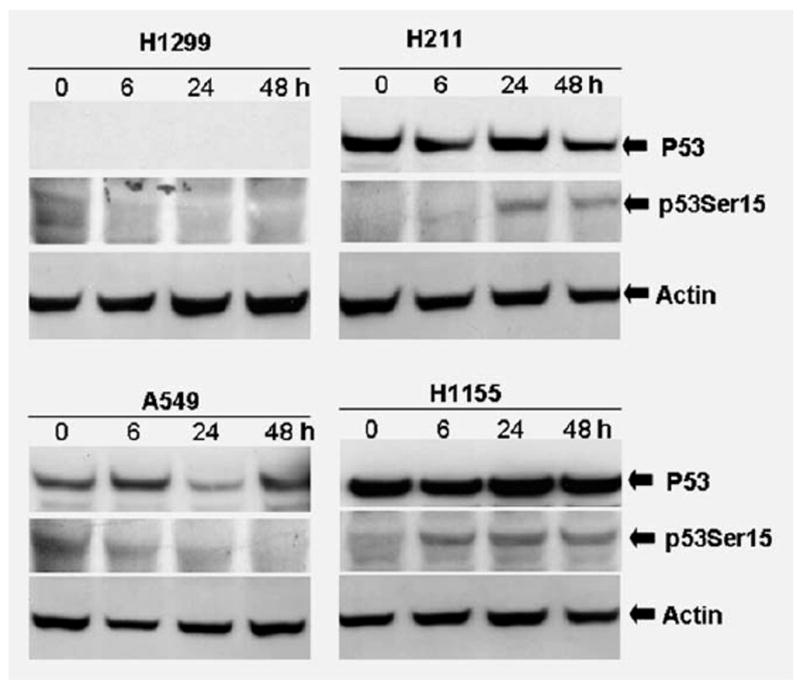
Analysis of p53 phosphorylation in lung cancer cell lines. Human lung cancer cells A549, H1299, H211 and H1155 were treated with PRIMA-1 (100 μM) and then harvested at 6, 24 and 48 hr posttreatment. Cell lysates were subjected to SDS-PAGE and probed with anti-p53 and anti-phospho-p53 antibodies against Ser15, respectively. PRIMA-1 exposure led to increase phosphorylation of p53 at Ser15. Loading control was β-actin.
Expression of microRNA-34a is regulated by PRIMA-1
To investigate whether PRIMA-1 regulates microRNA-34 family members in lung cancer cells, A549, H211, H1155 and H1299 cells were cultured with PRIMA-1 at a dose of 100 μM and harvested at 6, 24 and 48 h. We evaluated the expression of microRNA-34a, 34b and 34c in these lung cancer cells using the TaqMan microRNA assays. As shown in Figure 4a, the PRIMA-1 can upregulate microRNA-34a in the H211 and H1155 cells. The microRNA-34a was increased about 1.65- to 2-fold in the H211 and H1155 cells 24 and 48 hr posttreatment when compared with untreated controls. In contrast, microRNA-34b and 34c showed no change 24 and 48 hr posttreatment in the H211 and H1155 cells. No significant increase was found in microRNA-34 family members in the A549 and H1299 cells at any time point (Fig. 4a). These microRNA data are consistent with the apoptotic analysis evaluated by MTT and PARP cleavage analysis.
Figure 4.
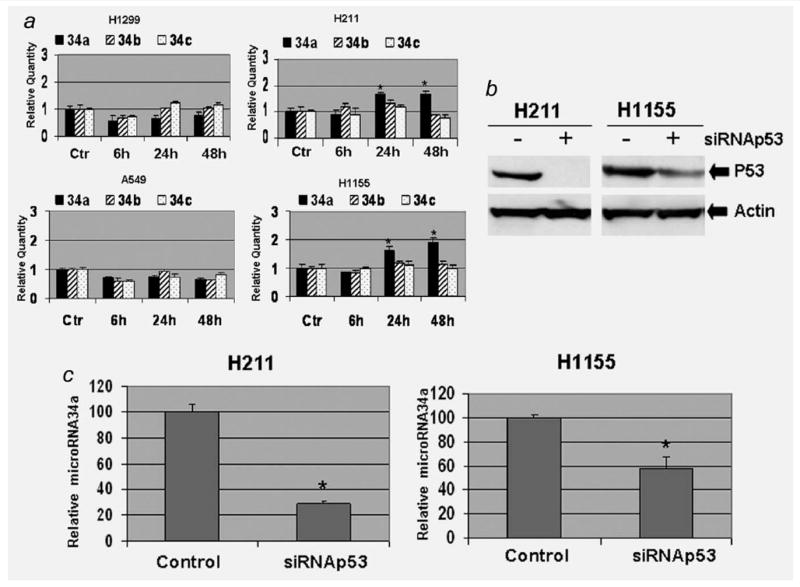
PRIMA-1 upregulates expression of microRNA-34a. (a) TaqMan microRNA real-time PCR analysis was used to evaluate the expression of microRNA-34a, b and c. Cells were treated with the PRIMA-1 (100 μM), and cells were harvested at 6, 24 and 48 hr posttreatment. Real-time PCR was carried out in a 96-well plate using an Applied Biosystems 7900HT Sequence Detection System. The relative quantitative method was used to determine expression of the microRNA-34 family members. The averaged microRNA-34 family member from the control (Ctr) group of each cell line was used as calibrator. For each cell line, the averaged values obtained from other time points were compared with the calibrator. Each value presented in this figure was an average value obtained from 3 amplifications. (b) Western blot analysis was used to confirm the knocking down of p53. (c) Expression levels of microRNA-34a were estimated with TaqMan microRNA real-time PCR in the anti-p53 siRNA transfected and control cells. The error bars show standard deviation.
* indicates p < 0.05, 2-tailed t-test.
To evaluate the relationship between mutant p53 and microRNA-34a expression posttreatment of PRIMA-1, we performed p53 knockdown experiments with siRNA transfection. The H211 and H1155 cells were treated with transfection of siRNA directed against p53 or negative control. Western blot analysis confirmed the reduction of p53 protein in both cell lines posttransfection (Fig. 4b). Real-time PCR results indicated that the level of microRNA-34a was reduced in the siRNA-tranfected H211 and H1155 cells post PRIMA-1 treatment (Fig. 4c). The expression level of PRIMA-1-induced microRNA-34a is associated with the p53 protein.
Knockdown microRNA-34a reduces PRIMA-1-induced apoptosis
As mentioned earlier, PRIMA-1 promotes expression of microRNA-34a in the H211 and H1155 cells. To investigate if the microRNA-34a regulates the PRIMA-1-induced apoptosis, we performed apoptosis analysis in H211 and H1155 cells transfected with miRIDIAN hairpin inhibitor anti-microRNA-34a or negative control. TaqMan microRNA real-time PCR showed that the microRNA-34a was reduced 8.7-and 2.5-fold, respectively, in the transfected (with miRIDIAN hairpin inhibitor anti-microRNA-34a) H211 and H1155 cells when compared with negative controls (Fig. 5a). After confirming the specific knockdown of microRNA-34a, the effect on PRIMA-1-induced apoptosis was assessed by FACS analysis of cells for annexin V and propidium iodide staining. The percentage of apoptotic cell in the control group was 76.5%, whereas the rate of apoptotic cell in the microRNA-34a knockdown group was 46.6% in the H211 cells. The apoptotic rate in the control group of H1155 cell was 22.3%; in contrast, the rate of apoptosis in the microRNA-34a knockdown group was only 16.5% (Fig. 5b). In addition, we also analyzed PARP cleavage with Western blotting analysis. The amount of cleaved PARP protein (PARP-89KD) was reduced in the microRNA-34a knockdown cells (Fig. 5c). Expression of miRIDIAN hairpin inhibitors to knockdown microRNA-34a decreases PRIMA-1-induced apoptosis in the H211 and H1155 cells.
Figure 5.
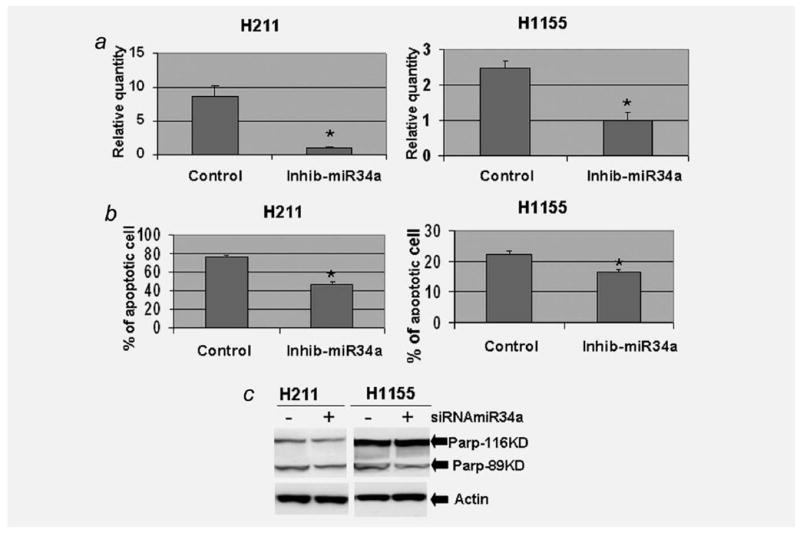
Knockdown microRNA-34a reduces PRIMA-1-induced apoptosis. H211 and H1155 cells were transfected with miRIDIAN hairpin inhibitor to inhibit expression of microRNA-34a or negative control. The transfected cells were treated with PRIMA-1 (25 μM for H211 and 100 μM for H1155) 24 hr posttransfection. After an additional incubation of 24 hr, the transfected and PRIMA-1-treated cells were harvested. TaqMan microRNA real-time PCR was used to confirm the specific knockdown of microRNA-34a in the H211 and H1155cells (a). The transfected and PRIMA-1-treated cells were then stained with Annexin V-FITC apoptosis kit and analyzed by fluorescence activated cell sorting (b). The apoptosis was also evaluated by Western blot analysis of PARP cleavage (c). Each value presented in this figure was an average value obtained from 3 independent analyses. The error bars show standard deviation. * indicates p < 0.05, 2-tailed t-test.
Discussion
Various strategies have been designed to restore the function of mutant p53 in cancer cells.16 Small molecules that can restore sequence-specific DNA-binding, wild-type conformation and transcriptional transactivation to mutant p53 have been identified.12,10 PRIMA-1 is a low-molecular-weight compound, which has been reported to restore wild-type function to mutant p53.13 In this study, we analyzed the ability of PRIMA-1 to induce apoptosis using 4 lung cancer cell lines containing wild-type, mutant or null p53. We showed that PRIMA-1 was able to induce apoptosis in the p53 mutant cells. The lung cancer cell line H1155 contains p53 with mutation in Codon 273 and H211 contains p53 with mutation in Codon 248. These findings may have significant clinical implications, because the IARC p53 mutation database (http://www-p53.iarc.fr/index.html) showed that 273 and 248 are 2 of the most frequently mutated sites in human lung cancers.
The mechanisms of apoptosis induced by PRIMA-1 are still unclear. Chipuk et al.15 reported PRIMA-1-induced Bax-dependent apoptosis involving cytochrome c release and caspase activation in the absence of transcription. However, it has also been reported that PRIMA-1-induced apoptosis involves the JNK pathway but not the Bcl-2 proteins.17 Clearly, the PRIMA-1-induced p53-dependent apoptosis is a complex process involving multiple genetic pathways.
MicroRNAs regulate target genes by a number of mechanisms that involves regulation of mRNA level and direct regulation of protein translation of target genes.18 MicroRNA-34 family members have been implicated in the regulation of cell proliferation and apoptosis.20-28 The involvement of microRNA-34 family members in the PRIMA-1-induced apoptosis is particular intriguing. We report here, for the first time, the ability of this drug to activate microRNA-34a. Importantly and consistent with its putative mechanism of action, this happened only with cells harboring p53 mutations. PRIMA-1 restores wild-type function to mutant p53, and the restored functional p53 proteins upregulate the microRNA-34a to induce apoptosis in the lung cancer cells containing mutant p53. We noticed the reduction of micro-RNA-34a in H1299 and A549 cells after 6, 24 and 48 hr of incubation with PRIMA-1. We think the reduction may associate with status of p53. However, we do not know the mechanism involved for the reduction. In addition, the expression level of microRNA-34a was not altered after the H211 and H1155 cells were treated with cisplatin at a dose of 5 μg/ml for 6, 24 and 48 hr (data not shown). This indicates that the PRIMA-1 has specific ability to restore p53 function to up-regulate microRNA-34a in these cells.
There were differences in the way PRIMA-1 causes micro-RNA-34 family members to activate. For example, micro-RNA-34a was upregulated in the H211 and H1155 cells post PRIMA-1 treatment, but microRNA-34b and 34c were not responsive to the treatment of PRIMA-1 in these cells (Fig. 4a). Thus, the apoptotic pathway induced by PRIMA-1 in the H211 and H1155 is associated exclusively with the micro-RNA-34a. Furthermore, by inhibition of microRNA-34a with miRIDIAN hairpin inhibitor, the PRIMA-1-induced apoptosis was reduced in the H211 and H1155 cells. These results indicate that the microRNA-34a plays an important role in the PRIMA-1-induced apoptosis in these cells. Further investigation in understanding the relationships among p53 mutations, expression of microRNA-34 family members and PRIMA-1-induced apoptosis in vitro and in vivo is necessary to develop effective strategies for p53-related pharmacological therapeutic intervention in human lung cancers.
It has been reported that p53 phosphorylation at Ser15 is related to activation of p53.32,33 In this study, we showed close correlations among p53 phosphorylation at Ser15, expression of microRNA34a and PRIMA-1-induced apoptosis. These results indicate that the microRNA-34a is one of the important components of the PRIMA-1-induced p53-dependent apoptotic network.
In summary, the above results suggest that PRIMA-1 promotes p53-dependent elevation of microRNA-34a to regulate apoptosis in the lung cancer cells containing mutant p53. Given the high prevalence of p53 mutations in Codon 273 and 248 in human lung cancer cells, development of related compounds for lung cancer therapy deserves consideration.
Acknowledgments
The authors thank the Real Time PCR Shared Resource and Flow Cytometry and Cell Sorting Analysis Core at the Ohio State University for their assistance. This work was supported in part by the Joan’s Legacy Foundation and the LUNGevity Foundation to (W.D).
Grant sponsors: Joan’s Legacy Foundation, LUNGevity Foundation
References
- 1.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 3.El-Hizawi S, Lagowski JP, Kulesz-Martin M, Albor A. Induction of gene amplification as a gain-of-function phenotype of mutant p53 proteins. Cancer Res. 2002;62:3264–70. [PubMed] [Google Scholar]
- 4.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607–14. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 5.Mitsudomi T, Steinberg SM, Nau MM, Carbone D, D’Amico D, Bodner S, Oie HK, Linnoila RI, Mulshine JL, Minna JD, Gazdar AF. p53 gene mutations in non-small-cell lung cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene. 1992;7:171–80. [PubMed] [Google Scholar]
- 6.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–55. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 7.Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–72. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Kim SH, El-Deiry WS. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci USA. 2006;103:11003–8. doi: 10.1073/pnas.0604507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–87. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 10.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 11.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286:2507–10. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- 13.Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–8. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 14.Tang X, Zhu Y, Han L, Kim AL, Kopelovich L, Bickers DR, Athar M. CP-31398 restores mutant p53 tumor suppressor function and inhibits UVB-induced skin carcinogenesis in mice. J Clin Invest. 2007;117:3753–64. doi: 10.1172/JCI32481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4:371–81. doi: 10.1016/s1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- 16.Selivanova G, Wiman KG. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene. 2007;26:2243–54. doi: 10.1038/sj.onc.1210295. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Mao Y, Brandt-Rauf PW, Williams AC, Fine RL. Selective induction of apoptosis in mutant p53 premalignant and malignant cancer cells by PRIMA-1 through the c-Jun-NH2-kinase pathway. Mol Cancer Ther. 2005;4:901–9. doi: 10.1158/1535-7163.MCT-04-0206. [DOI] [PubMed] [Google Scholar]
- 18.Kong YW, Cannell IG, de Moor CH, Hill K, Garside PG, Hamilton TL, Meijer HA, Dobbyn HC, Stoneley M, Spriggs KA, Willis AE, Bushell M. The mechanism of micro-RNA-mediated translation repression is determined by the promoter of the target gene. Proc Natl Acad Sci USA. 2008;105:8866–71. doi: 10.1073/pnas.0800650105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 20.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–93. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 22.Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007;67:8433–8. doi: 10.1158/0008-5472.CAN-07-1585. [DOI] [PubMed] [Google Scholar]
- 23.Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12:414–18. doi: 10.1016/j.ccr.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 24.Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, MacDougald OA, Cho KR, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 25.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53- mediated apoptosis. Mol Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 26.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017–22. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- 28.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421–6. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Körner H, Knyazev P, Diebold J, Hermeking H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 30.Duan W, Gao L, Wu X, Zhang Y, Otterson GA, Villalona-Calero MA. Differential response between the p53 ubiquitin-protein ligases Pirh2 and MdM2 following DNA damage in human cancer cells. Exp Cell Res. 2006;312:3370–8. doi: 10.1016/j.yexcr.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Duan W, Gao L, Jin D, Otterson GA, Villalona-Calero MA. Lung specific expression of a human mutant p53 affects cell proliferation in transgenic mice. Transgenic Res. 2008;17:355–66. doi: 10.1007/s11248-007-9154-3. [DOI] [PubMed] [Google Scholar]
- 32.Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R, Brady JN. Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem. 1998;273:33048–53. doi: 10.1074/jbc.273.49.33048. [DOI] [PubMed] [Google Scholar]
- 33.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–34. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]