

Abstract
Folate is an essential B vitamin required for the maintenance of AdoMet-dependent methylation. The liver is responsible for many methylation reactions that are used for post-translational modification of proteins, methylation of DNA, and the synthesis of hormones, creatine, carnitine, and phosphatidylcholine. Conditions where methylation capacity is compromised, including folate deficiency, are associated with impaired phosphatidylcholine synthesis resulting in non-alcoholic fatty liver disease and steatohepatitis. In addition, folate intake and folate status have been associated with changes in the expression of genes involved in lipid metabolism, obesity, and metabolic syndrome. In this review, we provide insight on the relationship between folate and lipid metabolism, and an outlook for the future of lipid-related folate research. © 2013 BioFactors, 40(3):277–283, 2014
Keywords: folate, one-carbon metabolism, choline, phospholipids, triacylglycerol, NASH, steatosis
1. Introduction
Folate is an essential vitamin that is synthesized in plants from 6-hydroxymethyldihydropterin, p-aminobenzoate, and glutamate. Almost all the B vitamins play a role in one-carbon metabolism; cobalamin (B12), folate (B9), and riboflavin (B2) serve as cofactors in one-carbon metabolism, while pyridoxine (B6) is essential for transsulfuration pathway [1]. However, folate differs from the other B vitamins in that it serves as a catalytic substrate for the transfer of one-carbon units. The role of folate in one-carbon metabolism has been studied extensively; for comprehensive reviews of folate-mediated one-carbon metabolism see Tibbetts and Appling [2] and Stover and Field [3]. As shown in Fig. 1, the methionine cycle is responsible for the synthesis of S-adenosylmethionine (AdoMet). AdoMet-dependant methylation reactions are required for post-translational modifications of proteins, methylation of DNA, as well as the synthesis of hormones and other small molecules including creatine, carnitine, and phosphatidylcholine (PC). Impaired methylation capacity can be caused by either a decrease in AdoMet or an increase in S-adenosylmethionine (AdoHcy), a competitive inhibitor of many methyltransferase reactions [4]. An important function of folate is in the maintenance of cellular AdoMet and AdoHcy concentrations. Tetrahydrofolate (THF) plays a crucial role in a number of reactions that generate methyl groups from the catabolism of sarcosine, serine, dimethylglycine, and glycine. These methyl groups are used for the remethylation of homocysteine thereby supporting AdoMet synthesis, AdoHcy removal, and hence maintaining methylation capacity.
FIG 1.
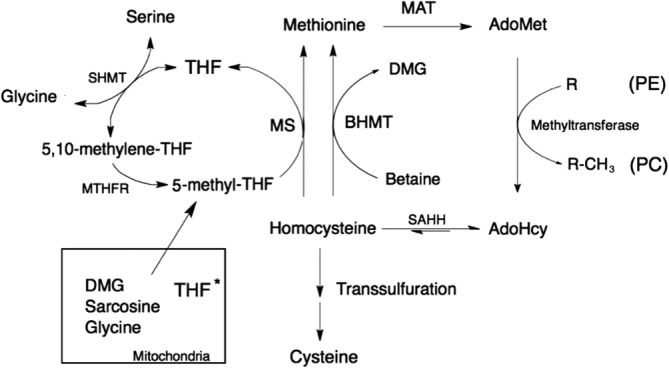
One-carbon and folate metabolism. Abbreviations: S-adenosylmethionine, AdoMet; S-adenosylhomocystine, AdoHcy; Betaine:homocysteine methyltransferase, BHMT; dimethylglycine, DMG; methionine adenosyltransferase, MAT; methionine tetrahydrofolate reductase, MTHFR; methionine synthase, MS; phosphatidylethanolamine, PE; phosphatidylcholine, PC; S-adenosylhomocysteine hydrolase, SAHH and serine hydroxymethyltransferase, SHMT. *Mitochondrial reactions require THF as a substrate.
Many methylation reactions occur in the liver and hepatic steatosis is commonly observed upon perturbation of one-carbon metabolism. Indeed, folate deficiency has been shown to result in the accumulation of triacylglycerol (TG) in the liver [5]. Alterations in both AdoMet availability and phospholipid metabolism have been implicated in the etiology of fatty liver. Furthermore, a fatty liver and changes in AdoMet and folate status are observed in obesity and metabolic syndrome. The purpose of this review is to highlight current research related to folate-mediated regulation of lipid metabolism and lipid-related diseases, such as, non-alcoholic fatty liver disease (NAFLD), obesity, and atherosclerosis.
2. One-Carbon and Lipid Metabolism
As mechanistic research has primarily focussed on investigating the link between altered folate status and hepatic lipid metabolism, we will restrict the majority of our discussion to this topic. Numerous reports have demonstrated a connection between folate, choline, and lipid metabolism [6–8]. Pogribny et al. showed that mouse strains with reduced expression of methionine adenosyltransferase, methionine synthase, and methylenetetrahydrofolate reductase (MTHFR) had significantly greater hepatic lipid accumulation when fed a choline:folate-deficient diet [9]. Christensen et al. observed that a folate-deficient diet alone was sufficient to induce hepatic steatosis in mice [10]. A separate study showed that folate deficiency decreases flux through phosphatidylethanolamine N-methyltransferase (PEMT), an enzyme that synthesizes PC via the methylation of phosphatidylethanolamine (PE) [11]. Together, these findings suggest that folate deficiency reduces de novo PC synthesis resulting in accumulation of hepatic TG.
PC is the most abundant phospholipid in mammalian cellular membranes, bile, and lipoproteins. All nucleated cells use dietary choline to synthesize PC via the CDP–choline pathway; flux through this pathway is regulated by CTP:phosphocholine cytidylyltransferase alpha (CTα). In liver, PC is also synthesized via PEMT which consumes a significant portion of AdoMet in the liver, estimated to be approximately 40% of all methylation reactions [12]. Alterations in PC synthesis affect hepatic lipid storage and secretion in rodent models [13–15]. In particular, pemt−/− mice develop steatosis when fed a high-fat diet and liver failure when fed a choline-deficient diet [13]. These observations can be explained by a reduction in hepatic secretion of VLDL particles in the pemt−/− mice [16]. Similarly, deletion of hepatic CTα impairs VLDL secretion and increases susceptibility to diet-induced steatosis [17,18]. These results and others highlight the role of PC synthesis in the maintenance of hepatic lipid metabolism.
Recently, Zeisel and colleagues generated betaine:homocysteine methyltransferase (BHMT) knockout mice that are incapable of completely catabolizing choline. The bhmt−/− mice have a 25-fold increase in hepatic betaine and develop severe choline deficiency [19]. BHMT-dependent remethylation is clearly important for regulating one-carbon metabolism, as bhmt−/− mice have elevated plasma homocysteine that is not influenced by the level of dietary folate [20]. The bhmt−/− mice also have impaired methylation potential, as illustrated by a significant reduction in the hepatic AdoMet:AdoHcy ratio [19,20]. The presence of reduced de novo choline synthesis via PEMT together with elevated choline dehydrogenase activity in the bhmt−/− mice leads to dramatic decreases in choline-containing metabolites. Moreover, severe choline deficiency results in steatohepatitis and early formation of hepatocarcinoma in bhmt−/− mice [19]. These knockout animals emphasize the importance of maintaining sufficient levels of choline and one-carbon metabolites for lipid homeostasis.
Steatosis is also observed in glycine N-methyltransferase (GNMT) deficiency [21]. GNMT is the most abundant and active methyltransferase enzyme, which serves as an “overflow” to regulate methylation in the cell [22,23]. Gnmt−/− mice have a 40-fold increase in hepatic AdoMet that is associated with steatosis and oxidative stress [24]. Pharmacological treatment of gnmt−/− mice with nicotinamide (a compound that is methylated) normalized AdoMet levels and markedly reduced hepatic steatosis [24]. More recently, Martinez-Una et al. provided evidence that super-physiological levels of AdoMet increase flux through PEMT, with the excess PC being converted to TG [25]. When gnmt−/− mice were fed a methionine-deficient diet, hepatic AdoMet, PC, and TG levels were normalized suggesting that the increased TG deposition is caused by increased conversion of PEMT-derived PC to TG.
To date, we do not know whether PEMT-derived PC contributes to hepatic TG during physiological changes in AdoMet concentration. Stable isotopic tracer studies in humans may provide insight into the potential for TG synthesis from PEMT-derived PC. The average whole-body transmethylation rate has been estimated to be approximately 20 mmoles/day in a 70 kg male [26], and the maximum consumption of AdoMet by PEMT was calculated to be 40% or 8 mmoles/day. If all PEMT-derived PC molecules were converted to TG (assuming no PC was use for lipoprotein or bile secretion) it would amount to approximately 2.67 mmoles or 2.27 g of TG per day (based on an average molecular weight of 850 g/mole per TG molecule). Hepatic TG content can vary considerably, but in humans with healthy liver function measurements range between 6 and 17 g/kg liver [27]. Assuming a liver weighs approximately 3% of bodyweight, the total liver TG content would range between 12 and 36 g. In addition, isotopic tracers have been used to measure the turnover of TG within liver of fasted humans giving a turnover of 15 g/hr and turnover of VLDL TG of 0.8 g/hr [28]. Considering the data and calculations presented here, it is clear that further research is required to determine whether PEMT-derived PC can contribute significantly to hepatic TG accumulation.
Thus far we have limited our discussion to links between folate, choline, and PC metabolism in the liver. However, there is another metabolite involved in lipid metabolism that requires AdoMet-dependent methylation for its synthesis. Carnitine, synthesized from the precursor trimethyllysine, plays an important role in the transport of long chain fatty acids into mitochondria to undergo β-oxidation. The capacity to synthesize carnitine is thought to be large, but the rate of carnitine synthesis is limited by the supply of trimethyllysine. Carnitine deficiency in adults is thought to be relatively rare, apart from an in-born error in a carnitine transporter [29], occurring only when carnitine concentrations are below 10% of normal values [30]. However, a recent study showed patients with non-alcoholic steatohepatitis (NASH) supplemented with l-carnitine had improvements in clinical plasma markers and histological scoring of liver biopsies [31]. It is unclear whether a fatty liver can be attributed to an impaired methylation capacity resulting in reduced carnitine synthesis; however, rats fed a choline-deficient diet had 50% less total-body carnitine [32] and a higher rate of carnitine turnover [33]. Future work examining the potential role of carnitine in NAFLD is certainly on the horizon.
3. Dietary Folate and Hepatic Lipid Metabolism
It is clear that impaired methylation capacity promotes hepatic fat accumulation through an impairment of PC synthesis. Dietary folate deficiency diminishes choline and PC levels in the liver [34]. The drain of choline caused by folate deficiency is twofold: decreased synthesis of choline through the PEMT pathway, and increased use of choline (via BHMT) as a source of methyl groups. Folate deficiency also reduces PEMT activity and choline kinase expression in the liver [5,35]. Impaired PC production through both the CDP–choline and PEMT pathways is associated with accumulation of hepatic TG due to a reduction in VLDL secretion [15,36].
Folate deficiency induces the expression of genes involved in hepatic lipid synthesis that may contribute to hepatic steatosis. Microarray analysis of folate-deficient mice revealed increased expression of lipid biosynthetic genes, including acetyl CoA synthetase long-chain family member 1, lipin 1, diacylglycerol O-acyltransferase 2, elongation of very long chain fatty acid-like 3, and ATP citrate lyase, suggesting increased lipid biosynthesis in folate deficiency [35]. It is possible that these changes in gene expression are linked to a reduction in hepatic PC biosynthesis, which activates sterol regulatory element binding protein 1a to stimulate de novo fatty acid synthesis [37]. However, other studies suggest that fatty acid oxidation is enhanced by short-term folate deficiency. Expression of cpt1a, carnitine O-octanoyltransferase, and acetyl-coenzyme A dehydrogenase was increased in folate-deficient mice [35]. Maternal folate deficiency during gestation increases cpt1a mRNA in fetal liver, another finding that supports an increase in fatty acid oxidation [38]. However, it should be noted that a major limitation of these studies is that hepatic lipid content was not measured and so it is impossible to make conclusions regarding the consequence of the observed changes in gene expression.
Although low dietary folate is associated with hepatic steatosis, there is conflicting evidence regarding the efficacy of using supplemental folic acid for the prevention of fatty liver disease. In humans, dietary folic acid is effectively integrated into the pool of active folate metabolites at low levels of intake (∼100 µg folic acid or 170 µg dietary folate equivalents [DFE]/day) [39]. As a result, the level of folic acid fortification of flours and cereal grain products in Canada and the United States was based on an estimated intake of 100 µg/day. However, estimates of folic acid intake after fortification began in 1998 suggest that actual intakes may double the predicted value [40]. Intake above 200 µg/day is the threshold at which folic acid begins to appear in the blood stream in an un-metabolized form [41]. In addition to fortified foods, excessively high intakes of folic acid can be achieved through intake of vitamin supplements containing up to 5 mg of folic acid. High plasma levels of un-metabolized folic acid can lead to a build-up of dihydrofolate in the cell, which has been shown to inhibit MTHFR [42] and thereby inhibiting remethylation of homocysteine. Thus, it has been postulated that high plasma concentrations of folic acid may lead to a “functional” folate deficiency [43,44]. Folate supplementation in the form of 5-methylTHF has been shown to effectively raise plasma folate levels without leading to an elevation in un-metabolized folic acid [45,46].
Excess macronutrient availability together with high intake of folic acid may contribute to development of obesity. Supplementation of 5 mg/kg folic acid to Sprague Dawley rats was initially found to reduce lipid content of the liver [47], suggesting folic acid has a lipotropic effect. However, Burdge et al. report increased weight gain and hepatic lipid accumulation in rats fed high-fat diet containing 5 mg/kg folic acid, compared with 1 mg/kg folic acid [48]. Under high fat feeding, excessive folic acid supplementation may promote hepatic lipid accumulation by impairing fatty acid oxidation in the liver through decreased expression of cpt1a [48]. These contradictory findings may be a consequence of differences in the fat content of the diet.
Studies dealing with dietary folic acid have are varied in life stage, animal model, duration, amount of folic acid supplemented, and dietary composition. Alterations in methylation status of key genes involved in lipid metabolism may be more likely during certain periods of development rather than later in life. The duration of exposure to folate is also important. In adult models, 4 weeks of folate deficiency may not be long enough to reduce folate status to a level at which lipid metabolism is affected, but 4–5 months folate deficiency yielded a marked hepatic lipid accumulation in rats [5,49]. Another significant factor when interpreting the effect of folate on lipid metabolism is the overall composition of the diet. The consequences of folate deficiency are more evident when methionine and choline are limiting in the diet [49]. For instance, the ability of folate deficiency to suppress PE methylation may be compensated by an ample supply of choline or betaine. In addition, the effects of folic acid supplementation may only become apparent in the presence of a high-fat diet. Finally the strain of rodent may impact severity of the phenotype. Both Pogribny and Tryndyak et al. have shown that the strain of animal also influences the severity of the development of NAFLD [9,39]. Thus, it is imperative that experimental design and choice of animal model be taken into consideration when investigating and interpreting the effects of folate on lipid metabolism.
4. Folate, Obesity, and Atherosclerosis
There is increasing evidence supporting a connection between folate and lipid metabolism. Recently, a potential link between folate status and obesity has garnered attention. For example, serum folate levels have been shown to be significantly lower in individuals with higher body fat mass [50,51], independent of dietary intake [52]. Gallistl et al. reported that serum folate is inversely correlated with body mass index (BMI) in Austrian adolescents [51]. Similarly, obese Thai subjects were found to have lower circulating folate than normal weight individuals [53]. In postmenopausal women, adiposity was associated with lower folate levels [52]. These studies show a relationship between folate status and obesity. However, it is unclear whether reduced folate status is exacerbating weight gain or, conversely, whether obesity is influencing folate metabolism.
A large body of research on folate status in pregnancy and development began due to the integral role of folate in neural tube closure and this field has been spurred on by more recent developments in the concepts of epigenetic regulation and metabolic imprinting. In fact, the importance of obtaining adequate folate in the diet has led to mandatory fortification of a number of staple foods with folic acid in Canada, United States, Australia, and many countries in South America, Africa, and the Middle East. Apart from the well-established role of folate in neural tube closure, low folate status during pregnancy has also been related to lower birth weight [54]. However, evidence linking folate status to obesity through changes in “metabolic imprinting” is mixed. In rats, folic acid supplementation during pregnancy increases total body fat of the offspring [55]. In humans, higher plasma folate concentration at 28 weeks gestation was associated with greater fat mass in children assessed at 6 years of age [56]. High folate intake in the presence of vitamin B12 deficiency exacerbated this weight gain, and was associated with increased risk to developing insulin resistance [56]. These results suggest that altered folate status may affect energy metabolism during development. However, in another human study, no association between childhood adiposity and estimated maternal folate intake at either 18 or 32 weeks of gestation was found in a follow-up of children 9 years of age [57].
Results from human epidemiological studies strongly correlate folate deficiency to increased cardiovascular disease (CVD) risk and incident [58–61]. In addition folate deficiency was shown to induce atherosclerosis development in apoe−/− mice [62–65], while folate supplementation in the same model decreased atherosclerotic lesions [66]. When fed high-fat-folate-deficient diet apoe−/− mice have increased plasma homocysteine and enhanced plaque lesion formation [65], and accumulation of pro-atherogenic lipoproteins in the aorta [64]. Similarly, apoe−/− mice fed methionine-rich, folate-deficient diet presented with an increased atherosclerotic lesion area [63]. However, supplementation with folate and other B vitamins failed to reduce the CVD risk in several large scale randomized controlled intervention trials in humans, despite successfully lowering plasma homocysteine levels [67–71]. It is worthwhile noting that these trials have been secondary prevention trials in patients that had a prior CVD history. However, these results from these studies clearly indicate that folate supplementation does not constitute an added benefit against CVD progression in the folate-sufficient population during folate fortification era [72]. Future studies on targeted populations including primary prevention trials with B-vitamins will be helpful in furthering our understanding of these phenomena.
5. Folate and Epigenetics
The effect of folate status on epigenetic mechanisms has mainly been investigated during key developmental stages, such as pregnancy and lactation. However, folate and AdoMet status have been shown to influence the expression of genes involved in fatty acid synthesis in adult mice. Hypermethylation of the promoter region of fads2, a gene that encodes delta-6 fatty acid desaturase (D6D), was associated with reduced fads2 mRNA expression and lower D6D activity in cystathionine β synthase (CBS) heterozygous mice [73]. These changes led to a reduction in the longer chain unsaturated fatty acids arachidonic acid and docosahexanoic acid, the end products of the D6D metabolism [73]. Exploring links between folate and epigenetic changes that impact essential fatty acid metabolism is a nascent field of study.
Peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear transcription factor that interacts with peroxisome proliferator response elements upon ligand binding, to regulate target gene expression. PPARγ is a key regulator of adipogenesis and has been shown to promote lipid storage and contribute to hepatic steatosis [74]. Folic acid supplementation during in utero and postnatal periods, or after weaning decreases the methylation of the pparg promoter in rat liver [75]. Promoter methylation is inversely related to gene expression, which suggests increased pparg expression in response to folic acid supplementation. In adipose, pparg is nearly tripled upon folic acid supplementation [47]. Therefore, increased body weight in folic acid supplemented animals may be due to enhanced lipid storage via PPARγ-dependent mechanisms.
New research suggests that folate status may be influencing microRNA expression linked to the severity of fatty liver disease. MicroRNAs (miRNAs) are small, non-coding transcripts, of approximately 22 nucleotides in length. They belong to a regulatory class of RNAs that repress expression of target mRNA. Folate supply influences the expression of miRNAs possibly through changes in methylation levels of promoter regions in the genome [76]. The severity of NAFLD induced by a choline- and folate-deficient diet in mice is associated with altered expression of hepatic miRNAs, including miR-181a, miR-34a, miR-200b, and miR-221 [39]. Future research should be directed towards characterizing the mechanisms by which folate supply alters miRNA expression and which miRNAs are important in modulating lipid metabolism.
6. Summary and Future Directions
We are beginning to understand the importance of folate supply in regulating hepatic lipid metabolism (summarized in Fig. 2). The availability of new knockout models that have allowed for a much deeper understanding of how lipid metabolism in the liver might be affected by altered folate metabolism. It is clear that adequate dietary folate is required to support PEMT-dependent de novo PC synthesis in the liver and that disruptions in the metabolism of PC negatively impact hepatic lipid homeostasis via impaired lipoprotein secretion. In addition the maintenance of AdoMet concentration is crucial for liver function and the synthesis of other methylated compounds like carnitine, which influence hepatic lipid homeostasis.
FIG 2.
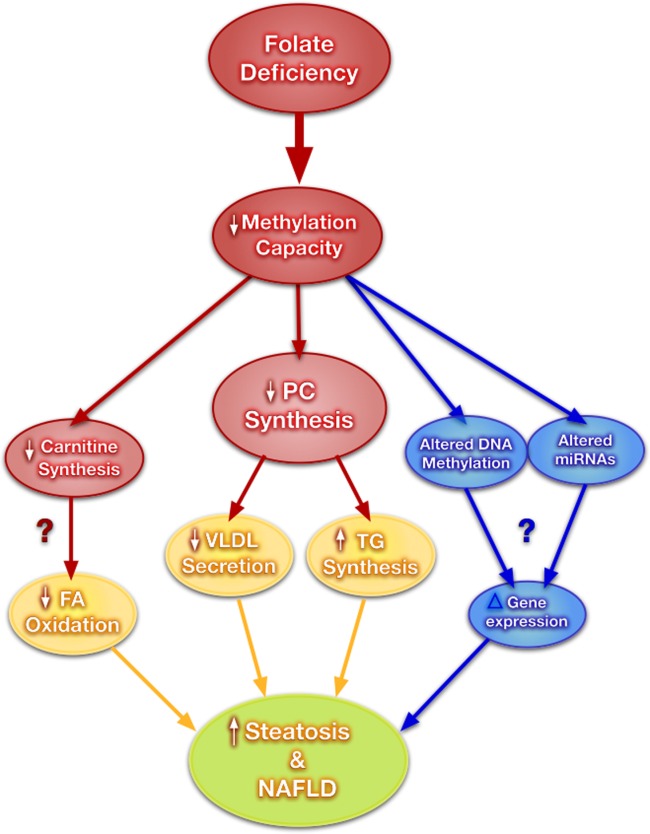
Potential mechanisms linking folate deficiency to fatty liver disease. A reduction in methylation capacity (a reduction in AdoMet/AdoHcy) inhibits the synthesis of carnitine and PC, and alters miRNA and DNA methylation patterns. Together these changes can promote the development of NAFLD through modulation of pathways involved in TG metabolism.
In conclusion, the institution of mandatory folic acid fortification has brought forth the need to understand the impact of high dietary folate intake. We have discussed literature that associates folate intake to obesity as well as the evidence linking folate intake to epigenetic changes that impact the expression of genes involved in lipid metabolism. However, the mechanisms that are responsible for these observations are unclear. Indeed, we do not know whether observed folate status in obesity is a cause or an effect, and we know very little about how epigenetic changes are modulated. Given the role of folate in one-carbon metabolism, the effects of dietary folate on epigenetics, and the current mandatory fortification of this vitamin moves to the importance of furthering our understanding in this field.
Acknowledgments
This work is supported by grants from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council. The authors would like to thank Dr. Catherine Field for helpful comments during the final revision of this manuscript.
References
- 1.Brosnan JT, Brosnan ME. The sulfur-containing amino acids: an overview. J. Nutr. 2006;136:1636S–1640S. doi: 10.1093/jn/136.6.1636S. [DOI] [PubMed] [Google Scholar]
- 2.Tibbetts AS, Appling DR. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010;30:57–81. doi: 10.1146/annurev.nutr.012809.104810. [DOI] [PubMed] [Google Scholar]
- 3.Stover PJ, Field MS. Trafficking of intracellular folates. Adv. Nutr. 2011;2:325–331. doi: 10.3945/an.111.000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clarke S, Banfield K. S-adenosylmethionine-dependent methyltransferases. In: Carmel R, Jacobsen DW, editors. In Homocysteine in Health and Disease. Cambridge: Cambridge University Press; 2001. pp. 63–78. [Google Scholar]
- 5.Akesson B, Fehling C, Jagerstad M. Stenram U. Effect of experimental folate deficiency on lipid metabolism in liver and brain. Br. J. Nutr. 1982;47:505–520. doi: 10.1079/bjn19820063. [DOI] [PubMed] [Google Scholar]
- 6.Koteish A, Diehl AM. Animal models of steatosis. Semin. Liver Dis. 2001;21:89–104. doi: 10.1055/s-2001-12932. [DOI] [PubMed] [Google Scholar]
- 7.Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 2004;40:47–51. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 8.Henkel AS, Dewey AM, Anderson KA, Olivares S. Green RM. Reducing endoplasmic reticulum stress does not improve steatohepatitis in mice fed a methionine- and choline-deficient diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;303:G54–59. doi: 10.1152/ajpgi.00052.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pogribny IP, Kutanzi K, Melnyk S, de Conti A, Tryndyak V, et al. Strain-dependent dysregulation of one-carbon metabolism in male mice is associated with choline- and folate-deficient diet-induced liver injury. FASEB J. 2013;27:2233–2243. doi: 10.1096/fj.12-227116. [DOI] [PubMed] [Google Scholar]
- 10.Christensen KE, Wu Q, Wang X, Deng L, Caudill MA. Rozen R. Steatosis in mice is associated with gender, folate intake, and expression of genes of one-carbon metabolism. J. Nutr. 2010;140:1736–1741. doi: 10.3945/jn.110.124917. [DOI] [PubMed] [Google Scholar]
- 11.Chew TW, Jiang X, Yan J, Wang W, Lusa AL, et al. Folate intake, MTHFR genotype, and sex modulate choline metabolism in mice. J. Nutr. 2011;141:1475–1481. doi: 10.3945/jn.111.138859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stead LM, Brosnan JT, Brosnan ME, Vance DE. Jacobs RL. Is it time to reevaluate methyl balance in humans? Am. J. Clin. Nutr. 2006;83:5–10. doi: 10.1093/ajcn/83.1.5. [DOI] [PubMed] [Google Scholar]
- 13.Vance DE, Li Z. Jacobs RL. Hepatic phosphatidylethanolamine N-methyltransferase, unexpected roles in animal biochemistry and physiology. J. Biol. Chem. 2007;282:33237–33241. doi: 10.1074/jbc.R700028200. [DOI] [PubMed] [Google Scholar]
- 14.Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, et al. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453:657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobs RL, Lingrell S, Zhao Y, Francis GA. Vance DE. Hepatic CTP:phosphocholine cytidylyltransferase-alpha is a critical predictor of plasma high density lipoprotein and very low density lipoprotein. J. Biol. Chem. 2008;283:2147–2155. doi: 10.1074/jbc.M706628200. [DOI] [PubMed] [Google Scholar]
- 16.Noga AA, Vance DE. A gender-specific role for phosphatidylethanolamine N-methyltransferase-derived phosphatidylcholine in the regulation of plasma high density and very low density lipoproteins in mice. J. Biol. Chem. 2003;278:21851–21859. doi: 10.1074/jbc.M301982200. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs RL, Devlin C, Tabas I. Vance DE. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J. Biol. Chem. 2004;279:47402–47410. doi: 10.1074/jbc.M404027200. [DOI] [PubMed] [Google Scholar]
- 18.Niebergall LJ, Jacobs RL, Chaba T. Vance DE. Phosphatidylcholine protects against steatosis in mice but not non-alcoholic steatohepatitis. Biochim. Biophys. Acta. 2011;1811:1177–1185. doi: 10.1016/j.bbalip.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 19.Teng YW, Mehedint MG, Garrow TA. Zeisel SH. Deletion of betaine-homocysteine S-methyltransferase in mice perturbs choline and 1-carbon metabolism, resulting in fatty liver and hepatocellular carcinomas. J. Biol. Chem. 2011;286:36258–36267. doi: 10.1074/jbc.M111.265348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teng YW, Cerdena I. Zeisel SH. Homocysteinemia in mice with genetic betaine homocysteine S-methyltransferase deficiency is independent of dietary folate intake. J. Nutr. 2012;142:1964–1967. doi: 10.3945/jn.112.166835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Chantar ML, Vazquez-Chantada M, Ariz U, Martinez N, Varela M, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luka Z. Methyltetrahydrofolate in folate-binding protein glycine N-methyltransferase. Vitam. Horm. 2008;79:325–345. doi: 10.1016/S0083-6729(08)00411-1. [DOI] [PubMed] [Google Scholar]
- 23.Jencks DA, Mathews RG. Allosteric inhibition of methylenetetrahydrofolate reductase by adenosylmethionine. Effects of adenosylmethionine and NADPH on the equilibrium between active and inactive forms of the enzyme and on the kinetics of approach to equilibrium. J. Biol. Chem. 1987;262:2485–2493. [PubMed] [Google Scholar]
- 24.Varela-Rey M, Martinez-Lopez N, Fernandez-Ramos D, Embade N, Calvisi DF, et al. Fatty liver and fibrosis in glycine N-methyltransferase knockout mice is prevented by nicotinamide. Hepatology. 2010;52:105–114. doi: 10.1002/hep.23639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Una M, Varela-Rey M, Cano A, Fernandez-Ares L, Beraza N, et al. Excess S-adenosylmethionine reroutes phosphatidylethanolamine towards phosphatidylcholine and triglyceride synthesis. Hepatology. 2013;58:1296–1305. doi: 10.1002/hep.26399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mudd SH, Brosnan JT, Brosnan ME, Jacobs RL, Stabler SP, et al. Methyl balance and transmethylation fluxes in humans. Am. J. Clin. Nutr. 2007;85:19–25. doi: 10.1093/ajcn/85.1.19. [DOI] [PubMed] [Google Scholar]
- 27.Mavrelis PG, Ammon HV, Gleysteen JJ, Komorowski RA. Charaf UK. Hepatic free fatty acids in alcoholic liver disease and morbid obesity. Hepatology. 1983;3:226–231. doi: 10.1002/hep.1840030215. [DOI] [PubMed] [Google Scholar]
- 28.Farquhar JW, Gross RC, Wagner RM. Reaven GM. Validation of an incompletely coupled two-compartment nonrecycling catenary model for turnover of liver and plasma triglyceride in man. J. Lipid Res. 1965;6:119–134. [PubMed] [Google Scholar]
- 29.Magoulas PL, El-Hattab AW. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 2012;7:68. doi: 10.1186/1750-1172-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strijbis K, Vaz FM. Distel B. Enzymology of the carnitine biosynthesis pathway. IUBMB Life. 2010;62:357–362. doi: 10.1002/iub.323. [DOI] [PubMed] [Google Scholar]
- 31.Malaguarnera M, Gargante MP, Russo C, Antic T, Vacante M, et al. L-carnitine supplementation to diet: a new tool in treatment of nonalcoholic steatohepatitis–a randomized and controlled clinical trial. Am. J. Gastroenterol. 2010;105:1338–1345. doi: 10.1038/ajg.2009.719. [DOI] [PubMed] [Google Scholar]
- 32.Cox RA, Hoppel CL. Biosynthesis of carnitine and 4-N-trimethylaminobutyrate from lysine. Biochem. J. 1973;136:1075–1082. doi: 10.1042/bj1361075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehlman MA, Therriault DG. Tobin RB. Carnitine-14C metabolism in choline-deficient, alloxan-diabetic choline-deficient and insulin-treated rats. Metabolism. 1971;20:100–107. doi: 10.1016/0026-0495(71)90063-1. [DOI] [PubMed] [Google Scholar]
- 34.Kim YI, Miller JW, da Costa KA, Nadeau M, Smith D, et al. Severe folate deficiency causes secondary depletion of choline and phosphocholine in rat liver. J. Nutr. 1994;124:2197–2203. doi: 10.1093/jn/124.11.2197. [DOI] [PubMed] [Google Scholar]
- 35.Champier J, Claustrat F, Nazaret N, Fevre Montange M. Claustrat B. Folate depletion changes gene expression of fatty acid metabolism, DNA synthesis, and circadian cycle in male mice. Nutr. Res. 2012;32:124–132. doi: 10.1016/j.nutres.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y, Su B, Jacobs RL, Kennedy B, Francis GA, et al. Lack of phosphatidylethanolamine N-methyltransferase alters plasma VLDL phospholipids and attenuates atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2009;29:1349–1355. doi: 10.1161/ATVBAHA.109.188672. [DOI] [PubMed] [Google Scholar]
- 37.Walker AK, Jacobs RL, Watts JL, Rottiers V, Jiang K, et al. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell. 2011;147:840–852. doi: 10.1016/j.cell.2011.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNeil CJ, Hay SM, Rucklidge GJ, Reid MD, Duncan GJ, et al. Maternal diets deficient in folic acid and related methyl donors modify mechanisms associated with lipid metabolism in the fetal liver of the rat. Br. J. Nutr. 2009;102:1445–1452. doi: 10.1017/S0007114509990389. [DOI] [PubMed] [Google Scholar]
- 39.Tryndyak VP, Latendresse JR, Montgomery B, Ross SA, Beland FA, et al. Plasma microRNAs are sensitive indicators of inter-strain differences in the severity of liver injury induced in mice by a choline- and folate-deficient diet. Toxicol. Appl. Pharmacol. 2012;262:52–59. doi: 10.1016/j.taap.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewis CJ, Crane NT, Wilson DB. Yetley EA. Estimated folate intakes: data updated to reflect food fortification, increased bioavailability, and dietary supplement use. Am. J. Clin. Nutr. 1999;70:198–207. doi: 10.1093/ajcn.70.2.198. [DOI] [PubMed] [Google Scholar]
- 41.Choumenkovitch SF, Selhub J, Wilson PW, Rader JI, Rosenberg IH, et al. Folic acid intake from fortification in United States exceeds predictions. J. Nutr. 2002;132:2792–2798. doi: 10.1093/jn/132.9.2792. [DOI] [PubMed] [Google Scholar]
- 42.Matthews RG, Daubner SC. Modulation of methylenetetrahydrofolate reductase activity by S-adenosylmethionine and by dihydrofolate and its polyglutamate analogues. Adv. Enzyme Regul. 1982;20:123–131. doi: 10.1016/0065-2571(82)90012-7. [DOI] [PubMed] [Google Scholar]
- 43.Smith AD, Kim YI. Refsum H. Is folic acid good for everyone? Am. J. Clin. Nutr. 2008;87:517–533. doi: 10.1093/ajcn/87.3.517. [DOI] [PubMed] [Google Scholar]
- 44.Ulrich CM, Potter JD. Folate supplementation: too much of a good thing? Cancer Epidemiol. Biomarkers Prev. 2006;15:189–193. doi: 10.1158/1055-9965.EPI-152CO. [DOI] [PubMed] [Google Scholar]
- 45.Prinz-Langenohl R, Bramswig S, Tobolski O, Smulders YM, Smith DE, et al. [6S]-5-Methyltetrahydrofolate increases plasma folate more effectively than folic acid in women with the homozygous or wild-type 677C–>T polymorphism of methylenetetrahydrofolate reductase. Br. J. Pharmacol. 2009;158:2014–2021. doi: 10.1111/j.1476-5381.2009.00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fohr IP, Prinz-Langenohl R, Bronstrup A, Bohlmann AM, Nau H, et al. 5,10-Methylenetetrahydrofolate reductase genotype determines the plasma homocysteine-lowering effect of supplementation with 5-methyltetrahydrofolate or folic acid in healthy young women. Am. J. Clin. Nutr. 2002;75:275–282. doi: 10.1093/ajcn/75.2.275. [DOI] [PubMed] [Google Scholar]
- 47.Kelley B, Totter JR. Day PL. The lipotropic effect of folic acid on rats receiving various purified diets. J. Biol. Chem. 1950;187:529–535. [PubMed] [Google Scholar]
- 48.Burdge GC, Lillycrop KA, Phillips ES, Slater-Jefferies JL, Jackson AA, et al. Folic acid supplementation during the juvenile-pubertal period in rats modifies the phenotype and epigenotype induced by prenatal nutrition. J. Nutr. 2009;139:1054–1060. doi: 10.3945/jn.109.104653. [DOI] [PubMed] [Google Scholar]
- 49.McNeil CJ, Hay SM, Rucklidge GJ, Reid M, Duncan G, et al. Disruption of lipid metabolism in the liver of the pregnant rat fed folate-deficient and methyl donor-deficient diets. Br. J. Nutr. 2008;99:262–271. doi: 10.1017/S0007114507798999. [DOI] [PubMed] [Google Scholar]
- 50.Mojtabai R. Body mass index and serum folate in childbearing age women. Eur. J. Epidemiol. 2004;19:1029–1036. doi: 10.1007/s10654-004-2253-z. [DOI] [PubMed] [Google Scholar]
- 51.Gallistl S, Sudi K, Mangge H, Erwa W. Borkenstein M. Insulin is an independent correlate of plasma homocysteine levels in obese children and adolescents. Diabetes Care. 2000;23:1348–1352. doi: 10.2337/diacare.23.9.1348. [DOI] [PubMed] [Google Scholar]
- 52.Mahabir S, Ettinger S, Johnson L, Baer DJ, Clevidence BA, et al. Measures of adiposity and body fat distribution in relation to serum folate levels in postmenopausal women in a feeding study. Eur. J. Clin. Nutr. 2008;62:644–650. doi: 10.1038/sj.ejcn.1602771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tungtrongchitr R, Pongpaew P, Tongboonchoo C, Vudhivai N, Changbumrung S, et al. Serum homocysteine, B12 and folic acid concentration in Thai overweight and obese subjects. Int. J. Vitam. Nutr. Res. 2003;73:8–14. doi: 10.1024/0300-9831.73.1.8. [DOI] [PubMed] [Google Scholar]
- 54.Furness DL, Yasin N, Dekker GA, Thompson SD. Roberts CT. Maternal red blood cell folate concentration at 10–12 weeks gestation and pregnancy outcome. J. Matern. Fetal Neonatal Med. 2012;25:1423–1427. doi: 10.3109/14767058.2011.636463. [DOI] [PubMed] [Google Scholar]
- 55.Engeham SF, Haase A. Langley-Evans SC. Supplementation of a maternal low-protein diet in rat pregnancy with folic acid ameliorates programming effects upon feeding behaviour in the absence of disturbances to the methionine-homocysteine cycle. Br. J. Nutr. 2010;103:996–1007. doi: 10.1017/S0007114509992662. [DOI] [PubMed] [Google Scholar]
- 56.Yajnik CS, Joglekar CV, Lubree HG, Rege SS, Naik SS, et al. Adiposity, inflammation and hyperglycaemia in rural and urban Indian men: coronary risk of insulin sensitivity in Indian subjects (CRISIS) study. Diabetologia. 2008;51:39–46. doi: 10.1007/s00125-007-0847-1. [DOI] [PubMed] [Google Scholar]
- 57.Lewis SJ, Leary S, Davey Smith G. Ness A. Body composition at age 9 years, maternal folate intake during pregnancy and methyltetrahydrofolate reductase (MTHFR) C677T genotype. Br. J. Nutr. 2009;102:493–496. doi: 10.1017/S0007114509231746. [DOI] [PubMed] [Google Scholar]
- 58.Boushey CJ, Beresford SA, Omenn GS. Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA. 1995;274:1049–1057. doi: 10.1001/jama.1995.03530130055028. [DOI] [PubMed] [Google Scholar]
- 59.Verhaar MC, Stroes E. Rabelink TJ. Folates and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2002;22:6–13. doi: 10.1161/hq0102.102190. [DOI] [PubMed] [Google Scholar]
- 60.Robinson K, Arheart K, Refsum H, Brattstrom L, Boers G, et al. Low circulating folate and vitamin B6 concentrations: risk factors for stroke, peripheral vascular disease, and coronary artery disease. European COMAC Group. Circulation. 1998;97:437–443. doi: 10.1161/01.cir.97.5.437. [DOI] [PubMed] [Google Scholar]
- 61.Morrison HI, Schaubel D, Desmeules M. Wigle DT. Serum folate and risk of fatal coronary heart disease. JAMA. 1996;275:1893–1896. doi: 10.1001/jama.1996.03530480035037. [DOI] [PubMed] [Google Scholar]
- 62.Alessio AC, Santos CX, Debbas V, Oliveira LC, Haddad R, et al. Evaluation of mild hyperhomocysteinemia during the development of atherosclerosis in apolipoprotein E-deficient and normal mice. Exp. Mol. Pathol. 2011;90:45–50. doi: 10.1016/j.yexmp.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 63.Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J. Clin. Invest. 2001;107:675–683. doi: 10.1172/JCI10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McNeil CJ, Beattie JH, Gordon MJ, Pirie LP. Duthie SJ. Nutritional B vitamin deficiency disrupts lipid metabolism causing accumulation of proatherogenic lipoproteins in the aorta adventitia of ApoE null mice. Mol. Nutr. Food Res. 2012;56:1122–1130. doi: 10.1002/mnfr.201100694. [DOI] [PubMed] [Google Scholar]
- 65.McNeil CJ, Beattie JH, Gordon MJ, Pirie LP. Duthie SJ. Differential effects of nutritional folic acid deficiency and moderate hyperhomocysteinemia on aortic plaque formation and genome-wide DNA methylation in vascular tissue from ApoE-/- mice. Clin. Epigenetics. 2011;2:361–368. doi: 10.1007/s13148-011-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carnicer R, Navarro MA, Arbones-Mainar JM, Acin S, Guzman MA, et al. Folic acid supplementation delays atherosclerotic lesion development in apoE-deficient mice. Life Sci. 2007;80:638–643. doi: 10.1016/j.lfs.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 67.Toole JF, Malinow MR, Chambless LE, Spence JD, Pettigrew LC, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA. 2004;291:565–575. doi: 10.1001/jama.291.5.565. [DOI] [PubMed] [Google Scholar]
- 68.Bonaa KH, Njolstad I, Ueland PM, Schirmer H, Tverdal A, et al. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N. Engl. J. Med. 2006;354:1578–1588. doi: 10.1056/NEJMoa055227. [DOI] [PubMed] [Google Scholar]
- 69.Lonn E, Yusuf S, Arnold MJ, Sheridan P, Pogue J, et al. Homocysteine lowering with folic acid and B vitamins in vascular disease. N. Engl. J. Med. 2006;354:1567–1577. doi: 10.1056/NEJMoa060900. [DOI] [PubMed] [Google Scholar]
- 70.Bazzano LA, Reynolds K, Holder KN. He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. JAMA. 2006;296:2720–2726. doi: 10.1001/jama.296.22.2720. [DOI] [PubMed] [Google Scholar]
- 71.Ebbing M, Bleie O, Ueland PM, Nordrehaug JE, Nilsen DW, et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA. 2008;300:795–804. doi: 10.1001/jama.300.7.795. [DOI] [PubMed] [Google Scholar]
- 72.Yang Q, Cogswell ME, Hamner HC, Carriquiry A, Bailey LB, et al. Folic acid source, usual intake, and folate and vitamin B-12 status in US adults: National Health and Nutrition Examination Survey (NHANES) 2003–2006. Am. J. Clin. Nutr. 2010;91:64–72. doi: 10.3945/ajcn.2009.28401. [DOI] [PubMed] [Google Scholar]
- 73.Devlin AM, Singh R, Wade RE, Innis SM, Bottiglieri T, et al. Hypermethylation of Fads2 and altered hepatic fatty acid and phospholipid metabolism in mice with hyperhomocysteinemia. J. Biol. Chem. 2007;282:37082–37090. doi: 10.1074/jbc.M704256200. [DOI] [PubMed] [Google Scholar]
- 74.Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- 75.Sie KK, Li J, Ly A, Sohn KJ, Croxford R, et al. Effect of maternal and postweaning folic acid supplementation on global and gene-specific DNA methylation in the liver of the rat offspring. Mol. Nutr. Food Res. 2013;57:677–685. doi: 10.1002/mnfr.201200186. [DOI] [PubMed] [Google Scholar]
- 76.Iizuka K, Horikawa Y. ChREBP: a glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr. J. 2008;55:617–624. doi: 10.1507/endocrj.k07e-110. [DOI] [PubMed] [Google Scholar]