

Abstract
Context:
The optimal approach to estrogen replacement in girls with Turner syndrome has not been determined.
Objective:
The aim of the study was to assess the effects of an individualized regimen of low-dose ethinyl estradiol (EE2) during childhood from as early as age 5, followed by a pubertal induction regimen starting after age 12 and escalating to full replacement over 4 years.
Design:
This study was a prospective, randomized, double-blind, placebo-controlled clinical trial.
Setting:
The study was conducted at two US pediatric endocrine centers.
Subjects:
Girls with Turner syndrome (n = 149), aged 5.0–12.5 years, were enrolled; data from 123 girls were analyzable for pubertal onset.
Intervention(s):
Interventions comprised placebo or recombinant GH injections three times a week, with daily oral placebo or oral EE2 during childhood (25 ng/kg/d, ages 5–8 y; 50 ng/kg/d, ages >8–12 y); after age 12, all patients received escalating EE2 starting at a nominal dosage of 100 ng/kg/d. Placebo/EE2 dosages were reduced by 50% for breast development before age 12 years, vaginal bleeding before age 14 years, or undue advance in bone age.
Main Outcome Measures:
The main outcome measures for this report were median ages at Tanner breast stage ≥2, median age at menarche, and tempo of puberty (Tanner 2 to menarche). Patterns of gonadotropin secretion and impact of childhood EE2 on gonadotropins also were assessed.
Results:
Compared with recipients of oral placebo (n = 62), girls who received childhood low-dose EE2 (n = 61) had significantly earlier thelarche (median, 11.6 vs 12.6 y, P < 0.001) and slower tempo of puberty (median, 3.3 vs 2.2 y, P = 0.003); both groups had delayed menarche (median, 15.0 y). Among childhood placebo recipients, girls who had spontaneous breast development before estrogen exposure had significantly lower median FSH values than girls who did not.
Conclusions:
In addition to previously reported effects on cognitive measures and GH-mediated height gain, childhood estrogen replacement significantly normalized the onset and tempo of puberty. Childhood low-dose estrogen replacement should be considered for girls with Turner syndrome.
Turner syndrome (TS), which results from partial or complete X-chromosome monosomy, occurs in approximately 1/2000 live female births (1). Ovarian dysgenesis, reported in approximately 90% of affected individuals (2, 3), results in estrogen deficiency that begins in infancy (4–7). Healthy prepubertal ovaries secrete low but measurable amounts of estradiol (8–10), and estrogens have wide-ranging physiological effects on numerous tissues (11–16). Thus, prolonged estrogen deficiency in girls with TS throughout the critical phases of childhood growth and development may have detrimental effects across many body systems.
Based on different dose-response characteristics for growth vs vaginal maturation in our early studies (17, 18), we postulated that ultralow-dose, physiological estrogen replacement during childhood might have potential benefits in TS, such as optimizing growth response to supplemental GH, normalizing pubertal timing, and improving cognition and behavior. We therefore conducted a randomized, double-blind, placebo-controlled clinical trial of GH and low-dose ethinyl estradiol (EE2) initiated during childhood (CLDE) in a large cohort of girls with TS. The effects of this regimen on the primary endpoint of adult height have been published (19). In the present report we describe the effects of individualized childhood estrogen replacement starting as early as 5 years of age, followed by an escalating EE2 pubertal induction regimen, on the timing and tempo of puberty and gonadotropin secretion in girls with TS. Because of its unique placebo-controlled childhood phase, this study provides data for two distinct estrogen regimens, one that had a childhood replacement component, and one that began pubertal induction after age 12, without childhood replacement. We also determined the prevalence of spontaneous breast development before age 12 in the childhood-placebo recipients in this cohort.
Sex steroid replacement for girls with TS remains an area of active investigation (20–24), and there is no consensus regarding optimal approaches in terms of dosage, type, and route of administration (oral, im, transdermal), or age of initiation. Our data provide novel insights into important aspects of childhood estrogen deficiency and replacement in TS.
Patients and Methods
Patients
Study entry criteria included karyotype diagnosis of TS (without Y-chromosome material), chronological age 5–12 years, bone age ≤12 years, height ≤10th percentile (25), Tanner breast stage 1–2 (B1–B2), adequate thyroid hormone replacement for ≥3 months in patients with hypothyroidism, absence of clinically relevant systemic illness, no recent or concurrent treatment that might influence growth, and written informed consent from parent(s)/guardian(s).
Methods
The study design has been reported in detail (19). Methods relevant to the current analyses are provided here.
Study treatments and procedures
All patients received a daily oral liquid (either placebo or EE2)1,2 and sc injected placebo or GH (Humatrope; Eli Lilly and Company), 0.1 mg/kg/injection, three times per week (0.3 mg/kg/wk). Thus, the four treatment groups in this 2 × 2 factorial design were: 1) placebo injection with childhood oral placebo (P/P); 2) placebo injection with childhood oral low-dose EE2 (P/E); 3) GH injection with childhood oral placebo (GH/P); and 4) GH injection with childhood oral low-dose EE2 (GH/E).
Protocol-specified dosages of EE2 (or its placebo equivalent, ages 5–12) were: 5–8 years, 25 ng/kg/d; >8–12 years, 50 ng/kg/d; >12–14 years, 100 ng/kg/d; >14–15 years, 200 ng/kg/d; >15–16 years, 400 ng/kg/d; and >16 years, 800 ng/kg/d. The 25- and 50-ng/kg/d doses represented the childhood phase of the study and were not intended to induce pubertal development. The escalating estrogen regimen for pubertal induction began at the first visit after the 12th birthday (all groups).
To individualize EE2 dosages for appropriate clinical reasons, the oral medication could be reduced (without unblinding the treatment group) by 50% at 6-month intervals for breast development ≥B2 before age 12, vaginal bleeding before age 14, bone age advancement of 2 years/chronological year, or bone age greater than chronological age before age 14. Reduced dosages were doubled at each subsequent transition; thus, dose reductions carried forward until attainment of menarche. Cyclic therapy with EE2 and progestin (medroxyprogesterone acetate for 10 d/mo or an oral contraceptive containing 30 μg of EE2) was introduced after menarche.
Evaluation at each 6-month study visit included pubertal assessment (26), bone age x-ray, and single measurements of LH and FSH between 8 am and noon.
Analysis populations and treatment groups
Baseline demographic and postbaseline safety data were analyzed for all 149 girls randomized in the study (Randomized Population; Supplemental Figure 1). The prospectively defined population to assess pubertal effects of childhood estrogen exposure comprised girls who were <12 years of age and prepubertal at study entry and had at least one postbaseline breast stage assessment (Pubertal Analysis Population).
In background analyses to assess the potential effect of GH on pubertal development using Cox proportional hazards models with baseline age as covariate, we found no significant effect of GH treatment vs placebo on age at Tanner breast stage ≥2 (P = .87). Similarly, in a comparison among the four randomized treatment groups using Cox models with effects for GH treatment, estrogen treatment, and the interaction between GH and estrogen, statistical significance was found only for the effect of estrogen (P < .05). Therefore, to assess effects of CLDE vs childhood oral placebo (COP), the two groups randomized to CLDE were pooled (combining patients who received placebo and GH injections [P/E+GH/E]) and compared with the pooled group of patients who received COP (combining patients who received placebo and GH injections [P/P+GH/P]).
Analysis variables and time points
The main developmental variables analyzed were ages at thelarche (defined as breast development ≥B2), each subsequent breast stage (B3–B5) and menarche, and tempo of puberty (B2 to menarche). Because some girls had early breast development that regressed after study drug dose reduction, age of thelarche was analyzed in two ways—first, as the earliest recorded observation of ≥B2, irrespective of subsequent regression to B1; and, second, for the girls in whom transient early breast development had regressed, as the age of sustained development ≥B2.
Bone age was determined according to the standards of Greulich and Pyle (27) by a central reader blinded to patient treatment. Serum LH and FSH concentrations were measured by RIA (Hazelton/Covance Laboratories). Values >15 IU/L for LH and >20 IU/L for FSH (∼2 SD above normative means) were designated as (28).
Gonadotropin data were analyzed at three time points: 1) baseline (pretreatment; all patients pooled), reflecting the natural history of gonadotropin secretion in TS at ages 5–12; 2) postbaseline during the childhood phase (COP vs CLDE), reflecting effects of CLDE at ages 5–12; and 3) during pubertal development after age 12, when all participants received the pubertal induction regimen.
Statistical methods
Data are reported as median (range) for continuous variables and as frequency [number (percentage)] for categorical variables. Age-related EE2 dosages are reported as mean ± SD, and EE2 dosage reductions are reported as frequency [number (percentage)] at each dosage level. Because all continuous outcome variables were normally distributed, P values are based on comparisons between pooled groups (COP vs CLDE) using ANOVA for baseline variables and analysis of covariance for postbaseline variables, with baseline age as covariate. Differences between median gonadotropin values were analyzed using Wilcoxon rank-sum tests. Between-group frequency differences were assessed by χ2 tests. Time-to-thelarche and tempo of puberty (thelarche-to-menarche) were estimated using Kaplan-Meier analyses adjusted for baseline age; between-group differences for time-to-event variables were assessed using Cox regression models with baseline age as covariate. Two-sided statistical tests were conducted with significance set at 0.05. Analyses were performed using SAS version 8.2 (SAS Institute, Inc).
Results
Baseline characteristics
Of 149 girls randomized, 123 fulfilled criteria for analysis of puberty (COP, n = 62; CLDE, n = 61). The 26 randomized patients excluded from the Pubertal Analysis Population comprised 10 girls who were >B1 at baseline; 11 girls who, although prepubertal, were ≥12 years old and therefore bypassed the childhood phase of the study; and five girls who discontinued without a postbaseline visit.
In the Pubertal Analysis Population, the CLDE group was somewhat older at baseline than the COP group (∼0.7 y chronological age [P = .035]; ∼0.8 y bone age [P = .077]; Table 1). There were no other significant baseline differences between treatment groups.
Table 1.
Baseline Clinical Characteristics of Patients in the Randomized Population and the Pubertal Analysis Population
| Pooled Treatment Group | Randomized Population, n = 149 |
Pubertal Analysis Population, n = 123 |
||||
|---|---|---|---|---|---|---|
| Childhood Oral Placebo (n = 74) | Childhood Low-Dose Estrogen (n = 75) | P Valuea | Childhood Oral Placebo (n = 62) | Childhood Low-Dose Estrogen (n = 61) | P Valuea | |
| Variable | ||||||
| Chronological age, y | 7.5 (5.0 to 12.5) | 8.5 (5.0 to 12.5) | .013 | 7.2 (5.0 to 11.9) | 7.9 (5.0 to 11.9) | .035 |
| Bone age, y | 6.8 (2.0 to 13.0) | 7.8 (2.5 to 12.0) | .076 | 6.0 (2.0 to 10.0) | 6.8 (2.5 to 11.5) | .077 |
| Bone age delay, y | −1.2 (−4.3 to 1.6) | −1.4 (−4.1 to 1.5) | .639 | −1.3 (−3.7 to 1.6) | −1.3 (−4.1 to 1.5) | .957 |
| Height SDS | −2.6 (−5.2 to −1.0) | −2.8 (−5.1 to −1.3) | .153 | −2.5 (−5.2 to −1.0) | −2.7 (−4.6 to −1.4) | .426 |
| Weight SDS | −1.4 (−5.0 to 2.9) | −1.6 (−4.2 to 2.1) | .270 | −1.4 (−5.0 to 2.9) | −1.5 (−4.2 to 2.1) | .395 |
| Body mass index SDS | 0.6 (−4.0 to 3.3) | 0.3 (−1.2 to 2.7) | .531 | 0.5 (−4.0 to 3.3) | 0.3 (−1.2 to 2.7) | .640 |
| 45,X karyotype, % | 76 | 71 | .547 | 82 | 77 | .663 |
Values shown are median (minimum to maximum). Childhood oral placebo group = oral placebo/placebo injection group + oral placebo/GH injection group; childhood low-dose estrogen group = childhood low-dose estrogen/placebo injection group + childhood low-dose estrogen/GH injection group; bone age delay = bone age minus chronological age; SDS = SD score.
P values are based on ANOVA models for continuous variables, and χ2 test for percentage 45,X karyotype.
Pubertal development
Of 123 girls in the Pubertal Analysis Population, 101 (82%) attained ≥B2 during the study (COP, n = 49; CLDE, n = 52). Of 49 girls in the COP group for whom ≥B2 was documented, thelarche was observed before age 12 (before exogenous estrogen exposure) in 14 (29%); four of the 14 had chromosomal mosaicism (45,X/46,XX). Median (range) serum FSH values over the course of the study were significantly lower for girls who had spontaneous breast development than for those who did not (39 [1–454] vs 58 [1–420] IU/L, respectively; P = .003). However, median LH values were not significantly different (10 [1–103] vs 13 [0–160]; P = .09). Transient development ≥B2 between 6.0 and 11.4 years of age, followed by regression to B1 after dosage reduction, was documented for 18 girls (COP, 4 of 49; CLDE, 14 of 52). The first pubertal breast stage recorded was ≥B3 for 22 of 101 girls (22%).
In general, CLDE-treated girls attained thelarche earlier than those who received COP. In the analysis that included girls who had initial transient breast development, median ages at ≥B2 were 12.6 and 11.1 years for the COP vs CLDE groups, respectively (P < .001; Table 2). Median ages at sustained ≥B2 were 12.6 and 11.6 years, respectively (P < .001, Table 2 and Figure 1A), with corresponding median bone ages 12.0 and 10.0 years (P = .003; Table 2). Of girls in the CLDE group, 14 of 52 (27%) achieved sustained ≥B2 by 10.4 years of age (median age of B2 in US non-Hispanic white girls [Ref. 29]), compared with two of 49 (4%) of the COP group.
Table 2.
Onset and Progression of Puberty for Patients in Pubertal Analysis Population
| Pooled Treatment Group | Childhood Oral Placebo (n = 62)a | Childhood Low-Dose Estrogen (n = 61)a | P Valueb |
|---|---|---|---|
| Variable, y | |||
| Chronological age at study entry | 7.22 (5.00–11.89) | 7.93 (5.02–11.91) | .035 |
| Bone age at study entry | 6.00 (2.00–10.00) | 6.83 (2.50–11.50) | .077 |
| Age at transient Tanner breast stage ≥2 | 12.56 (5.95–14.36) | 11.07 (6.14–15.39) | <.001 |
| Age at sustained Tanner breast stage ≥2 | 12.62 (9.69–14.44) | 11.64 (6.73–15.39) | <.001 |
| Bone age at Tanner breast stage ≥2 | 12.00 (8.83–14.50) | 10.00 (6.83–14.00) | .003 |
| Age at Tanner breast stage ≥3 | 13.29 (10.44–15.69) | 13.15 (8.70–15.90) | .086 |
| Age at Tanner breast stage ≥4 | 14.00 (11.69–15.91) | 14.27 (8.70–17.06) | .463 |
| Age at Tanner breast stage 5 | 14.96 (11.69–16.62) | 15.20 (11.63–17.05) | .997 |
| Age at menarche | 15.00 (11.00–16.00) | 15.00 (10.40–17.00) | .986 |
| Time from baseline to Tanner breast stage ≥2 | 5.08 (0.51–8.50) | 1.95 (0.44–8.55) | .040 |
| Time from baseline to menarche | 7.20 (3.11–10.00) | 5.58 (2.16–10.02) | .222 |
| Time from Tanner breast stage ≥2 to menarche | 2.16 (0.25–5.61) | 3.33 (0.92–6.42) | .003 |
| Time from Tanner breast stage ≥2 to Tanner breast stage 5 | 2.54 (0.49–4.03) | 3.01 (1.51–6.59) | .104 |
Values are expressed as median (minimum-maximum) in years. Age at Tanner breast stage ≥2 reflects the fact that the precise timing of Tanner 2 was not observed for some girls, as the first recorded post-baseline stage was ≥B3; the analyses were performed in the same fashion for Tanner breast stages 3 and 4.
Numbers shown for pooled treatment groups are the maximum numbers; numbers of patients with data available at each specific stage are provided in the patient flow diagram (Supplemental Figure 1).
P values for baseline variables were obtained from ANOVA; P values for comparison of ages at specific stages were obtained from analysis of covariance with baseline age as covariate; P values for time to event variables were obtained from Cox regression models with baseline age as covariate.
Figure 1.
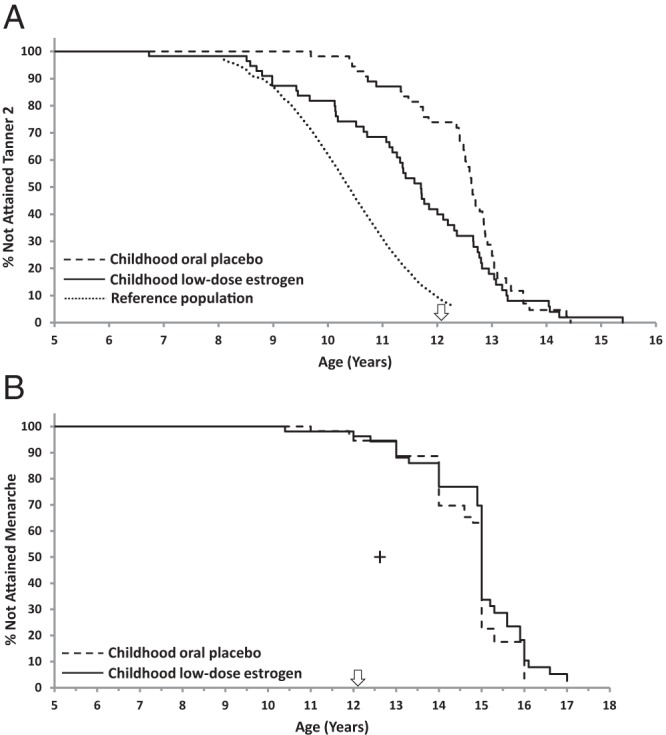
Kaplan-Meier analysis of ages at Tanner stage ≥2 breast development and menarche. A, Kaplan-Meier plot of age at attainment of Tanner stage ≥2 breast development. Starting at approximately 8.5 years of age, the curve for girls who received childhood low-dose estrogen is shifted to the left, indicating significantly earlier thelarche (P < .001), compared with that for girls who received oral placebo during childhood with initiation of pubertal estrogen replacement at the first study visit after age 12. All girls received escalating dosages of EE2 for pubertal induction from the first visit after the 12th birthday (arrow). The dotted line represents the curve for general population standards for attainment of Tanner breast stage 2, based on data from the US National Health and Nutrition Examination Survey III (29). B, Kaplan-Meier analysis of age at attainment of menarche; the curves for the two groups are not significantly different (P = .986). All girls received escalating dosages of EE2 for pubertal induction from the first visit after the 12th birthday (arrow). Median age at menarche for girls in the US general population (12.6 years [Ref. 51]) is shown by the cross.
By Kaplan-Meier analysis of age at thelarche, there was no divergence between groups before 8.5 years of age (Figure 1A), indicating minimal breast effect of the lowest EE2 dosage of 25 ng/kg/d (childhood phase; mean daily EE2 dose, ∼0.4 μg; Figure 2). However, from ages 8–12 years, when only the CLDE group received EE2 (nominally 50 ng/kg/d; mean daily EE2 dose, ∼0.7 μg), the patterns diverged, with the CLDE curve shifted to the left, closer to that of the reference population, indicating earlier thelarche. The curves converged again after age 13, when all girls received escalating EE2 for pubertal induction (Figure 1A).
Figure 2.
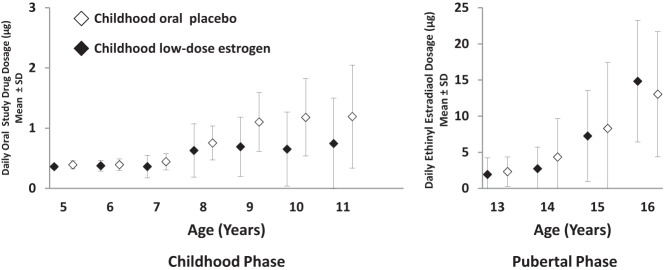
Oral study drug dosages in micrograms per day (mean ± SD) by integer age for patients receiving oral placebo during childhood or low-dose EE2 during childhood, and escalating dosages of EE2 during pubertal induction. During the childhood phase of the study (left), patients in the oral placebo group (open symbols) received no active oral medication, whereas those in the low-dose estrogen group (closed symbols) received EE2 at the mean daily dosages indicated on the vertical axis. The protocol-specified dosages in ng/kg/d were 5–8 years, 25 ng/kg/d; >8–12 years, 50 ng/kg/d. During the pubertal induction phase (right) all girls received escalating dosages of EE2 based on age and weight: protocol-specified dosages in ng/kg/d were >12–14 years, 100 ng/kg/d; >14–15 years, 200 ng/kg/d; >15–16 years, 400 ng/kg/d; >16 years, 800 ng/kg/d. Note that the scale on the vertical axis differs for the childhood and pubertal phases of the study.
Although the CLDE group had earlier thelarche, the median age at menarche, ascertained for 81 (COP, n = 40; CLDE, n = 41) of 101 girls (80%) for whom age at thelarche was available, was 15 years for both groups (Table 2 and Figure 1B) at EE2 dosages of 8.3 ± 9.1 μg/d and 7.3 ± 6.3 μg/d for the pooled COP and CLDE groups, respectively (Figure 2). Earliest ages at menarche were 11.0 years in the COP group and 10.4 years in the CLDE group. Because B2 was earlier for the CLDE group, but age at menarche was the same for both groups, the duration of puberty was longer (ie, tempo of puberty was slower) for the CLDE group: 3.3 vs 2.2 years (P = .003 [Cox]; Table 2).
Median ages at ≥B3, ≥B4, and B5 were not significantly different between groups (Table 2 and Supplemental Figure 2).
Oral study drug dosage reductions
Overall, 69 of 123 girls (56%) underwent one or more oral study drug dosage reductions (COP, 31 of 62 [50%]; CLDE, 38 of 61 [62%]; P = .21). Because the intent of estrogen therapy was different for the childhood and pubertal study phases, the dose individualization data are described separately for each phase.
Childhood phase
During the placebo-controlled childhood phase (ages 5–12; nominally EE2 25 or 50 ng/kg/d [less after dose individualization]), more girls in the CLDE group (33 of 61 [54%]) than in the COP group (21 of 62 [34%]; P = .030) underwent dosage reductions for protocol-specified reasons, primarily premature breast development at the 50-ng/kg/d EE2 dosage (21 of 46 [46%] CLDE vs 6 of 55 [11%] COP; P < .001; Table 3). One girl in the CLDE group underwent dosage reduction for premature vaginal bleeding (5- to 8-y age group, EE2 25 ng/kg/d; Table 3). Five girls in each group underwent EE2 reductions for non-protocol-specified reasons such as headache, emotional/behavior change, or other reason. Notably, eight of 62 (13%) girls in the COP group (seven of whom had 45,X karyotype) underwent reduction of their placebo dosage for early breast development before they had received exogenous estrogen treatment.
Table 3.
Summary of Reasons for Oral Study Drug Dosage Reductions: Number (Percentage) of Patients With Dosage Reductions by Treatment Group in Pubertal Analysis Population
| Reason for Dosage Reduction | Childhood Oral Placebo (n = 62)a | Childhod Low-Dose Estrogen (n = 61)a | P Value |
|---|---|---|---|
| Premature breast developmentb | 10 (16) | 26 (43) | .001 |
| 5–8 y (25 ng/kg/d) | 2/40 (5) | 5/31 (16) | .227 |
| >8–12 y (50 ng/kg/d) | 6/55 (11) | 21/46 (46) | <.001 |
| >12–14 y (100 ng/kg/d) | 2/29 (7) | 0/17 (0) | .253 |
| >14–15 y (200 ng/kg/d) | 0/12 (0) | 0/11 (0) | 1.000 |
| Premature vaginal bleedingb | 1 (2) | 6 (10) | .062 |
| 5–8 y (25 ng/kg/d) | 0/40 (0) | 1/31 (3) | .437 |
| >8–12 y (50 ng/kg/d) | 0/55 (0) | 0/46 (0) | 1.000 |
| >12–14 y (100 ng/kg/d) | 1/29 (3) | 4/17 (24) | .055 |
| >14–15 y (200 ng/kg/d) | 0/12 (0) | 1/11 (9) | .478 |
| Bone age advanceb | 20 (32) | 6 (10) | .004 |
| 5–8 y (25 ng/kg/d) | 2/40 (5) | 2/31 (7) | 1.000 |
| >8–12 y (50 ng/kg/d) | 11/55 (20) | 4/46 (9) | .161 |
| >12–14 y (100 ng/kg/d) | 7/29 (24) | 0/17 (0) | .036 |
| >14–15 y (200 ng/kg/d) | 0/12 (0) | 0/11 (0) | 1.000 |
| Non-protocol-specified reasonsc | |||
| Headache | 3 (5) | 4 (7) | .717 |
| Emotion/mood/behavior change | 1 (2) | 3 (5) | .365 |
| Other | 5 (8) | 2 (3) | .439 |
| Overall patients who had dose reduced for protocol-specified reasons, by age/dosage group | |||
| 5–8 y (25 ng/kg/d) | 4/40 (10) | 8/31 (26) | .112 |
| >8–12 y (50 ng/kg/d) | 17/55 (31) | 25/46 (54) | .025 |
| >12–14 y (100 ng/kg/d) | 10/29 (35) | 4/17 (24) | .520 |
| >14–15 y (200 ng/kg/d) | 0/12 (0) | 1/11 (9) | .478 |
| Total | 31/62 (50) | 38/61 (62) | .205 |
Values are expressed as number (percentage) or number/number in age group (percentage). Childhood oral placebo = P/P + GH/P groups; childhood low-dose estrogen = P/E + GH/E groups. The following were prespecified in the protocol as reasons for oral study drug dosage reduction: breast development ≥ Tanner stage 2 before age 12 years; vaginal bleeding before age 14 years; bone age advancement of 2 years per chronological year up to age 14 years. Other events that led to dosage reductions (not prespecified) included headache and emotion, mood, or behavior changes. The reasons for dose reduction presented in this analysis were the primary reasons noted by the investigators on the case report forms; however, in some cases, additional reasons were reported in the comments. Only the first event leading to dosage reduction is reported so that each patient is counted only once. Patients who underwent a dosage reduction at any time typically continued to receive lower than protocol-specified dosages thereafter. P values are derived from Fisher exact tests; data are not shown for patients in age groups older than 15 years because there were no significant differences between treatment groups for any of the events of interest.
Numbers represent maximum for each pooled treatment group.
Denominators (number in age group) represent number of patients who received the specified dose without prior dosage reduction, so the total across age groups is greater than total patient number within the pooled treatment group.
Total number of patients in each group with reductions for non-protocol-specified reasons was five because some patients had multiple reasons reported.
Pubertal phase
During the pubertal phase (escalating EE2 dosages for all patients), the pattern of dosage reductions was the converse of that seen in the childhood phase, with most reductions in the COP group, who had transitioned from oral placebo to EE2 after age 12.
Most dosage reductions in this group were made for accelerated bone maturation during the nominal 100 ng/kg/d EE2 period (COP, 7 of 29 [24%] vs CLDE, 0 of 17 [0%]; P = .036). Dosage reductions for vaginal bleeding before age 14 were implemented for four of 17 (24%) girls in the CLDE group vs one of 29 (3%) girls in the COP group (P = .055).
Bone age progression
Baseline bone age was delayed compared with chronological age by a median of 1.3 years in both groups. Median bone age remained commensurate with chronological age, although slightly delayed, in both treatment groups throughout, with minor divergence between the CLDE and COP groups from ages 6 to 10 and nearly identical median values from ages 11 to 16 (Supplemental Figure 3).
Gonadotropins3
Effects of childhood estrogen replacement on gonadotropins
During the childhood phase, there were few elevated LH values in either group before age 10 (Figure 3). At ages 10–12, LH values were elevated for 30–82% of girls, with trends toward greater proportions of elevated values in the COP group (Figure 3B). The pattern was more striking for FSH, with significantly greater proportions of elevated values for COP vs CLDE from ages 8–11. Overall, gonadotropins were elevated in most girls in both treatment groups from age 10 (FSH) or 11 (LH).
Figure 3.

Effect of low-dose estrogen on gonadotropins. A, Scatter plots of LH values (left, circles) and FSH values (right, diamonds) by age during the childhood phase of the study (multiple study visits and values per patient). Upper panels (open symbols) display values for girls who received oral placebo during childhood; lower panels (closed symbols) display values for girls who received childhood low-dose estrogen. All girls received escalating dosages of EE2 for pubertal induction from the first visit after the 12th birthday. Solid lines represent lines of best fit for the data. To avoid compressing the vertical axis, FSH values above 200 IU/L are not shown. Dashed lines represent the designated upper limit of normal (2 SD values above the mean for the relevant assays). B, Proportions of values by treatment group above upper limit of normal at each integer age for LH (15 IU/L, upper graph) and FSH (20 IU/L, lower graph); P values <.1 for differences between pooled treatment groups are provided. At ages 8–11 years (childhood phase), a significantly greater proportion of FSH values were supranormal for girls who received oral placebo (open bars) vs those who received low-dose estrogen during childhood (filled bars). During the latter stages of the pubertal induction phase there was a nonsignificant trend toward greater proportions of supranormal LH and FSH values in the original childhood low-dose estrogen recipients, likely as a result of lower EE2 dosages in this group during the pubertal phase of the study, due to prior estrogen dose reductions during childhood. Because 80% of patients had 45,X karyotype, no separate analysis was performed for patients with chromosomal mosaicism.
Effects of pubertal estrogen replacement on gonadotropins
During pubertal induction, mean EE2 dosages were somewhat lower at ages 13–15 for girls who had received CLDE because dose reductions during the childhood phase carried into the pubertal phase (Figure 2). Consequently, in the older teenage years, median (interquartile range) values for LH and FSH trended higher for the CLDE group than for the former placebo recipients (Figure 3B, Supplemental Figure 5). Although LH and FSH values declined by ages 16–17, when nearly all girls had transitioned to cyclic estrogen-progestin treatment after menarche, median values for both gonadotropins remained elevated in both groups (Supplemental Figure 5).
Discussion
Ovarian failure is a cardinal feature of TS (2, 20), and whereas the primary clinical manifestation of ovarian dysgenesis is absent or incomplete secondary sexual development, the estrogen deficiency in TS begins in infancy (4–7) and likely has significant consequences for health and well-being during childhood. Estrogen replacement to induce pubertal development has been the standard of care for TS since the 1960s (30, 31). However, because of long-held concerns regarding the role of estrogen in advancement of skeletal maturation and cessation of linear growth, estrogen was often delayed until the mid-teen years (3, 32, 33). Furthermore, although it has been known since the mid-1990s that healthy prepubertal ovaries secrete measurable amounts of estradiol (8), the concept of childhood estrogen replacement has been largely unexplored. The current report provides novel information on the pubertal development of girls with TS who received a unique low-dose, individualized estrogen regimen beginning as early as age 5. Compared to girls who began estrogen after age 12, those who received childhood estrogen had significantly earlier thelarche (median age, 11.6 vs 12.6 y) and a correspondingly slower tempo of puberty (3.3 vs 2.2 y). However, there were no significant between-group differences for median ages at later breast stages, menarche, or for bone age progression overall.
Although childhood EE2 treatment was not intended to induce feminization, the 50-ng/kg/d EE2 dosage in fact did stimulate breast development because approximately half of the girls in the CLDE group attained thelarche before the pubertal induction phase of the protocol after age 12. While this finding might be interpreted as rationale for maintaining the 25 ng/kg/d dosage until age 12, we propose that the general principle of increasing the estrogen dosage at around age 8 remains physiologically appropriate, as long as breast and bone maturation are carefully monitored and such increases are carefully individualized. Furthermore, lacking data on outcomes of a persistent 25-ng/kg/d dosage until initiation of pubertal replacement, we have no evidence on which to base alternative recommendations. The median 1.2-year delay in thelarche vs median age of 10.4 years for typical US non-Hispanic white girls (29) reflects our intent to minimize breast development before age 12, as evidenced by dosage reductions in 46% of girls for breast development between ages 8 and 12. However, when analysis of thelarche included the earliest observations of B2 (transient B2 for 14 CLDE girls before dosage reduction), the median age of 11.1 years was closer to that of the general population. In addition, because the girls were examined only twice per year, and because the initial pubertal stage observed was ≥B3 for 22% of girls, actual ages at thelarche were likely somewhat earlier than our estimates. In the only prior study of childhood estrogen replacement as early as age 5 in TS, mean ages at B2 in girls who received approximately 50 to 75 ng/kg/d EE2 from age 5, with or without GH, were 11.3 and 11.7 years, respectively, compared to mean 12.5 years for GH-treated girls whose EE2 initiation was delayed until after age 12 (34).
Despite the somewhat delayed median age of thelarche, 27% of childhood estrogen recipients attained this milestone by the median age for typical US girls (29). The near-normalization of thelarche timing for many girls resulting from childhood estrogen treatment may have psychosocial benefits, given the reported associations between breast development and positive body image/social adjustment in typical girls (35) and in girls with TS (2, 20, 36, 37). Girls with TS are at increased risk for low self-esteem and social isolation (36, 38–43), and a large French study found that delayed puberty negatively influenced subsequent sexual function (36). Although our study did not evaluate the psychosocial effects of estrogen replacement during childhood, we previously demonstrated improved self-image and psychological well-being in 12- to 16-year-old girls during 3 years of EE2 treatment in the pubertal induction phase (44). In addition, prospectively planned interim analyses from this study demonstrated improved verbal and nonverbal memory in 7- to 9-year-old CLDE recipients (45) and improved nonverbal processing speed and motor performance in 10- to 12-year-olds (46). Taken together, previous observations of the negative impact of delayed feminization, our earlier findings of childhood estrogen-induced cognitive improvements (45, 46), modest (2.1 cm) enhancement of adult height (19), and near-normalization of age of thelarche in the current analysis, all support the potential benefit of childhood estrogen replacement therapy in girls with TS.
In addition to elucidating the effects of childhood estrogen replacement, the placebo-controlled study design allowed prospective evaluation of the prevalence of spontaneous breast development before age 12. Among childhood placebo recipients for whom ≥B2 was observed, 29% attained thelarche before exposure to exogenous estrogen, indicating a substantial prevalence of residual ovarian function, consistent with previous observations (36, 47–49).
The placebo-controlled childhood phase of the study was followed by a carefully designed pubertal induction regimen (with prespecified clinical parameters for dosage reduction) starting after age 12 at 100 ng/kg/d of EE2 (the dose previously shown to maximize growth stimulation [Ref. 17]), followed by successive dosage doublings from ages 14–16. This regimen stimulated breast development at somewhat later ages than typical girls (29, 50), and age at menarche, which was 15 years in both groups, was substantially delayed compared with median age of 12.6 years for US non-Hispanic white girls (51). This delay reflects, at least in part, our intent to restrain menarche until after age 14 to optimize estrogen dosage for growth (19).
Despite near-normalization of thelarche in CLDE-treated girls, bone age remained somewhat delayed relative to chronological age throughout childhood. In contrast, exposure to estrogen starting at 100 ng/kg/d after age 12 in the COP group was accompanied by advances in bone maturation and resultant dosage reductions for almost one-fourth of girls (vs no girls in the CLDE group). This difference may be partially explained by the fact that many CLDE recipients had earlier dosage reductions and by age 12 may have been individualized to a dosage that would avoid later bone age acceleration. Alternatively, low-dose estrogen during childhood may have conditioned the growth plate to mature at a more normal rate in response to the escalating estrogen dosages at puberty.
In addition to elucidating the effects of childhood estrogen on physical maturation, this study provides unique longitudinal data, beginning as early as age 5, regarding exogenous estrogen effects on gonadotropins in a large cohort of girls with TS. There was little effect of CLDE on LH secretion, as evidenced by the similar proportions of supranormal LH values in both treatment groups and the failure of CLDE to suppress the progressive rise in LH after age 10. In contrast, the proportion of girls with elevated FSH values was significantly greater for the placebo group than the CLDE group from ages 8–11 years, likely reflecting partial negative feedback of estrogen on FSH secretion. After age 12, when both treatment groups received escalating EE2 dosages (slightly lower in the CLDE group due to prior dose individualization), LH and FSH concentrations remained persistently elevated in both groups (albeit declining by about 50% at the highest EE2 dosages), suggesting inadequate negative feedback by inhibin, estrogen, or both.
This study has certain limitations. First, although CLDE was beneficial for the timing of thelarche, memory (45), motor performance (46), and adult height (19), the doubling of the dosage to 50 ng/kg/d at age 8, whereas consistent with evidence for a similar estrogen rise in typical prepubertal girls (10), resulted in dose reductions for almost half the girls. These childhood dose reductions, when carried over into the pubertal phase, unintentionally caused somewhat lower mean estrogen dosages after age 12 for the CLDE group compared to the COP group. Consequently, it is not possible to clearly distinguish the degree to which differences observed after age 12 resulted from estrogen exposure during childhood vs somewhat lower estrogen dosages during the pubertal age range in the CLDE group. Second, the EE2 tailoring regimen was reactive, rather than proactive, because changes were made on the basis of clinical signs of estrogen effect, rather than measured estrogen concentrations (not feasible when this study was conducted and unlikely to have been useful, given a lack of clinical correlation for serum EE2 concentrations). Third, the estrogen-mediated benefits for memory, nonverbal processing speed, and motor performance described in interim reports from this study were not re-examined during the pubertal phase, and the study contained no provision for longer-term assessment of cognitive function, quality-of-life or behavioral measures. Thus, whether the cognitive benefits observed during childhood estrogen treatment translated into long-term advantages is unknown. Fourth, some investigators have proposed that transdermal estrogen administration is more physiological than oral administration (20, 52–54). However, studies in children comparing outcomes of oral vs transdermal estrogens have yielded inconsistent results, likely due to differences in study design, formulations, dosages, treatment duration, and clinical endpoints (53, 55–57). A pilot study in 12 girls with TS demonstrated improved bone mineralization and uterine growth after 1 year in girls treated with transdermal 17-β-estradiol (the principal estrogen receptor ligand), compared with those who received conjugated equine estrogen (56). In contrast, a randomized study that titrated oral and transdermal 17-β-estradiol dosages to targeted serum estradiol concentrations in 40 girls found no differences in metabolic or clinical measures between oral and transdermal treatment (57). To date, this is the only study to have compared outcomes using the same form of estrogen after achieving equivalent serum concentrations with each route of administration.
Although the outcomes of the present study may provide sufficient reason to consider childhood estrogen replacement in TS, the full impact of childhood estrogen deficiency and its correction has only begun to be explored. Other areas for which estrogens may have potential physiological roles during childhood include glucose homeostasis (14), lipid metabolism (58), hypothalamic-pituitary function (13, 59–61), liver function (62), cardiovascular function (15), skeletal development and mineralization (12, 63–65), hearing (66), cognition (16, 67), social interactions (68, 69), and sexuality (36, 70, 71). Because of the broad range of estrogen-mediated physiological effects in adults and the demonstrated effects of childhood estrogen replacement on the endpoints studied thus far, future research will likely define additional areas in which estrogen has important actions during childhood.
In conclusion, our study demonstrates that low-dose estrogen replacement beginning in childhood has several advantages over a regimen designed to begin pubertal induction in the teen years, including more physiological onset of pubertal development, optimization of GH-stimulated height gain (19), and benefits for cognitive development and memory (45, 46). Our data provide novel insights into the effects of two specific EE2 regimens, one of which was begun as early as age 5, for the treatment of childhood hypoestrogenism in TS and may help inform the development of improved hormone replacement regimens for girls with hypogonadism. As others have suggested, transdermal estradiol may provide more of a physiological mechanism for estrogen replacement than oral administration by delivering estrogen into the systemic circulation and avoiding exposure of the liver to supraphysiological estrogen concentrations (20, 22, 52, 57). Individualization of estradiol dosing could be accomplished with the help of high-sensitivity gas chromatography/tandem mass spectrometry estradiol assays, which would allow dose titration to attain estradiol concentrations within the appropriate prepubertal, and subsequently pubertal, range for age. However, as with oral EE2, transdermal estradiol replacement, particularly at the very low dosages required in childhood, is currently hindered by a lack of commercially available childhood dosage forms. To support future research on childhood estrogen replacement, including a broader range of long-term outcomes, and to facilitate clinical translation of research findings, the development of commercial childhood dosage forms is needed.
Acknowledgments
We sincerely thank the young women and their families for their participation in this study; Dr John Chipman, for support during its execution; the staff at both the National Institutes of Health (Bethesda, MD) and Thomas Jefferson University (Philadelphia, PA), who helped in recruitment and care of patients in this study; and Rong (Amy) Qi for detailed and helpful statistical review. In addition, we acknowledge the important role of Dr Penelope Feuillan (deceased) for her care of many of the study participants.
This work was supported by the National Institute of Child Health and Human Development and Lilly Research Laboratories.
Clinical Trial Registry No. NCT00001221.
Disclosure Summary: C.A.Q., X.W., and G.B.C. are former employees of Eli Lilly and Company. S.G. is a former employee of PharmaNet/i3, a contract research organization paid by Eli Lilly and Company. J.L.R. has received consulting fees from Eli Lilly and Company, Novo Nordisk, Pfizer, and Insmed.
Davenport has proposed the following estrogen equivalence relationship: 20μg oral EE2 = 2 mg oral 17-β-estradiol = 1.2 mg conjgated equine estrogen = 100 μg transdermal 17-β estradiol (20).
The procedure for formulation of EE2 liquid was provided as a supplement to Ross et al (19).
Gonadotropin values at baseline before estrogen exposure for all patients are provided in Supplemental Figure 4. Because 80% of patients had 45,X karyotype, no separate analyses were performed for patients with chromosomal mosaicism.
- B1
- Tanner breast stage 1
- CLDE
- childhood low-dose EE2
- COP
- childhood oral placebo
- EE2
- ethinyl estradiol
- GH/E
- GH injection with oral low-dose EE2
- GH/P
- GH injection with oral placebo
- P/E
- placebo injection with oral low-dose EE2
- P/P
- placebo injection with oral placebo
- TS
- Turner syndrome.
References
- 1. Stochholm K, Juul S, Juel K, Naeraa RW, Gravholt CH. Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab. 2006;91(10):3897–3902 [DOI] [PubMed] [Google Scholar]
- 2. Bondy CA, Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab. 2007;92(1):10–25 [DOI] [PubMed] [Google Scholar]
- 3. Saenger P, Wikland KA, Conway GS, et al. Recommendations for the diagnosis and management of Turner syndrome. J Clin Endocrinol Metab. 2001;86(7):3061–3069 [DOI] [PubMed] [Google Scholar]
- 4. Jenner MR, Kelch RP, Kaplan SL, Grümbach MM. Hormonal changes in puberty. IV. Plasma estradiol, LH, and FSH in prepubertal children, pubertal females, and in precocious puberty, premature thelarche, hypogonadism, and in a child with a feminizing ovarian tumor. J Clin Endocrinol Metab. 1972;34(3):521–530 [DOI] [PubMed] [Google Scholar]
- 5. Winter JS, Faiman C. Serum gonadotropin in concentrations in agonadal children and adults. J Clin Endocrinol Metab. 1972;35(4):561–564 [DOI] [PubMed] [Google Scholar]
- 6. Wilson CA, Heinrichs C, Larmore KA, et al. Estradiol levels in girls with Turner's syndrome compared to normal prepubertal girls as determined by an ultrasensitive assay. J Pediatr Endocrinol Metab. 2003;16(1):91–96 [DOI] [PubMed] [Google Scholar]
- 7. Hagen CP, Main KM, Kjaergaard S, Juul A. FSH, LH, inhibin B and estradiol levels in Turner syndrome depend on age and karyotype: longitudinal study of 70 Turner girls with or without spontaneous puberty. Hum Reprod. 2010;25(12):3134–3141 [DOI] [PubMed] [Google Scholar]
- 8. Klein KO, Baron J, Colli MJ, McDonnell DP, Cutler GB., Jr Estrogen levels in childhood determined by an ultrasensitive recombinant cell bioassay. J Clin Invest. 1994;94(6):2475–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janfaza M, Sherman TI, Larmore KA, Brown-Dawson J, Klein KO. Estradiol levels and secretory dynamics in normal girls and boys as determined by an ultrasensitive bioassay: a 10 year experience. J Pediatr Endocrinol Metab. 2006;19(7):901–909 [DOI] [PubMed] [Google Scholar]
- 10. Courant F, Aksglaede L, Antignac JP, et al. Assessment of circulating sex steroid levels in prepubertal and pubertal boys and girls by a novel ultrasensitive gas chromatography-tandem mass spectrometry method. J Clin Endocrinol Metab. 2010;95(1):82–92 [DOI] [PubMed] [Google Scholar]
- 11. Bondy CA, Bakalov VK. Investigation of cardiac status and bone mineral density in Turner syndrome. Growth Horm IGF Res. 2006;16(suppl A):S103—S108 [DOI] [PubMed] [Google Scholar]
- 12. Cutler GB., Jr The role of estrogen in bone growth and maturation during childhood and adolescence. J Steroid Biochem Mol Biol. 1997;61:141–144 [PubMed] [Google Scholar]
- 13. Gravholt CH, Veldhuis JD, Christiansen JS. Increased disorderliness and decreased mass and daily rate of endogenous growth hormone secretion in adult Turner syndrome: the impact of body composition, maximal oxygen uptake and treatment with sex hormones. Growth Horm IGF Res. 1998;8(4):289–298 [DOI] [PubMed] [Google Scholar]
- 14. Mauvais-Jarvis F. Estrogen and androgen receptors: regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol Metab. 2011;22(1):24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mortensen KH, Andersen NH, Gravholt CH. Cardiovascular phenotype in Turner syndrome–integrating cardiology, genetics, and endocrinology. Endocr Rev. 2012;33(5):677–714 [DOI] [PubMed] [Google Scholar]
- 16. Sherwin BB. Estrogen and cognitive functioning in women. Endocr Rev. 2003;24(2):133–151 [DOI] [PubMed] [Google Scholar]
- 17. Ross JL, Cassorla FG, Skerda MC, Valk IM, Loriaux DL, Cutler GB., Jr A preliminary study of the effect of estrogen dose on growth in Turner's syndrome. N Engl J Med. 1983;309(18):1104–1106 [DOI] [PubMed] [Google Scholar]
- 18. Ross JL, Long LM, Skerda M, et al. Effect of low doses of estradiol on 6-month growth rates and predicted height in patients with Turner syndrome. J Pediatr. 1986;109(6):950–953 [DOI] [PubMed] [Google Scholar]
- 19. Ross JL, Quigley CA, Cao D, et al. Growth hormone plus childhood low-dose estrogen in Turner's syndrome. N Engl J Med. 2011;364(13):1230–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davenport ML. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab. 2010;95(4):1487–1495 [DOI] [PubMed] [Google Scholar]
- 21. Divasta AD, Gordon CM. Hormone replacement therapy and the adolescent. Curr Opin Obstet Gynecol. 2010;22(5):363–368 [DOI] [PubMed] [Google Scholar]
- 22. Kenigsberg L, Balachandar S, Prasad K, Shah B. Exogenous pubertal induction by oral versus transdermal estrogen therapy. J Pediatr Adolesc Gynecol. 2013;26(2):71–79 [DOI] [PubMed] [Google Scholar]
- 23. Mauras N, Torres-Santiago L, Taboada M, Santen R. Estrogen therapy in Turner syndrome: does the type, dose and mode of delivery matter? Pediatr Endocrinol Rev. 2012;9(suppl 2):718–722 [PubMed] [Google Scholar]
- 24. Rosenfield RL, Devine N, Hunold JJ, Mauras N, Moshang T, Jr, Root AW. Salutary effects of combining early very low-dose systemic estradiol with growth hormone therapy in girls with Turner syndrome. J Clin Endocrinol Metab. 2005;90(12):6424–6430 [DOI] [PubMed] [Google Scholar]
- 25. Hamill PW, Drizd TA, Johnson CL, Reed RB, Roche AF. Health examination survey data, NCHS growth charts, 1976. Monthly Vital Statistics Report. 1976;25(suppl 3):1–22 [Google Scholar]
- 26. Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. 1969;44(235):291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Greulich WW, Pyle SI. Radiographic atlas of skeletal development of the hand and wrist. 2nd ed Stanford, CA: Stanford University Press; 1959 [Google Scholar]
- 28. Foster CM, Ross JL, Shawker T, et al. Absence of pubertal gonadotropin secretion in girls with McCune-Albright syndrome. J Clin Endocrinol Metab. 1984;58(6):1161–1165 [DOI] [PubMed] [Google Scholar]
- 29. Sun SS, Schubert CM, Chumlea WC, et al. National estimates of the timing of sexual maturation and racial differences among US children. Pediatrics. 2002;110(5):911–919 [DOI] [PubMed] [Google Scholar]
- 30. Dewhurst CJ, de Koos EB, Haines RM. Replacement hormone therapy in gonadal dysgenesis. Br J Obstet Gynaecol. 1975;82(5):412–416 [DOI] [PubMed] [Google Scholar]
- 31. Lucky AW, Marynick SP, Rebar RW, et al. Replacement oral ethinyloestradiol therapy for gonadal dysgenesis: growth and adrenal androgen studies. Acta Endocrinol (Copenh). 1979;91(3):519–528 [DOI] [PubMed] [Google Scholar]
- 32. Rosenfeld RG, Attie KM, Frane J, et al. Growth hormone therapy of Turner's syndrome: beneficial effect on adult height. J Pediatr. 1998;132(2):319–324 [DOI] [PubMed] [Google Scholar]
- 33. Chernausek SD, Attie KM, Cara JF, Rosenfeld RG, Frane J. Growth hormone therapy of Turner syndrome: the impact of age of estrogen replacement on final height. Genentech, Inc., Collaborative Study Group. J Clin Endocrinol Metab. 2000;85(7):2439–2445 [DOI] [PubMed] [Google Scholar]
- 34. Johnston DI, Betts P, Dunger D, et al. A multicentre trial of recombinant growth hormone and low dose oestrogen in Turner syndrome: near final height analysis. Arch Dis Child. 2001;84(1):76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brooks-Gunn J, Warren MP. The psychological significance of secondary sexual characteristics in nine- to eleven-year-old girls. Child Dev. 1988;59(4):1061–1069 [DOI] [PubMed] [Google Scholar]
- 36. Carel JC, Elie C, Ecosse E, et al. Self-esteem and social adjustment in young women with Turner syndrome–influence of pubertal management and sexuality: population-based cohort study. J Clin Endocrinol Metab. 2006;91(8):2972–2979 [DOI] [PubMed] [Google Scholar]
- 37. Kanaka-Gantenbein C. Hormone replacement treatment in Turner syndrome. Pediatr Endocrinol Rev. 2006;3(suppl 1):214–218 [PubMed] [Google Scholar]
- 38. Amundson E, Boman UW, Barrenäs ML, Bryman I, Landin-Wilhelmsen K. Impact of growth hormone therapy on quality of life in adults with Turner syndrome. J Clin Endocrinol Metab. 2010;95(3):1355–1359 [DOI] [PubMed] [Google Scholar]
- 39. Kagan-Krieger S. Brief report: women with Turner syndrome: a maturational and developmental perspective. J Adult Dev. 1998;5:125–135 [Google Scholar]
- 40. Mambelli MC, Perulli L, Casella S, et al. Difficulties and experiences in dealing with the needs of patients with Turner syndrome and their families: the usefulness of a multidisciplinary approach. In: Rovet J, ed. Turner Syndrome: Across the Lifespan. Markam, ON: Klein Graphics; 1995:108–112 [Google Scholar]
- 41. McCauley E, Ito J, Kay T. Psychosocial functioning in girls with Turner's syndrome and short stature: social skills, behavior problems, and self-concept. J Am Acad Child Psychiatry. 1986;25(1):105–112 [DOI] [PubMed] [Google Scholar]
- 42. Rovet JF. The psychoeducational characteristics of children with Turner syndrome. J Learn Disabil. 1993;26(5):333–341 [DOI] [PubMed] [Google Scholar]
- 43. Boman UW, Möller A, Albertsson-Wikland K. Psychological aspects of Turner syndrome. J Psychosom Obstet Gynaecol. 1998;19(1):1–18 [DOI] [PubMed] [Google Scholar]
- 44. Ross JL, McCauley E, Roeltgen D, et al. Self-concept and behavior in adolescent girls with Turner syndrome: potential estrogen effects. J Clin Endocrinol Metab. 1996;81(3):926–931 [DOI] [PubMed] [Google Scholar]
- 45. Ross JL, Roeltgen D, Feuillan P, Kushner H, Cutler GB., Jr Use of estrogen in young girls with Turner syndrome: effects on memory. Neurology. 2000;54(1):164–170 [DOI] [PubMed] [Google Scholar]
- 46. Ross JL, Roeltgen D, Feuillan P, Kushner H, Cutler GB., Jr Effects of estrogen on nonverbal processing speed and motor function in girls with Turner's syndrome. J Clin Endocrinol Metab. 1998;83(9):3198–3204 [DOI] [PubMed] [Google Scholar]
- 47. Aso K, Koto S, Higuchi A, et al. Serum FSH level below 10 mIU/mL at twelve years old is an index of spontaneous and cyclical menstruation in Turner syndrome. Endocr J. 2010;57(10):909–913 [DOI] [PubMed] [Google Scholar]
- 48. Conway GS. Considerations for transition from paediatric to adult endocrinology: women with Turner's syndrome. Growth Horm IGF Res. 2004;14(suppl A):S77–S84 [DOI] [PubMed] [Google Scholar]
- 49. Pasquino AM, Passeri F, Pucarelli I, Segni M, Municchi G. Spontaneous pubertal development in Turner's syndrome. Italian study group for Turner's syndrome. J Clin Endocrinol Metab. 1997;82(6):1810–1813 [DOI] [PubMed] [Google Scholar]
- 50. Marshall WA. Growth and sexual maturation in normal puberty. Clin Endocrinol Metab. 1975;4(1):3–25 [DOI] [PubMed] [Google Scholar]
- 51. Chumlea WC, Schubert CM, Roche AF, et al. Age at menarche and racial comparisons in US girls. Pediatrics. 2003;111;110–113 [DOI] [PubMed] [Google Scholar]
- 52. Ankarberg-Lindgren C, Elfving M, Wikland KA, Norjavaara E. Nocturnal application of transdermal estradiol patches produces levels of estradiol that mimic those seen at the onset of spontaneous puberty in girls. J Clin Endocrinol Metab. 2001;86(7):3039–3044 [DOI] [PubMed] [Google Scholar]
- 53. Jospe N, Orlowski CC, Furlanetto RW. Comparison of transdermal and oral estrogen therapy in girls with Turner's syndrome. J Pediatr Endocrinol Metab. 1995;8:111–116 [DOI] [PubMed] [Google Scholar]
- 54. Piippo S, Lenko H, Kainulainen P, Sipilä I. Use of percutaneous estrogen gel for induction of puberty in girls with Turner syndrome. J Clin Endocrinol Metab. 2004;89(7):3241–3247 [DOI] [PubMed] [Google Scholar]
- 55. Mauras N, Shulman D, Hsiang HY, Balagopal P, Welch S. Metabolic effects of oral versus transdermal estrogen in growth hormone-treated girls with Turner syndrome. J Clin Endocrinol Metab. 2007;92(11):4154–4160 [DOI] [PubMed] [Google Scholar]
- 56. Nabhan ZM, Dimeglio LA, Qi R, Perkins SM, Eugster EA. Conjugated oral versus transdermal estrogen replacement in girls with Turner syndrome: a pilot comparative study. J Clin Endocrinol Metab. 2009;94(6):2009–2014 [DOI] [PubMed] [Google Scholar]
- 57. Torres-Santiago L, Mericq V, Taboada M, et al. Metabolic effects of oral versus transdermal 17β-estradiol (E2): a randomized clinical trial in girls with Turner syndrome. J Clin Endocrinol Metab. 2013;98(7):2716–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ross JL, Feuillan P, Long LM, Kowal K, Kushner H, Cutler GB., Jr 1995 Lipid abnormalities in Turner syndrome. J Pediatr. 1995;126(2):242–245 [DOI] [PubMed] [Google Scholar]
- 59. Mauras N, Rogol AD, Veldhuis JD. Increased hGH production rate after low-dose estrogen therapy in prepubertal girls with Turner's syndrome. Pediatr Res. 1990;28(6):626–630 [DOI] [PubMed] [Google Scholar]
- 60. Weissberger AJ, Ho KK, Lazarus L. Contrasting effects of oral and transdermal routes of estrogen replacement therapy on 24-hour growth hormone (GH) secretion, insulin-like growth factor I, and GH-binding protein in postmenopausal women. J Clin Endocrinol Metab. 1991;72:374–381 [DOI] [PubMed] [Google Scholar]
- 61. Wit JM, Massarano AA, Kamp GA, et al. Growth hormone secretion in patients with Turner's syndrome as determined by time series analysis. Acta Endocrinol (Copenh). 1992;127(1):7–12 [DOI] [PubMed] [Google Scholar]
- 62. Simpson ER, Misso M, Hewitt KN, et al. Estrogen–the good, the bad, and the unexpected. Endocr Rev. 2005;26(3):322–330 [DOI] [PubMed] [Google Scholar]
- 63. Högler W, Briody J, Moore B, Garnett S, Lu PW, Cowell CT. Importance of estrogen on bone health in Turner syndrome: a cross-sectional and longitudinal study using dual-energy x-ray absorptiometry. J Clin Endocrinol Metab. 2004;89(1):193–199 [DOI] [PubMed] [Google Scholar]
- 64. Leung KC, Johannsson G, Leong GM, Ho KK. Estrogen regulation of growth hormone action. Endocr Rev. 2004;25(5):693–721 [DOI] [PubMed] [Google Scholar]
- 65. Bakalov VK, Bondy CA. Fracture risk and bone mineral density in Turner syndrome. Rev Endocr Metab Disord. 2008;9(2):145–151 [DOI] [PubMed] [Google Scholar]
- 66. Hultcrantz M, Simonoska R, Stenberg AE. Estrogen and hearing: a summary of recent investigations. Acta Otolaryngol. 2006;126(1):10–14 [DOI] [PubMed] [Google Scholar]
- 67. Marks SJ, Batra RR, Frishman WH. Estrogen replacement therapy for cognitive benefits: viable treatment or forgettable “senior moment”? Heart Dis. 2002;4(1):26–32 [DOI] [PubMed] [Google Scholar]
- 68. Christopoulos P, Deligeoroglou E, Laggari V, Christogiorgos S, Creatsas G. Psychological and behavioural aspects of patients with Turner syndrome from childhood to adulthood: a review of the clinical literature. J Psychosom Obstet Gynaecol. 2008;29(1):45–51 [DOI] [PubMed] [Google Scholar]
- 69. Kesler SR. Turner syndrome. Child Adolesc Psychiatr Clin N Am. 2007;16(3):709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hettmer E, Hoepffner W, Keller E, Brähler E. Studies on sexual development, sexual behavior and ability to experience sex of young women with Ullrich-Turner syndrome [in German]. Ther Umsch. 1995;52(2):146–149 [PubMed] [Google Scholar]
- 71. Brock O, Baum MJ, Bakker J. The development of female sexual behavior requires prepubertal estradiol. J Neuorsci. 2011;31:5574–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]