

Abstract
Context:
Hyperparathyroidism occurs frequently in X-linked hypophosphatemia (XLH) and may exacerbate phosphaturia, potentially affecting skeletal abnormalities.
Objective:
The objective of the study was to suppress elevated PTH levels in XLH patients.
Design:
This was a prospective, randomized, placebo-controlled, double-blind, 1-year trial of paricalcitol, with outcomes measured at entry and 1 year later.
Setting:
Patients were recruited from the investigators' clinics or referred from throughout the United States. Data were collected in an in-patient hospital research unit.
Patients:
Subjects with a clinical diagnosis of XLH and hyperparathyroidism were offered participation and were eligible if they were 9 years old or older and not pregnant, and their serum calcium level was less than 10.7 mg/dL, their 25-hydroxyvitamin D level was 20 ng/mL or greater, and their creatinine level was 1.5 mg/dL or less.
Intervention:
The intervention for this study was the use of paricalcitol or placebo for 1 year.
Main Outcome Measures:
Determined prior to trial onset was the change in PTH area under the curve. Secondary outcomes included renal phosphate threshold per glomerular filtration rate, serum phosphorus, serum alkaline phosphatase activity, and 99mTc-methylenediphosphonate bone scans.
Results:
PTH area under the curve decreased 17% with paricalcitol, differing (P = .007) from the 20% increase with placebo. The renal phosphate threshold per glomerular filtration rate increased 17% with paricalcitol and decreased 21% with placebo (P = .05). Serum phosphorus increased 12% with paricalcitol but did not differ from placebo. Paricalcitol decreased alkaline phosphatase activity in adults by 21% (no change with placebo, P = .04). Bone scans improved in 6 of 17 paricalcitol subjects, whereas no placebo-treated subject improved. Hypercalciuria developed in six paricalcitol subjects and persisted from baseline in one placebo subject.
Conclusions:
Suppression of PTH may be a useful strategy for skeletal improvement in XLH patients with hyperparathyroidism, and paricalcitol appears to be an effective adjunct to standard therapy in this setting. Although paricalcitol was well tolerated, urinary calcium and serum calcium and creatinine should be monitored closely with its use.
X-linked Hypophosphatemia (XLH) is a rachitic disorder characterized by low serum phosphorus, excessive phosphaturia, and inappropriately low circulating 1,25 dihydroxyvitamin D [1,25(OH)2D] levels (1, 2). Elevated circulating fibroblast growth factor 23 (FGF23) is considered the primary mediator of renal phosphate wasting; however, PTH may also contribute to this effect (3). Circulating PTH levels are often modestly elevated in XLH without medical intervention (4) and increase with the application of phosphate-based therapies (5, 6).
FGF23 and PTH have been proposed to act cooperatively to inhibit renal phosphate reabsorption (7), and PTH may amplify phosphaturic effects of FGF23. The observation that low serum phosphorus levels were corrected in a patient with XLH after parathyroidectomy suggests that PTH is necessary for effecting excessive phosphaturia (8). Indeed, an aberrant relationship between FGF23 and PTH is evident in XLH: FGF23 normally suppresses PTH secretion (9), but PTH levels are positively correlated to FGF23 levels (10). Hyperparathyroidism otherwise complicates XLH by incurring hypercalcemia and hypercalciuria and may contribute to heterotopic ossification and nephrocalcinosis.
Thus correction or prevention of hyperparathyroidism is an important goal in the chronic management of XLH. This creates an inherent problem with current therapy because the necessary chronic administration of phosphate stimulates PTH secretion (5, 11). Control or prevention of progressive hyperparathyroidism may avoid or diminish episodes of hypercalcemia or hypercalciuria, enhance renal phosphate retention, and may also improve skeletal outcomes. Furthermore, successful medical management of hyperparathyroidism would prevent the need for parathyroidectomy in severe cases.
These considerations support the development of a directed approach to manage hyperparathyroidism in XLH. We therefore initiated a prospective, randomized, double-blind trial using the vitamin D analog paricalcitol to suppress PTH secretion in XLH patients with previous evidence of hyperparathyroidism. Paricalcitol is thought to incur a lower increase in serum calcium than other vitamin D preparations (12) and is widely used to treat hyperparathyroidism accompanying chronic kidney disease (13).
Materials and Methods
Study protocol and human subjects
Thirty-three subjects (30 adults and three children; nine males and 24 females) with a clinical diagnosis of XLH and modest to moderate hyperparathyroidism were recruited from 2007 through 2011 by the investigators at clinic visits or by referring physicians. For local patients, serum PTH was required to be 40–150 nlEq/mL using our assay, which detects intact hormone and midmolecule fragments (normal, ≤ 25 nlEq/mL); elevated intact PTH was required for patients referred from out of state. Exclusion criteria included estimated creatinine clearance less than 60 cc/min and/or serum creatinine greater than 1.5 mg/dL, serum calcium 10.7 mg/dL or greater, serum 25-hydroxyvitamin D (25-OHD) less than 20 ng/mL, or treatment with medications potentially affecting skeletal metabolism, such as glucocorticoids and anticonvulsants. Pregnant subjects or those planning pregnancy during the study were excluded. Subjects remained on standard therapy (calcitriol and phosphate) if being treated for XLH at the time of consent and were instructed to stay on current doses throughout the study.
The study was approved by the Human Investigation Committee at the Yale University School of Medicine (Human Investigation Committee number 0607001637). Informed consent was obtained from all adult patients, and assent and parental permission were obtained for children/adolescents. The study was registered at clinicaltrials.gov (number NCT00417612). A safety officer appointed by the National Institutes of Health reviewed the protocol semiannually. Adverse events were reported to the safety officer and the institution's institutional review board.
Subjects underwent a 2-day in-patient admission at study entry and after 1 year of experimental therapy. Subjects were fed standardized diets prepared by a research metabolic kitchen with constant calcium and phosphate content during each admission. Blood was sampled at 4-hour intervals for PTH measures. Fasting serum calcium, phosphorus, 1,25(OH)2D, intact FGF23, total alkaline phosphatase activity, and renal phosphate threshold per glomerular filtration rate (TmP/GFR) were assessed, as were 24-hour urinary calcium and creatinine excretion.
Randomization and intervention
Subjects were stratified by age group and randomized using a random number generator in a 2:1 (active drug to placebo) ratio. Each subject randomized to active treatment received two 1-μg capsules of paricalcitol. Placebo subjects received two identically appearing placebo capsules. Subjects and investigators were blinded to randomization status. Both treatment groups were instructed to take the two capsules together each morning. Zemplar (paricalcitol) and placebo were manufactured and provided by Abbott Laboratories. All subjects were provided with coded cards that identified them as study participants and allowed for identification of randomization status in the event of emergency. Investigators and subjects remained blinded to all postintervention PTH values until the study was completed.
Dose titration
Doses of paricalcitol/placebo were titrated 1 and 2 months after beginning therapy. Local patients visited the Research Unit and distant patients underwent sampling near their residences. If the serum PTH level at the first titration point had not decreased by at least 25% from baseline, the dose of paricalcitol was increased by 1 μg to 3 μg daily. If serum calcium was 11.0 mg/dL or greater or the urinary calcium to creatinine ratio was greater than 0.4 mg/mg, the daily dose of paricalcitol was reduced from 2 μg per day to 1 μg per day (a decrement of one capsule, or 1 μg per day). This algorithm was followed again at the 2-month outpatient visit. Safety measures took precedent so that no patient underwent a PTH-based increase in paricalcitol dose if serum or urinary calcium values were elevated.
Subsequent sampling was performed at 4, 6, 8, and 10 months after beginning therapy. If a subject's serum calcium level increased to 11.0 mg/dL or greater, the dose of paricalcitol/placebo was reduced by 1 μg daily. Serum calcium was measured 2 weeks after all dose adjustments. If serum calcium level was 11.0 mg/dL or greater while receiving only 1 μg of drug, the subject was discontinued from the study.
Because we expected that only subjects assigned to active drug would demonstrate decrements in circulating PTH during treatment, the laboratory provided PTH values obtained at titration points to the pharmacist, who matched each placebo subject to a subject assigned to the active drug. A titration scheme that matched the previous titration course of an active drug subject to each placebo subject was devised. In this way, the subjects and investigators remained blinded to the treatment arm because the interval PTH levels and responses were not revealed, and the subjects receiving the active drug and those receiving the placebo underwent comparable dose adjustments.
Outcome variables
The primary outcome measure was percentage change in PTH levels expressed as the area under the curve. Area under the curve for PTH concentration (PTHauc) was calculated from the serial sampling values. PTHauc was chosen to optimally capture exposure to PTH; there is substantial circadian variance with nocturnal elevations in patients with XLH (4). The study was powered to this variable and has lesser variance than single PTH measures. Secondary outcomes included fasting serum phosphorus, 1,25(OH)2D, intact FGF23, total alkaline phosphatase activity, and 25-OHD at baseline and 1 year later. Twenty-four-hour urinary excretion of calcium, creatinine, and phosphorus was determined. Fasting morning blood samples and 2-hour urine collections were simultaneously collected for calculation of TmP/GFR. Severity of metabolic bone disease was assessed by a nuclear medicine physician (D.C.) blinded to the treatment arm using 99mTc-methyldiphosphonate bone scans. Twenty millicuries of isotope were administered to adults; the dose was proportionally reduced for children based on body weight. Anterior and posterior whole-body planar images were obtained 3 hours after isotope administration. A 5-point scale was established using normal appearing lumbar spine and the sacroiliac region as internal references to which all suspected lesions were compared and scored as follows: grade 0, normal scan without suspicious lesions; grade 1, lesion(s) less intense than normal lumbar spine; grade 2, lesion(s) similar in intensity to normal lumbar spine; grade 3, lesions more intense than normal lumbar spine but similar to the normal sacroiliac region; and grade 4, lesions more intense than the normal sacroiliac region.
Safety assessments
All dose adjustments were monitored with serum calcium and creatinine, and urinary calcium excretion was performed 2 weeks after discharge and dose changes and routinely at months 1, 2, 4, 6, 8, and 10 throughout the study.
Analytical methods
Serum phosphorus, alkaline phosphatase activity, and urinary phosphorus, calcium, and creatinine were measured using autoanalyzer technology in the Core Laboratory of the Yale Center for Clinical Investigation (10). Serum calcium and creatinine were measured using autoanalyzer methodology in the Yale-New Haven Clinical Laboratory. Quest Laboratories analyzed safety point serum calcium and creatinine levels for out-of-town subjects. Serum 25-OHD and 1,25(OH)2D levels were measured using an RIA kit from DiaSorin, Inc. Serum PTH was measured using a midregion assay (4). Serum FGF23 was measured using the Kainos intact ELISA kit (kindly provided by Kyowa Hakko Kirin Pharma, Inc).
Statistics and data analysis
Six subjects were excluded from the data analysis for the following reasons: one referred subject was found to be affected with a disorder other than XLH after the baseline evaluation; three subjects underwent major dose changes of standard medications through the treatment period; one subject developed hypercalcemia just prior to final evaluation and discontinued his prescribed therapy; and one subject rarely took any of the prescribed medication. An additional subject was discontinued from the study because of elevated serum creatinine levels during the treatment period, and one subject withdrew from the study after treatment assignment. Because of large baseline variability among subjects, biochemical data at 1 year were analyzed as percentage change from baseline. Comparisons were performed between treated and untreated XLH subjects with an ANOVA model using SAS software. Comparison of numbers of subjects demonstrating improved bone scans in each group was assessed by χ2 test and Fisher's exact test. Correlations between biochemical variables were examined using Pearson correlations.
Results
Subject profile
Twenty-three subjects had a family history consistent with XLH and 10 did not. The age of children ranged from 10 to 12 years and of adults from 18 to 69 years. Two subjects were of Hispanic ethnicity; all were white. Table 1 lists demographic and biochemical characteristics of the study population, and a flow chart of screening and randomization is shown in Figure 1. Mean age, height, and weight were comparable between treatment groups, and a similar percentage of males and females participated in each group. Half of the patients receiving placebo and 39% of those receiving paricalcitol were being treated with calcitriol and phosphate at enrollment. All subjects demonstrated low serum phosphorus levels (<2.1 mg/dL) and comparably low TmP/GFR. Mean serum PTH was similarly elevated in the two treatment arms (3.3-fold > upper limit of normal in the placebo and 2.5-fold > upper limit of normal in the active therapy group). PTHauc was slightly greater in the placebo (1449 ± 865) than in the active treatment group (1223 ± 426), but this difference was not statistically significant (P = .37). Serum calcium, phosphorus, creatinine, total alkaline phosphatase activity, 25-OHD, and 1,25(OH)2D levels were all comparable between groups. Serum FGF23 was elevated in all patients (excepting the excluded patient with an erroneous diagnosis) and slightly, but not significantly, greater in the group randomized to active therapy (283 ± 564 pg/mL vs 126 ± 103 pg/mL, P = .55). Both PTHauc (r = −0.56, P = .002) and serum FGF23 (r = −0.54, P = .004) correlated with TmP/GFR at baseline.
Table 1.
Clinical Characteristics and Biochemical Profile of the Study Subjects
| Characteristic | All | Placebo (n = 8) | Active Treatment (n = 19) |
|---|---|---|---|
| Age, y | 41 ± 19 | 41 ± 19 | 41 ± 19 |
| Gender, n, % female/n, % male | 21 (78)/6 (22) | 6 (75)/2 (25) | 15 (79)/4 (21) |
| Height, cm | 149 ± 8 | 149 ± 8 | 150 ± 9 |
| Height SDS | −2.18 ± 0.98 | −2.38 ± 0.74 | −2.09 ± 1.07 |
| Weight, kg | 69 ± 18 | 63 ± 14 | 71 ± 20 |
| Weight SDS | 1.26 ± 4.03 | 0.68 ± 1.52 | 1.76 ± 4.65 |
| BMI | 31 ± 9 | 28 ± 6 | 32 ± 10 |
| BMI SDS | 1.41 ± 0.83 | 1.25 ± 0.98 | 1.48 ± 0.79 |
| On standard therapy, n, %a | 12 (44) | 4 (50) | 8 (42) |
| Serum calcium, mg/dL | 9.2 ± 0.5 | 9.3 ± 0.5 | 9.2 ± 0.5 |
| PTH, nlEq/mL | 70 ± 43 | 83 ± 70 | 65 ± 27 |
| PTHauc | 1290 ± 582 | 1449 ± 865 | 1223 ± 426 |
| Phosphorus, mg/dL | 1.6 ± 0.3 | 1.5 ± 0.4 | 1.6 ± 0.3 |
| FGF23, pg/mLb | 112 (75–166) | 93 (46–188) | 120 (72–203) |
| Creatinine, mg/dL | 0.65 ± 0.18 | 0.69 ± 0.24 | 0.63 ± 0.16 |
| 1,25(OH)2D, pg/mL | 50 ± 16 | 47 ± 9 | 51 ± 18 |
| 25-OHD, ng/mL | 27 ± 9 | 25 ± 4 | 28 ± 10 |
| Alkaline phosphatase, IU/L | 167 ± 146 | 170 ± 213 | 166 ± 111 |
| TMP/GFR, mg/dL | 1.2 ± 0.5 | 1.2 ± 0.6 | 1.2 ± 0.4 |
| Urinary calcium, mg per 24 h | 129 ± 135 | 226 ± 210 | 93 ± 75 |
Abbreviations: SDS, SD score; PTHauc, measure for PTHauc samples taken every 4 hours over a 28-hour study period. Reference ranges for the listed laboratory values are as follows: serum calcium, 8.8–10.2 mg/dL; PTH, 10–25 nlEq/mL; phosphorus, 2.5–4.5 mg/dL; FGF23, 8–54 pg/mL; creatinine, 0.5–1.2 mg/dL; 1,25(OH)2D, 25–66 pg/mL; 25-OHD, 20–50 ng/mL; alkaline phosphatase, 50–480 IU/L in children; 30–130 IU/L in adults; TmP/GFR 2.5–4.5 mg/dL; urinary calcium, 100–300 mg per 24 hours (or < 4 mg/kg body weight per 24 h in children).
Oral phosphate salts and calcitriol.
Geometric mean and 95% CI.
Figure 1.
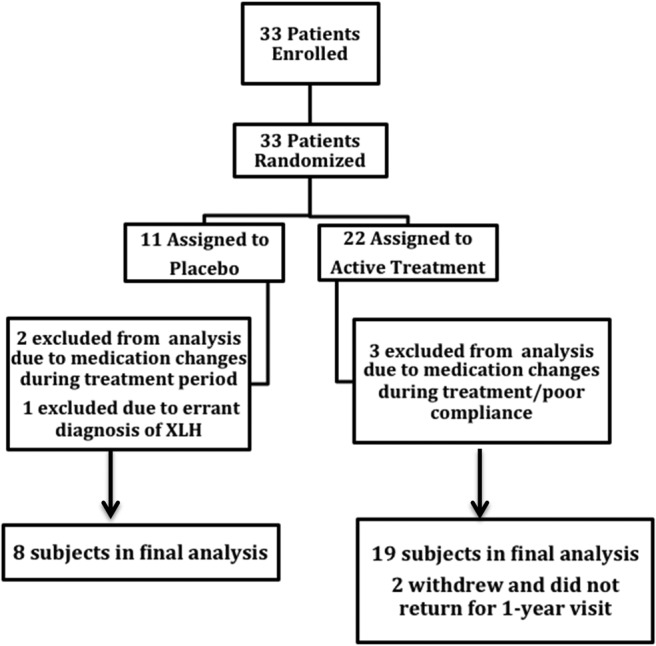
Flow diagram of the progress of study subjects through enrollment, recruitment, and retention.
Area under the curve for PTH concentration
Mean PTHauc decreased from 1223 ± 426 to 987 ± 372 (mean ± SD) or by 17% from baseline in subjects receiving paricalcitol (Figure 2A), significantly differing (P = .007) from the 20% percent increase observed in subjects receiving placebo (from 1449 ± 865 to 1616 ± 945) (Figure 2A). In the active treatment group, PTHauc decreased by 20%–29% in five subjects (29%), 30%–39% in one subject (6%), and 40% or more in three subjects (18%). In contrast, in the placebo group, PTHauc decreased by 20%–29% in one subject (13%), 30%–39% in one subject (13%), and no subject showed suppression of PTHauc by 40% or more. We considered a 20% or greater reduction in PTH as clinically significant; this degree of suppression was achieved in approximately twice the proportion of patients receiving paricalcitol (9 of 17; 53%) as compared with placebo (two of eight; 25%). We further evaluated the impact of standard therapy on the paricalcitol response. We identified a significant interaction between standard therapy and paricalcitol (P = .0009), although the magnitude of the additive effect of standard therapy was somewhat small (16% suppression of PTHauc in the group receiving paricalcitol alone and 19% suppression in the group receiving both standard therapy and paricalcitol).
Figure 2.

Changes in major outcome variables over the year of intervention as percentages of baseline values. Data are plotted as observed means and SEM of the respective groups. A, PTHauc. B, TmP/GFR. C, Fasting serum phosphorus (Pi) level. D, Fasting serum 1,25(OH)2D level. E, Fasting circulating intact FGF23 level (picograms per milliliter). F, Fasting circulating alkaline phosphatase activity in the adult patients. Active therapy subjects are shown in diagonal fill, and placebo-treated subjects are shown in white. *, Differs from placebo (P ≤ .05); **, differs from placebo (P < .01).
TmP/GFR and fasting serum phosphorus level
In subjects receiving paricalcitol, TmP/GFR increased by 17% differing from the 21% decrease in the placebo-treated group (P = .05) (Figure 2B). Fasting serum phosphorus increased by 12% in the treatment group (1.6–1.8 mg/dL, Figure 2C), whereas a 2% increase was observed in placebo-treated subjects, changes that did not significantly differ between groups (P = .37).
Serum 1,25(OH)2D
Mean serum 1,25(OH)2D did not change in subjects receiving paricalcitol (51 ± 18 pg/mL at baseline; 52 ± 17 pg/mL after therapy) or in those assigned to placebo (47 ± 9 pg/mL at baseline; 46 ± 14 pg/mL after therapy).
Serum alkaline phosphatase activity
Alkaline phosphatase activity was analyzed separately for children (less than 18 y of age) and adults as values varied considerably between these two groups. Mean serum alkaline phosphatase activity decreased by 21% (from 141 ± 96 IU/L to 112 ± 70 IU/L) in paricalcitol-treated adults (P = .003), whereas no change was evident in placebo-treated adults (96 ± 28 IU/L at baseline, and 98 ± 34 IU/L after therapy). The percentage change was significantly different between the groups (P = .04). In the two children receiving paricalcitol, baseline mean serum alkaline phosphatase activity (354 ± 2 IU/L) was no different from the posttreatment value (402 ± 211 IU/L).
Serum intact FGF23
The geometric mean for serum intact FGF23 level increased from 120 (95% confidence interval (CI) 72–203) pg/mL to 244 (95% CI 145–411) pg/mL or by 176% in paricalcitol-treated patients (P < .05, Figure 2D). There was a nonsignificant 15% increase in placebo-treated patients, from 93 (95% CI 46–188) pg/mL to 104 (95% CI 47–227); the difference between groups was significant (P < .05).
Serum and urinary calcium
Mean serum calcium increased slightly in paricalcitol-treated subjects (9.2 ± 0.5 mg/dL to 9.4 ± 0.5 mg/dL); however, this change was not different from the placebo group in whom serum calcium did not change from baseline (9.3 ± 0.5 mg/dL) (Figure 3A). Urinary calcium excretion was comparable between the 2 treatment groups at baseline, with the exception of one subject with hypercalciuria in the placebo group (Table 1). There was a substantial increase in mean urinary calcium excretion (from 93 mg per 24 h to 235 mg per 24 h) in patients receiving paricalcitol (253%, 95% CI 50–464), whereas only a modest change occurred in patients receiving placebo (from 226 mg per 24 h to 246 mg per 24 h) (9%, 95% CI 24–42) (Figure 3C). The difference between groups did not reach statistical significance (P = .09).
Figure 3.

Changes in safety measures and secondary outcome variables, as percentages of baseline values, at the 1-year evaluation. Data are plotted as observed means and SEM of the respective groups. A, Fasting serum calcium level. B, Fasting serum creatinine level. C, Twenty-four-hour urinary calcium excretion. Active therapy subjects are shown in diagonal fill, and placebo treated subjects are shown in white. **, Differs from placebo (P < .01).
99Tc-methyldiphosphonate bone scans
The mean bone scan severity score decreased slightly after 1 year of therapy (from 2.18 ± 1.19 to 1.76 ± 1.25) in the treatment arm and increased slightly in the placebo group (from 2.00 ± 1.51 to 2.12 ± 1.55), mean differences that were not statistically significant. The number of positive responders (6 of 17) was greater in the group receiving paricalcitol. In fact, none of the eight patients administered placebo improved (Fisher's exact test, P = .07).
PTH and FGF23 as determinants of TmP/GFR
Finally, correlative analyses assessing PTHauc and serum intact FGF23 levels as determinants of TmP/GFR were performed. At baseline, both PTHauc and FGF23 demonstrated significant inverse correlation with TmP/GFR (Figure 4, A and B). At the end of the study, PTHauc remained a significant determinant of TmP/GFR in the placebo-treated group (r = −0.82, P = .013), and serum FGF23 maintained a near-significant relationship with TmP/GFR (r = −0.67, P = .07). In contrast, TmP/GFR in subjects receiving active drug after the year of therapy correlated only with PTHauc (r = −0.42, P = .09). No significant relationship with FGF23 remained evident in those receiving paricalcitol (r = 0.04, P = .9) (Figure 4, C and D).
Figure 4.

A and C, Relationship of TmP/GFR (milligrams per deciliter) to PTHauc at baseline (r = −0.56; P = .002) (A) and after 1 year of therapy (placebo: r = −0.82, P = .013; paricalcitol, r = 0.42; P = .09) (C). B and D, Relationship of TmP/GFR to serum FGF23 level (picograms per milliliter) at baseline (r = −0.54; P = .004) (B) and after 1 year of therapy (placebo: r = −0.67, P = .07; paricalcitol, r = 0.04; P = .9) (D). The posttreatment data are shown by treatment group: solid symbols and solid lines represent the data from the active treatment group, and open symbols and broken lines represent the data from the placebo group.
Safety parameters and adverse events
Four subjects developed hypercalcemia, three of whom received active drug and one of whom received placebo. Two subjects had mild elevations in serum creatinine with upward titration of paricalcitol dose. In one subject serum creatinine (0.9 mg/dL at baseline) increased to 1.2 mg/dL at 3 months after receiving 4 μg of paricalcitol daily. Serum creatinine decreased to 0.9 mg/dL after a reduction in dose and was 1.1 mg/dL upon completion of the study. In a second patient, serum creatinine increased to 1.4 mg/dL by month 3 (0.7 mg/dL at baseline) while receiving 3 μg of paricalcitol daily. The patient was discontinued from the study and serum creatinine decreased to 1.15 mg/dL and was 1.20 mg/dL at final follow-up. Mean serum creatinine slightly increased in the paricalcitol group (0.63 ± 0.16 mg/dL to 0.74 ± 0.21 mg/dL), which differed (P < .001) from the placebo group (0.69 ± 0.24 mg/dL to 0.60 ± 0.21 mg/dL) (Figure 3B). Thirteen episodes of hypercalciuria (24 h urine calcium excretion > 300 mg or in children > 4 mg/kg body weight) were detected during the treatment course and occurred in six active treatment and one placebo subject. The placebo-treated patient demonstrated hypercalciuria at baseline, as described above. No other adverse events that were considered possibly or probably related to the treatment were reported, and there were no serious adverse events.
Discussion
Our study demonstrates that treatment with relatively modest doses of paricalcitol (mean dose 3 μg/d) suppressed serum PTH levels in patients with XLH and hyperparathyroidism. This was accompanied by a rise in TmP/GFR and occurred despite an increase in circulating FGF23 levels in subjects treated with paricalcitol.
Since the discovery that serum FGF23 levels are elevated in XLH (14) and that inhibition of FGF23 activity substantially improves disease in a murine model of XLH (15), attention has focused on understanding the molecular basis for this hormonal abnormality. However, it has been known for nearly 60 years that PTH acutely inhibits proximal tubular phosphate reabsorption. PTH-induced phosphaturia occurs within minutes of PTH exposure and is mediated by removal of sodium/phosphate cotransporters from the apical membrane of the tubular cell (16). FGF23, thought to act via different pathways, requires a longer time course (17), affecting transcription of cotransporter genes as well as membrane localization of the cotransporters (7).
The role of PTH in the pathogenesis of XLH has been previously studied (3, 4, 8, 18–21). Although no well-designed trials were conducted, case reports of parathyroidectomy in XLH patients with tertiary hyperparathyroidism indicated that correction of hyperparathyroidism is associated with an improvement in phosphate wasting (8). Consistent with this observation, several case reports and short-term studies have suggested that cinacalcet can suppress PTH in XLH with concomitant improvement in phosphate homeostasis (19–21). However, safety concerns regarding the use of cinacalcet in children have arisen, and no prospective, long-term studies with this drug in XLH are available (22).
Apart from its contribution to phosphate wasting in XLH, chronic elevations in PTH may contribute to the pathogenesis of nephrocalcinosis and enthesopathy (calcification of ligaments and tendons that leads to loss of mobility and chronic pain) in XLH. It is possible that such heterotopic mineralization could be influenced by excessive exposure to PTH. Cardiac calcification has been reported in XLH in association with hyperparathyroidism (23).
Paricalcitol was chosen for the current clinical trial because it has been studied extensively as treatment for hyperparathyroidism associated with renal insufficiency and failure (12). In that setting it has been shown to effectively lower PTH with less associated hypercalcemia and better overall survival when compared with calcitriol (13).
The current study enrolled subjects with significant hyperparathyroidism (mean serum PTH was more than twice the upper limit of normal at baseline). In addition, study subjects had elevated alkaline phosphatase values consistent with the presence of significant skeletal disease. Paricalcitol significantly lowered PTHauc in the paricalcitol-treated group; PTHauc decreased by 20% or greater in more than 50% of the treatment group, compared with only 25% of placebo-treated patients. At the same time, adult subjects treated with paricalcitol showed a significant decrease in serum alkaline phosphatase activity, but no change occurred in the placebo group, suggesting that the decrease in PTHauc was associated with improvement in skeletal disease. Moreover, 35% of paricalcitol treated subjects showed improvement in bone scans, whereas no subject in the placebo group improved. The potential for improvement of skeletal disease suggests that targeting PTH may be a useful strategy for management of XLH. Because these subjects had no changes in their doses of calcitriol and phosphorus it is likely that the paricalcitol is directly responsible for the observed changes.
As would be expected with calcitriol therapy, an increase in serum FGF23 occurred in the paricalcitol-treated subjects, likely mediated by paricalcitol's interaction with the vitamin D receptor at the osteocyte. Despite the rise in serum FGF23, TmP/GFR rose slightly in the paricalcitol group, whereas in the placebo-treated group TmP/GFR decreased from baseline such that after a year of therapy the difference in response of TmP/GFR between the two groups was statistically significant. This finding suggests that the effect of PTH suppression was more dominant than the increase in FGF23 with respect to regulation of TmP/GFR.
After 1 year of therapy, mean urinary excretion was 235 mg per 24 hours and 246 mg per 24 hours in the paricalcitol- and placebo-treated groups, respectively. Approximately one-third of the paricalcitol-treated patients experienced episodes of transient hypercalciuria throughout the year. Thus, paricalcitol treatment should be accompanied by monitoring of urinary calcium excretion to avoid prolonged hypercalciuria as is necessary when using calcitriol. Three patients in the paricalcitol arm and one in the placebo arm developed transient hypercalcemia. Serum creatinine levels increased in two patients in the treatment arm, one of whom had been titrated to receive 4 μg of paricalcitol daily and one who received 3 μg daily. These values decreased following discontinuation of medication; however, one patient was discontinued from the study on this basis.
Although our study was limited by a relatively small number of subjects, we demonstrated that paricalcitol suppressed elevated PTH levels in patients with XLH. Furthermore, despite increases in FGF23 levels, a rise in TmP/GFR occurs. Serum alkaline phosphatase activity, a measure of skeletal disease severity, was significantly decreased in paricalcitol-treated adults but not with placebo. Larger trials will be necessary to determine whether paricalcitol will fit into current or future treatment regimens for XLH, either with or without concomitant calcitriol. With monitoring of serum calcium and creatinine levels, as well as urinary calcium excretion, paricalcitol could be used as an adjunct to treatment with calcitriol and phosphate. Further study would be useful to identify an ideal dosing paradigm for these safety concerns. In the future with drugs to inhibit FGF23 on the horizon, it may be possible to treat any resultant hyperparathyroidism with paricalcitol without concern about concomitant rises in FGF23 levels.
Acknowledgments
We are grateful for the support of Abbott Pharmaceuticals, who kindly provided paricalcitol and placebo used in the study, and Osama Abdelghany (Investigational Drug Pharmacy at Yale-New Haven Hospital) for the local management of these materials. We acknowledge the dedicated commitment of the study participants, and the outstanding staff support of the Hospital Research Unit of the Yale Center for Clinical Investigation, supported by National Institutes of Health Clinical and Translational Science Awards from the National Center for Research Resources. We are also grateful to Kyowa Hakko Kirin, Inc for providing FGF23 assay materials and to Drs Dennis Carey, Ingrid Holm, Bernard Kaplan, Susan Ott, and Christine Resta for referring their interested patients.
This study was registered at clinicaltrials.gov with the number NCT00417612.
This work was supported by National Institutes of Health Center for Research Translation Award P50-AR054086 from the National Institute of Arthritis, Musculoskeletal, and Skin Disorders (to T.O.C.); and National Institutes of Health Clinical and Translational Science Awards Award UL1 TR000142 from the National Center for Research Resources.
Disclosure Summary: T.O.C. performs consulting duties for Kyowa Hakko Kirin and Ultragenyx. The other authors have nothing to declare.
Footnotes
- CI
- confidence interval
- FGF23
- fibroblast growth factor 23
- 25-OHD
- 25-hydroxyvitamin D
- 1,25(OH)2D
- 1,25 dihydroxyvitamin D3
- PTHauc
- area under the curve for PTH concentration
- TmP/GFR
- renal phosphate threshold per glomerular filtration rate
- XLH
- X-linked hypophosphatemia.
References
- 1. Burnett CH, Dent CE, Harper C, Warland BJ. Vitamin D-resistant rickets. Am J Med. 1964;36:222–232 [DOI] [PubMed] [Google Scholar]
- 2. Holm IA, Econs MJ, Carpenter TO. Familial hypophosphatemia and related disorders. In: Glorieux FH, Juppner H, Pettifor JM, eds. Pediatric Bone: Biology, Diseases. 2nd ed San Diego: Elsevier; 2012:699–726 [Google Scholar]
- 3. Alon U, Chan JC. Effects of parathyroid hormone and 1,25-dihydroxyvitamin D3 on tubular handling of phosphate in hypophosphatemic rickets. J Clin Endocrinol Metab. 1984;58:671–675 [DOI] [PubMed] [Google Scholar]
- 4. Carpenter TO, Mitnick MA, Smith C, Ellison A, Insogna KL. Nocturnal hyperparathyroidism: a frequent feature of X-linked hypophosphatemia. J Clin Endocrinol Metab. 1994;78:1378–1383 [DOI] [PubMed] [Google Scholar]
- 5. Firth RG, Grant CS, Riggs BL. Development of hypercalcemic hyperparathyroidism after long-term phosphate supplementation in hypophosphatemic osteomalacia. Report of two cases. Am J Med. 1985;78:669–773 [DOI] [PubMed] [Google Scholar]
- 6. Knudtzon J, Halse J, Monn E, et al. Autonomous hyperparathyroidism in X-linked hypophosphataemia. Clin Endocrinol (Oxf). 1995;42:199–203 [DOI] [PubMed] [Google Scholar]
- 7. Andrukhova O, Zeitz U, Goetz R, Mohammadi M, Lanske B, Erben RG. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone. 2012;51:621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lyles KW, Burkes EJ, Jr, McNamara CR, Harrelson JM, Pickett JP, Drezner MK. The concurrence of hypoparathyroidism provides new insights to the pathophysiology of X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 1985;60:711–717 [DOI] [PubMed] [Google Scholar]
- 9. Galitzer H, Ben-Dov I, Lavi-Moshayoff V, Naveh-Many T, Silver J. Fibroblast growth factor 23 acts on the parathyroid to decrease parathyroid hormone secretion. Curr Opin Nephrol Hypertens. 2008;17:363–367 [DOI] [PubMed] [Google Scholar]
- 10. Carpenter TO, Insogna KL, Zhang JH, et al. Circulating levels of soluble klotho and FGF23 in X-linked hypophosphatemia: circadian variance, effects of treatment and relationship to parathyroid status. J Clin Endocrinol Metab. 2010;95:E352–E357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bettinelli A, Bianchi ML, Mazzucchi E, Gandolini G, Appiani AC. Acute effects of calcitriol and phosphate salts on mineral metabolism in children with hypophosphatemic rickets. J Pediatr. 1991;118:372–376 [DOI] [PubMed] [Google Scholar]
- 12. Martin KJ, González EA, Gellens ME, Hamm LL, Abboud H, Lindberg J. Therapy of secondary hyperparathyroidism with 19-nor-1α,25-dihydroxyvitamin D2. Am J Kidney Dis. 1998;32:S61–S66 [DOI] [PubMed] [Google Scholar]
- 13. Robinson DM, Scott LJ. Paricalcitol: a review of its use in the management of secondary hyperparathyroidism. Drugs. 2005;65:559–576 [DOI] [PubMed] [Google Scholar]
- 14. Jonsson KB, Zahradnik R, Larsson T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;24:1656–1663 [DOI] [PubMed] [Google Scholar]
- 15. Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H, Urakawa I, Fujita T, Wada M, Yamashita T, Fukumoto S, Shimada T. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2009;24:1879–1888 [DOI] [PubMed] [Google Scholar]
- 16. Bergwitz C, Jüppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Farrow EG, White KE. Recent advances in renal phosphate handling. Nat Rev Nephrol. 2010;6:207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bai X, Miao D, Goltzman D, Karaplis AC. Early lethality in Hyp mice with targeted deletion of Pth gene. Endocrinology. 2007;148:4974–4983 [DOI] [PubMed] [Google Scholar]
- 19. Alon US, Levy-Olomucki R, Moore WV, Stubbs J, Liu S, Quarles LD. Calcimimetics as an adjuvant treatment for familial hypophosphatemic rickets. Clin J Am Soc Nephrol. 2008;3:658–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raeder H, Shaw N, Netelenbos C, Bjerknes R. A case of X-linked hypophosphatemic rickets: complications and the therapeutic use of cinacalcet. Eur J Endocrinol. 2008;159:S101–S105 [DOI] [PubMed] [Google Scholar]
- 21. Yavropoulou MP, Kotsa K, Gotzamani Psarrakou A, et al. Cinacalcet in hyperparathyroidism secondary to X-linked hypophosphatemic rickets: case report and brief literature review. Hormones. 2010;9:274–278 [DOI] [PubMed] [Google Scholar]
- 22. US Food and Drug Administration. Sensipar (cinacalcet hydrochloride): Drug Safety Communication. FDA suspends pediatric clinical trials after report of death. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm341255.htm Published February 26, 2013. July 19, 2014
- 23. Moltz KC, Friedman AH, Nehgme RA, Kleinman CS, Carpenter TO. Ectopic cardiac calcification associated with hyperparathyroidism in a boy with hypophosphatemic rickets. Curr Opin Pediatr. 2001;13:373–375 [DOI] [PubMed] [Google Scholar]