

Abstract
Context:
Adiponectin (adpN) production is down-regulated in several situations associated with insulin resistance. The hypoadiponectinemia, which develops in late pregnancy, suggests a role of adpN in pregnancy-induced insulin resistance.
Objective:
In obese pregnancy there is a decreased systemic adpN, which results from down-regulation of gene expression in adipose tissue.
Setting and Design:
One hundred and thirty-three women with uncomplicated pregnancies and a wide range in pre-gravid body mass index (18–62 kg/m2) were recruited at term for a scheduled cesarean delivery. Maternal blood, placenta, and sc abdominal adipose tissue were obtained in the fasting state. DNA methylation was analyzed by MBD-based genome-wide methylation sequencing and methyl-specific PCR of placenta and maternal adipose tissue. mRNA and protein expression were characterized by real-time RT-PCR and immunodetection. Plasma adpN, leptin, and insulin were assayed by ELISA.
Results:
Maternal adipose tissue was the prominent site of adpN gene expression with no detectable mRNA or protein in placenta. In obese women, adipose tissue adpN mRNA was significantly decreased (P < .01) whereas DNA methylation was significantly increased (P < .001) compared with lean women. The decreased adipose tissue expression resulted in normal-weight women having significantly greater plasma adpN compared with the severely obese (12.8 ± 4.3 ng/mL vs 8.6 ± 3.1, P < .001). Plasma adpN was negatively correlated with maternal body mass index (r = −0.28, P < .001) and homeostasis model assessment indices of insulin sensitivity (r = −0.32, P < .001) but not with gestational weight gain.
Conclusions:
Maternal adipose tissue is the primary source of circulating adpN during pregnancy. Further, based on our results, the placenta does not synthesize adiponectin at term. Obesity in pregnancy is associated with negative regulation of adpN adipose expression with increase in adpN DNA methylation associated with lower mRNA concentrations and hypoadiponectinemia. Maternal hypoadiponectinemia may have functional consequences in down-regulating biological signals transmitted by adpN receptors in various tissues, including the placenta.
Adiponectin is a master regulatory protein in metabolic homeostasis, which has insulin-sensitizing and anti-inflammatory properties (1). Plasma adiponectin (adpN) levels decrease in a variety of situations associated with loss of insulin sensitivity, such as obesity, metabolic syndrome, diabetes, and various cancers (2–4). Its role during pregnancy has been examined because of the necessity for decreasing maternal insulin sensitivity compared with pregravid to provide nutrients and ensure growth of feto-placental tissues (5). Circulating adpN levels are decreased during the third trimester when maternal insulin sensitivity is reduced compared with the first and second trimesters (6, 7). Plasma adpN is further decreased in pregnancies associated with preeclampsia and diabetes, when maternal insulin sensitivity is extremely low (8–11).
The human placenta is an active site for synthesis of multiple cytokines which can act locally as well as being released in both maternal and fetal circulations (12). Because adipose tissue and the placenta both have a similar cytokine profile, the question of whether the placenta also synthesizes adpN has been debated (13–15). Considering that decreasing adpN is associated with decreased insulin sensitivity and increased energy expenditure, both of which occur in late pregnancy, it is essential to understand its expression and mechanisms of regulation in various tissues. Structural modifications of DNA by methylation play an important role in repressing gene expression in a tissue-specific manner (16). Hence, the primary aim of this study was to investigate adpN DNA methylation, mRNA expression, and protein concentrations in adipose tissue and placenta in late pregnancy. Secondary aims were to examine the impact of obesity and DNA methylation in adipose tissue and the placenta of term pregnancies in women with various degrees of obesity.
Subjects and Methods
Human subjects
This study was approved by the Institutional Review Board of Metrohealth Medical Center, Case Western Reserve University (Cleveland, OH). A prospective cohort of healthy women with a wide range of pregravid body mass indices (BMI; 19.9–62.3) were recruited at 38.5–40 weeks pregnancy prior to a scheduled cesarean section. Subjects with multiple gestation, fetal anomalies, intrauterine growth restriction, preeclampsia, and diabetes (pre-existing and gestational) were excluded. All subjects provided written informed consent prior to collection of maternal blood, adipose tissue, and placenta. Maternal pregravid BMI was obtained from medical records. Anthropometric and metabolic parameters obtained from 133 women and neonates are presented in Table 1. Neonatal body composition measurements were obtained within 24 h of delivery. The variables included weight on a calibrated scale and length using a measuring board (17). Each placenta was weighed after the umbilical cord and fetal membranes were trimmed by research staff.
Table 1.
Anthropometric and Metabolic Parameters of the Study Cohort According to the World Health Organization BMI Category
| Characteristic | BMI <25 | BMI 25–30 | P Value Versus Lean | BMI 30–40 | P Value Versus Lean | BMI >40 | P Value Versus Lean |
|---|---|---|---|---|---|---|---|
| n | Lean 32 | 37 | 36 | 28 | |||
| HOMA-IR, mmol/L) | 2.48 ± 1.40 | 4.39 ± 2.82 | .0009 | 5.64 ± 7.68 | .0255 | 8.15 ± 3.49 | .0001 |
| Maternal age, y | 28.1 ± 5.7 | 27.3 ± 5.7 | .5797 | 28.4 ± 6.3 | .8244 | 28.9 ± 5.5 | .5525 |
| Gestational age, wk | 38.8 ± 0.4 | 38.9 ± 0.6 | .8685 | 38.6 ± 0.6 | .1353 | 38.8 ± 0.5 | .6366 |
| Pre-pregnancy BMI, kg/m2 | 22.33 ± 1.62 | 27.48 ± 1.42 | .0001 | 34.30 ± 3.26 | .0001 | 45.68 ± 5.47 | .0001 |
| Late-pregnancy BMI, kg/m2 | 28.74 ± 3.00 | 34.23 ± 3.08 | .0001 | 39.46 ± 4.02 | .0001 | 49.91 ± 5.61 | .0001 |
| Gestational weight gain, kg | 16.98 ± 8.20 | 18.06 ± 7.08 | .5621 | 13.74 ± 6.70 | .0770 | 11.02 ± 7.99 | .0061 |
| Maternal BP, mm Hg | 118/69 | 118/68 | ns | 121/71 | ns | 121/70 | ns |
| Maternal insulin, mU/mL | 12.46 ± 6.14 | 21.99 ± 12.23 | .0002 | 26.72 ± 27.06 | .0049 | 40.13 ± 18.06 | .0001 |
| Maternal glucose, mg/dL | 79 ± 18 | 78 ± 9 | .8241 | 78 ± 11 | .9222 | 83 ± 10 | .2717 |
| Maternal leptin, ng/mL | 34.02 ± 20.66 | 48.68 ± 17.47 | .0021 | 55.74 ± 25.58 | .0003 | 89.45 ± 40.33 | .0001 |
| Maternal adiponectin, mg/mL | 11.20 ± 4.55 | 9.80 ± 3.69 | .1640 | 9.85 ± 3.27 | .1631 | 8.17 ± 2.75 | .0033 |
| Ratio leptin/adiponectin | 3.05 ± 2.55 | 6.05 ± 2.82 | .000 | 7.73 ± 5.58 | .000 | 12.13 ± 5.55 | .000 |
| Placenta weight, g | 601 ± 137 | 659 ± 138 | .0873 | 674 ± 139 | .0349 | 709 ± 131 | .0034 |
| Neonatal weight, g | 3247 ± 445 | 3445 ± 554 | .0110 | 3415 ± 512 | .0157 | 3353 ± 447 | .1659 |
| Parity, 0–1/>1 | 15/17 | 21/16 | .6231 | 15/21 | .6111 | 13/15 | .8231 |
| Race, AA/Cauc/Hisp | 5/23/4 | 10/19/8 | 14/20/2 | 14/13/1 | |||
| Smoking Status, no/yes | 24/8 | 28/9 | 27/9 | 22/6 |
AA, African American; Cauc, Caucasian; Hisp, Hispanic; ns, not significant.
Results are means ± sd One-way ANOVA was used for assessing differences among the four BMI groups.
Biological specimen collection
Blood and tissue samples were obtained from 133 women following 8–10-h fast. Maternal venous blood (10 mL) samples were collected prior to placement of iv lines for hydration. A 5-ml aliquot was immediately processed to recover RNA from white blood cells. A 5-ml aliquot was drawn into EDTA tubes, and after separation, plasma was kept frozen at −20°C. At each cesarean delivery, sc white adipose tissue (1–3 g) was obtained from the lower abdominal sc region at the site of the incision. Total RNA was purified from white blood cells using RNeasy kit (Qiagen Inc). The placenta was obtained immediately after double clamping of the umbilical cord for venous fetal blood. Fragments of deep villous tissue (0.5 × 2 cm; ∼1-cm depth from the maternal surface) were removed from cotyledons proximal to the cord insertion. All placental fragments were washed three times in cold sterile PBS and were quickly blotted on sterile gauze to minimize the amount of intervillous blood trapped in the villous tissue. Placenta and adipose tissue fragments (∼300 mg) were flash frozen in liquid nitrogen within 10 min of delivery and were stored at −80°C for later molecular analysis. Primary trophoblast cells were isolated by sequential trypsin and DNase digestion followed by gradient centrifugation (18). Adipocytes and adipose stromal cells were isolated from adipose tissue fragments by collagenase digestion.
DNA methylation assays
DNA from placenta and sc white adipose tissue was purified using the DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer's instructions. Densely CpG-methylated regions of the genome were isolated using an MBD-isolated Genome Sequencing approach (19). Briefly, His-tagged recombinant MBD proteins and anti-α-His antibodies were bound to protein G beads. DNA was sonicated to obtain ∼200-bp DNA fragments, and samples were incubated with the MBD-coated beads at 4°C overnight. Unbound DNA was eluted from the beads and then was extracted and precipitated. Methylated DNA was used to prepare a sequencing library using the NEBNext DNA library prep kit (Illumina). After adenylation and ligation, 250-bp fragments were selected and enriched in an 18-cycle PCR. The DNA quality and quantity in the libraries were verified using an Agilent Bioanalyzer 2100. Each MBD library was sequenced on one lane of an Illumina Genome Analyzer II. All reads were mapped to the human genome (Hg18) using the Bowtie alignment.
Methylation-specific PCR
Genomic DNA (125 ng) was subjected to bisulphite conversion using the Cells-to-CpG bisulfite conversion kit (Applied Biosystems). This process allowed for the conversion of methylated cytosines into uracils (20). Completion of the bisulfite treatment was assessed by testing a cytosine outside a CG dinucleotide. For methylation-specific PCR (MSP) analysis, two primer pairs were designed for adpN gene (Integrated DNA technologies) using Methprimer software (21). The primers mapped the adpN gene spanning from 186560421–186562170 on chromosome 3. The first primer set recognized and annealed to methylated sequences only; the second set amplified unmethylated alleles. The sense and antisense primers for the bisulfite-converted methylated sequence were ADPN-MR (5′-aattacaaacacctaccatcacg-3′) and ADPN-ML (5′-taattttagtaatttgggagatcga-3′), respectively (product size, 140kb). The sense and antisense primers for the bisulfite-converted unmethylated sequence were ADPN-UR (5′-aaattacaaacacctaccatcacac-3′) and ADPN-UL (5′-gtaattttagtaatttgggagattga-3′), respectively (product size, 142 kb). The annealing temperature was experimentally determined using gradient PCR to ensure that only methylated templates were amplified. Real-time PCR was initiated with a denaturation step of 5 min at 95°C. The subsequent thermal profile for the ADPN-M and ADPN-U real-time MSPs was 95°C for 10 s, 50°C for 15 s, and 72°C for 10 s for 35 cycles. Data obtained following 35 cycles of amplification were analyzed. Human placental DNA treated in vitro with M. SssI methyltransferase (New England Biolabs) was used as a positive control for the methylated MSP. Genomic DNA from maternal blood was used as a control in the unmethylated reaction. Negative water blanks were included in every analysis.
Molecular studies
The study cohort was classified according to quartiles of prepregnancy BMI (<25, 25–30, 30–40, >40) and 20 samples of maternal adipose tissue and placenta from each quartile were processed for RNA and DNA methylation assays.
RNA preparation and real-time PCR
RNA was extracted from placenta and adipose tissue using TRIzol (Invitrogen). RNA was reversed-transcribed using a Superscript II RNase H–Reverse Transcriptase system (Invitrogen). AdpN, HPL, and actin gene expression levels were measured by real-time PCR using a LightCycler FastStart DNA Master SYBR Green I kit (Roche). Amplifications were performed in duplicate using 20-ng cDNA samples with the following primers pairs: adpN (NM_004797) forward 5′-gcagtctgtggttctgattcc-3′ and reverse 5′-ccagtggagccatcatagtg-3′; actin (NM_001101) forward 5′-ggacttcgagcaagagatgg-3′ and reverse 5′-agcactgtgttggcgtacag-3′; HPL (BC_057768) forward 5′-ccgttatccaggctttttga-3′ and reverse 5′-tggagggtgtcggaatagag-3′. Results were normalized for actin and expressed as copy number/ng RNA calculated against a standard curve of human term placental cDNA.
Protein detection by Western Blot and ELISA
Placental and adipose tissue homogenates were electrophoresed (80 μg/lane) under denaturing conditions and transferred to nitrocellulose membranes. Membranes were blocked for 1 h and incubated with antiactin primary antibody for 1 h at room temperature (Sigma) or with anti-adpN antibody overnight (ACRP302-A; Santa Cruz Biotechnology). Goat antirabbit IgG horseradish peroxidase–conjugated was used as secondary antibody. Immunocomplexes were visualized by chemiluminescence (Amersham).
Plasma assays
Plasma glucose was assessed by the glucose oxidase method. Plasma insulin was measured using ELISA (EMD Millipore Corporation). Leptin and adpN (R&D Systems) were measured using ELISA kits with intraassay coefficients of variation of 3.0–6.2% and 6.2–8.4%, respectively. High molecular weight (HMW) adiponectin multimers and HMW/total adpN were measured using ELISA as per manufacturer's instructions (Alpco). All plasma samples were assayed in duplicate. Insulin resistance was estimated using a homeostasis model assessment (HOMA-IR) as a ratio of fasting plasma insulin (micro units/mL) to fasting plasma glucose (mmol/l/22.5) (22).
Statistical analyses
Values are represented as mean ± SDs in Table 1 and as mean ± SEM in the figures. Differences between dependent variables were examined using one-way or two-way repeated-measures ANOVA. Statistical mean differences for real-time RT-PCR analysis were calculated by Kruskal-Wallis (for nonparametric data) two-way ANOVA (data normally distributed) followed by Bonferroni (Dunn's) multiple comparisons post hoc test. Differences in sequencing analyses were calculated using the Mann-Whitney U test. The relationships among maternal adpN, HOMA-IR, insulin, and BMI were estimated with univariate correlation analyses and were adjusted for potential confounders using partial correlations. Data were analyzed using the Statview II statistical package (Abacus Concepts) and Statistix 8.0 (Analytical Software). Statistical significance was set at P < .05.
Results
AdpN expression at the maternal-fetal interface
We measured adpN mRNA in maternal adipose tissue, circulating white blood cells and the placenta to assess the relative contribution of maternal and feto-placental tissue to adpN production (Figure 1). AdpN gene copy number in adipose tissue (1995 ± 66 copies/pg RNA) was 60-fold higher than in the white blood cells (33 ± 6 copies/pg RNA) and 600-fold higher than in the placenta (5.1 ± 0.2 copies/pg RNA). The human placenta at delivery retains a large volume of maternal and fetal blood even after extensively washing the villous tissue with saline buffer. We investigated the possibility that the small number of adpN transcripts found in placental tissue may be accounted for by adpN mRNA from residual maternal white blood cells. We thus compared adpN gene expression in placental tissue before and after wash in saline solution to minimize contamination with blood of maternal origin. The wash step decreased the concentration of the adpN transcripts to a level close to the limits of detection (Figure 2B). To determine whether washing affected expression of placental specific mRNA, we measured placental human placental lactogen (HPL) protein. The expression of HPL mRNA was not significantly different before or after placental tissue had been similarly washed and blood removed. Considering the low number of adpN gene copies in placenta, we tested the possibility that the adpN protein detected in placenta originated from circulating adpN rather than from in situ placental synthesis. A 30-kDa signal corresponding to the adpN monomer and a weaker signal migrating at ∼60 kDa, corresponding to the dimeric form, was detected in plasma, adipose tissue, and the placenta in Westhern blots performed under reducing condition (Figure 2A). Both adpN signals became undetectable after washing the placenta tissue with PBS containing 0.25% trypsin to remove external membrane bound proteins. In contrast, the actin protein signal was not abolished by the wash step.
Figure 1.
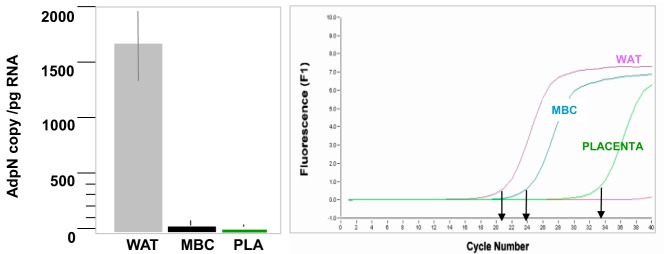
Expression of adpN in maternal blood and the placenta. Left: adpN mRNA expression in maternal adipose tissue, white blood cells, and the placenta was measured by real-time RT-PCR. Results calculated as gene copy numbers were normalized for actin mRNA levels. Data are shown as mean ± SE for tissues sampled from lean women (n = 9). Right: Amplification profile of a representative real-time analysis. WAT, maternal white adipose tissue; MBC, maternal blood cells; pla, placenta.
Figure 2.
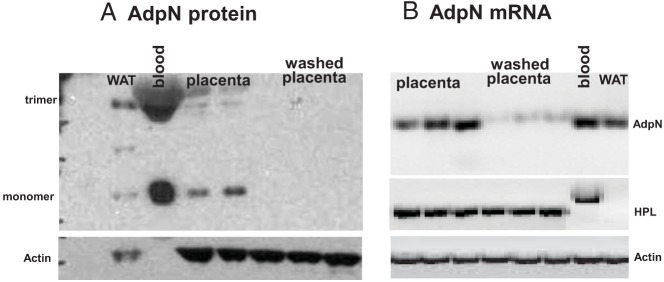
Effect of blood removal on adpN expression in the placenta. A) representative Western blot showing detection from tissues of normal-weight women at term delivery. The adpN monomer migrated as a 33-kDa apparent molecular weight signal, and the dimer migrated as a 60-kDa signal. Identical results were obtained from 4–6 independent experiments. B) RT-PCR analysis of adpN mRNA. Total RNA was extracted from human placenta before and after blood removal through serial PBS washes and was analyzed by RT-PCR. Maternal adipose tissue and blood were used as positive controls. The number of amplification cycles was 45 for adpN and 30 for β-actin. A nonspecific HPL amplification product was detected in white blood cells. Identical results were obtained from 4–6 independent experiments. WAT, white adipose tissue; bl, blood.
Methylation of the adpN gene
Structural modifications of DNA by methylation play an important role in repressing gene expression in a tissue-specific manner (16). Genome-wide DNA methylation patterns were examined in adipose tissue and the placenta (Figure 3). Methylation marks were detected in adipose tissue and none of the methylated regions overlapped with annotated CpG islands of the adpN gene. In contrast, DNA from placenta and isolated trophoblast cells did not exhibit methylation signals in adpN gene or flanking region (< 5kb). For comparison, in Figure 4A we show that in the same sample (where there is no methylation of adpN), leptin, exhibited dense methylation signals overlapping with one annotated CpG island in the proximal promoter (Figure 3).
Figure 3.

DNA methylation patterns at the adiponectin locus. Representative DNA methylation pattern in five tissues and cell types obtained from one normal-weight woman at term pregnancy. The x-axis represents > 60 kb of DNA sequence on chromosome 3, and the y-axis represents the number of reads. The brown histograms indicate the number of reads (y-axis, 0–78.5) obtained after MBD isolation in each 100-bp window (x-axis). Annotations from the UCSC genome browser are shown in blue. CpG islands are depicted in green at the bottom of the graphs. Right panel: methylation patterns at the leptin loci for cpmparison. Blue rectangle delineates the sequence spanned by the primers used in methyl-PCR analysis.
Figure 4.
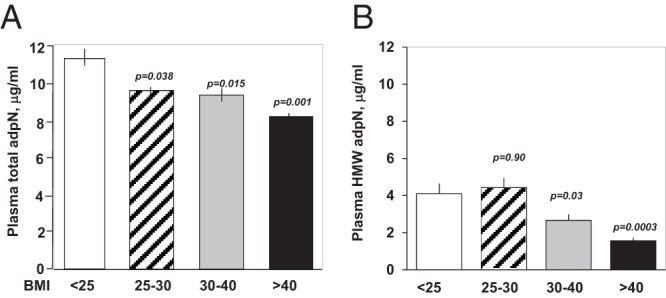
A, Total AdpN plasma concentrations were grouped according to the World Health Organization BMI category as reported in Table 1. B, HMW multimer distribution was expressed as percent of total adpN.
Impact of obesity on adpN expression and circulating levels
Overweight and obese women had significantly lower plasma adpN concentrations than normal-weight women (Figure 4A). The adpN concentrations was lowest (8.2 ± 2.7 vs 11.2 ± 4.5, P < .003) in the women with severe obesity (BMI >40). The plasma levels of the multimeric HMW AdpN, the most bioactive form of Adpn, closely followed that of total plasma adiponectin (Figure 4B). Consequently, plasma adpN concentrations were inversely correlated (r = −0.27, P < .001) with maternal late pregnancy BMI (Supplemental Figure 1). Adiponectin was also negatively correlated (r = −0.32, P < .0001) with HOMA-IR estimates of insulin sensitivity, which was significantly lower in obese women (Supplemental Figure 1 and Table 1). There was no association, however, between plasma adiponectin and gestational weight gain (Supplemental Figure 1). Maternal hypoadiponectinemia was associated with lower adipose tissue adpN mRNA (P < .001) and higher levels of DNA methylation (P < .01) in adipose tissue of severly obese women. Placental adpN gene expression and DNA methylation remained undetectable in obese as in lean women (Figure 5).
Figure 5.

Impact of obesity on adpN gene methylation and expression in adipose tissue and the placenta. DNA methylation patterns measured by methyl-specific PCR in adipose tissue A) and placenta B) were expressed as fold-change percent methylation in overweight (BMI, > 30), obese (BMI, 30–40), and severly obese (BMI, > 40) vs normal-weight (BMI, < 25) women. AdpN mRNA levels measured by real-time RT-PCR were expressed as copies/ng RNA in the four categories of BMI in the adipose tissue A) and placenta B). Data are shown as mean ± SEM of independent determination in adipose/placenta tissue from 20 women in each BMI quartile. Lean, open bars; overweight, hatched bars; obese, gray bars; severely obese, black bars.
Discussion
Adiponectin in pregnancy
Maternal adipose tissue and placenta represent the two primary sites for systemic cytokines during pregnancy. Because of the strong overlap between the adipose and placental cytokine gene expression profile (12) it has been proposed that adpN is expressed in both tissues. We report that adpN is expressed only in maternal adipose tissue, but not placenta. We showed that although adpN is detectable by Western blot in crude placental homogenates because of contamination with maternal blood, the protein is not synthesized de novo in placenta. Hence, the adpN present in the placenta is detected because of circulating adpN trapped in the placenta villous tissue. The human placenta retains a large amount of maternal blood in the intervillous space, which can account for up to 30% of the placenta volume at the end of pregnancy (23, 24). In addition, the high concentrations of adpN in plasma (several micrograms/mL) exceed those of most other cytokines by 1,000–100,000 fold. This may explain why several studies have reported expression of adpN in the human placenta (13, 25, 26). To place our results in context, late pregnancy is a condition of progressive decreasing maternal insulin sensitivity necessary for fetal growth. Given the potent insulin-sensitizing properties of adpN, increasing adpN production by placenta while decreasing concentrations in the blood would seem to be physiologically counterintuitive.
Why is Adpn not expressed in the placenta
The regulation of gene transcription and expression is a signature of cell specialization can be achieved through multiple levels of molecular strategies. One of the primary mechanisms for repressing gene expression in mammals is DNA methylation at CpG residues (16). We investigated whether changes in DNA methylation contributes to the absence of adpN gene transcription in the placenta. We searched for evidence of methylation around marks in a 100-kb region of chromosome 3 spanning the entire adpN gene in the placenta. Neither the proximal promoter of the adpN gene nor the closest CpG island (located 20 kb upstream of exon 1) showed evidence of methylation. This observation suggests that other regulatory mechanisms contribute to the lack of placental adpN gene expression. Whereas the human genome is highly methylated (>70%) in the placenta is globally hypomethylated (27), RNA-seq analysis has shown that genes in partially methylated domains, accounting for up to 40% of the placental genome, are repressed and less likely to be transcribed into proteins (28). Whether adpN falls under this type of regulation is unknown. Our results contrast with those of Bouchard et al (29), who reported significant methylation of three CpG islands at the placental adpN locus. The reasons for the discrepancy are unclear given that our analysis spanned the entire adpN gene.
Role of adpN in fetal-placental homeostasis
If the placenta is not itself a site of adpN-secreting cells, it still may play a role as a functional target of maternal adpN. Our findings that placental tissue retains large amount of adpN multimeric proteins (Figure 2B, 4B) supports the possibility that systemic adpN exert endocrine action in the placenta. The metabolic actions of adpN are mediated via binding to specific adpN receptors (30). Both R1 and R2 adpN receptors are expressed in the human placenta and can transduce biological signals (31, 32). For example, adpN regulates placental amino acid transporters via R2 receptor binding in placental cells (33). In vitro, adpN also decreases human chorionic gonadotropin and progesterone production by trophoblast cells (34). Taken together, these observations suggest that the decreased availability of maternal adpN may modify placental function in obese women.
Impact of obesity on adpN availability
Obesity in pregnancy is associated with hypoadiponectinemia, which is inversely correlated to maternal BMI but has no relationship with gestational weight gain. In the absence of placental adpN, the lower maternal plasma levels reflect fluctuations in the synthesis and secretion by all maternal adipose tissue depots including subcutanoeus and visceral. Because obese women have less gestational weight gain than their lean counterparts (mean, 11 vs 16 kg; Table 1) the ratio between maternal tissue weight gain and feto-placental weight decreases (4.3–2.7) with increasing BMI. This is also consistent with the observation that low plasma adiponectin adpN concentrations do not predict weight gain in humans (35). Given that HMW multimers represent 60–70% of total circulating adpN, the distribution of multimeric HMW species may contribute significantly to pregnancy-related regulations (6). The strong inverse association between adpN concentrations and insulin sensitivity is in line with the causal relationship reported in type 2 diabetes (36).
The transcriptional mechanisms of adpN regulation have been partially deciphered. cAMP response element-binding protein, nuclear factor of activated T-cells, and IGF binding protein-3, all act as transcriptional repressors of adpN (37). Alternatively, inflammatory stress and dietary factors such as a high-fat diet have act as epigenetic regulators of the adpN gene (38, 39). In this context, the hypermethylation of the adpN gene in adipose tissue of obese women may also contribute to decrease the mRNA expression. The renin-angiotensin system may also contribute to regulate local adiponectin content (40). Furthermore, the higher amounts of TNF-alpha and IL-6, which we have reported in adipose tissue of obese/insulin-resistant pregnant women (41, 42), are also potential regulatory candidates of adpN availability in pregnancy.
In conclusion, our study establishes that maternal adipose tissue is a prominent source of adpN in term pregnancy. Consequently, the hypoadiponectinemia associated with obesity reflects the decreased expression and methylation of the adipose adpN gene. Additional studies are warranted to establish the functional consequences of decreased adpN availability on placental function.
Acknowledgments
This work was supported by NIH RO1-HD22965 to P.M.C., and R01-DK088824 to D.S.
Author contributions: S.H.M. wrote the manuscript; S.B., M.H., and D.S. performed experiments; L.P. edited data; P.M.C. critically read and reviewed the manuscript. S.H.M. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AdpN
- plasma adiponectin
- BMI
- body mass index
- HMW
- high molecular weight
- HOMA-IR
- homeostasis model assessment
- MSP
- methylation specific polymerase chain reaction.
References
- 1. Turer AT, Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia. 2012;55:2319–2326 [DOI] [PubMed] [Google Scholar]
- 2. Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. 1996;271:10697–10703 [DOI] [PubMed] [Google Scholar]
- 3. Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83 [DOI] [PubMed] [Google Scholar]
- 4. Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes. 2003;52:1779–1785 [DOI] [PubMed] [Google Scholar]
- 5. Catalano PM, Tyzbir ED, Roman NM, Amini SB, Sims EA. Longitudinal changes in insulin release and insulin resistance in nonobese pregnant women. Am J Obstet Gynecol. 1991;165:1667–1672 [DOI] [PubMed] [Google Scholar]
- 6. Catalano PM, Hoegh M, Minium J, et al. Adiponectin in human pregnancy: implications for regulation of glucose and lipid metabolism. Diabetologia. 2006;29:1677–1685 [DOI] [PubMed] [Google Scholar]
- 7. Meyer BJ, Stewart FM, Brown EA, et al. Maternal obesity is associated with the formation of small dense LDL and hypoadiponectinemia in the third trimester. J Clin Endocrinol Metab. 2013;98:643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hendler I, Blackwell SC, Mehta SH, et al. The levels of leptin, adiponectin, and resistin in normal weight, overweight, and obese pregnant women with and without preeclampsia. Am J Obstet Gynecol. 2005;193:979–983 [DOI] [PubMed] [Google Scholar]
- 9. Ranheim T, Haugen F, Staff AC, Braekke K, Harsem NK, Drevon CA. Adiponectin is reduced in gestational diabetes mellitus in normal weight women. Acta Obstet Gynecol Scand. 2004;83:341–347 [DOI] [PubMed] [Google Scholar]
- 10. Mazaki-Tovi S, Romero R, Vaisbuch E, et al. Maternal serum adiponectin multimers in gestational diabetes. J Perinat Med. 2009;37:637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lacroix M, Battista MC, Doyon M, et al. Lower Adiponectin Levels at First Trimester of Pregnancy Are Associated With Increased Insulin Resistance and Higher Risk of Developing Gestational Diabetes Mellitus. Diabetes Care. 2013;36:1577–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hauguel-de Mouzon S, Guerre-Millo M. The placenta cytokine network and inflammatory signals. Placenta. 2006;27:794–798 [DOI] [PubMed] [Google Scholar]
- 13. Chen J, Tan B, Karteris E, Zervou S, Digby J, Hillhouse EW. Secretion of adiponectin by human placenta: differential modulation of adiponectin and its receptors by cytokines. Diabetologia. 2006;49:1292–1302 [DOI] [PubMed] [Google Scholar]
- 14. Pinar H, Basu S, Hotmire K, et al. High molecular mass multimer complexes and vascular expression contribute to high adiponectin in the fetus. J Clin Endocrinol Metab. 2008;93:2885–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Corbetta S, Bulfamante G, Cortelazzi D, et al. Adiponectin expression in human fetal tissues during mid- and late gestation. J Clin Endocrinol Metab. 2005;90:2397–2402 [DOI] [PubMed] [Google Scholar]
- 16. Bird A. DNA methylation patterns and epigenetic memory. Genes, Dev. 2002;16:6–21 [DOI] [PubMed] [Google Scholar]
- 17. Catalano PM, Thomas AJ, Avallone DA, Amini SB. Anthropometric estimation of neonatal body composition. Am J Obstet Gynecol. 1995;173:1176–1181 [DOI] [PubMed] [Google Scholar]
- 18. Kliman HJ, Nestler JE, Sermasi E, Sanger JM, Strauss JF. Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology. 1986;118:1567–1582 [DOI] [PubMed] [Google Scholar]
- 19. Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2011;38:391–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431 [DOI] [PubMed] [Google Scholar]
- 22. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Tracher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419 [DOI] [PubMed] [Google Scholar]
- 23. Barker G, Cunliffe N, Bardsley WG, D'Souza SW, Donnai P, Boyd RD. Fetal and maternal blood volumes in shed human placentae: discrepant results comparing morphometry to haemoglobin content. Placenta. 1988;9:289–296 [DOI] [PubMed] [Google Scholar]
- 24. Barker G, Boyd RD, D'Souza SW, Donnai P, Fox H, Sibley CP. Placental water content and distribution. Placenta. 1994;15:47–56 [DOI] [PubMed] [Google Scholar]
- 25. Lappas M, Yee K, Permezel M, Rice GE. Release and regulation of leptin, resistin and adiponectin from human placenta, fetal membranes, and maternal adipose tissue and skeletal muscle from normal and gestational diabetes mellitus-complicated pregnancies. J Endocrinol. 2005;186:457–465 [DOI] [PubMed] [Google Scholar]
- 26. Caminos JE, Nogueiras R, Gallego R, et al. Expression and regulation of adiponectin and receptor in human and rat placenta. J Clin Endocrinol Metab. 2005;90:4276–4286 [DOI] [PubMed] [Google Scholar]
- 27. Novakovic B, Saffery R. DNA methylation profiling highlights the unique nature of the human placental epigenome. Epigenomics. 2010;2:627–638 [DOI] [PubMed] [Google Scholar]
- 28. Schroeder DI, Blair JD, Lott P, et al. The human placenta methylome. Proc Natl Acad Sci U S A. 2013;110:6037–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouchard L, Hivert MF, Guay SP, St-Pierre J, Perron P, Brisson D. Placental adiponectin gene DNA methylation levels are associated with mothers' blood glucose concentration. Diabetes. 2012;61:1272–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamauchi T, Nio Y, Maki T, et al. Targeted disruption of adipoR1 and adipoR2 causes abrogation of adiponectin binding and metabolic actions. Nature Med. 2007;13:332–339 [DOI] [PubMed] [Google Scholar]
- 31. Tie W, Yu H, Chen J, Wang X, Chen W, Zhou R. Expressions of adiponectin receptors in placenta and their correlation with preeclampsia. Reprod Sci. 2009;16:676–684 [DOI] [PubMed] [Google Scholar]
- 32. Rosario FJ, Schumacher MA, Jiang J, Kanai Y, Powell TL, Jansson T. Chronic maternal infusion of full-length adiponectin in pregnant mice down-regulates placental amino acid transporter activity and expression and decreases fetal growth. J Physiol. 2012;590:1495–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones HN, Jansson T, Powell TL. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino Acid transport in human primary trophoblast cells. Diabetes. 2010;59:1161–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McDonald EA, Wolfe MW. Adiponectin attenuation of endocrine function within human term trophoblast cells. Endocrinology. 2009;150:4358–4365 [DOI] [PubMed] [Google Scholar]
- 35. Vozarova B, Stefan N, Lindsay RS, et al. Low plasma adiponectin concentrations do not predict weight gain in humans. Diabetes. 2002;51:2964–2967 [DOI] [PubMed] [Google Scholar]
- 36. Weyer C, Funahashi T, Tanaka S, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86:1930–1935 [DOI] [PubMed] [Google Scholar]
- 37. Liu B, Lee HY, Weinzimer SA, et al. Direct functional interactions between insulin-like growth factor-binding protein-3 and retinoid X receptor-alpha regulate transcriptional signaling and apoptosis. J Biol Chem. 2000;275:33607–33613 [DOI] [PubMed] [Google Scholar]
- 38. Bruun JM, Lihn AS, Verdich C, et al. Regulation of adiponectin by adipose tissue-derived cytokines: in vivo and in vitro investigations in humans. Am J Physiol Endocrinol Metab. 2003;285:E527–33 [DOI] [PubMed] [Google Scholar]
- 39. Karki S, Chakrabarti P, Huang G, Wang H, Farmer SR, Kandror KV. The multi-level action of fatty acids on adiponectin production by fat cells. PLoS One. 2011;6:e28146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vaidya A, Forman JP, Underwood PC, et al. The influence of body mass index and renin-angiotensin-aldosterone system activity on the relationship between 25-hydroxyvitamin D and adiponectin in Caucasian men. Eur J Endocrinol. 2011;164:995–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kirwan JP, Hauguel-De Mouzon S, Lepercq J, et al. TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes. 2002;51:2207–2213 [DOI] [PubMed] [Google Scholar]
- 42. Basu S, Haghiac M, Surace P, et al. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity. 2011;19:476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]