

Abstract
Context:
Visceral adipose tissue (VAT) is a key contributor to chronic inflammation in obesity. The 12/15-lipoxygenase pathway (ALOX) is present in adipose tissue (AT) and leads to inflammatory cascades that are causal for the onset of insulin resistance in rodent models of obesity.
Objective:
The pathophysiology of the ALOX 12/15 pathway in human AT is unknown. We characterized the ALOX pathway in different AT depots in obese humans with or without type 2 diabetes (T2D).
Design:
This study includes a cross-sectional cohort of 46 morbidly obese (body mass index >39 kg/m2) nondiabetic (n = 25) and T2D (n = 21) subjects.
Setting:
This study was conducted at Eastern Virginia Medical School (Norfolk, Virginia) in collaboration with Sentara Metabolic and Weight Loss Surgery Center (Sentara Medical Group, Norfolk, Virginia).
Patients:
Twenty-five obese (body mass index 44.8 ± 4.4 kg/m2) nondiabetic (hemoglobin A1c 5.83% ± 0.27%) and 21 obese (43.4 ± 4.1 kg/m2) and T2D (hemoglobin A1c 7.66% ± 1.22%) subjects were included in the study. The subjects were age matched and both groups had a bias toward female gender.
Main Outcomes and Measures:
Expression of ALOX isoforms along with fatty acid substrates and downstream lipid metabolites were measured. Correlations with depot-specific inflammatory markers were also established.
Results:
ALOX 12 expression and its metabolite 12(S)-hydroxyeicosatetraenoic acid were significantly increased in the VAT of T2D subjects. ALOX 15A was exclusively expressed in VAT in both groups. ALOX 12 expression positively correlated with expression of inflammatory genes IL-6, IL-12a, CXCL10, and lipocalin-2.
Conclusions:
ALOX 12 may have a critical role in regulation of inflammation in VAT in obesity and T2D. Selective ALOX 12 inhibitors may constitute a new approach to limit AT inflammation in human obesity.
The lipoxygenase (ALOX) family of enzymes is responsible for the oxidative metabolism of polyunsaturated fatty acids (1). 12/15-Lipoxygenase (LO) activity is required for the inflammation seen in high-fat diet-induced models of obesity in rodents (2), and 12/15-LO and its arachidonic acid (AA)-derived proinflammatory metabolites hydroperoxyeicosatetraenoic acid and hydroxyeicosatetraenoic acids (HETEs) are involved in insulin resistance (3). Sequential action of the 12-LO or 15-LO and 5-LO on docosahexanoic acid (DHA) leads to the formation of antiinflammatory lipid compounds such as maresins and resolvins (4). A recent paper described the presence and diversity of both the pro- and antiinflammatory lipid mediators derived from the lipoxygenase pathway in sc and perivascular fat in humans (5). We have previously demonstrated that in obese humans, ALOX 12, ALOX 15a, and ALOX 15b are more highly expressed in omental (OM) than in sc adipose tissue (AT) (6). Little is known about the upstream activators of the 12/15 ALOX enzymes in human AT. A potential candidate is lipocalin-2 (LCN2), a recently characterized proinflammatory molecule with positive correlation with obesity and insulin resistance (IR) (7) that was shown to stimulate activity of 12-LO in AT (8).
Here we characterize the ALOX12/15 family of enzymes and their pro- and antiinflammatory metabolites in the sc and OM depots of obese humans with and without type 2 diabetes (T2D). Results suggest a proinflammatory role for ALOX 12 and its metabolites and significant correlations between expression of LCN2, proinflammatory cytokines and ALOX 12 in OM fat in T2D.
Selective targeting of ALOX 12 may provide a novel therapeutic approach to reduce inflammation in visceral fat and resulting complications.
Materials and Methods
Research subjects
The study included a cross-sectional cohort of morbidly obese T2D (n = 21) and nondiabetic subjects (n = 25), aged 18–65 years, undergoing bariatric surgery at Sentara Metabolic and Weight Loss Surgery Center (Sentara Medical Group, Norfolk, Virginia).
Exclusion criteria included autoimmune disease including type 1 diabetes mellitus, conditions requiring chronic immunosuppressive therapy, antiinflammatory medications, thiazolinendiones, active tobacco use, chronic or acute infections, or a history of malignancy treated within the last 12 months. T2D was defined as a fasting plasma glucose of 126 mg/dL or greater, a glucose of 200 mg/dL or greater after a 2-hour glucose tolerance test, or use of antidiabetic medications. Informed consent was obtained, and the research project was approved by the Eastern Virginia Medical School Institutional Review Board.
Quantitative real-time PCR and gene arrays
These procedures were described previously (6). Results were normalized to 18S RNA and data were expressed as 1/Δ cycle threshold. The multiplex gene array PCR for AT was performed using the Cytokine and Chemokine RT2 Profiler PCR array from SA Bioscience.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue biopsies were incubated with antibodies for human leukocyte antigen (HLA-D-related [DR]; Dako; 1:100 dilution), CD68 (Dako; 1:100 dilution), or CD3 (Dako; 1:100 dilution) and detected using the avidin-biotin-peroxidase method. Quantification of the data was done using a MetaMorph software version 6.3 (Molecular Devices).
Lipid extraction and analysis by liquid chromatography and tandem mass spectrometry and gas chromatography
Lipids were extracted from liquid N2-pulverized AT samples using methanol/NaOH followed by the addition of the deuterated internal standards and hydrolysis. Fatty acids were extracted using borontrifluorid/methanol and pentadecanoic acid and further transesterified to corresponding methyl esters that were analyzed using a gas chromatograph equipped with a flame ionization detector. For lipid metabolites, a solid-phase extraction procedure using Varian Bond-Elut-Certify II was performed as described by Rivera et al (9). The liquid chromatography, electrospray ionization, and tandem mass spectrometry method for the determination of metabolites was described previously (10, 11).
Statistical analysis
The SAS MIXED analysis (repeated measures ANOVA) was used to evaluate the main and interactive effects of type of adipose tissue depot (omental vs sc) and diabetes status (diabetic vs nondiabetic), taking into account the clustering of data. Correlations were established using the linear regression analysis and Pearson's coefficient.
Results
Subjects
Twenty-five nondiabetic (three males, 22 females), aged 41.4 ± 9.2 years and 21 T2D subjects (six males, 15 females), aged 48.7 ± 6.8 years were included. The body mass index was 44.8 ± 4.4 kg/m2 for nondiabetic and 48.7 ± 6.8 kg/m2 for T2D subjects (P > .05). Hemoglobin A1c was significantly higher in the T2D vs nondiabetic subjects (7.66% ± 1.22% vs 5.83% ± 0.27%), whereas fasted insulin was not significantly different (6.21 ± 3.24 vs 7.05 ± 2.48 mU/L).
Expression and activity of ALOX 12/15 lipoxygenases
We examined the expression of ALOX 12, ALOX 15a, and ALOX 15b in sc and OM depots of T2D or nondiabetic subjects and found significantly higher expression of ALOX 12 in the OM vs sc depot independent of the presence of T2D. In addition, the effect of T2D on ALOX 12 expression is depot specific, with a significant increase in the OM depot only (Figure 1A). ALOX 15a expression was absent in the sc depot from both T2D and nondiabetic subjects, and we did find an increase in ALOX15B in the OM depot that was independent of T2D presence (Figure 1A).
Figure 1.

ALOX isoform gene expression, fatty acid substrates, and lipid metabolites in human adipose tissue. Gene expression (A) for ALOX 12, ALOX 15A, and ALOX 15B was measured using specific Taqman probes, and data were expressed as 1/Δ cycle threshold after normalization to 18S expression. Results represent mean ± SEM, and the null hypothesis was rejected for a value of P < .05. Fatty acid substrates (B) and metabolites (C) of the ALOX 12/15 pathway were measured in a subgroup of six diabetic and six nondiabetic subjects; the ALOX isoforms involved in formation of the various metabolites are illustrated based on the reported data. Data were analyzed using the SAS MIXED model and comparisons between depots (OM vs sc) and disease groups [nondiabetic (ND) vs diabetic (T2D)] as well as the interaction term (depot × disease) are illustrated below each of the graphs; the null hypothesis was rejected for a value of P < .05. Graphs show the median (solid line) and mean (dotted line) values for each group including the confidence interval and the outliers (dots).
Next, we profiled the 12/15 ALOX fatty acid substrates and metabolites in T2D and nondiabetic sc and OM (Figure 1, B and C). Linoleic acid (LA) showed 10-fold higher values compared with AA and 100-fold higher than DHA. The amount of AA, LA, and DHA did not differ between T2D and nondiabetic subjects in either of the depots (Figure 1B). Proportionally similar abundances were found for the major lipid metabolites originating from the three substrates (Figure 1C). Interestingly, the ALOX 12-derived metabolites followed the pattern of ALOX 12 expression. The expression of 12-HETE was independently increased in the OM depot and in T2D. In addition, the effect of diabetic status on 12-HETE expression is depot dependent, as suggested by the significant interaction effect (Figure 1C). This suggests that 12-HETE is a key inflammatory metabolite associated with T2D in the OM fat. Expression of 14(S)-HDHA [(S)-hydroxy-docosahexaenoic acid], the DHA-derived metabolite of ALOX 12 was also increased in the OM depot independent of the presence of T2D (Figure 1C). The levels of 13S-hydroxy-9Z,11E-octadecadienoic acid, the most abundant metabolite, were influenced only by the T2D presence and were depot independent.
The absence of ALOX 15A expression in the sc depot in all of the subjects (Figure 1A) was reflected by consistently lower abundance of the metabolites 15(S)-HETE and 17(S)-HDHA in sc compared with OM (Figure 1C). Interestingly, the antiinflammatory metabolite 17(S)-HDHA was significantly influenced in an independent manner by the depot type and T2D presence, whereas the interaction was not significant (Figure 1C). The increased levels of this antiinflammatory metabolite in the OM and T2D subjects suggest a protective compensatory mechanism in the setting of increased inflammation. 5-HETE was comparable with the other HETE levels; however, no differences in the abundance were found between depots or with T2D (not shown). This suggests that the formation of the antiinflammatory DHA-derived metabolites is primarily accounted for by differences in ALOX 12 and ALOX 15 expression.
Correlation between ALOX expression and AT inflammatory markers
The relationship of other AT inflammatory markers to changes in ALOX isoform expression was tested. We found an increased expression of IL-6, IL-12a, and chemokine ligand 10 (CXCL10) in the OM of T2D subjects compared with nondiabetic subjects as well as significant correlations between the expression of the latter and ALOX 12 expression (Figure 2A). Gene array analysis of 72 cytokines and chemokines indicated several additional proinflammatory molecules significantly up-regulated in the OM and sc of T2D subjects, notably the proinflammatory genes TNFRS11b and SPP1 (osteopontin) (Figure 2B).
Figure 2.


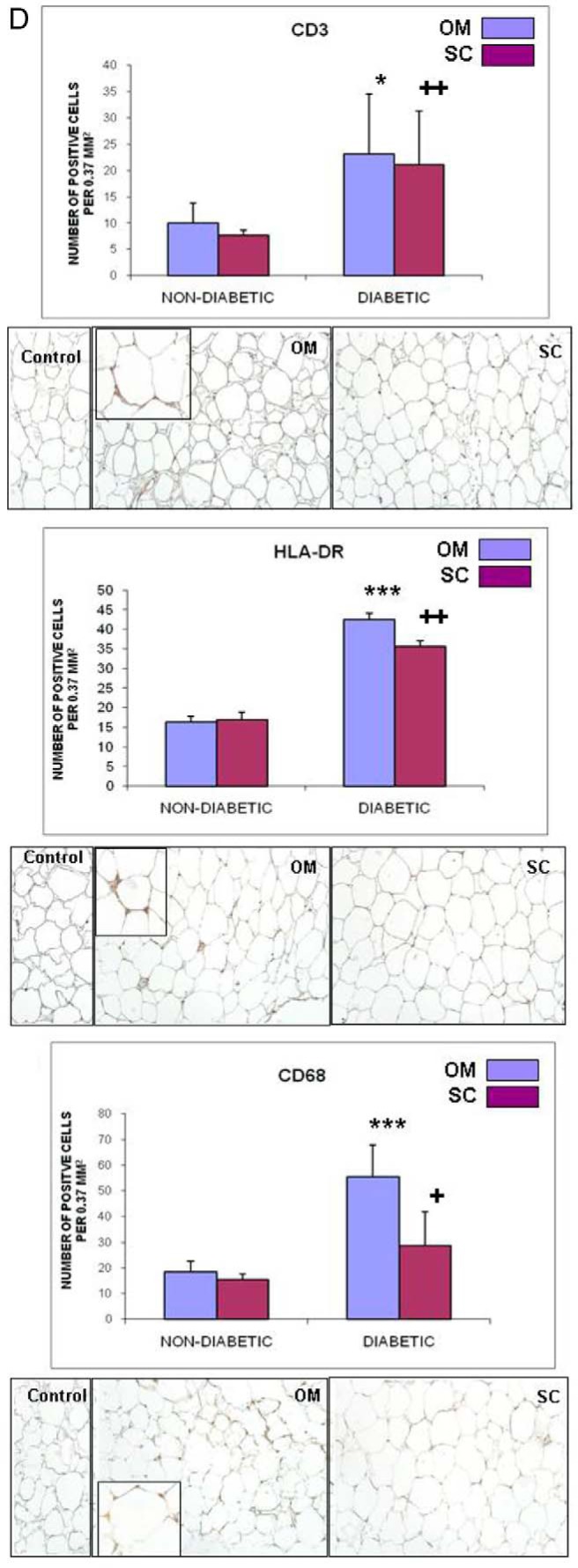
Inflammatory cytokines and immune infiltration in adipose tissue depots of diabetic and nondiabetic subjects. A, Gene expression of the proinflammatory cytokines IL-6, IL-12a, and chemokine CXCL10 was analyzed using the SAS MIXED model as described in Figure 1; linear regression analysis shows a significant correlation between the expression of all three of the cytokines and ALOX 12 expression in the OM depot of diabetic subjects. B, Plots representing genes significantly decreased (left quadrants) or increased (right quadrants) (P < .05) by at least 2-fold in OM and sc depots of diabetic compared with nondiabetic subjects (indicated by arrows in the upper quadrants of each panel); gene expression was measured using the SABioscience gene array in a subset of diabetic and nondiabetic subjects (n = 3). In addition to the cytokines in panel A, the following genes were found significantly changed: NODAL (nodal growth differentiation factor); SPP1 (osteopontin); TNFRSF11b (TNF receptor superfamily 11b); MSTN (myostatin); VEGFA (vascular endothelial growth factor A). C, Expression of LCN2 in human adipose tissue. Gene expression of LCN2 was measured in sc and OM depots in a subgroup of 10 diabetic (D) and 10 nondiabetic (ND) subjects and analyzed by the SAS MIXED model as described in Figure 1; a linear regression analysis shows significant correlation between LCN2 and ALOX 12 expression in OM fat from diabetic subjects; right panel shows semiquantitative analysis of immunohistochemical expression of LCN2 in OM fat of diabetic and nondiabetic subjects (n = 4); micrographs show representative pictures and method control; magnification, ×200; inset, ×400. D, Semiquantitative analysis of immunohistochemical staining for CD3, CD68, and HLA-DR in OM and sc adipose tissue in a subgroup of diabetic and nondiabetic subjects (n = 4/group); representative pictures are shown for paired OM and sc samples of diabetic subjects. Magnification, ×200; insets, ×400 magnification. *, P < .05 compared with nondiabetic group; ***, P < .01 compared with nondiabetic group; ++, P < .05 compared with OM depot.
We next examined the immune cell composition in AT and found an increase in the number and activation of the macrophages in the OM fat, reflected by an increase in CD68 and HLA-DR markers (Figure 2D). T2D had a significant effect on the number of macrophages and CD3+ cells in both of the OM and sc depots (Figure 2D).
LCN2, a proinflammatory neutrophil elastase, up-regulates the expression of 12-LO in murine models of obesity and IR. We found a significantly higher mRNA expression of LCN2 in OM vs sc that was influenced by T2D in a depot-independent manner Also, LCN2 protein expression was elevated in the OM of T2D vs nondiabetic subjects (Figure 2C). Importantly, LCN2 and ALOX12 expressions were significantly correlated in the OM depot of T2D subjects (Figure 2C). These results support LCN2 as a candidate molecule responsible for the up-regulation of ALOX 12 expression in T2D.
Discussion
Visceral AT inflammation is a major contributor to IR, T2D, and cardiovascular complications of obesity. The ALOX pathway was extensively described in visceral AT and vasculature in animal models and was causally related to the development of IR in obesity via generation of proinflammatory metabolites (2). Mice with a targeted deletion of 12-/15-LO in adipose tissue are protected from IR and pancreatic islet inflammation, suggesting a key role of AT 12-/15-LO for the onset of IR in obesity (12). We previously reported that ALOX 12, ALOX 15A and ALOX 15B have a depot- and cell-specific expression in human AT that differs from that of animal models of obesity (6), which emphasizes the importance of findings validation in humans (1).
One novelty of the current study is identifying ALOX 12 as the major isoform up-regulated in OM and increased by T2D in a depot-independent manner. A key feature of this study was that the fatty acid substrates for ALOXs 12/15 and the major classes of lipid metabolites were measured in parallel with ALOX expression in both sc and OM fat in T2D and nondiabetics. Despite the large difference in abundance between the AA, LA, and DHA, there were no differences between the depots or with T2D. This suggests equal substrate exposure but does not rule out possible differences in substrate preference as was previously seen for human ALOX 12, ALOX 15a, and ALOX 15b (13, 14).
In accordance with an earlier report (15), we found 13S-hydroxy-9Z,11E-octadecadienoic acid as the most abundant metabolite in AT. This LA metabolite was significantly increased by the presence of T2D in a depot-independent manner, suggesting a substantial contribution of the ALOX 12 isoform. The AA-derived metabolites, 12-HETE and 15-HETE, were more abundant in the OM vs sc depot. Also, 12-HETE was the only metabolite that was up-regulated in T2D in a depot-specific manner. The antiinflammatory resolvin D1 precursor 17(S)-HDHA, generated by ALOX 15a, was significantly increased by T2D in a depot-independent manner, possibly as an attempt to resolve persistent inflammation. These results suggest that in T2D subjects, ALOX 12 is producing predominantly proinflammatory metabolites, whereas the ALOX 15a produces more of the antiinflammatory metabolites. Future therapeutic development should focus on highly selective ALOX 12/15 inhibitors that target primarily the proinflammatory isoform.
Another key finding is a positive correlation between ALOX 12 expression and proinflammatory cytokines in the OM fat of T2D subjects. Macrophages abundantly infiltrate adipose tissue in obesity (16). We previously showed that ALOX 12 is mainly expressed in the CD34+ cellular component of the stromal vascular compartment including macrophages (6). Indeed, we find that the macrophage marker CD68 and the myeloid activation marker HLA-DR are both significantly increased in the OM fat of T2D subjects, suggesting that the increased macrophage infiltration and activation may contribute to the increased ALOX 12 expression. Also, IL-12 production by lipopolysaccharide-stimulated macrophages is dependent on 12-/15-LO (17), and therefore, increases in ALOX 12 in the OM depot may account for the increased IL-12 expression. We also found in OM of T2D subjects significant elevation of additional inflammatory molecules reportedly increased in obesity and vascular disease (osteopontin and TNFRSF11b). Similar increases in cytokines and inflammatory cell markers were reported recently in the visceral fat of obese subjects with a high correlation of IL6 expression with T2D (18). Importantly, ALOX variants in humans are associated with induced expression of IL-6, TNFα, and IL-1b, indicating a broad role for the enzyme in systemic inflammation (19). Therefore, the correlations found between ALOX 12 and proinflammatory cytokines support a role for ALOX 12 in the initiation and/or propagation of inflammation in visceral AT in T2D.
Reports show that LCN2, a neutrophil elastase produced by adipocytes, is elevated with aging and obesity in rodents and can stimulate the expression and activity of ALOX 12 in visceral fat (8). Our result showing a selective increase in LCN2 gene and protein expression in the OM depot of T2D subjects and significant positive correlation between expression of ALOX 12 and LCN2 supports a potential role of LCN2 as a positive regulator of ALOX 12 in human AT. Activation of LCN2 in OM may be due to increased nuclear factor-κB activation in adipocytes (20), whereas a role for insulin is less likely because no differences were found in fasted insulin between the T2D and nondiabetic groups.
In summary, data establish the depot-specific pattern of expression and activity of the 12/15 ALOX enzymes and identifies ALOX 12 as the main proinflammatory isoform increased by T2D in a depot-dependent fashion.
Acknowledgments
We thank Dr Qian Ma, research associate, John Lindsay, research assistant, and Norine Kuhn, research associate, for their excellent technical assistance. We also acknowledge Dr Michael Rothe (Lipidomix, Inc, Berlin, Germany) for the valuable technical advice on the measurement of lipid metabolites in human adipose tissue. In addition, we thank Ms Becky Marquez (clinical coordinator at Sentara Metabolic and Weight Loss Surgery Center, Norfolk, Virginia) for the valuable assistance with the study subject recruitment.
This work was supported by National Institutes of Health Grant R15HL114062 (to A.D.D.), National Institutes of Health Grant RO1HL112605 (to J.L.N.), and start-up funds (to J.L.N. and A.D.D.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AA
- arachidonic acid
- ALOX
- lipoxygenase
- AT
- adipose tissue
- CXCL10
- chemokine ligand 10
- DHA
- docosahexanoic acid
- DR
- D-related
- HETE
- hydroxyeicosatetraenoic acid
- HLA
- human leukocyte antigen
- IR
- insulin resistance
- LA
- linoleic acid
- LCN2
- lipocalin-2
- LO
- lipoxygenase
- OM
- omental
- (S)-HDHA
- (S)-hydroxy-docosahexaenoic acid
- T2D
- type 2 diabetes.
References
- 1. Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, Nadler JL. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res. 2010;50:115–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sears DD, Miles PD, Chapman J, et al. 12/15-Lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS One. 2009;4:e7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cole BK, Kuhn NS, Green-Mitchell SM, et al. 12/15-Lipoxygenase signaling in the endoplasmic reticulum stress response. Am J Physiol Endocrinol Metab. 2012;302:E654–E665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Claria J, Nguyen BT, Madenci A, Ozaki CK, Serhan CN. Diversity of lipid mediators in human adipose tissue depots. Am J Physiol Cell Physiol. 2013;304(12):C1141–C1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dobrian AD, Lieb DC, Ma Q, et al. Differential expression and localization of 12/15 lipoxygenases in adipose tissue in human obese subjects. Biochem Biophys Res Commun. 2010;403:485–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y, Lam KS, Kraegen EW, et al. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34–41 [DOI] [PubMed] [Google Scholar]
- 8. Law IK, Xu A, Lam KS, et al. Lipocalin-2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes. 2010;59:872–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rivera J, Ward N, Hodgson J, Puddey IB, Falck JR, Croft KD. Measurement of 20-hydroxyeicosatetraenoic acid in human urine by gas chromatography-mass spectrometry. Clin Chem. 2004;50:224–226 [DOI] [PubMed] [Google Scholar]
- 10. Gomolka B, Siegert E, Blossey K, Schunck WH, Rothe M, Weylandt KH. Analysis of ω-3 and ω-6 fatty acid-derived lipid metabolite formation in human and mouse blood samples. Prostaglandins Other Lipid Mediat. 2011;4:81–87 [DOI] [PubMed] [Google Scholar]
- 11. Arnold C, Markovic M, Blossey K, et al. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of ω-3 fatty acids. J Biol Chem. 285:32720–32733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cole BK, Morris MA, Grzesik WJ, Leone KA, Nadler JL. Adipose tissue-specific deletion of 12/15-lipoxygenase protects mice from the consequences of a high-fat diet. Mediators Inflamm. 2012;2012:851798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wecksler AT, Jacquot C, van der Donk WA, Holman TR. Mechanistic investigations of human reticulocyte 15- and platelet 12-lipoxygenases with arachidonic acid. Biochemistry. 2009;48:6259–6267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joshi N, Hoobler EK, Perry S, Diaz G, Fox B, Holman TR. Kinetic and structural investigations into the allosteric and pH effect on the substrate specificity of human epithelial 15-lipoxygenase-2. Biochemistry. 2013;52:8026–8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dandona P, Mohanty P, Ghanim H, et al. The suppressive effect of dietary restriction and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation, and protein carbonylation. J Clin Endocrinol Metab. 2001;86:355–362 [DOI] [PubMed] [Google Scholar]
- 16. Middleton MK, Rubinstein T, Pure E. Cellular and molecular mechanisms of the selective regulation of IL-12 production by 12/15-lipoxygenase. J Immunol. 2006;176:265–274 [DOI] [PubMed] [Google Scholar]
- 17. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lasselin J, Magne E, Beau C, et al. Adipose inflammation in obesity: relationship with circulating levels of inflammatory markers and association with surgery-induced weight loss. J Clin Endocrinol Metab. 2013;99(1):E53–E61 [DOI] [PubMed] [Google Scholar]
- 19. Fairfax BP, Vannberg FO, Radhakrishnan J, et al. An integrated expression phenotype mapping approach defines common variants in LEP, ALOX15 and CAPNS1 associated with induction of IL-6. Hum Mol Genet. 2010;19:720–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao P, Stephens JM. STAT1, NF-κB and ERKs play a role in the induction of lipocalin-2 expression in adipocytes. Mol Metab. 2013;2:161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]