

Abstract
In the present study, we aimed to determine whether mice with coronavirus-induced encephalomyelitis (CIE) develop neurogenic bladder dysfunction that is comparable with the neurogenic detrusor overactivity observed in patients with multiple sclerosis. Adult mice (C57BL/6J, 8 wk of age, n = 146) were inoculated with a neurotropic strain of mouse hepatitis virus (A59 strain) and followed for 4 wk. Inoculation with the virus caused a significant neural deficit in mice with an average clinical symptom score of 2.6 ± 0.5 at 2 wk. These changes were accompanied by 25 ± 5% weight loss at 1 and 2 wk postinoculation (P ≤ 0.001 vs. baseline) followed by a recovery phase. Histological analysis of spinal cord sections revealed multifocal sites of demyelinated lesions. Assessment of micturition patterns by filter paper assay determined an increase in the number of small and large urine spots in CIE mice starting from the second week after inoculation. Cystometric recordings in unrestrained awake animals confirmed neurogenic bladder overactivity at 4 wk postinoculation. One week after inoculation with the A59 strain of mouse hepatitis virus, mice became increasingly sensitive to von Frey filament testing with responses enhanced by 45% (n = 8, P ≤ 0.05 vs. baseline at 4 g); however, this initial increase in sensitivity was followed by gradual and significant diminution of abdominal sensitivity to mechanical stimulation by 4 wk postinoculation. Our results provide direct evidence showing that coronavirus-induced demyelination of the central nervous system causes the development of a neurogenic bladder that is comparable with neurogenic detrusor overactivity observed in patients with multiple sclerosis.
Keywords: neurogenic bladder, multiple sclerosis, animal model
lower urinary tract (LUT) dysfunction is a common complaint in patients with neurological disorders [e.g., multiple sclerosis (MS), Parkinson's disease, and Alzheimer's disease], characterized by significant structural and functional changes in the central nervous system (CNS). The development of therapies for these patients has proven a major challenge because of substantial LUT compromise at the time of clinical presentation. Therefore, understanding the pathophysiological mechanisms of the structural and functional interplay between the urological and nervous systems during the progression of neurodegenerative disorders is vital to facilitate the development of preventative and therapeutic approaches targeting the LUT and ensure their implementation earlier in the course of disease progression.
MS is a chronic autoimmune disorder of the CNS, which is predominantly diagnosed in young adults and has long-term implications for their personal and professional lives (7, 17, 35). This neurodegenerative disease is associated with complex neurogenic bladder dysfunction; the clinical progression of severe LUT symptoms often parallels the progression of MS. LUT symptoms are usually present in 32–97% of patients with MS (9, 17, 35) and include urinary urgency, incontinence, nocturia, urinary hesitancy, overflow incontinence, a sensation of incomplete emptying, urinary retention, and a weak urinary stream (38). The most frequent complaint is urgency of micturition followed by urinary frequency (9, 10, 17, 38). In ∼5–10% of patients, bladder symptoms are present at the onset of MS (38); however, patients with advanced MS regularly experience LUT symptoms (7, 41), which often correlate with bowel and sexual dysfunction (18, 33, 42).
Treatment modalities for LUT dysfunction in MS patients primarily consist of antimuscarinic drugs along with other supplemental approaches (11, 19, 50). However, given the unfavorable side effects of antimuscarinics and poor persistence with therapy, effective therapeutic approaches are desperately needed. The discovery of new pharmacological targets has been hampered by a lack of appropriate animal models of MS to study neurogenic bladder dysfunction (2). The development of animal models in which the clinical and histological pathology of the LUT is similar to that observed in the majority of MS patients is imperative to better understand the pathological mechanisms contributing to LUT dysfunction in MS. Prior work in this field has largely focused on the experimental autoimmune encephalomyelitis (EAE) rodent model of MS, which is induced by immunization with myelin basic protein (MBP) or its derived encephalitogenic peptides (30, 34). Animals with EAE develop pathological features of inflammation and demyelination in the CNS, characterized by infiltration of the spinal cord with inflammatory cells expressing proinflammatory cytokines (5, 36, 39). Several previous studies (5, 39, 52) have reported micturition abnormalities in the EAE model, including both detrusor areflexia and detrusor hyperactivity associated with spinal cord inflammation, and hindlimb paralysis. Although the EAE model has its merits, it is known as “a monophasic disease” characterized by chronic inflammation in the CNS, unclear onset, and limited neurodegeneration (30); these qualities have prevented researchers from determining the precise relationship between the onset and degree of neurological impairment and the level or nature of neurogenic bladder dysfunction.
The exact causes of the development of MS are still unknown. Epidemiological studies (6, 31) have postulate the multifactorial etiology of human MS, including the prominent role of environmental influences (such as viral infection). Viral models of demyelination in the CNS have been widely used for studying the mechanisms of MS pathogenesis (44, 51), but no model of neurogenic bladder dysfunction resulting from virus-induced demyelination of the CNS has been evaluated in animals. Among the available viral models of MS, several strains of mouse hepatitis virus (MHV) have been shown to cause inflammation and demyelination in the CNS, mediated by inflammatory T cells and macrophages, that were similar both clinically and histologically to human MS (8, 23). In the present study, we performed a comprehensive evaluation of neurogenic bladder dysfunction in a mouse model of coronavirus-induced encephalomyelitis (CIE) using inoculation with a neurotropic strain (A59) of MHV. Our data suggest that the CIE model of bladder dysfunction is suitable for studies of the pathological mechanisms driving neurogenic detrusor overactivity upon degeneration in the CNS. Future studies using this model may accelerate the development and testing of novel therapeutic approaches for the prevention and treatment of LUT symptoms in MS patients.
MATERIALS AND METHODS
Mouse model of CIE.
Adult male mice (C57BL/6J, 8 wks of age, n = 146, Jackson Laboratories) received a single intracranial inoculation of MHV (A59 strain, 5,000 plaque-forming units) in 20 μl PBS. For the production of the virus, we followed the method described in Ref. 29. Briefly, the virus was propagated in the 17Cl-1 mouse fibroblast cell line followed by three cycles of freezing-thawing. The large debris was spinned down, and the supernatant was used as a stock solution. The control group received sterile PBS. After inoculation, mice were observed daily for the evaluation of neurological signs of disease progression. Clinical symptoms were assessed daily for each animal, and a clinical symptom score (CSS) was assigned as per the following specifications: 0 = normal with no clinical signs, 1 = loss of tail tonicity/kyphosis, 2 = tail paralysis/severe kyphosis, 3 = partial hindlimb paralysis, 4 = complete hindlimb paralysis, and 5 = complete hindlimb paralysis and forelimb paresis/paralysis. Animals were weighed both before inoculation (baseline) and every subsequent week until the experimental end point. All mice had ad libitum access to food and water with special measures taken to accommodate neurologically compromised animals. To assess the degree and nature of LUT symptoms in mice with CIE, all experiments were performed at 4 wk postinoculation, corresponding to the development of demyelination in the spinal cord as previously established for the A59 strain of MHV (8, 23). All experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Histological visualization of CNS damage and demyelination in CIE mice.
Mice were anesthetized with pentobarbital (70 mg/kg) injected intraperitoneally. Animals were then transcardially perfused with physiological saline solution followed by 4% paraformaldehyde solution (pH 7.4). The brain, urinary bladder, spinal cord, and lumbosacral (L6-S2) dorsal root ganglia were isolated from both control and CIE mice and immediately immersed in 4% paraformaldehyde fixative on ice. Tissues were fixed overnight (4°C) and afterward cryoprotected in sucrose (20%) for 3 days. Next, specimens were sectioned on a cryotome at 10-μm increments. To assess the degree of myelination/demyelination in neural tissues, sections were rinsed in distilled water for 2 min and then incubated with luxol fast blue (0.1%, KTLFB, American MasterTech) for 2 h at 60°C to visualize myelin. After a subsequent wash in distilled water for 30 s, destained nonmyelinated areas were visualized by dipping the slides eight times for 1 s each in lithium carbonate (0.05%, catalog no. 62470, Sigma-Aldrich) followed by a wash with 70% reagent alcohol. Slides were rinsed in distilled water for 15 s and then differentiated by cresyl violet (0.25%, catalog no. C5042, Sigma-Aldrich) for 8 min. After the sections had been dipped in 70% reagent alcohol for 5–10 times, slides were dehydrated quickly through three changes of absolute alcohol (200 proof) and cleared by fresh xylene (3 times). To evaluate gross structural changes in the bladder wall, bladder sections were stained with hematoxylin and eosin (H&E Staining Kit, Richard-Allan Scientific, Kalamazoo, MI) and assessed for signs of edema and inflammation under a light microscope.
Immunofluorescent labeling was performed on spinal cord sections to evaluate the spatial distribution of MBP and glial fibrillary acidic protein (GFAP). Frozen samples of the cervical, thoracic, and lumbosacral spinal cord were washed in PBS for 5 min and blocked with blocking buffer containing 3% normal goat serum in PBS for 1 h at room temperature. Next, tissues were incubated overnight at 4°C with primary antibodies [anti-MBP (rabbit polyclonal antibody, ab40390, Abcam, 1:500 dilution) or anti-GFAP (rabbit polyclonal antibody, ab7260, Abcam, 1:4,000 dilution)] in dilution buffer containing 1% normal goat serum in PBS and 0.2% Triton X-100. After washes with PBS, sections were incubated with secondary antibodies [rabbit Alex fluor 555-conjugated antibody (no. 4413, Cell Signaling, 1:1,000 dilution)] for 1 h at room temperature in the dark followed by a wash with PBS. Images were collected using a Leica DM5000 microscope (Leica Microsystems, Wetzlar, Germany) and stored using ImagePro software (Media Cybernetics, Bethesda, MD).
Level of inflammation in the CNS and LUT assessed by multiplex ELISA.
Frozen brain, spinal cord, and urinary bladder tissues isolated from control (n = 5) and CIE mice at 1 wk (n = 3) and 4 wk (n = 6) postinoculation were homogenized using a PowerGen 500 homogenizer (Fisher Scientific, Rockford, IL) in ice-cold lysis buffer containing 25% glycerol, 62.5 mMTris·HCl, protease inhibitor cocktail tablets (1 tablet/10 ml lysis buffer, catalog no. 11 836 170 001, Roche Applied Science), and phosphatase inhibitor cocktail tablets (1 tablet/10 ml lysis buffer, catalog no. 04 906 837 001, Roche Applied Science). SDS (10%) was added to the samples, and samples were vortexed and boiled for 4 min. Extracts were centrifuged at 10,000 rpm for 15 min at 40°C, and supernatants with total protein were collected. The protein concentration in each sample was detected using the BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL).
To evaluate the level of active inflammation in the CNS and LUT, we performed ELISA in accordance with the manufacturer's instructions using the Cytokine Mouse 10-Plex Panel ELISA kit (Novex, LMC0001, Life Technologies). The Cytokine Mouse 10-Plex Panel for the Luminex platform quantifies the following in serum, plasma, and tissue extracts: mouse granulocyte-macrophage colony-stimulating factor, interferon (IFN)-γ, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12 (p40/p70), and TNF-α. Each sample and standards were run in duplicate. The concentration of total protein in each sample was 400 μg/ml. Briefly, a 96-well microplate was loaded with 25 μl diluted bead solution with antibodies for 30 s followed by supplementation with 200 μl wash solution for 30 s. Standard wash solution was used to wash plates twice followed by the addition of 50 μl incubation buffer to each well; 50 μl assay diluent and 50 μl sample were mixed in each of the assigned wells. Wells designated for the standard curve received 100 μl of the appropriate standard dilution. Plates were incubated for 2 h at room temperature on an orbital shaker to keep beads suspended during the incubation. After incubation, plates were washed two times. Subsequently, 100 μl biotinylated detector antibody was added, incubated for 1 h, and again washed two times; 100 μl of prepared 1× streptavidin-R-phycoerythrin was added to each occupied well except the blank, and samples were incubated for 30 min at room temperature and then washed (three times). After that, 100 μl wash solution was added to each well, and the plate was put on an orbital shaker (500–600 rpm) for 2–3 min to resuspend the beads. The plate was then inserted into the XY platform of the Luminex 200 instrument (Life Technologies) for sample reading and analysis.
Micturition patterns in CIE mice assessed by filter paper assay.
Mice scheduled for histological tissue collection in the experiments described above (n = 11) underwent evaluation of micturition patterns using filter paper assay. Animals were placed in individual cages containing elevated (by 1 in.) mesh flooring for 3 h; filter paper was placed on the bottom of the cage. Each mouse had free access to sterile water gel (HydroGel); however, no food was available during testing. Each animal underwent evaluation at the following time points: preinoculation (baseline) and weeks 1–4. Baseline testing provided an experimental structure in which each animal could serve as an internal control. The number of large (≥1.0 cm) and small (<1.0 cm) micturition spots on the filter paper were summed after each test.
Urodynamic evaluation of bladder function in unanesthetized animals.
The cohort selected for urodynamic evaluation of urinary bladder function (awake cystometry) consisted of both infected (n = 4 mice/CIE group with a CSS of 2 or 3) and control mice (n = 6). These animals underwent a survival surgical procedure with the insertion of bladder catheters 4 days before the experimental endpoint (4 wk postinoculation). Bladder catheter placement required animal anesthetization with isoflurane (VEDCO, St. Joseph, MO). Next, a PTF catheter with a blunted end (polyethylene-50, Clay Adams) was sutured in place at the bladder dome and tunneled out of the abdomen to the nape of the neck, where it was then inserted into the end of an angiocath catheter, as previously described (26). The angiocath was first tested with a gentle saline infusion to reveal no leak at the bladder and then anchored to the fascia and skin of the neck using two to three 5-0 Vicryl sutures. The abdominal incision was also closed in layers. Animals were allowed to recover from anaesthesia and subsequently transferred to individual cages to avoid possible catheter damage by coinhabitants. Mice were given 4 days to recover from surgery before the initiation of cystometric evaluation of bladder function.
For urodynamic assessment of bladder function, conscious mice were placed in cystometry cages (length: 24 cm, width: 16 cm, and height: 12 cm) without restraint and allowed to acclimate for 30 min. The tip of the exteriorized bladder catheter, located at the base of the mouse neck, was connected to a pressure transducer and the infusion pump of the cystometry station (Small Animal Laboratory Cystometry, Catamount Research and Development, St. Albans, VT) using a T-shaped valve. Room temperature saline solution (0.9% NaCl) was infused into the bladder at a rate of 10 μl/min. Voided urine was collected in a tray connected to a force displacement transducer, which actively integrated results into the data-acquisition system. Each animal was observed for approximately six to eight voiding cycles. Urodynamic values were continuously recorded using data-acquisition software (Small Animal Laboratory Cystometry, Catamount Research and Development). The following cystometric parameters were recorded and analyzed in this study: bladder capacity, pressure at the start of micturition, micturition rate, continuous intravesical pressure, intermicturition interval, and number of nonmicturition contractions. Nonmicturition contractions were defined as increased values of detrusor pressure from the baseline that were not associated with voiding. These contractions had amplitudes of at least a third of maximal pressure during a single micturition event.
Cystometric parameters were uploaded from the acquisition software into analysis software (SOF-552 Cystometry Data Analysis, version 1.4, Catamount Research and Development). Maximum pressure at micturition, bladder capacity, micturition volume, number of nonmicturition contractions, intermicturition interval, and micturition rate indexes were calculated. The results were statistically analyzed using one-way repeated-measures ANOVA between groups followed by Bonferroni's posttest as appropriate (Systat Software, San Jose, CA).
Behavioral experiments and assessment of pelvic sensitivity using von Frey filaments.
Pathophysiological changes in the innervation of the pelvic viscera and surrounding somatic structures (e.g., pelvic floor muscle) can affect abdominal sensitivity since both visceral and somatic peripheral inputs converge at the dorsal horn of the spinal cord. Alterations in abdominal sensitivity could be measured using mechanical stimulation with von Frey filaments on the lower abdominal/pelvic area. Mice (n = 8) were tested before (baseline) and every week after inoculation with the virus during a 4-wk period. During testing, mice were placed in individual Plexiglas chambers (6 × 10 × 12 cm) containing a stainless steel wire grid floor (acclimation period was 30 min before testing). The frequency of withdrawal responses was tested using five individual fibers with forces of 0.04, 0.16, 0.40, 1.00, and 4.00 g (Stoelting). Each filament was applied for 1–2 s with an interstimulus interval of 5 s for a total of 10 times, and the hairs were tested in ascending order of force. Stimulation was confined to the lower abdominal area in the general vicinity of the bladder, and care was taken to stimulate different areas within this region to avoid desensitization or “wind up” effects. Three types of behavior signified a positive response to filament stimulation: 1) sharp retraction of the abdomen, 2) immediate licking or scratching of the area of filament stimulation, or 3) jumping, as previously described (46).
Data analysis and statistical comparisons.
All data are expressed as means ± SE. Results were statistically analyzed using one-way repeated-measures ANOVA between groups followed by Bonferroni's posttest as appropriate (Systat Software). Data with P ≤ 0.05 were considered statistically significant.
RESULTS
Behavioral phenotype and neurological deficit development in the CIE model.
Inoculation of mice with MHV-A59 triggered significant changes in animal behavior, which corresponded to the development of acute inflammation in the CNS. Animals experienced the loss of tail tonicity/tail paralysis, kyphosis, and partial/complete hindlimb paralysis over the course of the disease process with CSS values ranging from 1 to 5 (n = 84). The loss of tail tonicity was observed as early as 2 days postinoculation in all CIE mice followed by a progressive diminution of motor reflexes. The control group, injected with PBS alone, showed no impairment throughout the experimental period. The average CSS in the CIE group of mice was 2.3 ± 0.5 at 1 wk postinoculation and 2.6 ± 0.5 at 2 wk postinoculation (P ≤ 0.001 vs. the control group; Fig. 1A). Among infected animals, 28% reached a CSS of 4 and 5, associated with the acute phase of encephalitis; none of these animals survived the acute phase of infection. The remaining mice with a CSS of 2 or 3 recovered from the acute phase by week 2 (n = 60) and had residual signs of neurological deficits, accompanied by the development of significant demyelination in the CNS by 4 wk postinfection. The majority of CIE mice reached their highest CSS within 1 wk after viral infection (Fig. 1B). Changes in the behavioral phenotype were accompanied by progressive weight loss (75.4 ± 5.1% and 77.6 ± 5.2% of baseline values at 1 and 2 wk, respectively, P ≤ 0.001; Fig. 1C). After 2 wk, the mice experienced a gradual increase in weight until they approached their baseline values at 4 wk postinfection. Weight fluctuations directly correlated with the severity of neurological impairment and CSS (Fig. 1D). Mice with a CSS of 2 or 3 experienced significant weight loss after inoculation with MHV-A59 compared with animals with a CSS of 1 (Fig. 1D).
Fig. 1.

Clinical phenotype of mice inoculated with the A59 strain of mouse hepatitis virus (MHV). A: timeframe of average clinical symptom score (CSS) progression through 4 wk postinoculation with MHV-A59 (n = 84). B: day of the highest CSS achieved in infected mice (n = 70). C: weight changes in mice with a CSS from 1 to 3 after inoculation with MHV (n = 24). D: CSS-dependent weight fluctuations in mice with coronavirus-induced encephalomyelitis (CIE) (n = 8 mice/group). *Statistical significance vs. baseline (P ≤ 0.05).
Distribution of demyelination and inflammation in CIE mice.
Histological analysis of spinal cord sections after staining with luxol blue/cresyl violet revealed multifocal, severe demyelination in the cervical (Fig. 2B), thoracic (Fig. 2C), and lumbosacral (Fig. 2D) spinal cord along with scattered inflammatory cell infiltration in the white matter of CIE mice. These features were absent in mice in the control group (Fig. 2A). The distribution of demyelinated lesions throughout the entire length of the spinal column did not follow any specific pattern; however, the cervical spinal cord seemed to be affected more severely compared with thoracic and lumbosacral segments, as evidenced from the analysis of normalized intensity of staining between the segments (Fig. 2E). While the intensity of staining was significantly different between normal spinal cord sections and lesion sites in CIE mice, there was no substantial difference in staining between nonlesion sites in CIE mice compared with control mice (Fig. 2E). Evaluation of luxol blue staining in the dorsal root ganglia did not reveal any significant changes in neuronal morphology or nerve fibers between control and CEI animals (Fig. 2F). To confirm that the formation of lesions was due to the loss of myelin, we performed immunohistochemical labeling of spinal cord sections with antibodies against MBP (Fig. 3A) and observed an ∼50% reduction in MBP labeling at lesion sites across all sections in CIE mice (P ≤ 0.05 vs. control mice; Fig. 3B). Additional immunohistochemical labeling with antibodies against GFAP (Fig. 3C) showed a twofold upregulation in GFAP expression at demyelinated sites postinoculation (P ≤ 0.05 vs. control mice; Fig. 3D).
Fig. 2.

Histological assessment of demyelination in the spinal cord of CIE mice. A: luxol fast blue-stained spinal cord section from a control (Ctrl) mouse showing no signs of demyelination or inflammation. The right image shows an increased magnification of the area selected on the left image. B–D: inoculation with MHV-A59 induced multifocal demyelination in the white matter of the cervical (B), thoracic (C), and lumbosacral (D) spinal cord. The sites of demyelination are indicated by arrows. E: quantification of luxol fast blue-stained spinal cord sections revealed decreased intensity of staining in demyelinated areas after inoculation. Automated digital imaging was used to normalize the intensity of staining in selected areas adjusted to background staining. Values are means ± SE of 5 mice/group. *P ≤ 0.001 vs. control mice. CIE-NL, areas with no lesion vs. lesion sites (CIE-L) evaluated in the same section. Scale bars in A–D = 200 μm for left images and 100 μm for right images. F: histological assessment of sensory (dorsal root) ganglia revealed no structural changes between inoculated and control (Ctr) animals. Scale bar = 50 μm.
Fig. 3.

Distribution of myelin basic protein (MBP) and glial fibrillary acidic protein (GFAP) in spinal cord sections of infected mice. A: representative image of a thoracic spinal cord section with immunohistochemical labeling for MBP. The site of demyelination (left) showed a significant loss of MBP labeling in the white matter. Scale bar = 200 μm. The right image is an enlarged area of the same demyelinated spot. Scale bar = 100 μm. B: normalized intensity of MBP staining combined for all analyzed spinal cord segments. *Significance level of P ≤ 0.05. C: representative image of the section with immunohistochemical labeling for GFAP in the cervical spinal cord. The site of demyelination (left) showed a significant increase in GFAP labeling at the lesion site. Scale bar = 200 μm. The right image is an enlarged area of the same demyelinated spot. Scale bar = 100 μm. D: normalized intensity of GFAP staining combined for all analyzed spinal cord segments. *Significance level of P ≤ 0.05.
Structural changes in the spinal cord of CIE mice were evaluated in parallel with potential changes in urinary bladders isolated from same animals. Gross analysis of hemotoxylin and eosin-stained sections from the urinary bladders of CIE mice did not reveal any substantial differences between control and infected mice (Fig. 4A). In addition, a cytokine multiplex ELISA was used to evaluate the level of inflammation in the CNS and urinary bladder at 1 wk (acute stage) and 4 wk (recovery phase) postinoculation. Several proinflammatory cytokines were significantly upregulated in the brains of infected mice at 1 wk postinoculation (Fig. 4B) followed by a gradual recovery to baseline values by 4 wk postinoculation. In the spinal cord, only IFN-γ levels were upregulated at 1 wk postinoculation (P ≤ 0.05; Fig. 4C), suggestive of a mild inflammatory reaction; however, this proved to be a transient response as normal levels were once again observed 4 wk postinfection. No changes in cytokine levels were noted in the urinary bladder during the entire course of investigation (Fig. 4D).
Fig. 4.

Evaluation of inflammation in the urinary bladder and central nervous system (CNS) after inoculation with MHV. A: hematoxylin and eosin staining of urinary bladder sections from a control mouse (top) and CIE mouse (bottom) showed no significant changes in the wall architecture. Scale bar = 200 μm. B: cytokine levels in the brain of control and CIE mice after 1 and 4 wk postinoculation as evaluated by multiplex ELISA. GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon. C: concentration of cytokines in the spinal cord of control and infected mice. D: multiplex ELISA revealed no changes in the cytokine profile in the urinary bladder. *P ≤ 0.05 vs. the control group.
Effects of demyelination on micturition patters and urodynamic parameters recorded in awake unrestrained mice.
To evaluate the effects of MHV-A59-induced demyelination on micturition patterns and urinary bladder function in vivo, we performed a filter paper assay in conscious mice (n = 11) at baseline and weekly thereafter after inoculation with the virus. This approach allowed us to observe dynamic changes in micturition patters in the same animals over a 4-wk period. Representative micturition patterns from one of the infected mice (CSS of 3) are shown in Fig. 5 and reflect the development of neurogenic bladder overactivity as evidenced by an increased number of both small (<1.0 cm in diameter; Fig. 5B) and large (≥1.0 cm in diameter; Fig. 5C) urine spots during the progression of demyelination. Increasing numbers of small and large urine spots correlated with the degree of clinical deficit; this relationship was observed from 2 wk postinoculation until 4 wk postinoculation in infected mice.
Fig. 5.

Micturition patterns in CIE mice as assessed by filter paper assay. A: representative images of voiding spots in a mouse with CIE (CSS of 3) taken every week for 4 wk. B: number of small size urine spots (<1.0 cm) in mice with a CSS of 2 or 3 during the progression of demyelination in the CNS. C: analysis of large urine spots (≥1.0 cm) in the same animals. *P ≤ 0.05 vs. baseline.
Cystometric assessment of urinary bladder function in conscious mice complemented the filter paper assay and was performed in control mice (n = 6) and MHV-A59-infected mice (n = 4 mice with a CSS of 2 and 4 mice with a CSS of 3) at 4 wk postinoculation, as described above in materials and methods. Demyelination of nerve fibers in the CNS after inoculation with the virus led to long-lasting changes in micturition patterns, which correlated with the level of developed neurological deficit. Figure 6A shows raw cystometric traces recorded in a control mouse (top) and in representative mice with a CSS of 2 (middle) and 3 (bottom). In the group of animals with a CSS of 2, the intermicturition interval (Fig. 6B) and pressure at voiding (Fig. 6C) were similar to the respective values in the control group. However, these mice developed voiding dysfunction, associated with an increased number of nonvoiding contractions (3.4 ± 0.5 vs. 1.0 ± 0.2 in control mice, P ≤ 0.05), lower bladder capacity (78.5 ± 5.2 vs 100.7 ± 10.1 μl, P ≤ 0.05), and decreased voided volume (0.03 ± 0.003 vs. 0.045 ± 0.005 ml, P ≤ 0.05; Fig. 6D). The progression of demyelination in the CNS (mice with a CSS of 3) caused more severe changes in urodynamic parameters, including shortened intermicturition intervals from 546 ± 51 s (in control mice) to 236 ± 14 s (P ≤ 0.05; Fig. 6B), decreased bladder capacity, and a reduced volume of voided urine (P ≤ 0.05; Fig. 6D). These results confirm the development of neurogenic bladder dysfunction in CIE mice and provide evidence of a distinct correlation between levels of neurological deficit and severity of the developed neurogenic bladder dysfunction.
Fig. 6.

Cystometric evaluation of bladder function in CIE mice during the development of neurogenic bladder dysfunction. A: representative traces of cystometrograms recorded in a freely moving control mouse (top) and inoculated mice with a CSS of 2 (middle) or 3 (bottom). Arrows indicate nonmicturition contractions. B: comparison of the duration of intermicturition intervals between control and CIE mice. C: analysis of bladder pressure at micturition before and after inoculation with MHV-A59. D: changes in the voided urine volume in control and experimental animals. *P ≤ 0.05 vs. the control group.
Virus-induced alterations in neural pathways induce phasic changes in abdominal sensitivity to mechanical stimulation.
To evaluate the relationships of sensory and motor reflexes with innervation of the pelvic viscera and surrounding somatic structures (e.g., pelvic floor muscle and skin), abdominal sensitivity was tested in a separate group of mice (n = 8) both pre- and postinoculation with MHV. Mice with a CSS of 2 or 3 were used for this experiment. Figure 7 shows the frequency of responses to von Frey filament testing in the lower abdominal area from baseline and every subsequent week postinoculation until the experimental end point. Baseline measurements demonstrated that the response frequency correlated with the applied force, reaching a plateau of 27.2 ± 4.2% with the maximal tested force of 4 g (Fig. 7). One week postinoculation, corresponding to the acute stage of inflammation in the CNS, mice became more sensitive to the filament testing with response rates reaching 45% upon application of higher filament forces (P ≤ 0.05 vs. baseline at 4 g; Fig. 7). Two and three weeks postinfection, the frequency of responses was completely eliminated for the lower ranges of the applied forces (from 0.04 to 0.4 g, P ≤ 0.05 vs. baseline for each force); in contrast, responses to higher force filaments did not differ from respective baseline levels. By 4 wk postinoculation, the significant neurological deficit was paralleled by a substantial diminution of abdominal sensitivity to mechanical stimulation of the lower pelvis (P ≤ 0.05). These results provide evidence showing that sensitivity of the pelvic region to mechanical stimulation during the development of chronic neurological impairment undergoes phasic changes; these changes are associated with increased sensitivity during acute inflammation of the CNS (1 wk postinoculation) followed by a gradual decrease in abdominal sensitivity response during the progression of neural deficit and demyelination.
Fig. 7.
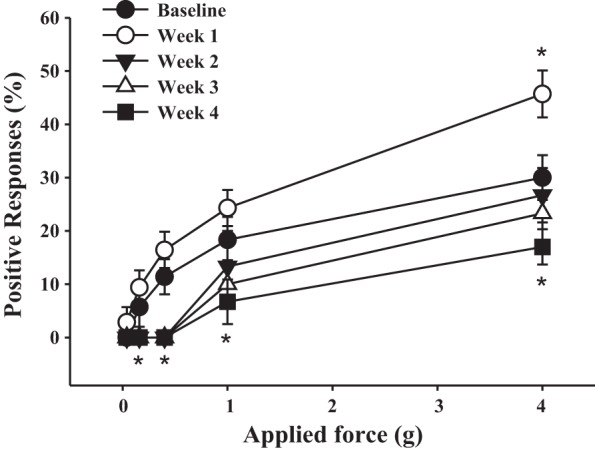
Virus-induced demyelination triggered dynamic changes in abdominal sensitivity in response to mechanical stimulation with von Frey filaments. Shown are effects of inoculation with MHV-A59 on the sensitivity of the lower pelvic region as measured by the response to mechanical stimulation using von Frey filaments. *P ≤ 0.05 vs. baseline.
DISCUSSION
In the present study, we evaluated the structural and functional pathophysiology of neurogenic bladder dysfunction using a coronavirus-induced mouse model of encephalomyelitis. Infection of mice with MHV is one of the two major viral models (along with Theiler virus) traditionally considered as an animal model of MS. This model has been previously used for the study of neurological demyelinating disease (8). After inoculation with MHV-A59, viral titers reach maximal values in the CNS at 5 days postinfection and then begin to decline after 7 days postinfection, coincident with the peak of the CD8T cell response. Subsequently, the infectious virus becomes undetectable ∼2 wk postinfection (32). However, despite the clearance of infectious virus, viral RNA (both genomic and mRNA) persist in the CNS; consequently, immune-mediated demyelination develops and plateaus at ∼4 wk postinfection (23, 25).
The clinical course of CIE progression depends on the amount of the virus introduced and the age of the mice at inoculation. It is characterized by weight loss and progressive ascending paralysis followed by spontaneous recovery (8). Our behavioral experiments established that the clinical signs of MHV infection ranged from the loss of tail tonicity and weak hindlimbs to almost total paralysis (in mice with a CSS of 4 or 5); the latter was associated with high mortality. Animals that survived the acute phase of the disease underwent an incomplete recovery from the initial attack with residual neuroparalytic signs (23, 25). The spinal cords of the affected mice exhibited myelin loss in the white matter of the spinal cord with axonal sparing, as previously detected by staining with luxol fast blue and/or electron microscopy (15, 25). The absence of gross structural changes in the sensory ganglia of CIE mice suggests that the effects of virus-induced demyelination seem to be confined to the CNS.
A key event in the formation of characteristic MS lesions in CIE mice is the infiltration into the CNS of myelin-specific CD4+ T helper (Th)1 cells capable of secreting proinflammatory Th1-type cytokines, such as IFN-γ and TNF-α (24). The process of transmigration of circulating immune cells to the urinary bladder is complex, involving their activation by cytokines and chemokines as well as the secretion of proteolytic enzymes. It has been previously established that TNF may promote differential trafficking of bladder mast cells in a model of neurogenic bladder induced by Bartha's strain of pseudorabies virus (12). Exposure of monocytes to IFN-γ induced proteolytic activity capable of degrading MBP (20). Additional studies have demonstrated that IFN-β can suppress T cell (27) and lymphocyte (45) migratory capacity. Future studies are warranted to determine if IFN or TNF perturbation would be a viable therapeutic approach for mitigating LUT symptoms in MS patients.
Our data correlate with previously published results from other animal models of MS, such as EAE. Experimental models of EAE include proteolipid protein-induced relapsing-remitting demyelinating disorder and chronic models of MS induced by either MBP (39) or oligodendrocyte glycoprotein (1). Rodents with EAE present CNS lesions with inflammation, demyelination, axonal loss, and gliosis (14) while displaying significant bladder dysfunction (2, 4, 5, 13, 39). A previous study (28) has reported a certain degree of hemorrhage, necrosis, and ulceration of the urothelium in the urinary bladders of rats with EAE; however, we did not detect any structural changes in the urinary bladder of CIE mice. This may be one of the distinguishing features of the CIE model from previously described EAE models.
In addition to extensive demyelination of the spinal cord, lesion sites in CIE mice were characterized by an upregulation of GFAP expression, suggestive of gliosis. The process of reactive gliosis is usually characterized by morphological changes (hypertrophy), functional alterations, and a profound increase in the expression of astrocyte-specific intermediate filament GFAP (37). Although increased GFAP expression is widely used as a marker for astrogliosis, the precise functional role of GFAP in astrocytes is not known, and the implications of increased GFAP expression for astrocyte-mediated functions in gliosis remain elusive (37, 48). On one hand, reactivation of astrocytes may have detrimental effects by increasing neurotoxic substances, exacerbating cell loss, and minimizing CNS repair by scar formation. On the other hand, reactive gliosis could be beneficial by enclosing the affected area, restricting inflammation, taking up excessive amounts of extracellular glutamate, and eliminating free radicals (3, 22, 47, 48). For instance, GFAP−/− mice have been demonstrated to be more sensitive to spinal cord injury (40) and to neurotoxicity (43), indicating a protective role for GFAP. The precise role of GFAP upregulation in the CIE model of MS and its effects on micturition pathways warrant future studies.
Analysis of voiding patterns with filter paper assays and micturition parameters using urodynamics in unanesthetized CIE mice confirmed the development of neurogenic detrusor overactivity in inoculated animals. The urodynamic profile of CIE mice matched the results of clinical studies that established that detrusor hyperreflexia associated with urinary frequency and urgency is the most common urodynamic abnormality in patients with MS (9, 10, 17, 38). A limitation of the cystometric evaluation in our study was the inability to catheterize mice preinoculation followed by weekly urodynamic evaluation of bladder function throughout the course of the demyelination process (until week 4). This approach was not feasible as bladder stones begin to develop 7–10 days after catheterization due to the narrow diameter of the catheters and the small size of mouse bladders; these features resulted in the initiation of local inflammation in the bladder, which precipitated to kidney failure due to outflow obstruction.
The neurogenic bladder overactivity detected in CIE mice aligns with the urodynamic findings of a variety of micturition abnormalities detected in EAE models of MS. Previous studies have determined that mice with EAE develop a significant increase in bladder size (2), shortened intermicturition intervals, decreased volume of voided urine, changes in morphology (5), and expression of molecular markers of fibrosis (4) in the urinary bladder. MBP-induced EAE in Lewis rats is primarily a monophasic inflammatory disease with relatively modest levels of demyelination and neurodegeneration (39, 49). Rats with MBP-induced EAE develop both detrusor areflexia and detrusor hyperactivity, which correlate with spinal cord inflammation and hindlimb paralysis (39). Using cystometric analysis, Mizusawa et al. (39) showed that EAE caused detrusor areflexia in 52% of female rats compared with 12% that developed detrusor overactivity. More recently, Vignes et al. (52) established that the majority of female rats presented symptoms of detrusor areflexia before the onset of EAE but that detrusor overactivity developed at the onset of the disease. Additionally, the occurrence of detrusor sphincter dyssynergia after the onset of EAE has also been reported in EAE rats (4, 5). The viral etiology of the LUT dysfunction in CIE mice and earlier experimental time point postinoculation (28 days compared with 70 days in EAE models) likely underlie the differences between cystometric findings in our study and the aforementioned EAE models. Future studies are warranted to follow CIE mice for longer periods of time to evaluate potential dynamic changes in the pathophysiology of CNS and LUT pathophysiology during disease progression.
Mice with CIE also experienced phasic changes in abdominal (somatic) sensitivity. During the acute phase of CNS inflammation, there was an increase in the response of the abdominal somatic field to mechanical stimulation with von Frey filaments. However, the development of a neurological deficit caused a progressive diminution of mechanosensitive responses, possibly due to general deterioration of motor reflexes. This is a novel observation, which has not been previously reported in EAE models of MS. The mechanisms underlying the loss of mechanical sensitivity in the lower pelvic area may include impaired synaptic activity in the lumbosacral spinal cord and modified neural plasticity of sensory and motor pathways due to inflammation, demyelination, axonal damage, and gliosis triggered by viral infection.
In summary, the CIE model of neurogenic bladder overactivity is a clinically relevant model of voiding dysfunction observed in patients with MS. Unlike other existing models of LUT abnormalities in MS, it is induced by an environmental agent (e.g., a virus), and, therefore, reflects the spontaneous nature of disease occurrence. This mouse model of MS is suitable for studying the relationship between voiding dysfunction and progressive neurological impairment over the course of virus-induced demyelinating disorder. Future testing and refinement of the CIE model may enable its use for the development and testing of novel therapeutic methods for the prevention and treatment of neurogenic bladder overactivity in MS patients.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-097819 (to A. P. Malykhina).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.T.M., X.-Q.P., and A.P.M. performed experiments; M.T.M., X.-Q.P., and A.P.M. analyzed data; M.T.M., A.L.S., D.K.N., S.R.W., and A.P.M. interpreted results of experiments; M.T.M. and A.P.M. drafted manuscript; M.T.M., X.-Q.P., A.L.S., D.K.N., S.R.W., M.R.R., and A.P.M. edited and revised manuscript; M.T.M., X.-Q.P., A.L.S., D.K.N., S.R.W., M.R.R., and A.P.M. approved final version of manuscript; X.-Q.P. and A.P.M. prepared figures; S.R.W., M.R.R., and A.P.M. conception and design of research.
ACKNOWLEDGMENTS
The authors thank Urology Scholars Manila Jinhal and Cullen O'Donnel for participation in the data collection and analysis. The authors are grateful to Ruth Elliott for help with the MHV-A59 mouse model. The authors acknowledge the Diabetes Research Center (National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-019525) and the services of the Radioimmunoassay and Biomarker Core in performing the cytokine Liminex ELISA.
REFERENCES
- 1.Aharoni R, Vainshtein A, Stock A, Eilam R, From R, Shinder V, Arnon R. Distinct pathological patterns in relapsing-remitting and chronic models of experimental autoimmune enchephalomyelitis and the neuroprotective effect of glatiramer acetate. J Autoimmun 37: 228–241, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Al-Izki S, Pryce G, Giovannoni G, Baker D. Evaluating potential therapies for bladder dysfunction in a mouse model of multiple sclerosis with high-resolution ultrasonography. Mult Scler 15: 795–801, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Allaman I, Belanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci 34: 76–87, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Altuntas CZ, Daneshgari F, Izgi K, Bicer F, Ozer A, Sakalar C, Grimberg KO, Sayin I, Tuohy VK. Connective tissue and its growth factor CTGF distinguish the morphometric and molecular remodeling of the bladder in a model of neurogenic bladder. Am J Physiol Renal Physiol 303: F1363–F1369, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altuntas CZ, Daneshgari F, Liu G, Fabiyi A, Kavran M, Johnson JM, Gulen MF, Jaini R, Li X, Frenkl TL, Tuohy VK. Bladder dysfunction in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol 203: 58–63, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ascherio A, Munger K. Epidemiology of multiple sclerosis: from risk factors to prevention. Semin Neurol 28: 17–28, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Awad SA, Gajewski JB, Sogbein SK, Murray TJ, Field CA. Relationship between neurological and urological status in patients with multiple sclerosis. J Urol 132: 499–502, 1984 [DOI] [PubMed] [Google Scholar]
- 8.Bender SJ, Weiss SR. Pathogenesis of murine coronavirus in the central nervous system. J Neuroimmune Pharmacol 5: 336–354, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Betts CD, D'Mellow MT, Fowler CJ. Urinary symptoms and the neurological features of bladder dysfunction in multiple sclerosis. J Neurol Neurosurg Psychiatry 56: 245–250, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borello-France D, Leng W, O'Leary M, Xavier M, Erickson J, Chancellor MB, Cannon TW. Bladder and sexual function among women with multiple sclerosis. Mult Scler 10: 455–461, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Chancellor MB, Anderson RU, Boone TB. Pharmacotherapy for neurogenic detrusor overactivity. Am J Phys Med Rehabil 85: 536–545, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Chen MC, Blunt LW, Pins MR, Klumpp DJ. Tumor necrosis factor promotes differential trafficking of bladder mast cells in neurogenic cystitis. J Urol 175: 754–759, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Ciancio SJ, Mutchnik SE, Rivera VM, Boone TB. Urodynamic pattern changes in multiple sclerosis. Urology 57: 239–245, 2001 [DOI] [PubMed] [Google Scholar]
- 14.Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 164: 1079–1106, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das Sarma J, Fu L, Tsai JC, Weiss SR, Lavi E. Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J Virol 74: 9206–9213, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Groat WC, Yoshimura N. Changes in afferent activity after spinal cord injury. Neurourol Urodyn 29: 63–76, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Seze M, Ruffion A, Denys P, Joseph PA, Perrouin-Verbe B. The neurogenic bladder in multiple sclerosis: review of the literature and proposal of management guidelines. Mult Scler 13: 915–928, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Fowler CJ. The cause and management of bladder, sexual and bowel symptoms in multiple sclerosis. Baillieres Clin Neurol 6: 447–466, 1997 [PubMed] [Google Scholar]
- 19.Fowler CJ. Systematic review of therapy for neurogenic detrusor overactivity. Can Urol Assoc J 5: S146–148, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galboiz Y, Shapiro S, Lahat N, Miller A. Modulation of monocytes matrix metalloproteinase-2, MT1-MMP and TIMP-2 by interferon-γ and -β: implications to multiple sclerosis. J Neuroimmunol 131: 191–200, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Kalsi V, Fowler CJ. Therapy insight: bladder dysfunction associated with multiple sclerosis. Nat Clin Pract Urol 2: 492–501, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, Verveer M, de Groot LR, Smith VD, Rangarajan S, Rodriguez JJ, Orre M, Hol EM. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLOS ONE 7: e42823, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavi E, Gilden DH, Highkin MK, Weiss SR. MHV-A59 pathogenesis in mice. Adv Exp Med Biol 173: 237–245, 1984 [DOI] [PubMed] [Google Scholar]
- 24.Lavi E, Gilden DH, Highkin MK, Weiss SR. The organ tropism of mouse hepatitis virus A59 in mice is dependent on dose and route of inoculation. Lab Anim Sci 36: 130–135, 1986 [PubMed] [Google Scholar]
- 25.Lavi E, Gilden DH, Wroblewska Z, Rorke LB, Weiss SR. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34: 597–603, 1984 [DOI] [PubMed] [Google Scholar]
- 26.Lei Q, Pan XQ, Villamor AN, Asfaw TS, Chang S, Zderic SA, Malykhina AP. Lack of transient receptor potential vanilloid 1 channel modulates the development of neurogenic bladder dysfunction induced by cross-sensitization in afferent pathways. J Neuroinflammation 10: 3, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leppert D, Waubant E, Burk MR, Oksenberg JR, Hauser SL. Interferon β-1b inhibits gelatinase secretion and in vitro migration of human T cells: a possible mechanism for treatment efficacy in multiple sclerosis. Ann Neurol 40: 846–852, 1996 [DOI] [PubMed] [Google Scholar]
- 28.Lifson JD, Oyasu R, Dreyer N, Carone FA, Williams RM. Acute hemorrhagic obstructive uropathy as a complication of experimental autoimmune encephalomyelitis. Arch Pathol Lab Med 107: 600–602, 1983 [PubMed] [Google Scholar]
- 29.Manaker RA, Piczak CV, Miller AA, Stanton MF. A hepatitis virus complicating studies with mouse leukemia. J Natl Cancer Inst 27: 29–51, 1961 [PubMed] [Google Scholar]
- 30.Mannie M, Swanborg RH, Stepaniak JA. Experimental autoimmune encephalomyelitis in the rat. Curr Protoc Immunol Chapter 15: Unit 15 12, 2009 [DOI] [PubMed] [Google Scholar]
- 31.Marrie RA. Environmental risk factors in multiple sclerosis aetiology. Lancet Neurol 3: 709–718, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Matthews AE, Weiss SR, Paterson Y. Murine hepatitis virus–a model for virus-induced CNS demyelination. J Neurovirol 8: 76–85, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattson D, Petrie M, Srivastava DK, McDermott M. Multiple sclerosis. Sexual dysfunction and its response to medications. Arch Neurol 52: 862–868, 1995 [DOI] [PubMed] [Google Scholar]
- 34.McCarthy DP, Richards MH, Miller SD. Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler's virus-induced demyelinating disease. Methods Mol Biol 900: 381–401, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCombe PA, Gordon TP, Jackson MW. Bladder dysfunction in multiple sclerosis. Expert Rev Neurother 9: 331–340, 2009 [DOI] [PubMed] [Google Scholar]
- 36.McCombe PA, Nickson I, Pender MP. Cytokine expression by inflammatory cells obtained from the spinal cords of Lewis rats with experimental autoimmune encephalomyelitis induced by inoculation with myelin basic protein and adjuvants. J Neuroimmunol 88: 30–38, 1998 [DOI] [PubMed] [Google Scholar]
- 37.Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol 93: 421–443, 2011 [DOI] [PubMed] [Google Scholar]
- 38.Miller H, Simpson CA, Yeates WK. Bladder dysfunction in multiple sclerosis. Br Med J 1: 1265–1269, 1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mizusawa H, Igawa Y, Nishizawa O, Ichikawa M, Ito M, Andersson KE. A rat model for investigation of bladder dysfunction associated with demyelinating disease resembling multiple sclerosis. Neurourol Urodyn 19: 689–699, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Nawashiro H, Messing A, Azzam N, Brenner M. Mice lacking GFAP are hypersensitive to traumatic cerebrospinal injury. Neuroreport 9: 1691–1696, 1998 [DOI] [PubMed] [Google Scholar]
- 41.Nicholas R, Young C, Friede T. Bladder symptoms in multiple sclerosis: a review of pathophysiology and management. Expert Opin Drug Saf 9: 905–915, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Nortvedt MW, Riise T, Frugard J, Mohn J, Bakke A, Skar AB, Nyland H, Glad SB, Myhr KM. Prevalence of bladder, bowel and sexual problems among multiple sclerosis patients two to five years after diagnosis. Mult Scler 13: 106–112, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Otani N, Nawashiro H, Fukui S, Ooigawa H, Ohsumi A, Toyooka T, Shima K, Gomi H, Brenner M. Enhanced hippocampal neurodegeneration after traumatic or kainate excitotoxicity in GFAP-null mice. J Clin Neurosci 13: 934–938, 2006 [DOI] [PubMed] [Google Scholar]
- 44.Pachner AR. Experimental models of multiple sclerosis. Curr Opin Neurol 24: 291–299, 2011 [DOI] [PubMed] [Google Scholar]
- 45.Prat A, Al-Asmi A, Duquette P, Antel JP. Lymphocyte migration and multiple sclerosis: relation with disease course and therapy. Ann Neurol 46: 253–256, 1999 [DOI] [PubMed] [Google Scholar]
- 46.Rudick CN, Chen MC, Mongiu AK, Klumpp DJ. Organ cross talk modulates pelvic pain. Am J Physiol Regul Integr Comp Physiol 293: R1191–R1198, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Salmina AB. Neuron-glia interactions as therapeutic targets in neurodegeneration. J Alzheimers Dis 16: 485–502, 2009 [DOI] [PubMed] [Google Scholar]
- 48.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol 119: 7–35, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swanborg RH. Experimental autoimmune encephalomyelitis in the rat: lessons in T-cell immunology and autoreactivity. Immunol Rev 184: 129–135, 2001 [DOI] [PubMed] [Google Scholar]
- 50.Tubaro A, Puccini F, De Nunzio C, Digesu GA, Elneil S, Gobbi C, Khullar V. The treatment of lower urinary tract symptoms in patients with multiple sclerosis: a systematic review. Curr Urol Rep 13: 335–342, 2012 [DOI] [PubMed] [Google Scholar]
- 51.van der Star BJ, Vogel DY, Kipp M, Puentes F, Baker D, Amor S. In vitro and in vivo models of multiple sclerosis. CNS Neurol Disord Drug Targets 11: 570–588, 2012 [DOI] [PubMed] [Google Scholar]
- 52.Vignes JR, Deloire MS, Petry KG, Nagy F. Characterization and restoration of altered inhibitory and excitatory control of micturition reflex in experimental autoimmune encephalomyelitis in rats. J Physiol 578: 439–450, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]