

Abstract
The inhibition of the central growth regulatory kinase TOR, which participates in two complexes, TORC1 and TORC2, has been a focus of metabolic and cancer studies for many years. Most studies have dealt with TORC1, the canonical target of rapamycin, and the role of this complex in autophagy, protein synthesis, and cell growth control. Recent work on TORC2 in budding and fission yeast species points to a conserved role of this lesser-known TOR complex in the survival of DNA damage. In budding yeast, TORC2 controls lipid biosynthesis and actin cytoskeleton through downstream AGC kinases, which are now, surprisingly, implicated in the survival of oxidative DNA damage. Preliminary data from mTORC2 modulation in cancer cells suggest that an extension to human chemotherapy is worth exploring.
Keywords: cancer therapies, DNA damage, mTOR, TORC1, TORC2
Introduction
TOR (target of rapamycin) is an atypical serine/threonine protein kinase that belongs to the family of phosphatidyl inositol-3 kinase related kinases (PI3K-like kinases or PIKKs). TOR proteins are best known for their roles in the nutrient-dependent signaling pathways underlying cell growth, proliferation, and survival. TOR was first identified in the budding yeast Saccharomyces cerevisiae as the molecular target of the immunosuppressive and anti-cancer drug rapamycin (Heitman et al, 1991). Later, TOR genes were isolated in all eukaryotes investigated. A breakthrough in our understanding of the TOR-dependent signaling pathway coincided with the identification of two distinct TOR-containing complexes, termed TOR complex 1 and TOR complex 2, or TORC1 and TORC2 (Loewith et al, 2002; Wedaman et al, 2003). Both complexes are conserved from yeast to man (reviewed in Wullschleger et al, 2006; Loewith & Hall, 2011).
TORC1 primarily regulates growth and is involved in the modulation of protein synthesis, ribosome biogenesis, and autophagy. Accordingly, disruption of TORC1 in many eukaryotes including S. cerevisiae, Schizosaccharomyces pombe, nematodes, flies, or mammals results in cellular phenotypes that resemble starvation (for examples, see Barbet et al, 1996; Noda & Ohsumi, 1998; Zhang et al, 2000; Meissner et al, 2004; Alvarez & Moreno, 2006; Matsuo et al, 2007; Weisman et al, 2007).
The cellular functions of TORC2 are less well understood. This is partly due to the fact that—in contrast to TORC1—there are no specific inhibitors of TORC2 (Sparks & Guertin, 2010). Rapamycin and a family of derivatives that function similarly (“rapalogs”) inhibit only TORC1 and have allowed in-depth analysis of its function in tissue culture cells and multicellular organisms. Rapalogs form a complex with the FK506 binding protein-12 (FKBP-12), which is bound by mTOR, and blocks mTORC1 activity. This in turn inhibits cell cycle progression, cell survival, and angiogenesis. Rapamycin derivatives have been successfully used to treat neurological and metabolic disorders, as well as some cancers, such as renal cell carcinoma, subependymal giant cell astrocytoma associated with tuberous sclerosis, pancreatic and neuroendocrine tumors, and ER+ breast cancer (Dienstmann et al, 2014; Porta et al, 2014). This success uncovered significant crosstalk between the TORC1 complex and the PI3K and Akt/PKB signaling pathways (Dazert & Hall, 2011; Laplante & Sabatini, 2012). Indeed, other pan-PIKK inhibitors that target collectively the related catalytic domains of mTOR and PI3K (examples being Novartis' NVP-BEZ235, and GDC-0980 from Roche/Genentech) have also had clinical success. These and related pan-PIKK/mTOR inhibitors are now being tested on advanced solid tumors, breast cancer, leukemias, and pancreatic and neuroendocrine tumors. Finally, there are also ATP-competitive inhibitors that inhibit only the two mTOR-containing complexes, mTORC1, and mTORC2, although have yet to yield positive clinical results (Dienstmann et al, 2014).
In contrast to the starvation-like phenotypes observed upon disruption or inhibition of TORC1, the loss of TORC2 generates diverse effects that often show species- or cell-type specificity. Remarkably, however, two recent reports showed that TORC2 plays a role in the maintenance of genome stability in face of oxidative or replicative stress, in both the fission yeast, S. pombe, and the budding yeast, S. cerevisiae (Schonbrun et al, 2013; Shimada et al, 2013). The similarity of these findings in such distantly related yeast reinforces the concept that TORC2 controls a network that might either directly or indirectly safeguard the genome from DNA damage. Here, we briefly review the cellular functions attributed to TORC2 in yeast and discuss the possibility that TORC2 helps ensure genomic stability in higher eukaryotes as well.
Conserved aspects of TORC1 and TORC2 complexes
Mammalian cells contain a single TOR gene (mTOR) that encodes the catalytic subunit of either TORC1 or TORC2, while in S. cerevisiae, there are two TOR genes, TOR1 and TOR2. The budding yeast Tor1 is found exclusively in the TORC1 complex, while Tor2 can serve as the catalytic subunit of either complex (Loewith et al, 2002; Loewith & Hall, 2011). Like budding yeast, the distantly related fission yeast also carries two TOR genes, tor1+ and tor2+. In S. pombe, Tor1 is the catalytic subunit of TORC2, while Tor2 is the catalytic subunit of TORC1, as the genes were named in order of discovery and not based on their function (Hayashi et al, 2007; Matsuo et al, 2007; Ikai et al, 2011). In all species, the two complexes are distinguished by conserved complex-specific subunits. Notably, the human subunits Rictor and Sin1 are TORC2-specific and are conserved in both yeast species, albeit with different names (Fig 1).
Figure 1. TORC2 complex signaling to AGC kinases.

Shown is a summary of TORC2 in human, S. cerevisiae and S. pombe. The human proteins Rictor and Sin1 are TORC2-specific subunits conserved in both yeast species, being known as Avo3 (Rictor) and Avo1 (Sin1) in S. cerevisiae (Loewith et al, 2002) or Ste20 (Rictor) or Sin1 in S. pombe (Hayashi et al, 2007; Matsuo et al, 2007). The target kinases of TORC2 are AKT1 (Sarbassov et al, 2005), SGK1 (Garcia-Martinez & Alessi, 2008), PKC-α (Facchinetti et al, 2008; Ikenoue et al, 2008), and PKC-ζ (Li & Gao, 2014) in higher eukaryotes, while they are Ypk1 and Ypk2 (Kamada et al, 2005), and the PKC ortholog Pkc1 (Facchinetti et al, 2008) in budding yeast. Fission yeast has only one identified AGC-kinase, Gad8 (Matsuo et al, 2003).
In all species studied to date, TOR complexes appear to work through a common mechanism: the phosphorylation of the protein kinase A/protein kinase G/protein kinase C (AGC) subfamily (Weisman, 2010). TOR kinase activates AGC kinases by phosphorylating their hydrophobic and turn motifs (Jacinto & Lorberg, 2008). As shown in Fig 1, the target kinases are AKT1, SGK1, PKC-α, and PKC-ζ in higher eukaryotes, Ypk1 and Ypk2 and the PKC ortholog Pkc1 in budding yeast, and Gad8 in S. pombe. The budding yeast TORC2 complex regulates cell wall integrity, actin polarity, endocytosis, membrane growth, and sphingolipid biosynthesis through Ypk1/2 (reviewed in Jacinto & Lorberg, 2008), while in fission yeast, Gad8 is thought to mediate many, if not most, of TORC2 functions (Fig 1).
Comparing phenotypes of budding and fission yeast TORC2 mutants
On a phenotypic level, the cellular activities of TORC2 complexes in fission and budding yeasts appear substantially different. It is unclear whether this reflects a true divergence in function or an incomplete understanding of the functions controlled by TORC2. Budding yeast TORC2 is essential for viability, while SpTORC2 is essential only under stress conditions, such as nutrient starvation. The essential cellular functions attributed to ScTORC2 are the regulation of actin polarization and sphingolipid biosynthesis (Schmidt et al, 1996; deHart et al, 2002; Aronova et al, 2008), reviewed in Loewith and Hall (2011). Because polarized actin filaments are needed for the transport of proteins and lipids to the growing daughter cell, actin regulation by the TORC2-Ypk1/2 pathway is crucial for growth in budding yeast. Sphingolipids, which have both structural and signaling functions, are core components of all lipid bilayers and are necessary for bud growth. Not only does ScTORC2 control sphingolipid biosynthesis, but the lipids themselves feedback to regulate ScTORC2 activity (Aronova et al, 2008). Furthermore, this complex is activated by plasma membrane stress, which stems from cell surface expansion during growth, or from chemical or mechanical stress on the plasma membrane (Berchtold et al, 2012). Finally, one study suggested that contacts between ScTORC2 and ribosomes activate the kinase, a mechanism possibly conserved in human cells (Oh et al, 2010; Zinzalla et al, 2011).
In S. pombe, disruption of SpTORC2 is non-lethal but results in a delayed entrance into mitosis and generates slightly elongated cells (Weisman & Choder, 2001; Ikeda et al, 2008; Ikai et al, 2011). The fission yeast TORC2 function is essential to execute the two main responses to starvation: sexual development and entrance into stationary phase (Kawai et al, 2001; Weisman & Choder, 2001) and is required for both amino acid uptake (Weisman et al, 2007) and growth on low glucose (Ikai et al, 2011). Loss of SpTORC2 activity rendered cells remarkably sensitive to a variety of environmental insults, including low or high temperature, osmotic, and/or oxidative stress. However, unlike the growth-inhibiting actin polarization defect observed in budding yeast, the S. pombe TORC2-Gad8 pathway currently appears to be only loosely connected with actin organization. Nonetheless, an abnormal distribution of actin cortical dots and excess actin polymerization at the cell equator do occur in S. pombe cells lacking functional TORC2 (Ikai et al, 2011). It may be that the differences in cell growth and actin phenotypes associated with loss of TORC2 in the two yeasts stem from their fundamentally different modes of cell growth and division. S. cerevisiae has unidirectional bud emergence, and its growth is strictly controlled by polarized actin filaments, while S. pombe growth occurs first at both ends and then shifts to a central cleavage furrow, where it promotes cell membrane and cell wall synthesis (Loewith, 2013).
Transcriptional profiles of S. pombe cells lacking the catalytic subunit of TORC2 resemble those of cells lacking histone de-acetylases or chromatin remodeling subunits (Schonbrun et al, 2009). Interestingly, disruption of gad8+ resulted in hypersensitivity to DNA-damaging agents, elongation of telomeres, and loss of gene silencing at the mating-type locus for unknown reasons (Schonbrun et al, 2009). Closer examination suggested that the complex was specifically required for cell survival under chronic DNA replication stress (Schonbrun et al, 2013).
Although the budding yeast TORC2 had never been directly implicated in the DNA damage response, a chemicogenetic screen carried out in budding yeast identified a novel imidazoquinoline NVP-BHS345 that targeted both ScTORC1 and ScTORC2. NVP-BHS345 conferred a synergistic lethality on cells exposed to oxidation and break-inducing agents like Zeocin or ionizing radiation (Shimada et al, 2013). The cells deficient for ScTORC2 showed no signs of DNA damage in the absence of exogenous agents, yet the combination of base-oxidizing damage with loss of either Tor2 or Ypk1/Ypk2 activity generated a genomewide fragmentation called yeast chromosome shattering (YCS), mimicking the effects of NVP-BHS345 and Zeocin (Shimada et al, 2013). To a lesser degree, the chemical inhibitor also enhanced sensitivity to replicative stress in strains lacking the RecQ helicase Sgs1, although these conditions did not lead to chromosome fragmentation (Shimada et al, 2013).
Response to DNA damage is crucial for cell survival, given that DNA is continuously assaulted by free radicals and damage arising from replication stress. Although many members of the PIKK family, which includes TOR kinases, play pivotal roles in DNA damage response (Carr, 2002; Friedel et al, 2009), neither ataxia telangiectasia mutated (ATM) nor ATR (ATM, SpRad3-related) kinases were responsible for the damage sensitivity observed in TORC2-deficient S. pombe or S. cerevisiae cells (Schonbrun et al, 2013; Shimada et al, 2013). Specifically, the ATR homologues (ScMec1 or SpRad3) and the effector kinases (ScRad53 or SpChk1) were fully functional in face of DNA damage, despite loss of TORC2 activity. Rad53 kinase was even hyperactivated by DNA damage upon TORC2 inhibition (Shimada et al, 2013). Nonetheless, disruption of the catalytic subunit of TORC2 or the downstream kinase, Gad8 in S. pombe cells, rendered cells sensitive to replication stress induced by hydroxyurea (HU), methyl-methane sulfonate (MMS), or camptothecin (CPT) (Schonbrun et al, 2013). Indeed, perturbation of SpTORC2-Gad8 signaling led to cell cycle arrest and elongation in response to DNA damage, consistent with activation of the DNA damage checkpoint response (Schonbrun et al, 2013).
The chemical identified in the budding yeast screen, NVP-BHS345, is not a general PIKK family inhibitor in yeast, but selectively inhibits S. cerevisiae TORC1 and TORC2. In human cells, on the other hand, the same compound is a broad spectrum inhibitor of the mammalian PIKK family (Shimada et al, 2013). The striking fragmentation of yeast chromosomes that occurs when inhibition of ScTORC2 is combined with low-level doses of Zeocin occurs rapidly (< 1 h) and without passage through the cell cycle. Treatment with rapamycin, which inhibits exclusively ScTORC1, did not have similar effects (Shimada et al, 2013). Genetic analysis showed that YCS arises specifically from the combination of ScTORC2 inhibition and low levels of oxidative damage. Indeed, a hyperactivating mutation in YPK2 rendered the cells resistant to the damage provoked by NVP-BHS345 and Zeocin. Collectively, chemicogenetic evidence proved that ScTORC2-dependent activation of Ypk1/Ypk2 participates in Zeocin-induced DNA damage survival (Shimada et al, 2013).
Its mechanism might entail inhibition of a repair pathway, as the essential homologues recombination (HR)-protein Rad52 accumulated in foci during the combined treatment. Perturbation of SpTORC2-Gad8 signaling also generated an accumulation of Rad52-containing repair foci. It remained unclear which repair pathway was impaired, although both HR and non-homologous end joining were ruled out (Shimada et al, 2013). Consistently, SpTORC2-Gad8 mutants were synthetic lethal with loss of either S. pombe Rad52 or Rad51, possibly indicating that SpTORC2-Gad8 mutants incur an enhanced level of DNA damage that renders them dependent on HR. In further support of this, negative synthetic interactions were scored between SpTORC2-Gad8 mutants and deletions of DNA repair or replication fork re-start genes, such as mus81, mms1, or mms22, even in the absence of damage-inducing agents (Schonbrun et al, 2013).
Although the fission yeast genetics suggested hypersensitivity to replication stress, the Zeocin-induced YCS in budding yeast did not require entry into S phase. Moreover, a similar sensitivity to low levels of oxidative damage was observed in budding yeast in the presence of the actin polymerization inhibitor, latrunculin A (LatA) or jasplakinolide, a cyclic peptide that stabilizes F-actin, in combination with low levels of Zeocin (Shimada et al, 2013). This suggested that either an actin-associated activity or actin filament turnover itself, which is indeed under TORC2-YPk1/2 control, impairs genome integrity under conditions of increased oxidative stress.
In the cytoplasm, actin forms a network of filaments that mediates macromolecular transport and maintains cell morphology. In the cell nucleus, monomeric globular (G) actin has been implicated in functions such as chromatin remodeling and transcription (Belin & Mullins, 2013), but it is unclear what role is played by filamentous (F) actin. There is an active system of actin import and export to and from the nucleus (Vartiainen, 2008), suggesting that nuclear actin concentration is under careful control. Nonetheless, it remains to be determined whether impaired G- to F-actin turnover impacts DNA repair, or otherwise protects the genome, and if that role is lost upon ScTORC2 inhibition.
Interestingly, in S. pombe, the loss of SpTORC2-Gad8 led to an increased level of Rad52 foci, yet did not activate the DNA damage checkpoint under normal growth conditions. This characteristic is shared with the brc1 mutant, which eliminates Brc1, a protein bearing six BRCT domains (Bass et al, 2012). This may indicate that the damage incurred by loss of TORC2 is not recognized by the DNA damage checkpoint or else is too low to activate the response. On the other hand, upon removal of DNA-damaging agents, the dephosphorylation of SpChk1 (termination of the checkpoint signal) and activation of SpCdc2, the central cyclin-dependent kinase, were delayed (Schonbrun et al, 2009, 2013). Further study is required to clarify whether this reflects an accumulation of unrepaired lesions or misregulation of a phosphatase required for SpChk1 dephosphorylation in the absence of SpTORC2.
Implications for human disease and therapies
As mentioned above, there is only one Tor kinase subunit in mammalian cells, mTOR, serving in both mTORC1 and mTORC2. It is therefore not surprising that there are no active-site inhibitors that distinguish the complexes, as strictly speaking, the active site is identical in both. Rapamycin and rapalogs do allow specific inhibition of mTORC1 signaling, while specific inhibition of mTORC2 requires shRNA of Rictor, an complex-specific co-regulator.
As mentioned above, a number of PIKK family kinase inhibitors inhibit mTOR along with PI3K and, in some cases, with other related PIKK family members (DNA-PK, ATR and ATM). These and the rapamycin derivative Affinitor® are well validated for combined chemotherapeutic protocols for solid tumors (Beuvink et al, 2005; Sparks & Guertin, 2010; Grunt & Mariani, 2013; Tentori et al, 2013). Nonetheless, rapalogs do not have broad anti-tumor activity. This may reflect the fact that they only inhibit certain cellular activities of mTORC1 (Shor et al, 2009) and can lead to activation of PI3 kinase as a consequence of inhibiting an important negative feedback loop (Wan et al, 2007).
The existence of mechanisms that downregulate mTORC1 upon DNA damage links mTORC1 to the DNA damage response in man (Zoncu et al, 2011; Hasty et al, 2013). Intriguingly, a recent study revealed physical interaction between the tandem BRCT domain of BRCA1 and Rictor, the mTORC2-specific subunit. BRCA1 is a protein with pleiotrophic effects on genome stability and is a major breast and ovarian susceptibility gene. A set of experiments suggested that BRCA1 binding to Rictor inhibits mTORC2-dependent phosphorylation of Akt-Ser473 (Woods et al, 2012). Whether the interactions between BRCA1 and Rictor affect DNA damage tolerance or genome integrity is unknown, although Akt, which is a downstream target of mTORC2 as well as DNA-PK, is linked to the survival of damage (Bozulic et al, 2008; Surucu et al, 2008). The notion arising from budding yeast, based on the fact that there are few effects of ScTORC2 inhibition alone, is that mTORC2 inhibition might sensitize mammalian cells to radiation or other oxidizing damage to DNA, thus improving standard DNA-damaging chemotherapies (Fig 2). Other more specific DNA-damaging agents, such as cis-platin, could also be combined with mTORC2 inhibition. Finally, there may be tumors deficient in specific repair pathways, for which inhibition of mTORC2 would lead to cell death (Fig 2).
Figure 2. Combinational therapies that impair multiple repair pathways can provoke synergistic lethality in “damage-addicted” cancer cells.
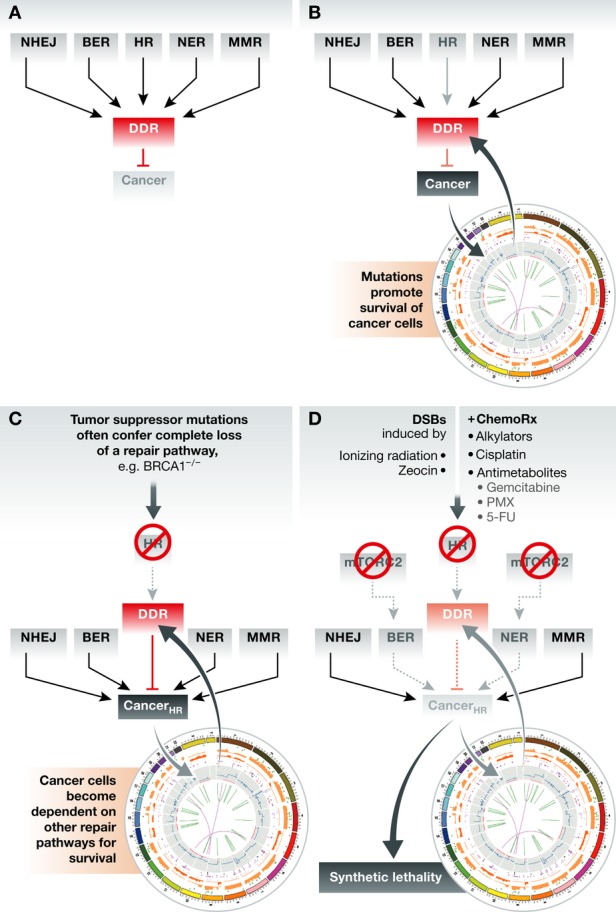
(A) Multiple repair pathways including non-homologous end joining (NHEJ), base-excision repair (BER), homologous recombination (HR), nucleotide excision repair (NER), and mismatch repair (MMR) protect the cellular genome from damage through conserved pathways that collectively activate the DNA damage response (DDR). (B) In precancerous lesions, overwhelming damage or weakened repair capacity (for example. loss of HR) can lead to selection for mutations that promote survival. Transformed cells often exhibit constitutively activated DDR and enhanced genomic instability. (C) Loss of a repair pathway (through inherited or incurred mutation, e.g. HR) can render cancer cells “addicted” or dependent on the other repair pathways (CancerHR). (D) Combined therapies that are effective against tumors bearing mutant repair pathways often inhibit a second repair pathway. This could be by targeting a general repair factor (e.g. PARP) or—as suggested here—the mTORC2 complex. The resulting accumulation of irreparable damage leads to cell death (synthetic lethality). DNA-damaging agents could enhance the synthetic lethality. Illustration partially adapted (with permission) from Forbes et al (2011).
The potential benefit of mTORC2 inhibition is suggested by studies that genetically manipulated the levels of Rictor. Deletion of Rictor in mouse models for prostate cancer, or a reduction of Rictor in cancer cell lines, was shown to inhibit tumor development and cell proliferation (Hietakangas & Cohen, 2007; Masri et al, 2007; Guertin et al, 2009). mTORC2 was also found to be activated in glioblastoma, where it promoted survival and resistance to chemotherapy in a NF-κB-dependent manner (Tanaka et al, 2011). To date, no analysis of genome stability under these conditions has been reported.
Second generation mTOR inhibitors that act as ATP-competitive inhibitors of both mTORC1 and mTORC2, or dual specificity inhibitors that target PI3K as well, indirectly suggest a role for mTORC2 in cancer development, possibly by rendering cancer cells more sensitive to replication stress (Yu et al, 2009; Falcon et al, 2011; Guo et al, 2014). In mouse models for T-cell leukemia (Molt-Luc2), treatment with pp242, an mTOR ATP-competitive inhibitor, enhanced DNA damage-induced apoptosis and delayed cancer development (Guo et al, 2014). Importantly, treatment of Molt-Luc2 cells with pp242, but not with rapamycin, led to downregulation of the expression of FANCD2, a component of the Fanconi anemia DNA repair complex, which responds to replication fork-associated damage. Consistently, treatment of Molt-Luc2 cells with pp242 (but again, not with rapamycin) sensitized cells to the inhibition of DNA synthesis by cytosine arabinoside (AraC), consistent with a perturbation of recovery from replication stress (Guo et al, 2014).
The benefits of simultaneous use of general mTOR inhibitors and DNA-damaging agents to enhance DNA damage-induced apoptosis are well documented (Beuvink et al, 2005; Grunt & Mariani, 2013; Tentori et al, 2013), reviewed by Hasty et al (2013) and Zoncu et al (2011). However, it remains to be explored whether the selective inhibition of mTORC2 can improve combined therapy, by sensitizing cancer cells to DNA-damaging agents. Based on the yeast results discussed here, one might expect that targeting mTORC2 will trigger a more selective and synergistic cell death, than pan-mTOR or pan-PIKK inhibitors. The need for more efficient combination therapies against metastatic cancer suggests that this pathway should, indeed, be tested.
Bridge the gap.
The gap
The mTOR kinase subunit is found in two distinct cytoplasmic complexes in mammals, TORC1, and TORC2. It has been difficult to study the second TOR complex in mammals due to the absence of complex-specific inhibitors. Studies in budding and fission yeast have now independently identified a role for the TORC2 complex in the survival of DNA damage. In fission yeast, survival of S phase-specific damage was compromised in strains lacking a functional TORC2 complex. In budding yeast, strong synthetic lethality with oxidizing damage from ionizing radiation or Zeocin was identified in strains treated with a TORC2 inhibitor. To date, there has been no link uniting these diverse results with the potential of using TORC2 as a chemotherapeutic target.
The bridge
Here, we explore the evidence arguing that the TORC2 signaling cascade may contribute to resistance or survival in the face of DNA damage in cancer cells, as it does in yeast. It is noteworthy that existing mTOR inhibitor studies are inconclusive with respect to TORC2 as a drug target, because the chemotherapeutic agents used to date are either selective for TORC1 (rapamycin-like inhibitors) or else inhibit not only TORC1 and TORC2, but also PI3 kinases. Given the striking conservation of DNA damage survival phenotypes in budding and fission yeasts, we argue that inhibition of TORC2 may be a novel and fruitful pathway for synthetic chemotherapy of repair-compromised cancers.
Acknowledgments
The Gasser laboratory is supported by the Friedrich Miescher Institute for Biomedical Research, the Swiss National Science Foundation, the Human Frontiers Science Program and the European Union Marie Curie initiative. SMG thanks Kenji Shimada for his work on this topic. RW acknowledges support from the Association for International Cancer Research (AICR) UK and from the Open University of Israel and thanks Miriam Schonbrun, Dana Laor and Masha Kolesnikov for their work on this topic.
Glossary
- Actin cytoskeleton
Filaments made from a chain of globular actin molecules that reside in the cytoplasm of eukaryotic cells and give the cell its shape and capacity for directed movement.
- AGC kinases
A subfamily of serine/threonine protein kinases, including the A, G, and C families of kinases (PKA, PKG and PKC). Other well known members of this family of proteins are Akt, S6K, RSK, and PDK1. A common and highly conserved feature of TOR-mediated signaling pathways is the phsophorylation and activation of specific subset of AGC kinases by TORC1 or TORC2.
- AKT kinase
AKT, also known as protein kinase B (PKB), is a member of the AGC kinases that plays a key role in protein synthesis, glucose metabolism, cell proliferation, and survival. AKT can be phosphorylated and activated by mTORC2.
- Combination therapy
The use of more than a single therapy or medication to treat a disease, generally cancer. Benefits of combination therapy include reduced rates of drug resistance or toxicity.
- DNA damage response (DDR)
DNA damage response entails activation of cell cycle checkpoints, DNA repair, and damage tolerance pathways. Failure of DDR causes genome instability that can lead to human syndromes and aging-associated diseases, in particular cancer.
- DNA replication stress
Inefficient DNA replication that causes DNA replication forks to progress slowly or stall. DNA replication stress can result from drugs such as hydroxyurea, which depletes the nucleotides pools of DNA intercalating or modifying drugs.
- Genome instability
Increased frequency of mutations, chromosomal rearrangements, or aneuploidy. Genome instability is often associated with cancer and can be indicative of a poor prognosis for some types of cancer.
- Homologous recombination (HR)
A major pathway for DNA damage repair that requires the exchange of DNA sequences between identical and similar (i.e. homologous) molecules of DNA.
- Rapamycin
A lipophilic macrolide produced by the soil bacterium Steptomyces hygroscopicous. Rapamycin can inhibit cell proliferation of a variety of cells types, an effect that results in anti-fungal, immunosuppressive, or anti-cancer activities.
- Rapalogs
Rapamycin and its derivatives, which act through a common pathway to inhibit mTORC1.
- Synthetic lethality
Cell death resulting from the combination of two or more mutations that are not lethal when present as single mutations
Conflict of interest
The authors declare that they have no conflict of interest.
For more information
List of current clinical anti-cancer trials that include pan-mTOR or pan-PIKK inhibitors
Roswell Park Cancer Institute: https://www.roswellpark.org/clinical-trials/list
National cancer Institute at the National Institutes for Health: http://www.cancer.gov/clinicaltrials
List of current clinical trials related to TSC (Tuberous Sclerosis Alliance)
http://www.tsalliance.org/pages.aspx?content=370
SGD—S. cerevisiae genome database
PombeBase—S. pombe genome database
References
- Alvarez B, Moreno S. Fission yeast Tor2 promotes cell growth and represses cell differentiation. J Cell Sci. 2006;119:4475–4485. doi: 10.1242/jcs.03241. [DOI] [PubMed] [Google Scholar]
- Aronova S, Wedaman K, Aronov PA, Fontes K, Ramos K, Hammock BD, Powers T. Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab. 2008;7:148–158. doi: 10.1016/j.cmet.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell. 1996;7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass KL, Murray JM, O'Connell MJ. Brc1-dependent recovery from replication stress. J Cell Sci. 2012;125:2753–2764. doi: 10.1242/jcs.103119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin BJ, Mullins RD. What we talk about when we talk about nuclear actin. Nucleus. 2013;4:291–297. doi: 10.4161/nucl.25960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold D, Piccolis M, Chiaruttini N, Riezman I, Riezman H, Roux A, Walther TC, Loewith R. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat Cell Biol. 2012;14:542–547. doi: 10.1038/ncb2480. [DOI] [PubMed] [Google Scholar]
- Beuvink I, Boulay A, Fumagalli S, Zilbermann F, Ruetz S, O'Reilly T, Natt F, Hall J, Lane HA, Thomas G. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell. 2005;120:747–759. doi: 10.1016/j.cell.2004.12.040. [DOI] [PubMed] [Google Scholar]
- Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203–213. doi: 10.1016/j.molcel.2008.02.024. [DOI] [PubMed] [Google Scholar]
- Carr AM. DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair (Amst) 2002;1:983–994. doi: 10.1016/s1568-7864(02)00165-9. [DOI] [PubMed] [Google Scholar]
- Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23:744–755. doi: 10.1016/j.ceb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;18:1021–1031. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011;71:1573–1583. doi: 10.1158/0008-5472.CAN-10-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel AM, Pike BL, Gasser SM. ATR/Mec1: coordinating fork stability and repair. Curr Opin Cell Biol. 2009;21:237–244. doi: 10.1016/j.ceb.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Grunt TW, Mariani GL. Novel approaches for molecular targeted therapy of breast cancer: interfering with PI3K/AKT/mTOR signaling. Curr Cancer Drug Targets. 2013;13:188–204. doi: 10.2174/1568009611313020008. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H, Sabatini DM. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–159. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Li J, Zhang S, Du W, Amarachintha S, Sipple J, Phelan J, Grimes HL, Zheng Y, Pang Q. mTOR kinase inhibitor sensitizes T-cell lymphoblastic leukemia for chemotherapy-induced DNA damage via suppressing FANCD2 expression. Leukemia. 2014;28:203–206. doi: 10.1038/leu.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deHart AK, Schnell JD, Allen DA, Hicke L. The conserved Pkh-Ypk kinase cascade is required for endocytosis in yeast. J Cell Biol. 2002;156:241–248. doi: 10.1083/jcb.200107135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Sharp ZD, Curiel TJ, Campisi J. mTORC1 and p53: clash of the gods? Cell Cycle. 2013;12:20–25. doi: 10.4161/cc.22912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Hatanaka M, Nagao K, Nakaseko Y, Kanoh J, Kokubu A, Ebe M, Yanagida M. Rapamycin sensitivity of the S. pombe tor2 mutant and organization of two highly phosphorylated TOR complexes by specific and common subunits. Genes Cells. 2007;12:1357–1370. doi: 10.1111/j.1365-2443.2007.01141.x. [DOI] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Hietakangas V, Cohen SM. Re-evaluating AKT regulation: role of TOR complex 2 in tissue growth. Genes Dev. 2007;21:632–637. doi: 10.1101/gad.416307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikai N, Nakazawa N, Hayashi T, Yanagida M. The reverse, but coordinated, roles of Tor2 (TORC1) and Tor1 (TORC2) kinases for growth, cell cycle and separase-mediated mitosis in Schizosaccharomyces pombe. Open Biol. 2011;1:110007. doi: 10.1098/rsob.110007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Morigasaki S, Tatebe H, Tamanoi F, Shiozaki K. Fission yeast TOR complex 2 activates the AGC-family Gad8 kinase essential for stress resistance and cell cycle control. Cell Cycle. 2008;7:358–364. doi: 10.4161/cc.7.3.5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Lorberg A. TOR regulation of AGC kinases in yeast and mammals. Biochem J. 2008;410:19–37. doi: 10.1042/BJ20071518. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Fujioka Y, Suzuki NN, Inagaki F, Wullschleger S, Loewith R, Hall MN, Ohsumi Y. Tor2 directly phosphorylates the AGC kinase Ypk2 to regulate actin polarization. Mol Cell Biol. 2005;25:7239–7248. doi: 10.1128/MCB.25.16.7239-7248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M, Nakashima A, Ueno M, Ushimaru T, Aiba K, Doi H, Uritani M. Fission yeast tor1 functions in response to various stresses including nitrogen starvation, high osmolarity, and high temperature. Curr Genet. 2001;39:166–174. doi: 10.1007/s002940100198. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gao T. mTORC2 phosphorylates protein kinase Czeta to regulate its stability and activity. EMBO Rep. 2014;15:191–198. doi: 10.1002/embr.201338119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R. Growth control: function follows form. Curr Biol. 2013;23:R607–R609. doi: 10.1016/j.cub.2013.05.048. [DOI] [PubMed] [Google Scholar]
- Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011;189:1177–1201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, Gera J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007;67:11712–11720. doi: 10.1158/0008-5472.CAN-07-2223. [DOI] [PubMed] [Google Scholar]
- Matsuo T, Kubo Y, Watanabe Y, Yamamoto M. Schizosaccharomyces pombe AGC family kinase Gad8p forms a conserved signaling module with TOR and PDK1-like kinases. EMBO J. 2003;22:3073–3083. doi: 10.1093/emboj/cdg298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo T, Otsubo Y, Urano J, Tamanoi F, Yamamoto M. Loss of the TOR kinase Tor2 mimics nitrogen starvation and activates the sexual development pathway in fission yeast. Mol Cell Biol. 2007;27:3154–3164. doi: 10.1128/MCB.01039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner B, Boll M, Daniel H, Baumeister R. Deletion of the intestinal peptide transporter affects insulin and TOR signaling in Caenorhabditis elegans. J Biol Chem. 2004;279:36739–36745. doi: 10.1074/jbc.M403415200. [DOI] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, Roux PP, Su B, Jacinto E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010;29:3939–3951. doi: 10.1038/emboj.2010.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014;4 doi: 10.3389/fonc.2014.00064. :eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Kunz J, Hall MN. TOR2 is required for organization of the actin cytoskeleton in yeast. Proc Natl Acad Sci U S A. 1996;93:13780–13785. doi: 10.1073/pnas.93.24.13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonbrun M, Kolesnikov M, Kupiec M, Weisman R. TORC2 is required to maintain genome stability during S phase in fission yeast. J Biol Chem. 2013;288:19649–19660. doi: 10.1074/jbc.M113.464974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonbrun M, Laor D, Lopez-Maury L, Bahler J, Kupiec M, Weisman R. TOR complex 2 controls gene silencing, telomere length maintenance, and survival under DNA-damaging conditions. Mol Cell Biol. 2009;29:4584–4594. doi: 10.1128/MCB.01879-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Filipuzzi I, Stahl M, Helliwell SB, Studer C, Hoepfner D, Seeber A, Loewith R, Movva NR, Gasser SM. TORC2 signaling pathway guarantees genome stability in the face of DNA strand breaks. Mol Cell. 2013;51:829–839. doi: 10.1016/j.molcel.2013.08.019. [DOI] [PubMed] [Google Scholar]
- Shor B, Gibbons JJ, Abraham RT, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8:3831–3837. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2010;29:3733–3744. doi: 10.1038/onc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surucu B, Bozulic L, Hynx D, Parcellier A, Hemmings BA. In vivo analysis of protein kinase B (PKB)/Akt regulation in DNA-PKcs-null mice reveals a role for PKB/Akt in DNA damage response and tumorigenesis. J Biol Chem. 2008;283:30025–30033. doi: 10.1074/jbc.M803053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B, Dang J, Zhu S, Yang H, De Jesus J, et al. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer Discov. 2011;1:524–538. doi: 10.1158/2159-8290.CD-11-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentori L, Lacal PM, Graziani G. Challenging resistance mechanisms to therapies for metastatic melanoma. Trends Pharmacol Sci. 2013;34:656–666. doi: 10.1016/j.tips.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Vartiainen MK. Nuclear actin dynamics–from form to function. FEBS Lett. 2008;582:2033–2040. doi: 10.1016/j.febslet.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- Wedaman KP, Reinke A, Anderson S, Yates J, 3rd, McCaffery JM, Powers T. Tor kinases are in distinct membrane-associated protein complexes in S. cerevisiae. Mol Biol Cell. 2003;14:1204–1220. doi: 10.1091/mbc.E02-09-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman R. Fission yeast TOR and rapamycin. In: Hall MN, Tamanoi F, editors. The Enzymes Structure, Function and Regulation of TOR Complexes from Yeast to Mammals Part A. Vol. 27. London: Elsevier; 2010. pp. 251–269. [Google Scholar]
- Weisman R, Choder M. The fission yeast TOR homolog, tor1+, is required for the response to starvation and other stresses via a conserved serine. J Biol Chem. 2001;276:7027–7032. doi: 10.1074/jbc.M010446200. [DOI] [PubMed] [Google Scholar]
- Weisman R, Roitburg I, Schonbrun M, Harari R, Kupiec M. Opposite effects of tor1 and tor2 on nitrogen starvation responses in fission yeast. Genetics. 2007;175:1153–1162. doi: 10.1534/genetics.106.064170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods NT, Mesquita RD, Sweet M, Carvalho MA, Li X, Liu Y, Nguyen H, Thomas CE, Iversen ES, Jr, Marsillac S, et al. Charting the landscape of tandem BRCT domain-mediated protein interactions. Sci Signal. 2012;5:rs6. doi: 10.1126/scisignal.2002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69:6232–6240. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000;14:2712–2724. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]