

Abstract
There is a progressive impairment of vascular repair mechanisms with advancing age concomitant with a steady decline in circulating androgen levels in men. Emerging evidence indicates androgens regulate angiogenesis; however, little research has focused on the impact of age upon androgen-mediated regulation of angiogenic mechanisms. Human dermal fibroblasts from young (<30 years) and older (>65 years) men were incubated with DHT, with or without androgen receptor antagonist hydroxyflutamide, or phosphoinositide 3-kinase inhibitor. Fibroblast-conditioned medium was used to stimulate angiogenic functions in human umbilical vein endothelial cells. Nuclear fractionation and fluorescence microscopy were used to study androgen receptor (AR) distribution. Conditioned medium from fibroblasts of young men, but not old men, treated with DHT produced a 3-fold increase in human umbilical vein endothelial cell tubulogenesis and 2-fold increase in migration via increased vascular endothelial growth factor (VEGF) expression and secretion, predominantly of VEGF145. DHT-induced VEGF secretion from fibroblasts of young men was AR-dependent and increased AKT phosphorylation, which was abrogated by phosphoinositide 3-kinase inhibition. By contrast, fibroblasts from older men were unresponsive to DHT and lacked androgen-mediated enhancement in VEGF production. These findings were associated with reduced AR nuclear translocation in old fibroblasts. The failure of DHT-induced paracrine stimulation of angiogenesis in fibroblasts from older men is likely due to defective nuclear translocation of AR. This first demonstration of androgen resistance (or insensitivity) acquired by human fibroblasts with aging suggests that pharmacological testosterone therapy for old men may be less effective in enhancing angiogenesis and facilitating tissue regeneration mechanisms reliant on paracrine release of VEGF.
Aging, a major risk factor for cardiovascular disease, is accompanied by a decline in cardiovascular repair mechanisms including blood vessel regeneration (1–3). Angiogenesis, the growth of new blood vessels, is fundamental to tissue regeneration and healing in adult tissue. It relies on a coordinated process of paracrine production of angiogenic growth factors and efficient response to them by endothelial cells (ECs) (4). Aged animals display marked impairment in angiogenesis and re-endothelialization after ischemic and vascular injuries (5, 6). These impairments are in part due to a decline in the production of proangiogenic growth factors and cytokines in aged animals, specifically vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and TGFβ1 (7–10). Restoration of VEGF signaling in aged animals has shown promising results by partially rescuing the age-dependent decline in angiogenesis (6, 11).
Male aging is also associated with a progressive decline in circulating testosterone concentrations of up to 1% annually from midlife onward (12). There is increasing evidence to suggest that circulating testosterone levels are associated with the prevalence and risk of cardiovascular diseases (13). Men with coronary artery disease have significantly lower levels of androgens (14, 15), whereas testosterone concentrations are inversely associated with mortality due to all causes and cardiovascular disease-related deaths (16, 17). Furthermore, recent studies have reported an association between androgen deprivation therapy for prostate cancer and increased risk of cardiovascular events, including myocardial infarction and cardiovascular mortality (18). These studies suggest that there is a relationship between testosterone levels and progression of cardiovascular disease. However, little is known about the role of androgens in the regulation of cardiovascular regeneration and repair.
Androgens exert their function by binding to the androgen receptor (AR), which primarily acts as a ligand-activated transcription factor (19). Androgen binding to the AR results in a conformational change, dimerization, and shedding of chaperone proteins, which allows for translocation of the receptor from the cytoplasm to the nucleus to subsequently interact with the DNA (20). ARs interact with hormone response elements on the promoter regions of target genes (21) that are modulated by coactivators, corepressors, and the recruitment of other transcription factors to their specific promoters (22).
Androgen-regulated genes include key mediators of angiogenesis (23, 24). There is mounting evidence for a role of androgens in angiogenesis through the production and secretion of VEGF. In vitro, adult human prostatic epithelial cells (PNT1), primary prostatic fetal fibroblasts, and human umbilical vein ECs (HUVECs) increase VEGF production with DHT treatment in a dose-dependent manner (25–27). Similar findings are seen in pathological cell lines (28). In vivo, androgen withdrawal leads to impaired vascularization of Matrigel plugs and blood vessel regeneration after hind limb ischemia in mice (27). In BALB/c mice inoculated with prostatic cancer cells (CWR22Rv1) and castrated, the progression of tumor growth was reduced with concomitant reduction in tumor VEGF levels (29). Thus, androgen treatment can stimulate both autocrine and paracrine production of VEGF in both physiological and pathological contexts; however, little is known about the mechanisms involved.
Given that circulating testosterone declines with age and that castration due to androgen deprivation therapy is widely used to treat prostate cancer, very little is known about how these differing degrees of androgen withdrawal influence angiogenesis and vascular regeneration. Additionally, the influence of aging on the role of androgens in angiogenesis remains unknown. Therefore, the current study investigates the effects of androgens on VEGF-mediated angiogenesis in fibroblasts isolated from young and old men. We report that androgen exposure stimulates VEGF production in cells from young but not old men, an effect culminating in enhanced EC function. VEGF production in cells from young men was dependent on phosphoinositide 3-kinase (PI3-kinase)/AKT activation and associated with ligand-dependent nuclear translocation of the AR, which was lacking in cells from old men. This study provides mechanistic insight into androgen-mediated angiogenesis through the regulation of VEGF and finds that androgen insensitivity in aging may limit the potential for effective pharmacological testosterone treatment in the stimulation of angiogenesis.
Materials and Methods
Cell culture, androgen treatment, and AR small interfering RNA knockdown
Primary human dermal fibroblasts were isolated from young donors 30 to 31 years of age (n = 4) and older donors aged 68 to 80 years (n = 4). This study had ethics committee approval in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines (Sydney South West Area Health Service Ethics Approval X11–0289, HREC/11/RPAH/444, and SSA/11/RPAH/619). All study participants provided written informed consent. Fibroblasts were isolated from the skin tissue as described (30). In brief, skin tissue was washed with Hanks' balanced salt solution (Sigma-Aldrich) containing 2% streptomycin and penicillin and digested with 0.07% Liberase III Blendzyme (Roche) in MesoEndo cell growth medium (Cell Applications) for 1.5 hours at 37°C. Cells were filtered through 70-μm mesh and centrifuged at 520g for 10 minutes. Fibroblasts were cultured in DMEM (Sigma) with 10% fetal bovine serum (FBS) (Sigma) at 37°C with 5% CO2. Fibroblasts (less than passage 10) were seeded for 24 hours and washed twice with PBS and changed to phenol red-free DMEM (Sigma) with 10% charcoal-stripped FBS (csFBS). DHT was added at 40nM concentration in absolute ethanol, and control cells were incubated with ethanol alone at 1 μL/mL. A concentration of 40nM DHT was chosen for all experiments because this yielded maximum VEGF secretion from young fibroblasts in a dose-response trial (Supplemental Figure 1). AR antagonist hydroxyflutamide (HF) or PI3-kinase inhibitor (LY294002) (Sigma) was added 1 hour before the addition of DHT at concentrations of 4μM and 10μM, respectively. Concentrations of LY294002 higher than 10μM resulted in more than 30% growth inhibition compared with untreated fibroblasts (Supplemental Figure 2). Hence, 10μM LY294002 was chosen as the maximum dose that does not inhibit baseline fibroblast function or cause cell death (31, 32). Cells were treated for 48 hours before RNA or protein extraction. For insulin stimulation experiments, fibroblasts from young and old men were serum-starved for 2 hours before 15 minutes insulin treatment (500nM). Fibroblast-conditioned medium (CM) was obtained in accordance with a previously published method (33). Fibroblasts were treated for 48 hours, washed with PBS, and changed to fresh phenol red-free DMEM with 10% csFBS for 24 hours before CM collection. Fibroblasts required for immunofluorescence or fractionation were serum-starved for 24 hours in phenol red-free, serum-free DMEM before treatment with 40nM DHT for 6 hours.
For AR small interfering RNA (siRNA) knockdown, fibroblasts were grown to 60% confluency in a 6-well plate and transfected for 24 hours with 250 pmol/well of either scrambled or AR siRNA in DMEM with 10% FBS. Cell transfection was performed using Lipofectamine 2000 (Invitrogen) at ratio of 1:1 of siRNA (micrograms) to Lipofectamine 2000 (microliters). Sequences used were as previously published (34): scrambled siRNA 5′-GAUAGCAAUGACGAAUGCGUA[dT][dT] and pan-AR-siRNA 5′-GACUCAGCUGCCCCAUCCA[dT][dT]. Fresh medium was replaced for another 48 hours followed by 48 hours treatment with 40nM DHT in phenol red-free DMEM with 10% csFBS. Successful knockdown of AR was confirmed by Western blot as a reduction >60%.
Transwell migration assay
HUVECs were cultured in MesoEndo until 70% confluent. A 24-well culture plate was filled with 600 μL fibroblast CM in duplicate per treatment and donor. Transwells with 8-μm pore size (Corning Inc) were then added, and 6000 HUVECs were plated onto the Transwell and cultured at 37°C with 5% CO2 for 24 hours. To determine the effect of recombinant human VEGF145 (rhVEGF145) (R&D Systems) on EC function, rhVEGF145 was added to phenol red-free DMEM with 10% csFBS at equivalent concentrations to that secreted by young fibroblasts with/without DHT treatment. Migration was assessed after 24 hours of incubation. Transwells were then removed, washed in cold PBS, and fixed in chilled 70% ethanol for 30 minutes. Cells were washed twice with PBS and stained with fluorescein isothiocyanate (FITC)-conjugated lectin from Ulex europaeus (Sigma) at 2 μg/mL in PBS for 30 minutes at room temperature. Membranes were placed on glass slides, coverslipped with Vectashield containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories), and imaged on an Olympus IX71 microscope (Olympus Corporation) at ×20 magnification. Five images were taken on each Transwell, 10 images per donor per treatment. Cells were counted using Image J software.
Tubulogenesis assay
HUVECs at 70% confluency were washed with PBS. Fibroblast CM or rhVEGF145 in phenol red-free DMEM plus10% csFBS was added to HUVECs and cultured for 24 hours. HUVECs were washed with PBS, detached with trypsin-EDTA, and plated onto phenol red-free, growth factor-reduced Matrigel (BD Biosciences) in a 96-well plate with 4000 cells per well. Four wells were plated per treatment per donor and incubated at 37°C with 5% CO2 for 4 hours. Cells were imaged using an Olympus IX71 microscope (Olympus Corporation) with phase contrast at ×20 magnification. Two images were taken per well, 8 images per treatment per donor. Cell-to-cell connections were quantified using Image J software.
Proliferation assay
HUVECs were plated at a seeding density of 6000 cells per well in a 24-well plate, left for 24 hours in MesoEndo medium, and then washed twice with PBS and grown for 24 hours in fibroblast CM obtained from DHT-, HF-, or LY294002-treated fibroblasts. To determine the appropriate concentration of LY294002, fibroblast cells were seeded at 5000 cells per well in a 24-well-plate and treated with LY294002 at concentrations 5, 10, 20, 50 and 100 μM in phenol red-free DMEM with 10% csFBS. Fibroblast cell proliferation was assessed after 2 days of growth. Wells containing known fibroblast cell numbers (625, 1250, 2500, 5000, 10 000, 20 000 cells per well; 3 wells per cell density) were grown for 2 days to establish a standard curve (Supplemental Figure 2). To quantify proliferation, cells were washed twice with PBS and fixed in cold 3.7% paraformaldehyde in PBS for 15 minutes. After washing in PBS, cells were stained with 1% Crystal Violet for 15 minutes at room temperature. Cells were then rinsed 3 times with distilled water and dissolved in 650 μL acetic acid per well. The colored acetic acid was then transferred into a 96-well plate (200 μL/well) in triplicate and read on a Flex Station 3 plate reader at an absorbance of 570 nm (Molecular Devices).
ELISA analysis
For the comparison of growth factors and cytokines in fibroblast CM, a human growth factor II ELISA kit (Signosis) was used. For the analysis of secreted VEGF in CM from fibroblasts from young men with additional HF and PI3-kinase inhibition, human VEGF Quanti-Glo ELISA kit was used (R&D Systems). VEGF145 secretion in CM from young fibroblasts was analyzed by VEGF145-specific ELISA (Cloud-Clone Corp), with 100 μl of undiluted fibroblast CM used per well in triplicate in each ELISA. Total VEGF and VEGF145 standard curves are included in Supplemental Figure 3. All ELISA kits were followed as per supplier instructions and measured on a Flex Station 3 plate reader at an absorbance of 450 nm (Molecular Devices).
RNA isolation and quantitative RT-PCR
RNA was isolated from fibroblasts using TRI reagent (Sigma) according to the manufacturer's protocol. cDNA synthesis was performed using the iScript cDNA synthesis kit (Bio-Rad Laboratories). Sequences for primers (Gene Works) were human VEGF, forward primer 5′-TGTGAATGCAGACCAAAGAAAGA-3′ and reverse 5′-TGCTTTCTCCGCTCTGAGC-3′; human AR, forward 5′-CAGCAGCAGCCCAAGCGATG-3′ and reverse 5′-GTACCACACTTCGGAGGCAGAGT-3′; and internal control 28S rRNA, forward primer 5′-GTTCACCCACTAATAGGGAACGTGA-3′ and reverse 5′-GGATTCTGACTTAGAGGCGTTCAGT-3′. VEGF primers for isoforms 145, 165, 121, 206, and 189 were as published previously (35). Samples were analyzed with CFX96 real-time PCR detection system (Bio-Rad). Quantitative RT-PCR was performed in triplicate for each treatment per donor.
Protein isolation and Western blot analysis
Cells were lysed with mammalian cell lysis buffer (50mM Tris-HCl, 1mM EDTA, 150mM sodium chloride, 0.1% sodium dodecyl sulfate [SDS], 0.5% deoxycholic acid sodium salt solution, 1% IGEPAL, 1% protease inhibitor cocktail, 1% phosphatase inhibitor cocktail) (Sigma) for 15 minutes on ice before freezing at −80°C. Protein estimation was performed using a standard bicinchoninic acid assay (Thermo Fisher Scientific) and read on a Flex Station 3 plate reader at an absorbance of 562 nm (Molecular Devices). Western blot analysis was performed on equal amounts of protein loaded onto 4% to 12% Bis-Tris precast gels (Invitrogen) and separated using SDS-PAGE electrophoresis. Protein was transferred to polyvinylidene difluoride membranes (Invitrogen) using a semidry iBlot gel transfer system (Invitrogen) and incubated for 1 hour at room temperature with 1% skim milk powder in Tris-buffered saline (TBS). Primary antibodies were applied overnight in TBS at 1:1000 dilution for rabbit anti-AKT, anti-phospho-AKT (Ser473 or Tyr308), and anti-LSD1; (Cell Signaling Technology), 0.3 μg/mL for rabbit anti-AR (Fisher Scientific); and 1 μg/mL for mouse antitubulin (Abcam). Membranes were washed in TBS and incubated in either antirabbit IgG secondary antibody (Invitrogen) conjugated with horseradish peroxidase at 0.8 μg/mL or antimouse IgG secondary antibody-horseradish peroxidase (Invitrogen) at 0.6 μg/mL for 2 hours at room temperature in 1% skim milk in TBS. Membranes were washed and imaged after 5 minutes incubation in ECL (GE Healthcare Biosciences) on a Bio-Rad ChemiDoc MP imaging system (Bio-Rad). Densitometric analysis of protein bands were quantified using Image Lab Software version 4.0.1 (Bio-Rad). Western blots for AKT were always performed after first blotting for phosphorylated AKT (pAKT), and then membranes were stripped in Restore stripping buffer (ThermoFisher Scientific) according to manufacturer's instructions before reprobing for total AKT and tubulin. Western blots were conducted in duplicate, and the densitometry readings for each of the treatment bands were normalized to the average of the control readings.
AR dual-luciferase reporter assay
Young and old fibroblasts were cultured in a 24-well plate to 60% confluency in DMEM with 10% FBS. Fibroblasts were transfected overnight with 100 ng/well of AR luciferase reporter (CCS-1019L) according to manufacturer's instruction (QIAGEN). Cell transfection was performed using Lipofectamine 2000 (Invitrogen) at ratio of 1:1 of reporter plasmid (micrograms) to Lipofectamine 2000 (microliters). Cells were then cultured in phenol red-free DMEM with 10% csFBS and treated with 40nM DHT for 24 hours. AR transcriptional activity was assessed using a dual-luciferase reporter assay as instructed (Promega). Firefly luciferase activities were normalized to those of renilla luciferase controls.
Immunofluorescence microscopy
Fibroblasts were seeded onto 15-mm glass coverslips in DMEM with 10% FBS for 48 hours and serum-starved for 24 hours in phenol red-free DMEM. Cells were treated with 40nM DHT for 6 hours, washed with PBS, and fixed in 3.7% formaldehyde for 10 minutes at room temperature. Cells were then washed with PBS and blocked in 0.05% BSA in PBS for 1 hour. Rabbit anti-AR was added at 3 μg/mL in 0.05% BSA in PBS for 2 hours, washed with PBS, and incubated with antirabbit IgG-FITC (Jackson ImmunoResearch Laboratory) at 3 μg/mL for 30 minutes. Cells were coverslipped with Vectashield containing DAPI (Vector Laboratories) and imaged on a Zeiss deconvolution microscope using the same exposure for control and DHT-treated cells (Carl Zeiss). Fluorescence intensity was measured by isolating nuclei into a new image. Nuclei were transformed into grayscale images, and a horizontal line was drawn through each nucleus. The corresponding plot profile gives an intensity value for each pixel crossed by the horizontal line, and subsequently the average intensity per nucleus was calculated.
Nuclear and cytoplasmic protein fractionation
Nuclear and cytoplasmic fractions were isolated from young and old fibroblasts. Cultured cells were detached with trypsin and washed twice with PBS, and 1 × 106 cells were placed into 100 μL hypotonic buffer (20mM Tris-HCL [pH 7.4], 10mM NaCl, 3mM MgCl2, 1% protease inhibitor cocktail, and 1% phosphatase inhibitor cocktail) (Sigma), passed 25 times through a 27-gauge needle, and incubated on ice for 15 minutes. After this, 0.5% IGEPAL (Sigma) was added to the homogenate and vortexed. Homogenates were centrifuged for 10 minutes at 845g at 4°C. The supernatant (cytoplasmic fraction) was collected and stored at −80°C. The pellet was washed with 50 μL hypotonic buffer and centrifuged for 10 minutes at 845g at 4°C. The supernatant was discarded, and the pellet was resuspended in 17 μL hypertonic buffer (50mM Tris-HCl, 1mM EDTA, 300mM sodium chloride, 0.1% SDS, 0.5% deoxycholic acid sodium salt solution, 1% IGEPAL, 1% protease inhibitor cocktail, 1% phosphatase inhibitor cocktail) (Sigma), vortexed and incubated on ice for 30 minutes. Samples were centrifuged for 30 minutes at 14 000g at 4°C. The supernatant (nuclear fraction) was collected and stored at –80°C. Western blotting was then performed as described above.
Statistical analysis
All statistics were conducted with n = 3 to 4 donors in triplicate experiments. Data are presented as mean ± SEM. Statistical analysis was performed using two-tailed unpaired Student's t test, one-way ANOVA, or two-way ANOVA. Data analyzed using ANOVA were subjected to Bonferroni post-test correction. Statistical significance was considered when P < .05.
Results
Age-related impairment in androgen-mediated paracrine regulation of angiogenesis
To investigate the effect of androgens on the paracrine promotion of angiogenesis by cells taken from young and old men, CM from DHT-treated and control fibroblasts was used to stimulate key angiogenic events, including EC migration and tubulogenesis. Fibroblast CM was collected and placed on the bottom of a Transwell with HUVECs seeded into the upper chamber. After 12 hours, HUVECs that had migrated to the bottom chamber were quantified. Compared with untreated controls, CM from DHT-treated fibroblasts from young men increased the migration of HUVECs 2-fold (Figure 1A). In contrast, the CM of DHT-treated fibroblasts from old men did not alter endothelial migration compared with controls.
Figure 1.

DHT mediates paracrine regulation of EC function. A, Relative number of migrated HUVECs per Transwell, toward conditioned media (CM) from untreated control vs 48 hours DHT (40nM) treatment of fibroblasts from young and old men. Statistical analysis was performed using 2-way ANOVA with Bonferroni correction. *, P < .05 vs young control; n = 3 donors for young and old performed in duplicate over 3 individual experiments with data presented as means ± SEM. B, Relative number of tubule branch points per 4000 HUVECs treated for 24 hours with CM from control and 40nM DHT-treated young and old male fibroblasts. Statistical analysis was performed using 2-way ANOVA with Bonferroni correction. *, P < .05 vs young control; n = 3 donors for young and old performed in quadruplicate over 3 individual experiments with data presented as means ± SEM. Representative micrographs of HUVEC tubule formation after 4 hours on Matrigel after 24 hours pretreatment with CM from control and 40nM DHT-treated fibroblasts from young and old men. Bar, 50 μm. C, Relative absorbance of HUVECs incubated in young and old DHT-treated fibroblast CM after 24 hours; n = 3 donors for young and old performed in quadruplicate over 3 individual experiments with data presented as means ± SEM.
For tubulogenesis assays, HUVECs pretreated with CM for 24 hours were seeded onto growth factor-reduced Matrigel and imaged after 4 hours for the presence of tubules. Fibroblast CM from young men treated with DHT induced a 3-fold increase in tubule branch points compared with untreated controls. Although the basal tubule branching of HUVECs treated with fibroblast CM from old men was slightly higher (not significant) than those treated with fibroblast CM from young men (Figure 1B), CM from DHT-treated old fibroblasts had no effect on endothelial tubule formation (Figure 1B). DHT-treated fibroblast CM from young and old fibroblasts did not alter HUVEC proliferation after 24 hours (Figure 1C). These results demonstrate that DHT increases the proangiogenic capacity of fibroblasts from young men, which can enhance EC functions such as migration and tubule formation but is not able to increase the proangiogenic capacity of fibroblasts from old men.
Androgen administration induces VEGF production and secretion in young but not in old human fibroblasts
To determine which growth factors or cytokines are involved in the DHT-mediated increase in EC function, mRNA and protein expression of known angiogenic factors in fibroblast CM was analyzed by ELISA. DHT treatment of fibroblasts from young men resulted in an increase in VEGF mRNA expression, which was not observed in DHT-treated cells from old men (Figure 2A). The CM collected from DHT-treated fibroblasts from young men contained a higher concentration of VEGF than nontreated controls (Figure 2B), whereas the concentration of VEGF in CM from DHT-treated fibroblasts from old men remained unchanged compared with controls (Figure 2B). The concentrations of secreted VEGF (picograms per milliliter) in the CM from fibroblasts isolated from each young donor are included in Supplemental Table 1. The mRNA expression levels of VEGF isoforms were analyzed in young male fibroblasts treated with DHT. A significant increase in VEGF145 mRNA expression was noted in DHT-treated cells compared with controls (Figure 2C). Similarly, a 1.25-fold increase in VEGF145 concentration was detected in the CM of young fibroblasts treated with DHT compared with those treated with vehicle control (Figure 2D). No change in isoforms VEGF121, VEGF165, VEGF189, and VEGF206 mRNA levels with DHT treatment was observed (Supplemental Figure 4).
Figure 2.

The effect of DHT treatment on fibroblast VEGF production. A, Quantitative RT-PCR on VEGF mRNA levels in fibroblasts from young and old men after 48 hours of 40nM DHT treatment. Statistical analysis was performed using 2-way ANOVA with Bonferroni correction. *, P < .05 vs young control. B, The level of VEGF protein in CM from fibroblasts after 48 hours of DHT treatment using ELISA. Statistical analysis was performed using 2-way ANOVA with Bonferroni correction. *, P < .05 vs young control. C, VEGF145 isoform mRNA levels in fibroblasts from young men after 48 hours of 40nM DHT treatment analyzed by quantitative RT-PCR. Statistical analysis was performed using Student's t test. *, P < .05 vs young control. D, The levels of VEGF145 protein in CM from young fibroblasts after 48 hours DHT treatment using ELISA. Statistical analysis was performed using Student's t test. ***, P < .001 vs young control. E, Relative number of tubule branch points per 4000 HUVECs treated for 24 hours with rhVEGF145 at indicated concentrations. Statistical analysis was performed using one-way ANOVA with Bonferroni correction. ###, P < .001 vs untreated control; ***, P < .001; 73.7 pg/mL vs 52.9 pg/mL rhVEGF145-treated HUVECs. n = 3 donors of HUVECs performed in quadruplicate. Data presented as means ± SEM. F, Relative fold increase of migrated HUVECs per transwell, toward rhVEGF145 after 24 hours treatment. Statistical analysis was performed using one-way ANOVA with Bonferroni correction. ###, P < .001, 73.7 pg/mL rhVEGF145-treated HUVECs vs untreated; #, P < .05, 500 pg/mL rhVEGF145-treated HUVECs vs untreated; *** P < .001, 73.7 pg/mL vs 52.9 pg/mL rhVEGF145-treated HUVECs; n = 3 donors of HUVECs performed in duplicate with data presented as means ± SEM.
To demonstrate that VEGF145 secreted by fibroblasts is a key mediator of EC function, HUVECs were treated with rhVEGF145 at concentrations equivalent to those found in the CM (Supplemental Table 2). Tubule formation and cell migration of HUVECs were assessed after 24 hours treatment with rhVEGF145. HUVECs treated with 52.9 pg/mL rhVEGF145 (equivalent to the secreted concentration in control young fibroblast CM) did not induce a significant difference in tubule formation compared with untreated HUVECs, whereas HUVECs treated with 73.7 pg/mL rhVEGF145 (equivalent to the secreted concentration from DHT-treated fibroblasts) showed a 2-fold increase in tubule branching compared with those treated with 52.9 pg/mL rhVEGF145 (Figure 2E). HUVEC migration increased 1.7-fold when treated with 73.7 pg/mL rhVEGF145 compared with those treated with 52.9 pg/mL rhVEGF145 (Figure 2F). Increasing the rhVEGF145 concentration (500 pg/mL) did not further increase HUVEC tubule branching or migration. This indicates that a subtle increase in VEGF145 is sufficient to induce HUVEC tubule branching and migration. Together, these results suggest that VEGF145 secreted by fibroblasts in response to androgen exposure is a key mediator of EC function.
Angiogenic factors, other than VEGF, were also determined in the fibroblast CM from young and old men after 48 hours DHT treatment using ELISA. Epidermal growth factor (Figure 3A), basic FGF (Figure 3B), platelet-derived growth factor (PDGF) (Figure 3C), nerve growth factor (Figure 3D), TNF-α (Figure 3E), and TGFβ (Figure 3F) remained unchanged with DHT treatment in all fibroblast CM.
Figure 3.

The effect of DHT treatment on cytokine production. A–F, The level of epidermal growth factor (EGF), basic FGF (FGFb), PDGF, nerve growth factor (NGF), TNF-α, and TGFβ, respectively in CM of fibroblasts from young and old men after 48 hours DHT treatment; n = 3 donors for young and old performed in triplicate over 3 individual experiments (means ± SEM).
Androgen modulation of paracrine angiogenic mechanisms is due to AR-dependent stimulation of VEGF production
To elucidate the roles of the AR and of VEGF in androgen-mediated paracrine regulation of angiogenesis, fibroblast CM obtained from DHT-treated cells with the addition of HF or VEGF-neutralizing antibody was added to HUVECs before functional experiments. AR antagonism with HF abrogated DHT stimulation of VEGF secretion by young fibroblasts (Figure 4A). Knockdown of AR in young male fibroblasts with specific AR siRNA inhibited the DHT-mediated increase in VEGF protein expression observed in cells transfected with scrambled nonspecific siRNA (Figure 4B). HUVEC migration toward DHT-treated young fibroblast CM increased by 2-fold, a finding that was inhibited by the presence of HF or the neutralization of VEGF (Figure 4C). HUVECs pretreated with CM from DHT-treated fibroblasts enhanced tubule formation by 2-fold, an effect that was also inhibited with the addition of HF or VEGF-neutralizing antibody (Figure 4D). These findings indicate that DHT-treated fibroblasts from young men enhance EC function by AR-dependent enhancement of VEGF secretion.
Figure 4.

DHT-mediated increase in paracrine regulation of EC function by fibroblasts from young men is via an AR-dependent increase in VEGF. A, Level of VEGF in fibroblast CM measured by ELISA after 48 hours treatment with 40nM DHT in the presence or absence of 4μM HF. Statistical analysis was performed using one-way ANOVA with Bonferroni correction. *, P < .05 vs control. B, Level of VEGF protein in fibroblasts after 24 hours transfection with 250 pmol of either control or AR siRNA, and after another 48 hours, cells were treated for 48 hours with 40nM DHT. Western blot demonstrating the level of AR knockdown 72 hours after transfection. *, P < .05 DHT vs control scrambled siRNA transfected cells. C, Relative level of HUVEC migration toward fibroblast CM after 48 hours DHT (40nM) treatment in the presence or absence of 4μM HF or 1 μg/mL VEGF-neutralizing antibody. Statistical analysis was performed using one-way ANOVA with Bonferroni correction. *, P < .05 vs control. D, Tubule branch points per 4000 HUVECs after 24 hours HUVEC pretreatment with fibroblast CM after fibroblasts were treated for 48 hours with 40nM DHT in the presence or absence of 4μM HF or 1 μg/mL VEGF-neutralizing antibody. Statistical analysis was performed using one-way ANOVA with Bonferroni correction. ***, P < .001 vs control; n = 4 donors performed in triplicate over 3 individual experiments (means ± SEM).
Androgen-mediated VEGF secretion is dependent on PI3-kinase pathway activation
Studies have shown that VEGF production is dependent on the PI3-kinase pathway (36); therefore, the current study investigated whether PI3-kinase activation is involved in DHT-mediated VEGF production. The DHT-mediated increase in secreted VEGF from fibroblasts from young men was suppressed by additional incubation of fibroblasts with PI3-kinase inhibitor LY294002 (Figure 5A). This was similarly observed for VEGF mRNA (Supplemental Figure 5). To demonstrate DHT activation of the PI3-kinase pathway, AKT phosphorylation was examined. DHT treatment of fibroblasts from young men increased AKT phosphorylation on Ser473 and not Tyr308, which also occurred in the presence of HF, but was blocked by PI3-kinase inhibitor LY294002 (Figure 5, B and C). To further elucidate the mechanism of DHT-induced AKT phosphorylation, the levels of pAKT(Ser473) were examined in young fibroblasts transfected with scrambled and AR siRNA (Figure 5D). DHT increased the ratio of pAKT(Ser473)/AKT in cells transfected with scrambled siRNA. However, DHT-induced AKT phosphorylation was abolished in cells transfected with AR siRNA. Together these results indicate that DHT-induced AKT phosphorylation is AR-dependent, but independent of the ligand binding domain of AR because DHT-induced AKT phosphorylation is not inhibited by the presence of HF. DHT-induced AKT(Ser473) phosphorylation was absent in fibroblasts from old men compared with DHT-treated young fibroblasts (Figure 5E). In addition, the basal levels of pAKT(Ser473)/AKT of old fibroblasts were significantly decreased when compared with young fibroblasts (Supplemental Figure 6A). To determine whether the lack of AKT activation in old fibroblasts was specific to androgen exposure or a general inability to phosphorylate AKT, cells were treated with 500nM insulin as an alternative means of activating the PI3-kinase/AKT pathway (37). Insulin induced a 2-fold increase of AKT phosphorylation in young male fibroblasts after 15 minutes exposure, but fibroblasts from old men were unresponsive (Supplemental Figure 6B), indicating that PI3-kinase/AKT activation may generally be impaired in old cells and not limited to androgen-dependent pathways.
Figure 5.

PI3-kinase pathway activation in DHT-mediated VEGF production. A, Level of VEGF in fibroblast CM measured by ELISA after 48 hours treatment with 40nM DHT in the presence or absence of 10μM PI3-kinase inhibitor (LY294002). Statistical analysis was performed using one-way ANOVA with Bonferroni correction. *, P < .01 vs untreated control; n = 3 donors performed in triplicate over 3 individual experiments (means ± SEM). B, The level of pAKT(Ser473)/AKT expressed in fibroblasts from young men after 48 hours treatment with 40nM DHT in the presence or absence of 4μM HF or 10μM PI3-kinase inhibitor (LY294002). Data are normalized to vehicle control; n = 3. Statistical analysis was performed using one-way ANOVA with Bonferroni correction. *, P < .05, DHT vs untreated control; #, P < .01, DHT plus HF vs untreated control plus HF. C, The level pAKT(Tyr308)/AKT expressed in fibroblasts from young men treated for 48 hours with DHT with or without the addition of LY294002. D, The level of pAKT(Ser473)/AKT expressed in young fibroblasts transfected with scrambled siRNA or AR siRNA after 48 hours treatment with 40nM DHT; n = 3. Data are normalized to untreated cells transfected with scrambled siRNA or AR siRNA. Statistical analysis was performed using one-way ANOVA with Bonferroni correction. *, P < .05, DHT vs control scrambled siRNA transfected cells. E, The level of pAKT(Ser473)/AKT expressed in fibroblasts from young and old men after 48 hours treatment with 40nM DHT. Statistical analysis was performed using 2-way ANOVA with Bonferroni correction. *, P < .05 vs young control; n = 3 donors performed in duplicate over 3 individual experiments (means ± SEM).
Fibroblasts from old men have comparable levels of AR protein but are defective in androgen-mediated AR transcriptional activity
To investigate the potential cause for the age-related impairment in androgen-mediated regulation of angiogenesis, the expression and function of AR was compared in cells from old and young men. Although the level of AR mRNA in fibroblasts from old men was 3-fold lower than that in fibroblasts from young men (Figure 6A), this was not translated to protein expression, and the levels of AR in young and old fibroblasts were the same (Figure 6B). Despite similar expression levels, the function of AR demonstrated age-related impairment. DHT treatment of young fibroblasts induced a 3-fold increase in AR transcriptional activity, as measured by luciferase reporter assay, whereas DHT treatment of fibroblasts from old men had no effect (Figure 6C).
Figure 6.
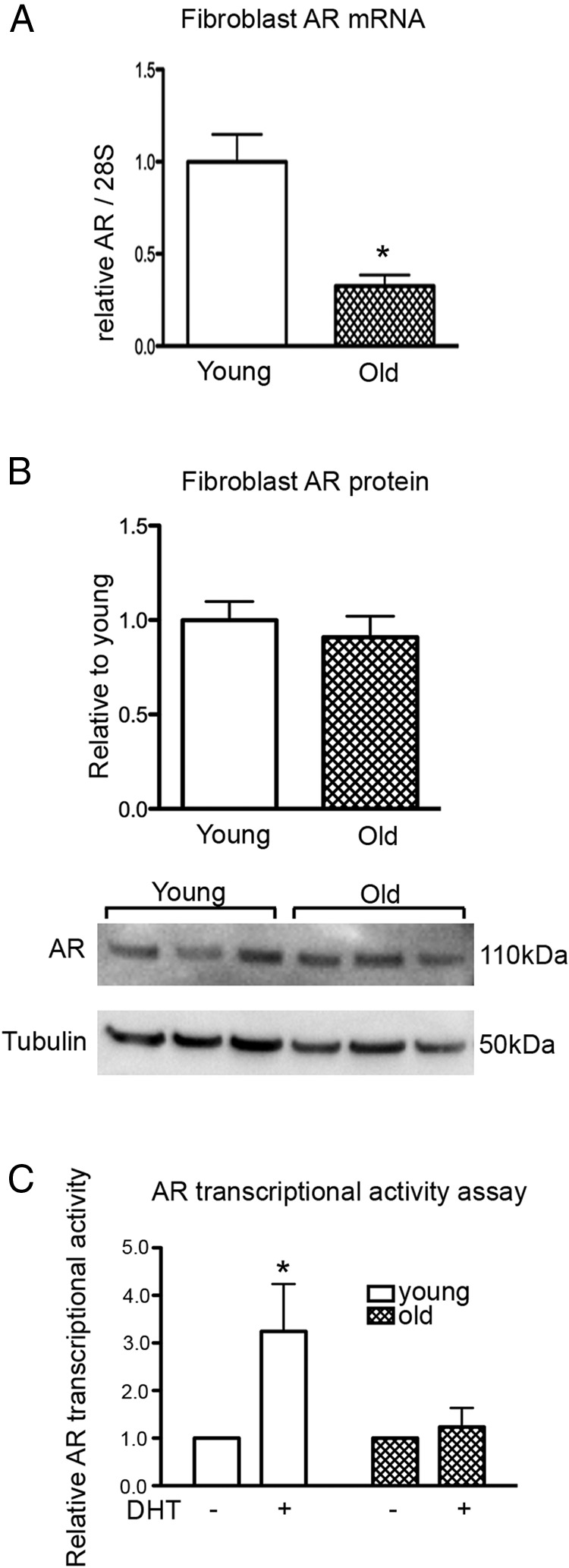
AR expression in fibroblasts from young and old men. A, AR mRNA expression in fibroblasts from young and old men. Statistical analysis was performed using Student's t test. *, P < .05 vs young. B, Protein expression of AR in fibroblasts from young and old men; n = 3 donors performed in triplicate over 3 individual experiments (means ± SEM). C, Relative AR transcriptional activities of young and old fibroblasts after overnight treatment with 40nM DHT. Statistical analysis was performed using 2-way ANOVA with Bonferroni correction. *, P < .05 vs young control; n = 3 donors performed in triplicate over 3 individual experiments (means ± SEM).
Nuclear AR increases with DHT treatment in fibroblasts from young men but not in fibroblasts from old men.
Cytoplasmic and nuclear subcellular fractions were separated after DHT treatment of fibroblasts to demonstrate changes in AR protein expression and subcellular localization. Cytoplasmic and nuclear markers LSD1 and tubulin, respectively, were used to show adequate separation of fractions. Nuclear marker lamin A/C confirmed a similar fractionation pattern to LSD1 (Supplemental Figure 7). In young fibroblasts, DHT treatment was associated with an increase in the total amount of AR protein that was located in the nuclear fraction, whereas the amount of AR in the cytoplasm remained stable between DHT-treated and control cells (Figure 7A and Supplemental Figure 7). By contrast, in cells from old men, AR expression in both the cytoplasmic and nuclear fractions remained unchanged with DHT treatment (Figure 7B).
Figure 7.

The effect of DHT treatment on AR localization in fibroblasts from young and old men. Panel A, Cytoplasmic (C) and nuclear (N) expression of AR after subcellular fractionation of fibroblasts from young men subjected to 6 hours treatment with 40nM DHT. Statistical analysis was performed using Student's t test. *, P < .01 vs N control; n = 4 donors performed in quadruplicate over 3 individual experiments (means ± SEM). LSD1 and tubulin were used to demonstrate equal separation of nuclear and cytoplasmic fractions, respectively. Panel B, Cytoplasmic (C) and nuclear (N) expression of AR after subcellular fractionation of fibroblasts from old men subjected to 6 hours treatment with 40nM DHT. Statistical analysis was performed using Student's t test; n = 4 donors performed in quadruplicate over 3 individual experiments (means ± SEM). Panel C, Immunofluorescence localization of AR labeled with FITC (green) in untreated and 6 hours DHT-treated (40nM) fibroblasts from young men. Nuclei are stained blue with DAPI. Panel D, Immunofluorescence localization of AR labeled with FITC (green) in untreated and 6 hours DHT-treated (40nM) fibroblasts from old men. Nuclei are stained blue with DAPI. Bar, 30 μm. Panel E, Pixel fluorescence intensity of nuclear AR in young and old fibroblasts after 6 hours treatment with 40nM DHT. Statistical analysis was performed using 2-way ANOVA with Bonferroni correction. *, P < .05 vs young control.
Immunofluorescence localization of AR (FITC/green) in control fibroblasts from young men confirmed that AR localized to the nucleus and faintly throughout the cytoplasm under basal conditions. After DHT treatment, ARs were concentrated in the perinuclear region of the cells in addition to intense staining within the nucleus (Figure 7C). Immunofluorescence localization of AR in control cells from old men displayed similar AR localization to young control cells, with staining in both the nucleus and dispersed throughout the cytoplasm; however, there was no change to AR localization upon DHT stimulation (Figure 7D). Nuclear AR was quantified from the average fluorescence intensity of the individual nuclei. Nuclear AR of young DHT-treated cells increased compared with control treated young fibroblasts, whereas nuclear AR after DHT treatment of fibroblasts from old men remained unchanged compared with control cells (Figure 7E and Supplemental Table 3). These results suggest that despite adequate levels of AR expression in cells from old men, the response to androgens is defective due to diminished AR nuclear translocation.
Discussion
Androgens have been implicated in the regulation of angiogenesis in both physiological and pathological settings (38). Given that aging is associated with a progressive decline in circulating testosterone levels (12) and with progressive impairment in cardiovascular repair mechanisms such as angiogenesis (3), the age-dependent mechanisms underlying androgen regulation of angiogenesis warrant investigation and are the focus of the current study. Fibroblasts from young and old men treated with DHT were used to assess the effect of androgens on the paracrine production of angiogenic factors. Previous studies have demonstrated that fibroblasts are effective mediators of angiogenesis through the production of VEGF (39–41). The current study demonstrates that androgens regulate angiogenesis in a paracrine manner by AR-dependent stimulation of VEGF expression and secretion from fibroblasts from young men. The increase in DHT-mediated VEGF secretion from young fibroblasts biologically enhanced key angiogenic events including EC migration and tubule formation. Additionally, the increased capacity for fibroblast-promoted angiogenesis with DHT treatment was blocked with a VEGF-neutralizing antibody confirming a VEGF-mediated response.
VEGF isoforms are generated by alternative splicing from a single VEGF gene, containing domains that are encoded by exons 1 to 5 and exon 8 of the gene but differing in the combination of coding sequence at exons 6 and 7 (42, 43). VEGF145 contains exon 6 but lacks exon 7 (44, 45). It predominantly binds to VEGF receptor KDR/flk-1, albeit with a lower affinity than that of VEGF165 (46, 47). VEGF145 mRNA has been previously found in placenta (44, 45), human blastocysts (48), and human epithelial ovarian carcinoma cells (40). In the present study, DHT-mediated VEGF145 mRNA upregulation in fibroblasts indicates that VEGF145 is also present as a secreted form of VEGF in fibroblasts and is specifically responsive to androgen stimulation.
The age-dependent impairment of androgen-mediated proangiogenic paracrine effects of fibroblasts was demonstrated by performing functional experiments on HUVECs cultured in fibroblast CM. Interestingly, a higher level of HUVEC tubule formation was observed with CM from untreated fibroblasts isolated from old men (compared with untreated young fibroblast CM) but was statistically insignificant (Figure 1B). This observation may be due, in part, to a compensatory mechanism in the aging cells, similar to what has been observed in aged murine kidneys whereby the basal erythropoietin levels are elevated to compensate for the chronic hypoxic conditions experienced with advanced aging (49). It is possible that fibroblasts from older donors have adapted to a higher level of basal hypoxic stress. In addition, aging is associated with a persistent inflammatory status (50, 51). Cross-talk between inflammation and angiogenesis has been demonstrated by Mor et al (52), who found that IL-2–activated T cells increase VEGF secretion, which in turn augments IFN-γ release. Inflammation-mediated angiogenesis is also well established in cancer models (53). In this study, the fibroblasts from elderly donors may have been exposed to higher levels of proinflammatory cytokines in circulating plasma, resulting in a higher basal level of paracrine proangiogenic capacity. Importantly, the present study demonstrates that aging is associated with impairment of androgen regulation of angiogenesis in that fibroblasts from old men lacked DHT-mediated enhancement in VEGF production and were therefore unable to stimulate angiogenesis in a paracrine manner.
Androgen regulation of both angiogenesis and VEGF production has been previously demonstrated in a number of young healthy and pathologic tissues. The androgen-mediated angiogenesis that is associated with prostate cancer progression and tumor growth has been shown to be in part due to androgen-mediated VEGF production from both normal prostate and prostate cancer cells (25, 26, 29). In vivo, androgen administration increased VEGF production in the rat prostate, and androgen withdrawal was associated with reduced VEGF production and decreased microvessel density in prostate cancer tumors. More recently, androgens have been implicated in the autocrine stimulation of cultured HUVECs and human aortic ECs, which increased VEGF production with DHT administration in a dose-dependent and AR-dependent manner (27, 54). We previously demonstrated a role for androgens in regulation of ischemia-mediated angiogenesis in a murine model of hind limb ischemia (27). Androgen withdrawal by orchiectomy significantly impaired ischemic recovery and tissue angiogenesis after femoral artery ligation, a finding rescued by androgen replacement with DHT. In a transplantation model where human foreskin tissue was subcutaneously placed in a nude rat and topically administered with testosterone gel, grafts treated with testosterone displayed significantly greater vascularity compared with placebo treatment (55). Together with the current study, these results suggest that androgens effectively promote angiogenesis and VEGF production in young tissues, both healthy and pathological.
The activation of the PI3-kinase pathway is integral for upregulation of VEGF by ECs in response to androgens, (27), PDGF (56), and insulin (57). Similarly, the current study demonstrated that androgen-mediated upregulation of VEGF secretion from young fibroblasts was dependent on a functional PI3-kinase pathway and was accompanied by androgen-mediated upregulation and phosphorylation of AKT, specifically on Ser473 and not Tyr308. The addition of a PI3-kinase inhibitor (LY294002) prevented DHT-mediated phosphorylation of AKT(Ser473); however, the concentration of 10μM did not significantly lower pAKT(Ser473) below baseline, which is consistent with previous studies (58). The inability of AR-antagonist HF to prevent DHT-induced AKT phosphorylation, which is abolished when AR is knocked down by siRNA transfection, suggests that this is an AR-dependent, but AR-ligand binding-independent event. Because ligand-bound AR induces genomic transcription, DHT-induced AKT phosphorylation is likely to be a nongenomic mechanism. Supporting this finding, it has also been demonstrated that androgens stimulate the formation of a triple complex of AR- p85α-Src to activate the PI3-kinase/AKT pathway without the involvement of the ligand binding domain of AR (59). In the study, androgen-induced PI3-kinase and AKT kinase activities were detected in HEK293 cells transfected with mutant AR lacking the C terminus where the ligand binding domain is situated. However, the activities were completely abolished in HEK293 transfected with mutant AR lacking a proline-rich region in the N terminus. Taken together, the results of the current study demonstrate that both genomic AR function and AR-ligand binding-independent, nongenomic PI3-kinase activation are collectively necessary for DHT-mediated VEGF production.
In contrast to young fibroblasts, we found that fibroblasts from old men failed to activate the PI3-kinase pathway in response to androgens, lacking DHT-mediated enhancement in AKT production and phosphorylation. Hence, a loss of PI3-kinase activation by androgens in aged cells may contribute to the impairment in androgen-mediated enhancement of VEGF production with aging. Because fibroblasts from old men also lacked PI3-kinase activation upon stimulation with insulin (Supplemental Figure 6), the inability to activate the PI3-kinase/AKT pathway in aged cells is likely to be a general impairment. Similarly, a failure in AKT activation with increased age has been observed in hepatocytes after exposure to oxidative stress (60). Hydrogen peroxide exposure significantly increased the phosphorylation of AKT in hepatocytes isolated from young rats, but only minimally in hepatocytes from old rats, leading to decreased ability for old cells to survive oxidative stress. In addition, a study performed in young mice that underwent partial pancreatectomy demonstrated that the remaining pancreatic acini increased AKT phosphorylation and cell proliferation necessary for pancreatic regeneration, a phenomenon that was blocked by the PI3-kinase inhibitor LY294002. By contrast, old mice that underwent the same procedure displayed markedly attenuated AKT phosphorylation and pancreatic regeneration (61). Furthermore, in rats subjected to carotid artery balloon injury, young adult rats showed increased AKT phosphorylation and smooth muscle cell proliferation, whereas AKT phosphorylation was barely present in carotid arteries from old animals, a finding associated with reduced cell proliferation (62). Together with the results of the current study, these findings are consistent with a general age-related impairment in PI3-kinase activation and subsequent AKT phosphorylation in response to a variety of stimuli.
We sought to investigate whether the age-related impairment in androgen-induced PI3-kinase activation was a result of aging alone or as a result of increased androgen insensitivity with age. We therefore compared the transcriptional function, expression, and cellular distribution of AR in response to androgen treatment in young and old cells. The current study found that, unlike cells from young men, cells from old men treated with DHT were unable to increase AR transcriptional activity. To adequately function as a transcription factor, ligand-bound AR must translocate to the cell nucleus to interact with the hormone response element directly, a process that involves the recruitment of coactivators, corepressors, and other transcription factors (19, 20). The results from the current study demonstrate that young fibroblasts effectively increase AR translocation to the nucleus in response to androgen exposure. In contrast, cells from old men were unresponsive to DHT treatment, failing to translocate AR to the nucleus in response to androgens. Given that the baseline AR mRNA in old cells is greatly reduced compared with young, it is not unexpected that AR protein did not increase in old fibroblasts treated with DHT. However, reduced AR mRNA in old cells did not translate into a reduced level of AR protein, likely due to slower protein turnover in aged cells (63). It is particularly interesting that old male fibroblasts were unable to translocate existing AR to the nucleus with DHT treatment and hence are unable to initiate AR-mediated transcriptional activity. These latter findings suggest an age-related impairment in the nuclear translocation of AR.
Nuclear translocation of ligand-bound AR is a complex multifactorial process (64, 65). AR has at least 16 phosphorylation sites that regulate protein stabilization, activation, and subcellular localization (64, 65), although the exact mechanistic processes attributed to each of these phosphorylation sites are largely unknown and vary greatly among cell types and in the presence of androgens. More well-known are the roles of chaperone proteins in AR stabilization and nuclear translocation. Unbound AR remains in the cytoplasm bound to heat-shock proteins (HSPs) HSP70, HSP90, and HSP56, which act to stabilize AR, and androgen binding causes HSP dissociation (66, 67). Also upon ligand binding, AR induces the phosphorylation of HSP27 by p38 and AKT, which results in AR nuclear translocation chaperoned by HSP27 (68). Knockdown or inhibition of the phosphorylation of HSP27 inhibited AR nuclear translocation, leading to AR degradation. Additionally, PI3-kinase inhibition leads to decreased AR protein expression in androgen-sensitive LNCaP and LAPC-4 cells, and constitutively active myr-Akt1 increased AR mRNA and protein levels, demonstrating that PI3-kinase activation is intertwined with the regulation of AR levels (69). Therefore, the inability of cells from old men to activate the PI3-kinase pathway may contribute to reduced AR mRNA with age.
Despite much research focusing on the decline of serum testosterone with aging, very few studies have focused on tissue changes in AR level or function with age. AR mRNA and protein expression declines in the liver of old rats when compared with young adult rats, and interestingly, caloric restriction inhibits the age-related decline in AR mRNA (70, 71). Similar to the current study, investigations into AR sensitivity in the brain of rats and mice have suggested that despite adequate AR expression and AR-positive cells in the brain, old animals are less responsive to testosterone administration and replacement. Testosterone replacement in gonadectomized young males demonstrated a greater increase in nuclear AR in brain cells than in old males, despite comparable cytosolic AR and serum testosterone concentration (72). Copulatory activity and circulating testosterone levels decline with age, but sexual activity cannot be restored by exogenous testosterone in aged male rats (73, 74). Furthermore, the prevention of circulating testosterone decline with age via long-term (6 month) testosterone administration could not prevent the age-related decline in copulatory activity. Together with the current study, these results suggest that male aging is associated with reduced responsiveness to androgen exposure in some tissues as a result of age-related changes in expression and nuclear translocation of the AR. Hence in the age-related decline in testosterone-mediated processes, a decline in androgen responsiveness due to AR insensitivity with increased age may be at least as significant as any decline in circulating testosterone levels.
In conclusion, this study demonstrates that androgens regulate angiogenesis in a paracrine manner by AR-dependent stimulation of VEGF secretion. Aging is associated with marked impairment of DHT-mediated regulation of VEGF-dependent angiogenesis, a finding characterized by androgen insensitivity with age-related defective nuclear translocation of the AR with androgen exposure. The findings in this study suggest that age-related androgen insensitivity, due to reduced AR responsiveness, may subsequently diminish androgen modulation of paracrine-mediated angiogenesis.
Additional material
Supplementary data supplied by authors.
Acknowledgments
We acknowledge Dr Louise Cole (Core Facilities Manager, Bosch Institute Advanced Microscopy Facility, The University of Sydney) for her support and assistance. We also thank those from the Melanoma Institute Australia Bio-specimen Bank for Melanoma Research, specifically John Thompson, Robyn Saw, Andrew Spillane, Jonathan Stretch, Richard Scolyer, Chitra De Silva, Jessica Hyman, Benafsha Yosufi, and Valerie Jakrot, for their assistance with the collection of human dermal fibroblasts.
This study was supported by National Health and Medical Research Council 1011111 (to M.K.C.N.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AR
- androgen receptor
- CM
- conditioned medium
- csFBS
- charcoal-stripped FBS
- DAPI
- 4′,6-diamidino-2-phenylindole
- EC
- endothelial cell
- FBS
- fetal bovine serum
- FGF
- fibroblast growth factor
- FITC
- fluorescein isothiocyanate
- HF
- hydroxyflutamide
- HSP
- heat-shock protein
- HUVEC
- human umbilical vein EC
- pAKT
- phosphorylated AKT
- PDGF
- platelet-derived growth factor
- PI3-kinase
- phosphoinositide 3-kinase
- rhVEGF145
- recombinant human VEGF145
- SDS
- sodium dodecyl sulfate
- siRNA
- small interfering RNA
- TBS
- Tris-buffered saline
- VEGF
- vascular endothelial growth factor.
References
- 1. Reed MJ, Edelberg JM. Impaired angiogenesis in the aged. Sci Aging Knowl Environ. 2004;2004: pe7. [DOI] [PubMed] [Google Scholar]
- 2. Ballard VL, Edelberg JM. Stem cells and the regeneration of the aging cardiovascular system. Circ Res. 2007;100:1116–1127. [DOI] [PubMed] [Google Scholar]
- 3. Lähteenvuo J, Rosenzweig A. Effects of aging on angiogenesis. Circ Res. 2012;110:1252–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nicosia RF, Zorzi P, Ligresti G, Morishita A, Aplin AC. Paracrine regulation of angiogenesis by different cell types in the aorta ring model. Int J Dev Biol. 2011;55:447–453. [DOI] [PubMed] [Google Scholar]
- 5. Gennaro G, Ménard C, Michaud SE, Rivard A. Age-dependent impairment of reendothelialization after arterial injury: role of vascular endothelial growth factor. Circulation. 2003;107:230–233. [DOI] [PubMed] [Google Scholar]
- 6. Rivard A, Fabre JE, Silver M, et al. Age-dependent impairment of angiogenesis. Circulation. 1999;99:111–120. [DOI] [PubMed] [Google Scholar]
- 7. Ahluwalia A, Narula J, Jones MK, Deng X, Tarnawski AS. Impaired angiogenesis in aging myocardial microvascular endothelial cells is associated with reduced importin alpha and decreased nuclear transport of HIF1 alpha: mechanistic implications. J Physiol Pharmacol. 2010;61:133–139. [PubMed] [Google Scholar]
- 8. Sadoun E, Reed MJ. Impaired angiogenesis in aging is associated with alterations in vessel density, matrix composition, inflammatory response, and growth factor expression. J Histochem Cytochem. 2003;51:1119–1130. [DOI] [PubMed] [Google Scholar]
- 9. Swift ME, Kleinman HK, DiPietro LA. Impaired wound repair and delayed angiogenesis in aged mice. Lab Invest. 1999;79:1479–1487. [PubMed] [Google Scholar]
- 10. Rivard A, Berthou-Soulie L, Principe N, et al. Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J Biol Chem. 2000;275:29643–29647. [DOI] [PubMed] [Google Scholar]
- 11. Wang H, Keiser JA, Olszewski B, et al. Delayed angiogenesis in aging rats and therapeutic effect of adenoviral gene transfer of VEGF. Int J Mol Med. 2004;13:581–587. [PubMed] [Google Scholar]
- 12. Yeap BB, Alfonso H, Chubb SA, et al. Reference ranges and determinants of testosterone, dihydrotestosterone, and estradiol levels measured using liquid chromatography-tandem mass spectrometry in a population-based cohort of older men. J Clin Endocrinol Metab. 2012;97:4030–4039. [DOI] [PubMed] [Google Scholar]
- 13. Liu PY, Death AK, Handelsman DJ. Androgens and cardiovascular disease. Endocr Rev. 2003;24:313–340. [DOI] [PubMed] [Google Scholar]
- 14. English KM, Mandour O, Steeds RP, Diver MJ, Jones TH, Channer KS. Men with coronary artery disease have lower levels of androgens than men with normal coronary angiograms. Eur Heart J. 2000;21:890–894. [DOI] [PubMed] [Google Scholar]
- 15. Araujo AB, Dixon JM, Suarez EA, Murad MH, Guey LT, Wittert GA. Clinical review: Endogenous testosterone and mortality in men: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011;96:3007–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khaw KT, Dowsett M, Folkerd E, et al. Endogenous testosterone and mortality due to all causes, cardiovascular disease, and cancer in men: European prospective investigation into cancer in Norfolk (EPIC-Norfolk) Prospective Population Study. Circulation. 2007;116:2694–2701. [DOI] [PubMed] [Google Scholar]
- 17. Hyde Z, Norman PE, Flicker L, et al. Low free testosterone predicts mortality from cardiovascular disease but not other causes: the Health in Men Study. J Clin Endocrinol Metab. 2012;97:179–189. [DOI] [PubMed] [Google Scholar]
- 18. Levine GN, D'Amico AV, Berger P, et al. Androgen-deprivation therapy in prostate cancer and cardiovascular risk: a science advisory from the American Heart Association, American Cancer Society, and American Urological Association: endorsed by the American Society for Radiation Oncology. Circulation. 2010;121:833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–3015. [DOI] [PubMed] [Google Scholar]
- 20. Kemppainen JA, Lane MV, Sar M, Wilson EM. Androgen receptor phosphorylation, turnover, nuclear transport, and transcriptional activation. Specificity for steroids and antihormones. J Biol Chem. 1992;267:968–974. [PubMed] [Google Scholar]
- 21. Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995;270:7341–7346. [DOI] [PubMed] [Google Scholar]
- 22. Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. [DOI] [PubMed] [Google Scholar]
- 23. Death AK, McGrath KC, Sader MA, et al. Dihydrotestosterone promotes vascular cell adhesion molecule-1 expression in male human endothelial cells via a nuclear factor-κB-dependent pathway. Endocrinology. 2004;145:1889–1897. [DOI] [PubMed] [Google Scholar]
- 24. Ng MK, Quinn CM, McCrohon JA, et al. Androgens up-regulate atherosclerosis-related genes in macrophages from males but not females: molecular insights into gender differences in atherosclerosis. J Am Coll Cardiol. 2003;42:1306–1313. [DOI] [PubMed] [Google Scholar]
- 25. Sordello S, Bertrand N, Plouët J. Vascular endothelial growth factor is up-regulated in vitro and in vivo by androgens. Biochem Biophys Res Commun. 1998;251:287–290. [DOI] [PubMed] [Google Scholar]
- 26. Levine AC, Liu XH, Greenberg PD, et al. Androgens induce the expression of vascular endothelial growth factor in human fetal prostatic fibroblasts. Endocrinology. 1998;139:4672–4678. [DOI] [PubMed] [Google Scholar]
- 27. Sieveking DP, Lim P, Chow RW, et al. A sex-specific role for androgens in angiogenesis. J Exp Med. 2010;207:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joseph IB, Nelson JB, Denmeade SR, Isaacs JT. Androgens regulate vascular endothelial growth factor content in normal and malignant prostatic tissue. Clin Cancer Res. 1997;3:2507–2511. [PubMed] [Google Scholar]
- 29. Cheng L, Zhang S, Sweeney CJ, Kao C, Gardner TA, Eble JN. Androgen withdrawal inhibits tumor growth and is associated with decrease in angiogenesis and VEGF expression in androgen-independent CWR22Rv1 human prostate cancer model. Anticancer Res. 2004;24:2135–2140. [PubMed] [Google Scholar]
- 30. Chang EI, Loh SA, Ceradini DJ, et al. Age decreases endothelial progenitor cell recruitment through decreases in hypoxia-inducible factor 1alpha stabilization during ischemia. Circulation. 2007;116:2818–2829. [DOI] [PubMed] [Google Scholar]
- 31. Jiang H, Fan D, Zhou G, Li X, Deng H. Phosphatidylinositol 3-kinase inhibitor(LY294002) induces apoptosis of human nasopharyngeal carcinoma in vitro and in vivo. J Exp Clin Cancer Res. 2010;29:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Conte E, Fruciano M, Fagone E, et al. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: the role of class I P110 isoforms. PLoS ONE. 2011;6:e24663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Newman AC, Nakatsu MN, Chou W, Gershon PD, Hughes CC. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol Biol Cell. 2011;22:3791–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Compagno D, Merle C, Morin A, et al. SIRNA-directed in vivo silencing of androgen receptor inhibits the growth of castration-resistant prostate carcinomas. PLoS One. 2007;2:e1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zygalaki E, Stathopoulou A, Kroupis C, et al. Real-time reverse transcription-PCR quantification of vascular endothelial growth factor splice variants. Clin Chem. 2005;51:1518–1520. [DOI] [PubMed] [Google Scholar]
- 36. Pagès G, Pouyssegur J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene–a concert of activating factors. Cardiovasc Res. 2005;65:564–573. [DOI] [PubMed] [Google Scholar]
- 37. Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J Biol Chem. 2001;276:30392–30398. [DOI] [PubMed] [Google Scholar]
- 38. Sieveking DP, Chow RW, Ng MK. Androgens, angiogenesis and cardiovascular regeneration. Curr Opin Endocrinol Diabetes Obes. 2010;17:277–283. [DOI] [PubMed] [Google Scholar]
- 39. Berthod F, Germain L, Tremblay N, Auger FA. Extracellular matrix deposition by fibroblasts is necessary to promote capillary-like tube formation in vitro. J Cell Physiol. 2006;207:491–498. [DOI] [PubMed] [Google Scholar]
- 40. Kellouche S, Mourah S, Bonnefoy A, et al. Platelets, thrombospondin-1 and human dermal fibroblasts cooperate for stimulation of endothelial cell tubulogenesis through VEGF and PAI-1 regulation. Exp Cell Res. 2007;313:486–499. [DOI] [PubMed] [Google Scholar]
- 41. Martin TA, Harding KG, Jiang WG. Regulation of angiogenesis and endothelial cell motility by matrix-bound fibroblasts. Angiogenesis. 1999;3:69–76. [DOI] [PubMed] [Google Scholar]
- 42. Tischer E, Mitchell R, Hartman T, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- 43. Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol. 1991;5:1806–1814. [DOI] [PubMed] [Google Scholar]
- 44. Charnock-Jones DS, Sharkey AM, Rajput-Williams J, et al. Identification and localization of alternately spliced mRNAs for vascular endothelial growth factor in human uterus and estrogen regulation in endometrial carcinoma cell lines. Biol Reprod. 1993;48:1120–1128. [DOI] [PubMed] [Google Scholar]
- 45. Cheung CY, Singh M, Ebaugh MJ, Brace RA. Vascular endothelial growth factor gene expression in ovine placenta and fetal membranes. Am J Obstet Gynecol. 1995;173:753–759. [DOI] [PubMed] [Google Scholar]
- 46. Poltorak Z, Cohen T, Sivan R, et al. VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J Biol Chem. 1997;272:7151–7158. [DOI] [PubMed] [Google Scholar]
- 47. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 48. Krüssel JS, Behr B, Milki AA, et al. Vascular endothelial growth factor (VEGF) mRNA splice variants are differentially expressed in human blastocysts. Mol Hum Reprod. 2001;7:57–63. [DOI] [PubMed] [Google Scholar]
- 49. Benderro GF, LaManna JC. Kidney EPO expression during chronic hypoxia in aged mice. Adv Exp Med Biol. 2013;765:9–14. [DOI] [PubMed] [Google Scholar]
- 50. Franceschi C, Bonafè M, Valensin S, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. [DOI] [PubMed] [Google Scholar]
- 51. Salminen A, Huuskonen J, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev. 2008;7:83–105. [DOI] [PubMed] [Google Scholar]
- 52. Mor F, Quintana FJ, Cohen IR. Angiogenesis-inflammation cross-talk: vascular endothelial growth factor is secreted by activated T cells and induces Th1 polarization. J Immunol. 2004;172:4618–4623. [DOI] [PubMed] [Google Scholar]
- 53. Ono M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008;99:1501–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cai J, Hong Y, Weng C, Tan C, Imperato-McGinley J, Zhu YS. Androgen stimulates endothelial cell proliferation via an androgen receptor/VEGF/cyclin A-mediated mechanism. Am J Physiol Heart Circ Physiol. 2011;300:H1210–H1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stern JM, Chen J, Peters SB, et al. Testosterone treatment of human foreskin in a novel transplant model. Urology. 2004;63:999–1003. [DOI] [PubMed] [Google Scholar]
- 56. Wang D, Huang HJ, Kazlauskas A, Cavenee WK. Induction of vascular endothelial growth factor expression in endothelial cells by platelet-derived growth factor through the activation of phosphatidylinositol 3-kinase. Cancer Res. 1999;59:1464–1472. [PubMed] [Google Scholar]
- 57. Jiang BH, Zheng JZ, Aoki M, Vogt PK. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci U S A. 2000;97:1749–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Maeda T, Kawane T, Horiuchi N. Statins augment vascular endothelial growth factor expression in osteoblastic cells via inhibition of protein prenylation. Endocrinology. 2003;144:681–692. [DOI] [PubMed] [Google Scholar]
- 59. Sun M, Yang L, Feldman RI, et al. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J Biol Chem. 2003;278:42992–43000. [DOI] [PubMed] [Google Scholar]
- 60. Ikeyama S, Kokkonen G, Shack S, Wang XT, Holbrook NJ. Loss in oxidative stress tolerance with aging linked to reduced extracellular signal-regulated kinase and Akt kinase activities. FASEB J. 2002;16:114–116. [DOI] [PubMed] [Google Scholar]
- 61. Watanabe H, Saito H, Rychahou PG, Uchida T, Evers BM. Aging is associated with decreased pancreatic acinar cell regeneration and phosphatidylinositol 3-kinase/Akt activation. Gastroenterology. 2005;128:1391–1404. [DOI] [PubMed] [Google Scholar]
- 62. Torella D, Leosco D, Indolfi C, et al. Aging exacerbates negative remodeling and impairs endothelial regeneration after balloon injury. Am J Physiol Heart Circ Physiol. 2004;287:H2850–H2860. [DOI] [PubMed] [Google Scholar]
- 63. Rattan SI. Synthesis, modifications, and turnover of proteins during aging. Exp Gerontol. 1996;31:33–47. [DOI] [PubMed] [Google Scholar]
- 64. van der Steen T, Tindall DJ, Huang H. Posttranslational modification of the androgen receptor in prostate cancer. Int J Mol Sci. 2013;14:14833–14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gioeli D, Paschal BM. Post-translational modification of the androgen receptor. Mol Cell Endocrinol. 2012;352:70–78. [DOI] [PubMed] [Google Scholar]
- 66. Veldscholte J, Berrevoets CA, Zegers ND, van der Kwast TH, Grootegoed JA, Mulder E. Hormone-induced dissociation of the androgen receptor-heat-shock protein complex: use of a new monoclonal antibody to distinguish transformed from nontransformed receptors. Biochemistry. 1992;31:7422–7430. [DOI] [PubMed] [Google Scholar]
- 67. Pratt WB. Interaction of hsp90 with steroid receptors: organizing some diverse observations and presenting the newest concepts. Mol Cell Endocrinol. 1990;74:C69–C76. [DOI] [PubMed] [Google Scholar]
- 68. Zoubeidi A, Zardan A, Beraldi E, et al. Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res. 2007;67:10455–10465. [DOI] [PubMed] [Google Scholar]
- 69. Ha S, Ruoff R, Kahoud N, Franke TF, Logan SK. Androgen receptor levels are upregulated by Akt in prostate cancer. Endocr Relat Cancer. 2011;18:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shi L, Ko S, Kim S, et al. Loss of androgen receptor in aging and oxidative stress through Myb protooncoprotein-regulated reciprocal chromatin dynamics of p53 and poly(ADP-ribose) polymerase PARP-1. J Biol Chem. 2008;283:36474–36485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Song CS, Rao TR, Demyan WF, Mancini MA, Chatterjee B, Roy AK. Androgen receptor messenger ribonucleic acid (mRNA) in the rat liver: changes in mRNA levels during maturation, aging, and calorie restriction. Endocrinology. 1991;128:349–356. [DOI] [PubMed] [Google Scholar]
- 72. Chambers KC, Thornton JE, Roselli CE. Age-related deficits in brain androgen binding and metabolism, testosterone, and sexual behavior of male rats. Neurobiol Aging. 1991;12:123–130. [DOI] [PubMed] [Google Scholar]
- 73. Hsu HK, Hsu C, Yu JY, Peng MT. Effects of long-term testosterone replacement on copulatory activity in old male rats. Gerontology. 1986;32:10–17. [DOI] [PubMed] [Google Scholar]
- 74. Taylor G, Bardgett M, Farr S, Humphrey W, Womack S, Weiss J. Aging of the brain-testicular axis: reproductive systems of healthy old male rats with or without endocrine stimulation. Proc Soc Exp Biol Med. 1996;211:69–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.