

Abstract
Carotid intima formation is a significant risk factor for cardiovascular disease. C3H/FeJ (C3H/F) and SJL/J (SJL) inbred mouse strains differ in susceptibility to immune and vascular traits. Using a congenic approach we demonstrated that the Intima modifier 2 (Im2) locus on chromosome 11 regulates leukocyte infiltration. We sought to determine whether inflammation was due to changes in circulating immune cells or activation of vascular wall cells in genetically pure Im2 (C3H/F.SJL.11.1) mice. Complete blood counts showed no differences in circulating monocytes between C3H/F and C3H/F.SJL.11.1 compared with SJL mice. Aortic vascular cell adhesion molecule-1 (VCAM-1) total protein levels were dramatically increased in SJL and C3H/F.SJL.11.1 compared with C3H/F mice. Immunostaining of aortic endothelial cells (EC) showed a significant increase in VCAM-1 expression in SJL and C3H/F.SJL.11.1 compared with C3H/F under steady flow conditions. Immunostaining of EC membranes revealed a significant decrease in EC size in SJL and C3H/F.SJL.11.1 vs. C3H/F in regions of disturbed flow. Vascular permeability was significantly higher in C3H/F.SJL.11.1 compared with C3H/F. Our results indicate that Im2 regulation of leukocyte infiltration is mediated by EC inflammation and permeability. RNA sequencing and pathway analyses comparing genes in the Im2 locus to C3H/F provide insight into candidate genes that regulate vascular wall inflammation and permeability highlighting important genetic mechanisms that control vascular intima in response to injury.
Keywords: SJL, C3Heb/Fe, congenics, inflammation, endothelial cell, permeability
alterations in blood flow result in vessel wall remodeling as a compensatory mechanism to maintain hemodynamic homeostasis (27, 29, 30, 38), which presents as intima-media thickening (IMT) in humans (36). Carotid IMT is a significant risk factor for cardiovascular disease including atherosclerosis, myocardial infarction, stroke, and peripheral vascular disease (2, 41). In humans IMT is largely governed by genetic factors (6, 32, 37). Empirical evidence using genome-wide association studies and quantitative trait locus (QTL) analyses showed that subclinical cardiovascular disease has a significant genetic component and that IMT is a highly predictive clinical measurement (6, 32, 37). Intimal thickening is governed by genetic susceptibility as demonstrated by animal models (16, 28, 39, 53). We identified three QTLs for intimal thickening in a cross of C3HeB/FeJ (C3H/F) and SJL/J (SJL) mice. These intima modifier (Im) loci are on chromosomes (chrs) 2, 11, and 18, termed Im1, Im2, and Im3, respectively (28). We utilized this genetic cross and found a QTL for carotid inflammation on chr11 that overlaps with the Im2 locus (42). We created several congenic mouse lines for the Im2 locus and showed that C3H/F.SJL.11.1 mice exhibited leukocyte (CD45+) infiltration into the vascular wall similar to SJL mice in response to low blood flow (42). Inflammation associated with intimal thickening can be a result of either alterations in circulating immune cells or dysfunction in endothelial cells (EC). We reported that SJL mice have dysfunction in endothelium-dependent aortic vasorelaxation compared with C3H/F (8), while others found more circulating white blood cells (WBC) in SJL compared with C3H/F (26). In the current study we investigated whether the increased inflammation in the Im2 locus was due to variations in circulating cells or if the CD45+ trait was a result of increased inflammation in resident vascular wall cells.
MATERIALS AND METHODS
Animals
For all in vivo experiments male mice were 8–12 wk of age. Mice were anesthetized with a mixture of ketamine (130 mg/kg), xylazine (8.8 mg/kg), and saline or isoflurane. The University of Rochester Animal Care Committee approved all animal procedures, which were conducted according to guidelines of the National Institutes of Health and American Heart Association for the Use of Laboratory Animals.
Congenic Single Nucleotide Polymorphism Mapping
Introgression of the chr11 Im2 locus from SJL into the C3H/F background to create C3H/F.SJL.11.1 congenic mice was previously published (42). We further genotyped mice by whole genome analysis using Jackson Laboratory Mouse Diversity Panel and single nucleotide polymorphism (SNP) genotyping on an Affymetrix Platform. There were 103,761 polymorphic SNPs between SJL and C3H/F. There is a polymorphic SNP between the strains at every ∼5 kb. This provides a 10,000-fold greater resolution of the locus compared with our previously published analysis using microsatellite markers, which spans ∼50 Mb per marker (28).
Complete Blood Counts
Mice were anesthetized, and blood was drawn via retro-orbital eye bleeds with heparinized capillary tubes. Blood was collected in EDTA tubes, and hematological parameters were measured (16 parameters in total) using a fully automated five-part differential cell counter (VetScan HM5, Abaxis).
Western Blotting for Vascular Cell Adhesion Molecule-1
Mice were anesthetized and perfused with heparinized saline. Aortas were cleaned of fat and snap-frozen in liquid nitrogen. Tissues were sonicated on ice in cell lysis buffer (1×, Cell Signaling) with protease inhibitor (1:1,000, Sigma) for 3–5 s bursts three times. Homogenates were centrifuged at 12,000 rpm for 10 min at 4°C. Bradford protein assay (Bio-Rad) was performed, and equal amounts of protein were separated on 8% sodium dodecyl sulfate gels. Proteins were transferred to nitrocellulose membrane and blocked in 5% milk-phosphate buffered saline (PBS) for 1 h at room temperature followed by overnight incubation in 2% BSA/PBS/Tween containing vascular cell adhesion molecule-1 (VCAM-1) (1:1,000, H-276, Santa Cruz Biotechnology) and glyceraldehyde phosphate dehydrogenase (1:1,000, Cell Signaling). Blots were washed in PBS/Tween and incubated with secondary horseradish peroxidase-conjugated rabbit antibodies for 2 h at room temperature. Protein was visualized by enhanced chemiluminescence.
En Face VCAM-1 and Cell Size Staining and Quantification
Mice were anesthetized and perfusion fixed with heparinized saline and then with 10% neutral buffered formalin (NBF) to preserve vessel morphology. An incision was made ventrally, and the aorta was removed postfixation and allowed to continue to fix in 10% NBF overnight. After adipose tissue was removed, aortas were cut longitudinally and permeabilized with PBS containing 0.1% Triton X-100 and blocked by Tris-buffered saline containing 10% goat serum and 2.5% Tween 20 for 30 min. Aortas were incubated with 10 μg/ml rabbit anti-VCAM-1 (H-276, Santa Cruz Biotechnology). Rabbit IgG as a control and either 10 μg/ml rat anti-CD31 or 5 μg/ml of anti-CD144 (both EC membrane markers, BD Biosciences) were used in the blocking solution overnight at 4°C. After PBS wash, anti-rabbit IgG and anti-rat IgG (1:1,000 dilution, Alexa Fluor 546 and 488, respectively; Invitrogen) were applied for 1 h at room temperature.
Images were acquired under a confocal laser-scanning microscope (Fluoview 300, Olympus) equipped with krypton-argon-HeNe laser lines and 20 × 0.70 numerical aperture (NA), ×40 1.0 NA, and ×60 1.4 NA objectives. Images in figures were captured with the ×40 objective. For quantification, 10–15 optical sections were collected at 0.3–0.5 μm increments so that Z stacks of ∼4 μm thick cell blocks from the luminal surface were obtained. Images were collected with the same confocal settings. A maximum intensity Z projection was made. The channels were then split and analyzed in the following manner.
VCAM-1.
Images from areas of the aorta exposed to steady flow were thresholded, and an average threshold was calculated as previously published (7). This average threshold was then applied to all images, and a binarized “mask” created for each image. This mask was multiplied by the original image to remove areas below threshold, and the intensity of areas above threshold only was calculated and recorded. Furthermore, as the mask corresponds to the total VCAM-1-positive (above threshold) area in the image, this value was also recorded.
CD31 (cell size).
An existing ImageJ program used to quantify vascular networks (http://rsb.info.nih.gov/) was modified. In brief, the program segments the plasma membrane fluorescent image, resulting in a binary image. This binary image is then reduced to the minimal structural level (skeletonization). Areas inside closed loops (continuous cell membrane) of this skeletonized network (areas corresponding to the interior of cells) are calculated.
Miles Assay for Vascular Permeability
Mice were anesthetized and injected with 100 μl of 1% Evans blue dye (Sigma) via tail vein. After 30 min, mice were perfused with heparinized saline followed by 10% NBF for 5 min. Aortas were immediately harvested, placed in 10% NBF, dried, and weighed. Evans blue dye was extracted from the aortas by placing the vessels in 200 μl formamide (Amresco) at 55°C for 24 h. Samples were centrifuged at 12,000 rpm for 10 min. Optical density (OD) was obtained with a FLUOstar OPTIMA (BMG Labtech) plate reader at 620 nm. Sample ODs were extrapolated to a linearized standard and converted to mg/μl and normalized to weight of the dried aorta.
RNA Sequencing
Mice were saline perfused, and carotid arteries were harvested and cleaned of all connective tissue with sterile autoclaved instruments. Left and right carotids from each animal were pooled together and placed into RNase-free tubes and immediately snap-frozen in liquid nitrogen and kept on dry ice until RNA isolation was performed. RNA isolation and sequencing (RNA-Seq) was performed by the Genomics Research Center (GRC) at the University of Rochester Medical Center. Total RNA was isolated using the RNeasy Plus Kit (Qiagen, Valencia, CA) per manufacturer's recommendations. Residual genomic DNA was removed from purified RNA with TURBO DNASE (Life Technologies, Grand Island, NY). In brief, total RNA was diluted in 1× TURBO DNase buffer along with 1 μl of the TURBO DNase enzyme in a final volume of 50 μl. The reaction was incubated at 37°C for 30 min followed by DNase inactivation with addition of 0.1 μl volume of DNase inactivation reagent. The inactivation was carried out for 5 min at room temperature (22–26°C). Total RNA quantity was determined with the NanoDrop 1000 spectrophotometer (NanoDrop, Wilmington, DE), and RNA quality was assessed with the Agilent Bioanalyzer (Agilent, Santa Clara, CA). Illumina compatible library construction was performed using the TruSeq Stranded mRNA Sample Preparation Kit (Illumina, San Diego, CA) per manufacturer's protocols. In brief, mRNA was purified from 100 ng total RNA with oligo-dT magnetic beads and chemically fragmented. First-stand cDNA synthesis was performed by random hexamer priming followed by second-strand cDNA synthesis with dUTP. End repair and 3′ adenylation were performed on the double-stranded cDNA. Illumina adaptors were ligated to both ends of the cDNA, purified by gel electrophoresis, and amplified with PCR primers specific to the adaptor sequences to generate amplicons of ∼200–500 bp in size. The amplified libraries were hybridized to the Illumina single end flow cell and amplified using the cBot (Illumina) at a concentration of 8 pM per lane. Single end reads of 100 nucleotides were generated for each sample and aligned to the organism-specific reference genome.
Differential Expression Analysis
Sequenced reads were cleaned according to a rigorous preprocessing workflow: Trimmomatic-0.32 (http://www.usadellab.org/cms/?page=trimmomatic). The Trimmomatic parameters used were SLIDINGWINDOW:4:20 TRAILING:13 LEADING:13 ILLUMINACLIP:trimmomatic_adapters.fasta:2:30:10 MINLEN:25. We then mapped reads to the Mus musculus reference genome with SHRiMP2.2.3 (http://compbio.cs.toronto.edu/shrimp) followed by analysis using software cufflinks2.0.2 [cuffdiff2 - Running Cuffdiff (http://cufflinks.cbcb.umd.edu/manual.html#cuffdiff)] to perform differential expression analysis with an false discovery rate (FDR) cutoff of 0.05 (95% confidence interval and additional parameters ′-u -b′). A Perl script was used after differential expression analysis to make the result files more human readable. We selected differentially expressed genes based genomic position between the following groups: C3H/F.SJL.11.1 Im2 locus vs. C3H/F and C3H/F.SJL.11.1 outside the locus vs. C3H/F. Two group Student's t-test was used for this analysis. Benjamini-Hochberg procedure (4) was used to control the FDR at 0.05 level.
Gene Ontology Pathway Analysis
To identify how genes within the Im2 locus affect biological pathways we performed Gene Ontology (GO) analyses on the differentially expressed genes identified with RNA-Seq from C3H/F.SJL.11.1 and C3H/F. In the differential expression analysis we identified 65 differentially expressed genes for the comparison between C3H/F.SJL.11.1 and C3H/F out of 1,746 genes on chr11. We performed GO pathway analysis to better understand the collective effect of these differentially expressed genes. The hyperGTest function from the BioConductor package GOstats (14) was used to find GO categories that are significantly overrepresented in these differentially expressed genes compared with all genes on chr11. The significance level used in this analysis was P value < 0.001.
Statistical Analysis
Data are presented as means ± SE. Statistical analysis for en face quantification was done as described above. We used JMP5.1.2 software for evaluation of statistically significant changes. Differences between groups were analyzed by one-way ANOVA. We followed up on these analyses with post hoc comparisons of all means. The level of P < 0.05 was regarded as significant.
RESULTS
Whole Gnome SNP Mapping of C3H/F.SJL.11.1 Congenic Line
We previously published that the Im2 locus on chr11 regulates inflammation in response to low blood flow (42). We generated the C3H/F.SJL.11.1 congenic with conventional microsatellite marker-assisted genotyping (42). In the current study we performed whole genome dense SNP genotyping at Jackson Laboratories to confidently assure introgression of the Im2 locus from SJL into the C3H/F background. Figure 1 confirms that the C3H/F.SJL.11.1 congenic mouse line only contains genetic material from SJL within the Im2 region. Increased peak height indicates SJL (donor) SNP detection along the chrs (x-axes). There are no detectable peaks above background (C3H/F, recipient) for the Im1 locus on chr2 (Fig. 1A). However, as expected there are increased peaks along chr11 only between 17 and 63 Mb, which covers the Im2 locus (Fig. 1B). Along chr18 (Im3) there are no detectable donor SNPs above background (Fig. 1C). We also confirmed that there are no other SJL genomic regions introgressed throughout the C3H/F genome (data not shown). In summary, we confirmed the purity of our C3H/F.SJL.11.1 congenic and can, with confidence, deduce that any observed phenotypic differences in the C3H/F.SJL.11.1 mouse relative to C3H/F are due solely to the SJL contribution of the chr11 Im2 locus.
Fig. 1.
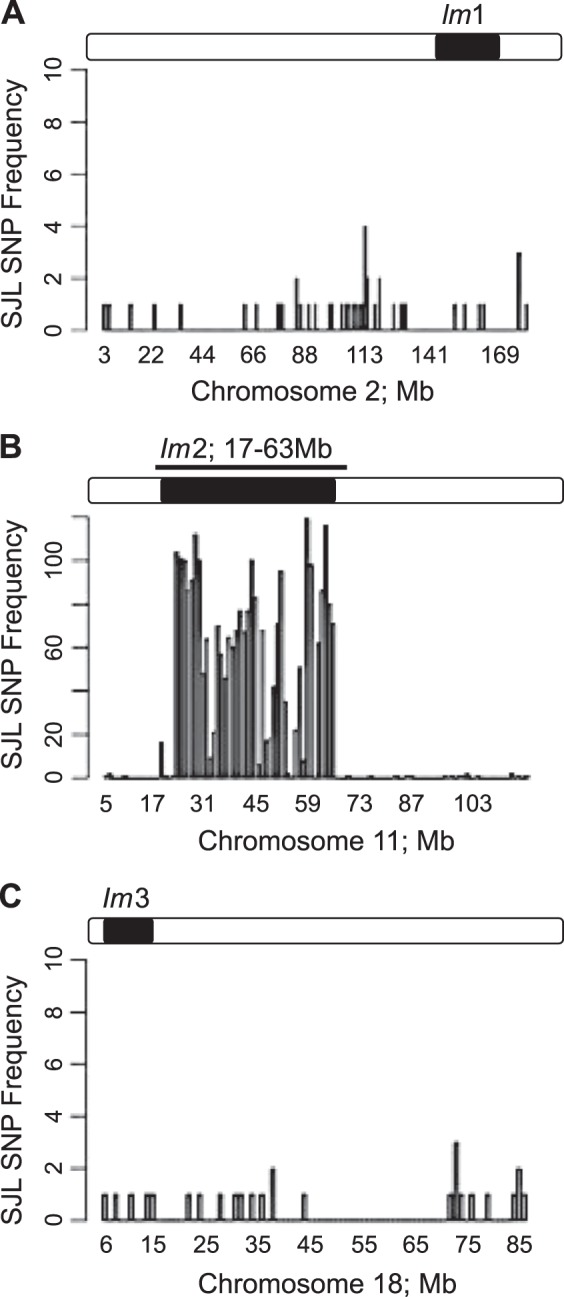
Dense single nucleotide polymorphism (SNP) mapping of C3H/F.SJL.11.1 congenic mouse. There were no detectable SJL SNPs expressed above background for chromosome 2 (A) and chromosome 18 (C). Chromosome 11 expressed SJL SNPs only in the region of 17–63 Mb (B). SNP frequency of SJL alleles (y-axis) are plotted against chromosome position (x-axis). Cartoon of the Im loci are drawn above each panel. Open bar = C3H/F; black bar = SJL genomic material.
Im2 Locus Does Not Affect Levels of Circulating Immune Cells
We demonstrated that the Im2 locus controls leukocyte (CD45+)-mediated inflammation (42). However, significant variation in WBC was also found between SJL and C3H/F mouse strains (26). To determine if the strain-dependent increased inflammation was due to variances in circulating cell profiles or vascular wall cells we measured complete blood counts in SJL, C3H/F, and C3H/F.SJL.11.1 mice. SJL mice were equally different compared with C3H/F and C3H/F.SJL.11.1 in total WBC (Table 1). There were no significant differences between C3H/F and C3H/F.SJL.11.1 in all measured counts. Therefore, the presence of inflammatory cells in the vascular wall in the C3H/F.SJL.11.1 mice must be due to resident vascular cells promoting inflammation.
Table 1.
Hematologic differences between congenic and inbred mice
| Strain Parameter | SJL, n = 10 | C3H/F, n = 14 | SJL.C3H/F.11.1, n = 14 |
|---|---|---|---|
| White blood cells, × 109/l | 6.6 ± 0.7 | 4.3 ± 0.6* | 4.6 ± 0.6 |
| Lymphocytes, × 109/l | 5.8 ± 0.6 | 3.4 ± 0.5* | 3.6 ± 0.5* |
| Neutrophils, × 109/l | 0.58 ± 0.17 | 0.77 ± 0.15 | 0.73 ± 0.15 |
| Monocytes, × 109/l | 0.15 ± 0.05 | 0.15 ± 0.04 | 0.22 ± 0.04 |
| Red blood cells, × 109/l | 6.7 ± 0.9 | 6.9 ± 0.7 | 7.4 ± 0.7 |
| Platelets, × 109/l | 346 ± 67 | 377 ± 57 | 445 ± 57 |
| Hematocrit, % | 30 ± 4 | 35 ± 4 | 38 ± 4 |
| Hemoglobin, g/dl | 10.3 ± 1.5 | 11.4 ± 1.3 | 12.4 ± 1.3 |
| Plateletcrit, % | 0.26 ± 0.06 | 0.30 ± 0.05 | 0.37 ± 0.05 |
| Mean corpuscular volume | 44.2 ± 0.5 | 50.7 ± 0.4* | 50.9 ± 0.4* |
| Mean corpuscular hemoglobin | 14.9 ± 0.6 | 16.1 ± 0.5 | 16.7 ± 0.5* |
| Mean corpuscular hemoglobin concentration, g/dl | 34 ± 1 | 32 ± 1 | 34 ± 1 |
| Red blood cell distribution | 17.9 ± 0.3 | 16.4 ± 0.3* | 16.3 ± 0.3* |
| Mean platelet volume | 7.1 ± 0.3 | 7.9 ± 0.2* | 8.2 ± 0.2* |
| Platelet distribution | 33.1 ± 1.0 | 33.7 ± 0.9 | 34.5 ± 0.9 |
P < 0.05 compared with SJL (ANOVA).
n, Number of mice.
Activation of EC Inflammation Is Increased in Im2 Congenic Mice
SJL mice exhibited dysfunction in endothelium-dependent vasorelaxation of aortic rings compared with C3H/F (8). Since there were no changes in circulating monocytes between SJL, C3H/F, and C3H/F.SJL.11.1 mice, we next wanted to investigate EC-mediated inflammation. We isolated aortas from SJL, C3H/F, and C3H/F.SJL.11.1 mice and measured basal levels of VCAM-1 (Fig. 2). Total VCAM-1 protein expression was dramatically increased in SJL and C3H/F.SJL.11.1 aortas relative to C3H/F (Fig. 2A). To determine if aortic EC were activated we performed VCAM-1 en face immunostaining. It is well established that endothelial inflammation varies in response to blood flow patterns in the aortic arch (50) and that there is a genetic component to this pattern (24). Figure 2B depicts the areas of the aorta that were stained for VCAM-1. We imaged areas of steady flow (S-flow, greater curvature) and disturbed flow (D-flow, lesser curvature). Consistent with the total aortic VCAM-1 expression there was significantly more VCAM-1 expression in SJL and C3H/F.SJL.11.1 compared with C3H/F in regions of S-flow. Although it did not reach statistical significance, there was also a slightly higher level of VCAM-1 expression in D-flow in SJL and C3H/F.SJL.11 compared with C3H/F (Fig. 2, C and D). Interestingly, in D-flow areas there was qualitatively greater redistribution of VCAM-1 expression from the nucleus to the membrane in SJL and C3H/F.SJL.11.1 compared with C3H/F (Fig. 2C). These findings indicate a concomitant activation of inflammation with increased VCAM-1 expression in endothelium, which is largely controlled by the Im2 locus.
Fig. 2.

Vascular cell adhesion molecule-1 (VCAM-1) expression in SJL, C3H/F, and C3H/F.SJL.11.1 mice. A: representative Western blot image for VCAM-1 in whole aorta. VCAM-1 expression was increased in SJL and C3H/F.SJL.11.1 compared with C3H/F. B: cartoon depicting aortic arch dissection for en face VCAM-1 and CD31 immunostaining. S-flow, greater curvature; D-flow, lesser curvature. C: representative en face VCAM-1 immunostaining at regions of S-flow and D-flow across strains. IgG control is shown in bottom right panel. D: quantified en face immunostaining shows that VCAM-1 expression was significantly increased in endothelial cells (EC) from SJL and C3H/F.SJL.11.1 compared with C3H/F. White scale bar = 20 μm. Data presented as means ± SE. *P < 0.05 vs. C3H/F; n = 3 per strain.
EC Size Is Decreased in SJL and C3H/F.SJL.11.1 Congenic Mice
It has been shown that changes in blood flow patterns affect EC morphology (49). We measured differences in aortic EC size between SJL, C3H/F, and C3H/F.SJL.11.1 mice. Aortas were immunostained for CD31 and CD144 to outline EC membranes (Fig. 3). We observed no differences in EC size between the mouse strains in regions of S-flow (Fig. 3, A and B). However, in regions of D-flow SJL and C3H/F.SJL.11.1 EC size was significantly decreased compared with C3H/F mice (Fig. 3, A and B). Thus, the Im2 locus determines EC size in areas of D-flow in addition to increased EC activation.
Fig. 3.

EC size in SJL, C3H/F, and C3H/F.SJL.11.1 mice. A: representative en face membrane CD144 immunostaining in mouse strains in S-flow and D-flow regions. IgG control is shown in bottom right panel. B: quantified en face immunostaining shows that under conditions of D-flow EC size is significantly decreased in SJL and C3H/F.SJL.11.1 compared with C3H/F. White scale bar = 20 μm. Data presented as means ± SE. *P < 0.05 vs. C3H/F; n = 3 per strain.
Vascular Permeability Is Elevated in SJL and C3H/F.SJL.11.1 Congenic Mice
Our current findings showing increased VCAM-1 and decreased EC area in SJL and C3H/F.SJL.11.1 compared with C3H/F might explain increased vascular inflammation (42). It is possible that these factors lead to increased EC permeability, which would allow for enhanced extravasation of inflammatory cells. We measured permeability by injecting Evans blue dye via tail vein in the studied mouse strains (Fig. 4). Evans blue binds to circulating albumin, and therefore wherever albumin permeates into a tissue it will be detected by blue coloration (17, 20). There was globally greater permeability in SJL and C3H/F.SJL.11.1 mice compared with C3H/F as demonstrated by qualitatively increased blue tissue coloration upon dissection (Fig. 4A, insets). As evident by footpad coloration, SJL mice showed signs of increased permeability within ∼5 min of Evans blue dye injection (not shown). Upon dissection all internal organs were saturated with the blue dye (Fig. 4A, left panel inset). C3H/F mice exhibited little to no blue tissue coloration (Fig. 4A, middle panel inset). Similar to SJL, the C3H/F.SJL.11.1 mice exhibited increased permeability with greater blue coloration of the footpads soon after injection (∼10 min, not shown) and greater penetrance of the dye into tissues (Fig. 4A, right panel inset). To quantify vascular permeability we harvested aortas and extracted the Evans blue dye. SJL had significantly greater OD measurements per dry weight of aorta compared with C3H/F (Fig. 4B). The C3H/F.SJL.11.1 mice exhibited some variance between animals. However, permeability in C3H/F.SJL.11.1 mice was comparable to SJL and significantly increased compared with C3H/F. The data indicate that the Im2 locus regulates vascular permeability.
Fig. 4.

Permeability measured by Evans blue dye in SJL, C3H/F, and C3H/F.SJL.11.1 mice. A: representative images of aortas (from arch attached to heart to iliac bifurcation) in mouse strains following 30 min of Evans blue dye injection via tail vein. Insets: chest cavity and internal organs following injection and complete perfusion. B: quantified data form Evans blue dye in aortas. Permeability is graphed as μg of dye permeated into mg of dried tissue. There was a significant increase in permeability in C3H/F.SJL.11.1 compared with C3H/F. Individual measurements in SJL are shown by black squares (■), in C3H/F by open circles (○), and in C3H/F.SJL.11.1 by black circles (●). *P < 0.05 vs. C3H/F; n ≥ 5 per strain.
RNA-Seq and GO Pathway Analyses
To identify genes within the Im2 locus that cause inflammation and EC dysfunction we performed RNA-Seq on mouse carotid arteries from C3H/F.SJL.11.1. We found 22 genes within the Im2 locus that were significantly different compared with C3H/F: 20 genes had increased expression, and two genes had decreased expression (Fig. 5A). However, more genes were upregulated in C3H/F.SJL.11.1 compared with C3H/F outside the Im2 locus on chr11 (Fig. 5B). We used GO pathway analysis to bin the differentially expressed genes. Immunological categories differed between C3H/F.SJL.11.1 and C3H/F among biological processes in GO terms (Table 2). Importantly, most of the differentially regulated pathways involved genes outside of the Im2 locus except gas/oxygen transport (Table 2). Several GO cellular component terms were affected by the locus in the C3H/F.SJL.11.1 congenics (Table 3). However, only one gene, obscurin (OBSCN), in the Im2 locus was found in this term. Thus, introgression of the Im2 locus affected a limited number of genes involved in immunological processes and cellular component terms, while a majority of differentially expressed genes in the GO pathways were outside the Im2 locus.
Fig. 5.
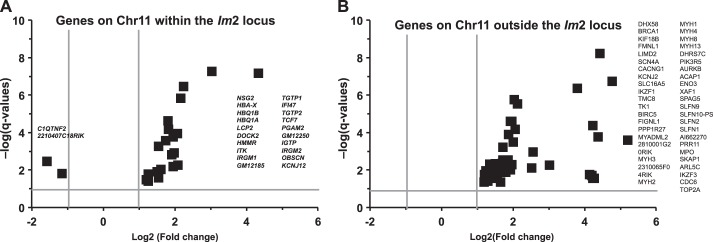
RNA sequence profiles in the arteries from C3H/F.SJL.11.1 vs. C3H/F mice for chromosome 11. A: gene differences within the Intima modifier 2 (Im2) locus. B: gene differences outside of the Im2 locus on chromosome 11. Individual genes are abbreviated and represented as squares on the volcano plots. x-Axes show log2-transformed fold changes in gene expression: negative values, downregulated; positive, upregulated. y-Axes show −log(q values). Two gray vertical lines: 2-fold cut-offs. A horizontal gray line represents a level of significance at P = 0.05. The RNA-Seq expression data were obtained from 4 mice per strain.
Table 2.
List of biological processes that are different between C3H/F vs. 11.1 arteries based on GO terms
| GOBPID | P Value | GO Term | Genes |
|---|---|---|---|
| GO:0002376 | 4.19E-05 | immune system process | DOCK2, HBA-X, IRGM1, IRGM2, ITK, LCP2, TCF7, TGTP1, DHX58, IKZF1, IKZF3, MPO, SKAP1, SLFN1 |
| GO:0015669 | 0.0001962 | gas transport | HBA-X, HBQ1A, HBQ1B |
| GO:0015671 | 0.0001962 | oxygen transport | HBA-X, HBQ1A, HBQ1B |
| GO:0046631 | 0.0003187 | alpha-beta T cell activation | DOCK2, ITK, TCF7, IKZF1 |
| GO:0007059 | 0.0003708 | chromosome segregation | BIRC5, BRCA1, CDC6, SPAG5, TOP2A |
| GO:0010564 | 0.0004296 | regulation of cell cycle process | AURKB, BIRC5, BRCA1, CDC6, SLFN1, TOP2A |
| GO:0035456 | 0.0004774 | response to interferon-beta | IGTP, TGTP1, XAF1 |
| GO:0090068 | 0.0007099 | positive regulation of cell cycle process | AURKB, BIRC5, BRCA1, CDC6 |
GO, Gene Ontology; GOBPID, Gene Ontology Biological Process Identification number. Abbreviated genes within the Intima modifier 2 locus are shown in italics/boldface.
Table 3.
List of cellular components that are different between C3H/F vs. 11.1 arteries based on GO terms
| GOCCID | P Value | GO Term | Genes |
|---|---|---|---|
| GO:0032982 | 6.83E-08 | myosin filament | OBSCN, MYH1, MYH3, MYH4, MYH8 |
| GO:0016459 | 1.88E-06 | myosin complex | OBSCN, MYH1, MYH13, MYH2, MYH3, MYH4, MYH8 |
| GO:0043292 | 2.13E-05 | contractile fiber | OBSCN, MYH1, MYH13, MYH2, MYH3, MYH4 |
| GO:0005859 | 5.28E-05 | muscle myosin complex | OBSCN, MYH1, MYH2 |
| GO:0030016 | 0.0001047 | myofibril | OBSCN, MYH1, MYH13, MYH2, MYH4 |
| GO:0015629 | 0.0004844 | actin cytoskeleton | OBSCN, MYH1, MYH13, MYH2, MYH3, MYH4, MYH8 |
| GO:0016460 | 0.0004992 | myosin II complex | OBSCN, MYH1, MYH2 |
| GO:0031672 | 0.0009713 | A band | OBSCN, MYH1, MYH2 |
GOCCID, Gene Ontology Cellular Component Identification number. An abbreviated gene within the Intima modifier 2 locus is shown in italics/boldface.
DISCUSSION
The main finding of this study is that the Im2 locus on mouse chr11 increases vascular inflammation via permeability and endothelial cell activation. We reported that the Im2 locus regulates leukocyte infiltration in mouse carotids in response to low blood flow (42). In the present study we expand our findings by demonstrating four important results regarding the Im2 locus: 1) The purity of our congenic mouse was confirmed by dense SNP mapping; 2) The inflammatory phenotype is attributed to EC activation and not to the changes in circulating immune cells; 3) There is increased vascular permeability related to EC activation and changes in EC morphology; 4) RNA-Seq on the Im2 congenic pinpoints candidate genes in pathways that regulate immune processes and cellular components. Thus, our findings in congenic mice signify the importance of resident vascular wall cells vs. circulating blood cells in intima formation. The latter suggest more complex than expected interpretation of the contribution of the circulating cells in human genetic studies on vascular inflammation traits.
Traditional congenic studies use marker-assisted selection, originally proposed by Lande and Thompson (31). This conventional approach for congenic strain development follows the average proportion of heterozygous donor-recipient genome that is eliminated with each round of breeding and predicts that after 10 generations of backcrossing one should obtain a strain carrying 99.9% of the recipient genome (15, 34). We used a modified breeding scheme, termed speed congenics, where we choose the best male breeder from each generation and backcross to the same female for five generations (42). By calculations discussed in Markel et al. (34) this strategy yields a homozygous recipient strain. We developed the C3H/F.SJL.11.1 mouse by conventional microsatellite marker-assisted genotyping using this speed congenic method (1, 34, 42). However, given the sparse mapping available with this approach (every ∼50 Mb) we wanted to confirm that the C3H/F.SJL.11.1 congenic genome was not contaminated with SJL material other than on chr11. By performing dense SNP sequencing we finely genotyped the C3H/F.SJL.11.1 genome at every ∼5 kb and confirmed that the Im2 locus was the only SJL genome introgressed into the C3H/F background. This method of congenic confirmation is not commonly reported in the literature, but since the likelihood of unintended genomic contamination cannot be easily detected with microsatellite (or less dense SNP) mapping, it is a necessary endpoint to confirm congenic purity. Thus, by verifying chr11 congenic purity we can confidently make additional conclusions regarding Im2 regulation of inflammation.
We hypothesized that the Im2 locus mediates inflammation via either changes in circulating immune cells or endothelial dysfunction. In our previous study we had not determined what mediated CD45+ cell infiltration in the carotid wall. A previous study by Kile et al. (26) showed strain-dependent differences in circulating immune cells. Consistent with this study we show a significant increase in WBC and lymphocytes in SJL compared with C3H/F. However, we found no detectable differences in circulating immune cell profiles between C3H/F and C3H/F.SJL.11.1 congenic mice. Contribution of the blood-derived cells to vascular remodeling has now proven rather weak (12, 23). Major pitfalls were related to extend of the vascular injury and detection methods (21, 22). Our data indicate that the Im2 locus does not control circulating immune cell populations but rather vascular wall activation and presumably inflammation. Findings in C3H/F.SJL.11.1 congenics further support a central role for the vascular wall in intima formation in response to injury.
The endothelium is the primary sensor of changes in blood flow patterns. Inflammatory cell adherence and extravasation is the result of monocyte binding to the endothelium (10, 51). It is well known that increased expression of VCAM-1 is a marker of inflammation, which correlates with increased EC attraction of monocytes (3, 13). It is also known that changes in VCAM-1 expression are regulated by flow patterns (35, 47, 48, 52). Under S-flow conditions inflammation and VCAM-1 expression are typically low, but under D-flow VCAM-1 expression increases (19, 46, 51). Interestingly, we found that VCAM-1 expression in ECs is significantly elevated in SJL and C3H/F.SJL.11.1 aortas compared with C3H/F under S-flow, where it should normally be low. These new findings could explain our previous report on dysfunction in endothelium-dependent vasorelaxation in SJL vs. C3H/F mice (8). Others have shown that 129P3 and 129X1/Sv strains have impaired acetylcholine-dependent vasorelaxation (40). Interestingly, the apolipoprotein E-null mutation (ApoE−/−) on the 129 background resulted in significantly different aortic arch geometry and progression of atherosclerosis compared with ApoE−/− on the C57BL/6 background (33). A genetic cross between ApoE−/− on 129 and C57BL/6 backgrounds identified several QTLs for curvature of the aortic arch and atherosclerosis (45). Even though none of these loci overlapped with the Im2 locus, more recent data suggest that arch geometry and local hemodynamics are critical for atherosclerotic plaque formation and determined by 129 background in ApoE−/− mice (45, 54). It is possible that both SJL and 129 backgrounds contribute to endothelial dysfunction via different intermediate phenotypes. Thus, the Im2 locus from SJL promotes an inflammatory environment in the endothelium even in homeostatic conditions. Presumably, this altered homeostasis primes the response to pathogenic blood flow conditions (D-flow) and inflammatory stimuli.
In addition to changes in inflammatory cell adhesion markers, changes in EC morphology are also important factors mediating an inflammatory response. Alterations in EC structure and alignment occur in response to multiple stimuli including vascular endothelial growth factor (9, 18, 43) and G protein coupled receptor agonists (44). However, changes in blood flow can also induce EC structural alterations (49). When exposed to D-flow, EC round up and change cell size and volume (11, 25). Experiments in cultured bovine aortic EC showed changes in permeability that were attributed to changes in EC shape and attachment to the basement membrane and extracellular matrix (25). We found decreased EC size in response to D-flow and a dramatic increase in vascular permeability in SJL and C3H/F.SJL.11.1 compared with C3H/F. Since permeability is affected by both flow patterns and EC morphology we conclude that the increased leukocyte infiltration in the SJL and C3H/F.SJL.11.1 aortas is likely regulated by genes within the Im2 locus. Using RNA-Seq we identified some candidate genes that may explain the increased permeability in the context of EC morphological changes. Of the annotated genes that had significantly higher expression in the Im2 locus there are a couple that are likely candidates to affect endothelial dysfunction. The GO biological pathway analysis for immune system process identified immunity-related GTPase family, M member 1 (IRGM1). This gene has been shown to have proinflammatory properties that disrupt the blood-brain barrier and increase permeability to lymphocytes (49). OBSCN was the most differentially expressed gene compared with C3H/F. OBSCN is a cytoskeletal protein, which was identified in all GO cellular component categories. Although OBSCN has not been described in EC function, it has been shown to play a role in adaptive remodeling of myofibrils in cases of hypertrophic cell growth in response to aortic constriction (5). Presumably, these candidate genes can induce changes in vascular cells and alterations in the structural matrix of the vascular wall that will result in altered shape and behavior of the endothelium. However, we also very clearly show that genes outside the Im2 locus on chr11 are also important for immune activation and structural homeostasis. The interaction of these gene networks likely work to promote EC dysfunction. It will be interesting to explore the potential role of these genes in future studies, because genes that regulate vascular wall structural integrity and modulate EC function are potential therapeutic targets for managing inflammation and vessel wall pathology.
In summary, we developed a pure Im2 congenic mouse, which is a valuable tool with which to study vascular inflammation. We now have new insight into the mechanism by which Im2 promotes leukocyte infiltration in response to changes in blood flow. We demonstrated that this locus does not affect immune cell profiles but does control EC morphology and vascular permeability. Our data strongly suggest that EC size changes may alter vascular wall structural integrity, allowing areas of enhanced permeability. Our RNA-Seq and GO analyses point to candidate genes and pathways that may regulate vascular inflammation. Our findings suggest that human genetic studies on vascular inflammation should be carefully evaluated and not too centrally focused on the effect of circulating immune cells in vascular inflammation.
GRANTS
This study was supported in part by funds from National Heart, Lung, and Blood Institute Grants HL-105623 (V. A. Korshunov), HL-62826 (B. C. Berk), and HL-102746 and by American Heart Association Grant 11GRNT5850001 (K. Fujiwara).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: E.M.S., K.F., B.C.B., and V.A.K. conception and design of research; E.M.S., R.M.B., C.W., T.T., S.N.B., and M.Z. performed experiments; E.M.S., R.M.B., C.W., T.T., S.N.B., X.Q., M.Z., and V.A.K. analyzed data; E.M.S., R.M.B., C.W., T.T., S.N.B., X.Q., M.Z., K.F., and V.A.K. interpreted results of experiments; E.M.S., R.M.B., C.W., and T.T. prepared figures; E.M.S. drafted manuscript; E.M.S., R.M.B., C.W., T.T., S.N.B., X.Q., M.Z., K.F., B.C.B., and V.A.K. approved final version of manuscript; C.W., S.N.B., X.Q., K.F., B.C.B., and V.A.K. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Michelle Zanche, Dr. John Ashton, and Jason Myers (GRC) for genotyping, gene profiling, and RNA sequencing assistance. We also thank Dr. Dietrich Machleder for support with congenic genotyping, Dr. Craig Morrell for help with hematological assays, and Amy Mohan and Christine Christie for animal assistance.
Current address for S. N. Batchu: St. Michaels Hospital, University of Toronto, Toronto, ON Canada.
REFERENCES
- 1.Armstrong E, Postiglioni A, Martinez A, Rincon G, Vega-Pla JL. Microsatellite analysis of a sample of Uruguayan Creole bulls (Bos taurus). Genet Mol Biol 29: 267–272, 2006 [Google Scholar]
- 2.Baldassarre D, Veglia F, Hamsten A, Humphries SE, Rauramaa R, de Faire U, Smit AJ, Giral P, Kurl S, Mannarino E, Grossi E, Paoletti R, Tremoli E. Progression of carotid intima-media thickness as predictor of vascular events: results from the IMPROVE study. Arterioscler Thromb Vasc Biol 33: 2273–2279, 2013 [DOI] [PubMed] [Google Scholar]
- 3.Behm CZ, Kaufmann BA, Carr C, Lankford M, Sanders JM, Rose CE, Kaul S, Lindner JR. Molecular imaging of endothelial vascular cell adhesion molecule-1 expression and inflammatory cell recruitment during vasculogenesis and ischemia-mediated arteriogenesis. Circulation 117: 2902–2911, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B Met 57: 289–300, 1995 [Google Scholar]
- 5.Borisov AB, Raeker MO, Kontrogianni-Konstantopoulos A, Yang K, Kurnit DM, Bloch RJ, Russell MW. Rapid response of cardiac obscurin gene cluster to aortic stenosis: differential activation of Rho-GEF and MLCK and involvement in hypertrophic growth. Biochem Biophys Res Commun 310: 910–918, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Bots ML, Hoes AW, Koudstaal PJ, Hofman A, Grobbee DE. Common carotid intima-media thickness and risk of stroke and myocardial infarction: the Rotterdam Study. Circulation 96: 1432–1437, 1997 [DOI] [PubMed] [Google Scholar]
- 7.Burke RM, Madden KS, Perry SW, Zettel ML, Brown EB., 3rd Tumor-associated macrophages and stromal TNF-alpha regulate collagen structure in a breast tumor model as visualized by second harmonic generation. J Biomed Optics 18: 86003, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C, Korshunov VA, Massett MP, Yan C, Berk BC. Impaired vasorelaxation in inbred mice is associated with alterations in both nitric oxide and super oxide pathways. J Vasc Res 44: 504–512, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, Lim ST, Tomar A, Tancioni I, Uryu S, Guan JL, Acevedo LM, Weis SM, Cheresh DA, Schlaepfer DD. VEGF-induced vascular permeability is mediated by FAK. Dev Cell 22: 146–157, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiu JJ, Chen CN, Lee PL, Yang CT, Chuang HS, Chien S, Usami S. Analysis of the effect of disturbed flow on monocytic adhesion to endothelial cells. J Biomechan 36: 1883–1895, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Chiu JJ, Wang DL, Chien S, Skalak R, Usami S. Effects of disturbed flow on endothelial cells. J Biomechan Engineer 120: 2–8, 1998 [DOI] [PubMed] [Google Scholar]
- 12.Daniel JM, Bielenberg W, Stieger P, Weinert S, Tillmanns H, Sedding DG. Time-course analysis on the differentiation of bone marrow-derived progenitor cells into smooth muscle cells during neointima formation. Arterioscler Thromb Vasc Biol 30: 1890–1896, 2010 [DOI] [PubMed] [Google Scholar]
- 13.Dansky HM, Barlow CB, Lominska C, Sikes JL, Kao C, Weinsaft J, Cybulsky MI, Smith JD. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler Thromb Vasc Biol 21: 1662–1667, 2001 [DOI] [PubMed] [Google Scholar]
- 14.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics 23: 257–258, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Falconer D. Introduction to Quantitative Genetics. Harlow, UK: Pearson Education, 1989 [Google Scholar]
- 16.Fox CS, Cupples LA, Chazaro I, Polak JF, Wolf PA, D'Agostino RB, Ordovas JM, O'Donnell CJ. Genomewide linkage analysis for internal carotid artery intimal medial thickness: evidence for linkage to chromosome 12. Am J Hum Genet 74: 253–261, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fry DL, Herderick EE, Johnson DK. Local intimal-medial uptakes of 125I-albumin, 125I-LDL, and parenteral Evans blue dye protein complex along the aortas of normocholesterolemic minipigs as predictors of subsequent hypercholesterolemic atherogenesis. Arterioscler Thromb 13: 1193–1204, 1993 [DOI] [PubMed] [Google Scholar]
- 18.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 8: 1223–1234, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Harry BL, Sanders JM, Feaver RE, Lansey M, Deem TL, Zarbock A, Bruce AC, Pryor AW, Gelfand BD, Blackman BR, Schwartz MA, Ley K. Endothelial cell PECAM-1 promotes atherosclerotic lesions in areas of disturbed flow in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 28: 2003–2008, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Himburg HA, Grzybowski DM, Hazel AL, LaMack JA, Li XM, Friedman MH. Spatial comparison between wall shear stress measures and porcine arterial endothelial permeability. Am J Physiol Heart Circ Physiol 286: H1916–H1922, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Hoglund VJ, Dong XR, Majesky MW. Neointima formation: a local affair. Arterioscler Thromb Vasc Biol 30: 1877–1879, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoofnagle MH, Thomas JA, Wamhoff BR, Owens GK. Origin of neointimal smooth muscle: we've come full circle. Arterioscler Thromb Vasc Biol 26: 2579–2581, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Iwata H, Manabe I, Fujiu K, Yamamoto T, Takeda N, Eguchi K, Furuya A, Kuro-o M, Sata M, Nagai R. Bone marrow-derived cells contribute to vascular inflammation but do not differentiate into smooth muscle cell lineages. Circulation 122: 2048–2057, 2010 [DOI] [PubMed] [Google Scholar]
- 24.Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med 203: 2073–2083, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kajimura M, O'Donnell ME, Curry FE. Effect of cell shrinkage on permeability of cultured bovine aortic endothelia and frog mesenteric capillaries. J Physiol 503: 413–425, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kile BT, Mason-Garrison CL, Justice MJ. Sex and strain-related differences in the peripheral blood cell values of inbred mouse strains. Mamm Genome 14: 81–85, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Korshunov VA, Berk BC. Flow-induced vascular remodeling in the mouse: a model for carotid intima-media thickening. Arterioscler Thromb Vasc Biol 23: 2185–2191, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Korshunov VA, Berk BC. Genetic modifier loci linked to intima formation induced by low flow in the mouse carotid. Arterioscler Thromb Vasc Biol 29: 47–53, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korshunov VA, Schwartz SM, Berk BC. Vascular remodeling: hemodynamic and biochemical mechanisms underlying Glagov's phenomenon. Arterioscler Thromb Vasc Biol 27: 1722–1728, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol 17: 2238–2244, 1997 [DOI] [PubMed] [Google Scholar]
- 31.Lande R, Thompson R. Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics 124: 743–756, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lange LA, Bowden DW, Langefeld CD, Wagenknecht LE, Carr JJ, Rich SS, Riley WA, Freedman BI. Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke 33: 1876–1881, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Maeda N, Johnson L, Kim S, Hagaman J, Friedman M, Reddick R. Anatomical differences and atherosclerosis in apolipoprotein E-deficient mice with 129/SvEv and C57BL/6 genetic backgrounds. Atherosclerosis 195: 75–82, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Markel P, Shu P, Ebeling C, Carlson GA, Nagle DL, Smutko JS, Moore KJ. Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains. Nat Genet 17: 280–284, 1997 [DOI] [PubMed] [Google Scholar]
- 35.Matharu NM, McGettrick HM, Salmon M, Kissane S, Vohra RK, Rainger GE, Nash GB. Inflammatory responses of endothelial cells experiencing reduction in flow after conditioning by shear stress. J Cell Physiol 216: 732–741, 2008 [DOI] [PubMed] [Google Scholar]
- 36.Mathiesen EB, Johnsen SH, Wilsgaard T, Bonaa KH, Lochen ML, Njolstad I. Carotid plaque area and intima-media thickness in prediction of first-ever ischemic stroke: a 10-year follow-up of 6584 men and women: the Tromso Study. Stroke 42: 972–978, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Melton PE, Carless MA, Curran JE, Dyer TD, Goring HH, Kent JW, Jr, Drigalenko E, Johnson MP, Maccluer JW, Moses EK, Comuzzie AG, Mahaney MC, O'Leary DH, Blangero J, Almasy L. Genetic architecture of carotid artery intima-media thickness in Mexican Americans. Circ Cardiovasc Genet 6: 211–221, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyashiro JK, Poppa V, Berk BC. Flow-induced vascular remodeling in the rat carotid diminishes with age. Circ Res 81: 311–319, 1997 [DOI] [PubMed] [Google Scholar]
- 39.Nestor Kalinoski AL, Ramdath RS, Langenderfer KM, Sikanderkhel S, Deraedt S, Welch M, Park JL, Pringle T, Joe B, Cicila GT, Allison DC. Neointimal hyperplasia and vasoreactivity are controlled by genetic elements on rat chromosome 3. Hypertension 55: 555–561, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryan MJ, Didion SP, Davis DR, Faraci FM, Sigmund CD. Endothelial dysfunction and blood pressure variability in selected inbred mouse strains. Arterioscler Thromb Vasc Biol 22: 42–48, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Sacco RL, Blanton SH, Slifer S, Beecham A, Glover K, Gardener H, Wang L, Sabala E, Juo SH, Rundek T. Heritability and linkage analysis for carotid intima-media thickness: the family study of stroke risk and carotid atherosclerosis. Stroke 40: 2307–2312, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smolock EM, Machleder DE, Korshunov VA, Berk BC. Identification of a genetic locus on chromosome 11 that regulates leukocyte infiltration in mouse carotid artery. Arterioscler Thromb Vasc Biol 33: 1014–1019, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun Z, Li X, Massena S, Kutschera S, Padhan N, Gualandi L, Sundvold-Gjerstad V, Gustafsson K, Choy WW, Zang G, Quach M, Jansson L, Phillipson M, Abid MR, Spurkland A, Claesson-Welsh L. VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAd. J Exp Med 209: 1363–1377, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thennes T, Mehta D. Heterotrimeric G proteins, focal adhesion kinase, and endothelial barrier function. Microvasc Res 83: 31–44, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomita H, Zhilicheva S, Kim S, Maeda N. Aortic arch curvature and atherosclerosis have overlapping quantitative trait loci in a cross between 129S6/SvEvTac and C57BL/6J apolipoprotein E-null mice. Circ Res 106: 1052–1060, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsao PS, Buitrago R, Chan JR, Cooke JP. Fluid flow inhibits endothelial adhesiveness. Nitric oxide and transcriptional regulation of VCAM-1. Circulation 94: 1682–1689, 1996 [DOI] [PubMed] [Google Scholar]
- 47.Tsou JK, Gower RM, Ting HJ, Schaff UY, Insana MF, Passerini AG, Simon SI. Spatial regulation of inflammation by human aortic endothelial cells in a linear gradient of shear stress. Microcirculation 15: 311–323, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang C, Baker BM, Chen CS, Schwartz MA. Endothelial cell sensing of flow direction. Arterioscler Thromb Vasc Biol 33: 2130–2136, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang C, Wang C, Dong H, Wu XM, Wang C, Xia F, Li G, Jia X, He S, Jiang X, Li H, Xu H. Immune-related GTPase Irgm1 exacerbates experimental auto-immune encephalomyelitis by promoting the disruption of blood-brain barrier and blood-cerebrospinal fluid barrier. Mol Immunol 53: 43–51, 2013 [DOI] [PubMed] [Google Scholar]
- 50.Wang S, Zhang H, Wiltshire T, Sealock R, Faber JE. Genetic dissection of the Canq1 locus governing variation in extent of the collateral circulation. PLoS One 7: e31910, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang XQ, Nigro P, World C, Fujiwara K, Yan C, Berk BC. Thioredoxin interacting protein promotes endothelial cell inflammation in response to disturbed flow by increasing leukocyte adhesion and repressing Kruppel-like factor 2. Circ Res 110: 560–568, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamawaki H, Pan S, Lee RT, Berk BC. Fluid shear stress inhibits vascular inflammation by decreasing thioredoxin-interacting protein in endothelial cells. J Clin Invest 115: 733–738, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan Z, Pei H, Roberts DJ, Zhang Z, Rowlan JS, Matsumoto AH, Shi W. Quantitative trait locus analysis of neointimal formation in an intercross between C57BL/6 and C3H/HeJ apolipoprotein E-deficient mice. Circ Cardiovasc Genet 2: 220–228, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu H, Zhang J, Shih J, Lopez-Bertoni F, Hagaman JR, Maeda N, Friedman MH. Differences in aortic arch geometry, hemodynamics, and plaque patterns between C57BL/6 and 129/SvEv mice. J Biomechan Engineer 131: 121005, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]