

Abstract
Our laboratory shows that acid-sensing ion channel 1 (ASIC1) contributes to the development of hypoxic pulmonary hypertension by augmenting store-operated Ca2+ entry (SOCE) that is associated with enhanced agonist-induced vasoconstriction and arterial remodeling. However, this enhanced Ca2+ influx following chronic hypoxia (CH) is not dependent on an increased ASIC1 protein expression in pulmonary arterial smooth muscle cells (PASMC). It is well documented that hypoxic pulmonary hypertension is associated with changes in redox potential and reactive oxygen species homeostasis. ASIC1 is a redox-sensitive channel showing increased activity in response to reducing agents, representing an alternative mechanism of regulation. We hypothesize that the enhanced SOCE following CH results from removal of an inhibitory effect of hydrogen peroxide (H2O2) on ASIC1. We found that CH increased PASMC superoxide (O2·−) and decreased rat pulmonary arterial H2O2 levels. This decrease in H2O2 is a result of decreased Cu/Zn superoxide dismutase expression and activity, as well as increased glutathione peroxidase (GPx) expression and activity following CH. Whereas H2O2 inhibited ASIC1-dependent SOCE in PASMC from control and CH animals, addition of catalase augmented ASIC1-mediated SOCE in PASMC from control rats but had no further effect in PASMC from CH rats. These data suggest that, under control conditions, H2O2 inhibits ASIC1-dependent SOCE. Furthermore, H2O2 levels are decreased following CH as a result of diminished dismutation of O2·− and increased H2O2 catalysis through GPx-1, leading to augmented ASIC1-dependent SOCE.
Keywords: superoxide dismutase, di-(4-carboxybenzyl) hyponitrite-1, glutathione peroxidase, catalase, pulmonary hypertension, degenerin/epithelial sodium channel
the nonselective cation channel, acid-sensing ion channel 1a (ASIC1a), belongs to the degenerin/epithelial sodium channel family and is the major ASIC subtype with Ca2+ permeability (53, 59). Our laboratory has recently demonstrated that ASIC1 contributes to the development of hypoxic pulmonary hypertension (32). Following exposure to chronic hypoxia (CH), ASIC1-mediated store-operated Ca2+ entry (SOCE) is augmented in pulmonary arterial smooth muscle cells (PASMC), resulting in enhanced agonist-induced vasoconstriction, vascular smooth muscle remodeling, and right ventricular hypertrophy (23, 32). Despite the requirement of ASIC1 for enhanced Ca2+ influx, these responses are not dependent on an increased ASIC1 protein expression (32). Therefore, the goal of the present study was to examine alternative regulatory mechanisms of ASIC1 function.
Cellular redox status affects the function of various proteins, including ASIC1a. ASIC subunits are cysteine rich, and the cellular redox status can modify both extracellular and intracellular cysteine residues (11, 60). Reducing agents potentiate ASIC1 activity, acid-induced membrane depolarization, and Ca2+ influx. More specifically, reducing agents increase ASIC1 current amplitude and pH sensitivity while decreasing channel inactivation (2, 10, 11, 60). In addition, oxidizing agents like hydrogen peroxide (H2O2) may introduce intersubunit disulfide bonds, thereby decreasing the amount of ASIC1a present on the cell surface and reducing acid-evoked currents (60). Together, these studies suggest that redox regulation of ASIC1 is an important factor in determining the overall physiological function of these channels. Furthermore, because cellular redox status can change dramatically under pathological states, this represents a potential mechanism by which ASIC1 contributes to the progression of many diseases (5, 12, 57).
It is well documented that hypoxic pulmonary hypertension is associated with alterations in the cellular redox environment. However, whether the generation of reactive oxygen species (ROS) is enhanced or diminished during hypoxia is still debated (34, 42, 54). Furthermore, the relative importance of changes in ROS levels vs. redox status is unclear. Numerous studies show that ROS, in particular superoxide (O2·−), and subsequent oxidative stress contribute to hypoxic pulmonary hypertension (34). Indeed, our laboratory has consistently shown that pulmonary arterial O2·− levels increase following exposure to CH (7, 24, 33). In contrast, CH has been shown to decrease H2O2 levels in several different species (6, 13, 40). Various models of pulmonary hypertension are associated with a dysregulation of superoxide dismutase (SOD), and possibly other antioxidant pathways, that could lead to lowering of H2O2 levels (4, 6, 13, 15, 40). However, neither the mechanisms by which CH mediates this decrease in H2O2 nor the importance of this response to regulation of pulmonary arterial smooth muscle calcium homeostasis are clearly defined. Consequently, the present study examined effects of CH on both O2·− dismutation and H2O2 degradation pathways in pulmonary arterial smooth muscle. In addition, we evaluated the importance of H2O2 in regulation of ASIC1, testing the hypothesis that enhanced SOCE following CH results from removal of an inhibitory effect of H2O2 on ASIC1.
MATERIALS AND METHODS
Animals and Chronic Hypoxic Exposure
All protocols employed in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine (Albuquerque, NM). Male Wistar rats (∼12 wk old, Harlan Industries) designated for exposure to CH were housed in a hypobaric chamber with barometric pressure maintained at ∼380 mmHg for 4 wk. Age-matched control rats were housed at ambient barometric pressure (∼630 mmHg in Albuquerque, NM). We have previously shown that exposure of rats and mice to CH causes pulmonary hypertension, along with the associated right ventricular hypertrophy, arterial remodeling, and polycythemia (32, 44). Male and female SOD1−/− and SOD+/+ mice were obtained from Jackson Laboratory (Sod1 tm1Leb/J, no.002972) and bred on a C56B6/J background. Previous reports from our laboratory indicate that SOD1−/− mice develop spontaneous pulmonary hypertension (40).
Isolation of Pulmonary Arteries and Generation of PASMC Culture
Rats were anesthetized with pentobarbital sodium (200 mg/kg ip), and the heart and lungs were exposed by midline thoracotomy. The left lung was removed and immediately placed in physiological saline solution (PSS) [pH adjusted to 7.4 with NaOH containing (in mM) 130 NaCl, 4 KC1, 1.2 MgSO4, 4 NaHCO3, 1.8 CaC12, 10 HEPES, 1.18 KH2PO4, and 6 glucose]. Intrapulmonary arteries (∼2nd-5th order) were dissected from surrounding lung parenchyma and either snap-frozen in liquid N2 or utilized to generate short-term PASMC cultures. To isolate PASMCs, intrapulmonary arteries were enzymatically digested in reduced Ca2+ HBSS containing papain (9.5 U/ml), type-I collagenase (1,750 U/ml), and dithiothreitol (1 mM) at 37°C for 30 min. The cell suspension was placed on 25-mm glass coverslips and cultured in Ham's F-12 media supplemented with 5% fetal bovine serum and 1% penicillin/streptomycin for 3–4 days in a humidified atmosphere of 5% CO2-95% air at 37°C.
Assessment of PASMC Superoxide Levels
To determine the effect of CH on O2·− levels, PASMC from control and CH rats were incubated with dihydroethidium (DHE; 5 μM in 0.05% pluronic acid) and TO-PRO-3 (1:2,000, Molecular Probes) at 37°C for 15 min. Cells were subsequently fixed with 2% paraformaldehyde for 10 min at room temperature. To verify the specificity of DHE in this preparation to detect O2·−, experiments were repeated with preincubation of either polyethylene glycol (PEG)-SOD (50 U/ml) or PEG-catalase (250 U/ml, Sigma-Aldrich) for 1 h at 37°C. Fluorescence images were acquired with a ×63 objective on a confocal microscope (Leica, TCS SP5). Mean fluorescence intensity was taken per field from 10 images and was averaged from 3–5 animals per group. Each image was thresholded using MetaMorph Imaging software (Molecular Devices) to select for positive-stained areas with fluorescence intensity values above background (i.e., of cells not treated with DHE).
Determination of Pulmonary Arterial H2O2 Levels
The effect of CH on pulmonary arterial H2O2 levels was determined by an Amplex Red Hydrogen Peroxide/Peroxidase Assay according to manufacturer's protocol (Life Technologies). The left descending intrapulmonary artery from control and CH rats was dissected into 2-mm segments in ice-cold PSS. Pulmonary arterial segments were incubated with Amplex Red reagent for 30 min at 37°C. The supernatant was transferred to a 96-well plate, and Amplex Red fluorescence was measured with a fluorescence microplate reader (Tecan Infinite M200) using excitation at 550 nm and fluorescence detection at 590 nm. Protein concentration was determined for each segment following the assay to verify that equal amounts of sample were used for each group. Additional experiments were performed to evaluate H2O2 levels following pretreatment for 30 min with di-(4-carboxybenzyl) hyponitrite (SOTS-1; 10 μM; Cayman Chemicals), PEG-SOD (50 U/ml), the SOD mimetic tiron (10 mM), or the catalase inhibitor 3-amino-1,2,4-triazole (AT; 5 mM) and glutathione peroxidase (GPx) inhibitor mercaptosuccinic acid (3 mM).
Pulmonary arterial antioxidant capacity.
The Amplex Red assay was used to examine the antioxidant capacity of tissue as previously described (50). Pulmonary arteries from control and CH rats were incubated with or without H2O2 (1 μM) for 1 h at 37°C. The rate of H2O2 catalyzed (Rcat) was determined by Rcat = [H2O2 − (SampleH2O2 − Sampleuntreated)]/1 h, where H2O2 is the fluorescence value of 1 μM H2O2 with no tissue, SampleH2O2 is the tissue sample incubated with 1 μM H2O2, and Sampleuntreated is an untreated tissue sample. Sampleuntreated was negligible, as it was not detected above background fluorescence (see Fig. 3A, inset).
Fig. 3.

CH decreases intrapulmonary arterial H2O2 levels. A: Amplex Red relative fluorescence units (RFU) from untreated control and CH intrapulmonary arteries were below the detectable linear range of the background-subtracted standard curve (inset). SOTS-1 increased H2O2 levels in a dose-dependent manner. B: summary data of H2O2 levels in intrapulmonary arteries from control and CH rats in the absence or presence of SOTS-1 (0.01 mM) and SOTS-1 plus PEG-catalase (250 U/ml). Values are means ± SE; n = 3–6 animals per group; τP < 0.05 vs. untreated pulmonary arteries; *P < 0.05 vs. pulmonary arteries from control rats; #P < 0.05 vs. treatment with 0.01 mM SOTS-1.
Western Blotting
Snap-frozen intrapulmonary arteries were homogenized in 10 mM Tris·HCl containing 255 mM sucrose, 2 mM EDTA, 12 μM leupeptin, 4 μM pepstatin A, 1 μM aprotinin (Sigma) and centrifuged at 10,000 g at 4°C to remove insoluble debris. Supernatant was collected, and sample protein concentrations were determined by the Bradford method (Bio-Rad) or spectrophotometer (Nano Drop 2000, Thermo Scientific). Equal protein concentration of pulmonary artery lysates were separated by SDS-PAGE (Tris·HCl gels, Bio-Rad) and transferred to polyvinylidene difluoride membranes. Blots were blocked for 1 h at room temperature with 5% milk and 0.05% Tween 20 (Bio-Rad) in Tris-buffered saline containing 10 mM Tris·HCl and 50 mM NaCl (pH 7.4) before incubation with primary antibody overnight. For immunochemical labeling, blots were incubated for 1 h at room temperature with goat anti-rabbit IgG-horseradish peroxidase (1:3,000, Bio-Rad). After chemiluminescence labeling (Pierce Thermo Scientific), bands were detected by exposure of the blots to chemiluminescence-sensitive film (Bio-Express). Bands were quantified by densitometric analysis using ImageJ (NIH). Blots were normalized to total protein determined by dividing the target protein by the intensity of the corresponding lane in the Coomassie-stained blots because exposure to CH significantly elevates both mRNA and protein expression of the traditional loading controls, β-actin and GAPDH, in pulmonary arterial homogenates (unpublished observations).
SOD Expression, Activity, and Modulation of SOCE
Protein expression.
To determine the effect of CH on SOD expression, Western blot analysis was performed in pulmonary artery lysates from control and CH rats. Blots were incubated with rabbit anti-SOD1 (1:5,000), rabbit anti-SOD2 (1:5,000), and rabbit anti-SOD3 (1:500, Abcam) primary antibodies.
Activity.
The effect of CH on SOD activity was determined by an SOD Assay kit (Cayman Chemical). Intrapulmonary arteries were homogenized in 5 μl buffer (20 mM HEPES pH 7.2 containing 1 mM EGTA, 210 mM mannitol, and 70 mM sucrose) per milligram of tissue and centrifuged at 1,500 g for 5 min at 4°C. Supernatant was collected, diluted 1:25, and transferred to a 96-well plate, where all samples were incubated in the presence or absence of NaCN (1.8 mM) to inhibit Cu/Zn SOD (SOD1 and SOD3) for 20 min at room temperature. Absorbance was detected at 450 nm using an absorbance microplate reader (ELx800, BioTek Instruments). Cu/Zn SOD activity was calculated by subtracting absorbance from the NaCN-treated samples (Mn SOD activity) from total SOD activity.
Modulation of SOCE.
To determine the influence of SOD1 expression and activity on ASIC1-dependent SOCE, small intrapulmonary arteries from SOD1+/+ and SOD1−/− mice were cannulated and pressurized for measurement of [Ca2+]i as described previously (32). Briefly, mice were anesthetized with pentobarbital sodium (200 mg/kg ip). The left lung was removed, and small intrapulmonary arteries (4th-5th order) were dissected free and transferred to a vessel chamber (Living Systems) and secured to tapered glass pipettes with a single strand of silk ligature. After cannulation, the artery was pressurized with a servo-controlled peristaltic pump (Living Systems) to 12 mmHg. Any artery that failed to maintain pressure upon switching off of the servo-controller was discarded. The vessel chamber was superfused with HEPES-based PSS (5 ml/min at 37°C). Arteries were incubated abluminally with fura 2-AM (2 μM and 0.05% pluronic acid in PSS, Molecular Probes) for 45 min at room temperature. Fura 2-loaded arteries were alternately excited at 340 and 380 nm at a frequency of 1 Hz with an IonOptix Hyperswitch dual-excitation light source (IonOptix), and the respective 510-nm emissions were detected with a photomultiplier tube. Following baseline fura 2 measurements, arteries were superfused (5 ml/min at 37°C) with Ca2+-free PSS containing 3 mM EGTA to chelate extracellular Ca2+, 50 μM diltiazem to prevent Ca2+ entry through L-type voltage-gated Ca2+ channels, and 10 μM cyclopiazonic acid (CPA) to deplete intracellular Ca2+ stores and prevent Ca2+ reuptake through the sarcoplasmic reticulum Ca2+-ATPase. SOCE was determined by measuring the changes in [Ca2+]i (area under curve) upon repletion of PSS containing 1.8 mM CaCl2 in the continued presence of diltiazem and CPA. Parallel experiments were performed in the presence of the specific ASIC1 inhibitor, psalmotoxin 1 (PcTX1; 20 nM).
ASIC1 protein expression.
To determine the effect of SOD1 on ASIC1 expression, Western blot analysis was performed in pulmonary artery lysates from SOD1+/+ and SOD1−/− mice. Blots were incubated with rabbit anti-ASIC1 (1:500, Millipore) or rabbit anti-SOD1 (1:5,000, Abcam).
Catalase Expression and Activity
Protein expression.
To determine the effect of CH on catalase expression, Western blot analysis was performed as described above in pulmonary artery lysates from control and CH rats. Blots were incubated with rabbit anti-catalase (1:2,000, Pierce Thermo Scientific) primary antibody.
Activity.
The effect of CH on catalase activity was determined by a catalase assay kit (Cayman Chemical) according to the manufacturer's directions. Intrapulmonary arteries were homogenized in 5 μl buffer (50 mM potassium phosphate pH 7.0 containing 1 mM EDTA) per milligram of tissue and centrifuged at 10,000 g for 15 min at 4°C. Supernatant was collected, diluted 1:5, and transferred to a 96-well plate. Absorbance was detected at 540 nm using an absorbance microplate reader (SpectraMax Plus384, Molecular Devices).
GPx Expression and Activity
Protein expression.
To determine the effect of CH on GPx expression, Western blot analysis was performed as described above in pulmonary artery lysates from control and CH rats. Blots were incubated with rabbit anti-GPx-1 (1:5,000, Abcam).
Activity.
The effect of CH on GPx activity was determined by an assay kit (Cayman Chemical). Intrapulmonary arteries were homogenized in 5 μl buffer (50 mM potassium phosphate pH 7.0 containing 1 mM EDTA) per milligram of tissue and centrifuged at 10,000 g for 15 min at 4°C. Supernatant was collected, diluted 1:1 and 1:5, and transferred to a 96-well plate. Absorbance was detected at 340 nm using an absorbance microplate reader (SpectraMax Plus384, Molecular Devices).
H2O2 Regulation of ASIC1-dependent SOCE
To assess the role of H2O2 on ASIC1-dependent SOCE, a similar protocol to that described above was used to measure SOCE in PASMC from control and CH rats following fura 2 loading (2 μM and 0.05% pluronic acid for 30 min at 32°C). PASMC were treated with PEG-catalase (250 U/ml); H2O2 (25 μM); GPx mimetic, ebselen (30 μM); or GPx inhibitor, mercaptosuccinic acid (3 mM) throughout the experiment. The contribution of ASIC1 to SOCE was determined in paired experiments by subsequently repeating the SOCE response following 20-min incubation with PcTX1 (20 nM). Data are expressed as percentage of control vehicle taken as an average of the control vehicle responses.
Calculations and Statistics
All data are expressed as means ± SE. Values of n refer to number of animals in each group unless otherwise stated. A t-test or two-way ANOVA was used to make comparisons when appropriate. If differences were detected by ANOVA, individual groups were compared with the Student-Newman-Keuls test. A Mann-Whitney rank sum t-test or Kruskal-Wallis one-way ANOVA was used to make comparisons for data converted to percentage. A probability of P < 0.05 was accepted as significant for all comparisons.
RESULTS
CH Increases O2·− and Decreases H2O2 Levels
Exposure to CH resulted in a significant increase in right ventricular hypertrophy expressed as right ventricular/left ventricular plus septum weight as an index of pulmonary hypertension (control: 0.28 ± 0.01 and CH: 0.53 ± 0.02; P < 0.001). Consistent with previous studies from our laboratory in isolated pressurized vessels (7, 24, 33), DHE fluorescence was increased in PASMC from CH rats compared with controls (Fig. 1). Dismutation of O2·− by PEG-SOD reduced fluorescence in both groups. DHE fluorescence was not altered by PEG-catalase in PASMC from control animals, suggesting that DHE selectively detects O2·− over H2O2. However, the augmented DHE fluorescence in PASMC from CH rats was diminished by preincubation with PEG-catalase.
Fig. 1.

Chronic hypoxia (CH) increases pulmonary artery smooth muscle cell (PASMC) O2·−. Representative images (A) and summary data (B) showing background-subtracted mean fluorescence intensity (MFI) of dihydroethidium (DHE) in PASMC from control and CH rats treated with vehicle, polyethylene glycol-superoxide dismutase (PEG-SOD) (50 U/ml), or PEG-catalase (250 U/ml). Fluorescence images were digitally inverted to provide better contrast and visibility of immunofluorescence. Values are means ± SE; n = 3–5 animals per group; *P < 0.05 vs. control, #P < 0.05 vs. vehicle.
In initial measurements of H2O2 in untreated pulmonary artery segments, detection of Amplex Red fluorescence was below the linear range of the standard curve (Fig. 3, A, inset, and B). Therefore, O2·− and subsequent H2O2 production was stimulated with SOTS-1 in experiments described by Figs. 2 and 3. SOTS-1 is an azo-compound that can be thermally decomposed in aqueous solution to generate O2·− at physiological pH and exhibits a half-life of ∼90 min (22). Because there is limited information regarding the use of SOTS-1 to generate O2·− in biological systems, we examined DHE and Amplex Red fluorescence to increasing concentrations of SOTS-1 (0.01, 0.1, 1.0 mM). SOTS-1 resulted in a concentration-dependent increase in DHE fluorescence that was abolished by coincubation with PEG-SOD but not PEG-catalase (P = 0.073; Fig. 2). In addition to increasing cellular O2·− levels, SOTS-1 caused a concentration-dependent increase in H2O2 as measured by Amplex Red fluorescence (Fig. 3). SOTS-1 at 1 mM caused profound PASMC contraction and saturated the Amplex Red fluorescence signal. Therefore, experiments in Fig. 3B were performed using the lowest concentration (0.01 mM SOTS-1) that resulted in a detectable increase in H2O2 production. In contrast to effect of CH on O2·− levels, CH caused a decrease in pulmonary arterial SOTS-1-induced H2O2 production (Fig. 3). PEG-catalase significantly reduced H2O2 levels in both groups.
Fig. 2.
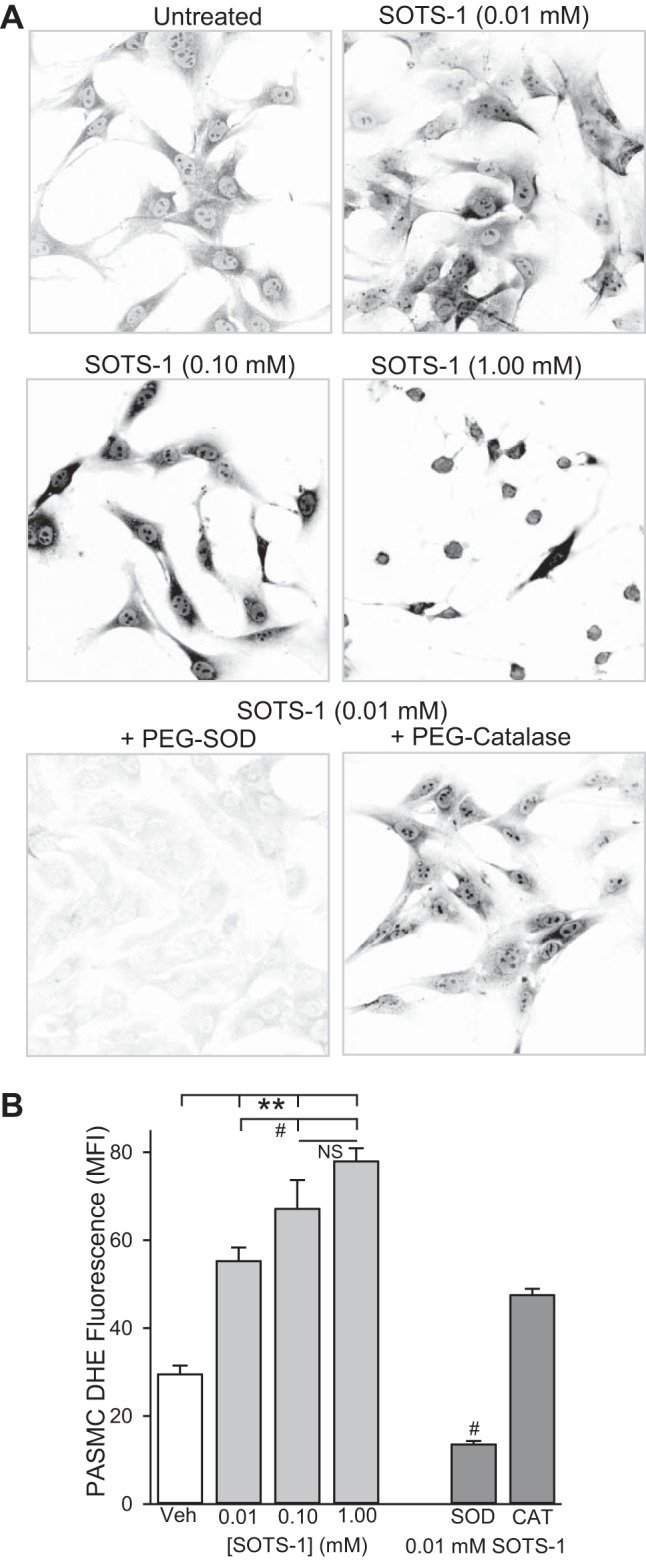
Di-(4-carboxybenzyl) hyponitrite (SOTS-1) increases PASMC O2·− levels. Representative images (A) and summary data (B) showing background-subtracted MFI of DHE in PASMC from control rats treated with increasing concentrations of SOTS-1 (0.01, 0.1, 1.0 mM) or SOTS-1 (0.01 mM) plus PEG-SOD (50 U/ml) or PEG-catalase (250 U/ml). Fluorescence images were digitally inverted to provide better contrast and visibility of immunofluorescence. Values are means ± SE; n = 3–5 animals per group. **P < 0.001 vs. vehicle; #P < 0.05 vs. 0.01 mM SOTS-1 treatment.
CH Decreases SOD Expression and Activity
One possibility for the reciprocal levels of O2·− and H2O2 following CH is dysregulation of SOD, which has been shown in several models of pulmonary hypertension (4, 13, 15, 26, 35, 40). Consistent with this possibility, CH decreased Cu/Zn SOD1 (cytosolic) and Cu/Zn SOD3 (extracellular) protein expression (Fig. 4, A and B). This correlated with diminished total SOD and Cu/Zn SOD (SOD1 and SOD3) activity (Fig. 4C). There was no detectable difference in Mn SOD2 (mitochondrial) protein expression or activity between groups.
Fig. 4.
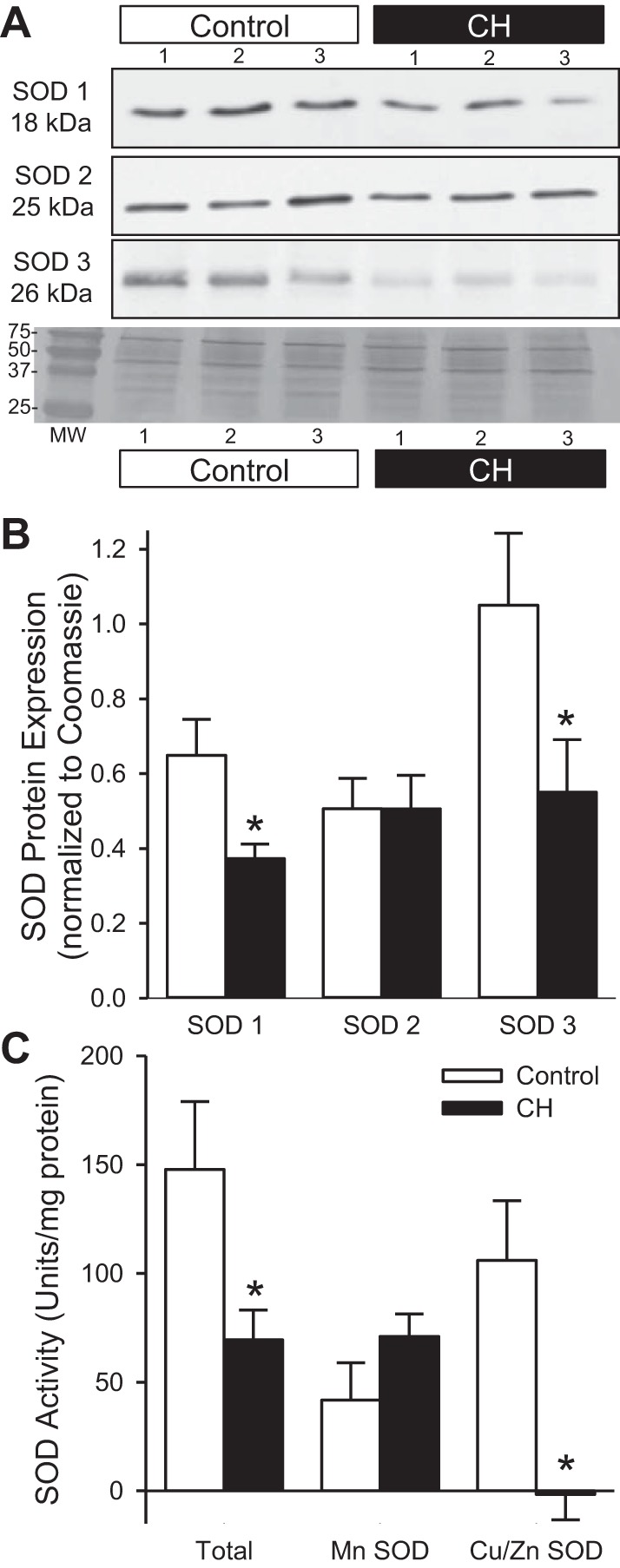
CH decreases pulmonary arterial SOD expression and activity. Representative Western blots (A) and summary data (B) for SOD1 (18 kDa), SOD2 (25 kDa), and SOD3 (26 kDa) protein expression in isolated pulmonary arteries from control and CH rats. SOD expression was normalized to total protein using the corresponding Coomassie-stained blot. C: summary data for total SOD, MnSOD (SOD 2), and Cu/ZnSOD (SOD 1 and 3) activity in isolated pulmonary arteries from control and CH rats. Values are means ± SE; n = 6–9 animals per group; *P < 0.05 vs. control. MW, molecular weight.
ASIC1-depedent SOCE is Increased in SOD1−/− Mice
Our laboratory has previously demonstrated that SOD1−/− mice develop spontaneous pulmonary hypertension (40). To determine whether the expression and activity of SOD1 influences ASIC1 activity, we examined SOCE in isolated-pressurized pulmonary arteries (average inner diameter: 138 ± 17 μm). SOCE was greater in pulmonary arteries from SOD1−/− compared with SOD1+/+ mice (Fig. 5). The specific ASIC1a inhibitor, PcTX1, attenuated SOCE in both groups, and SOCE was not different between groups in the presence of PcTX1. Expression of ASIC1 was similar in pulmonary arteries from SOD1+/+ and SOD−/− mice (Fig. 5, B and C). Expression of SOD1 was only present in pulmonary arteries from SOD1+/+ mice, confirming knockout of this protein (Fig. 5B). These data suggest that a decrease in SOD1 expression and activity is sufficient to increase ASIC1-mediated SOCE, and this is further independent of a change in ASIC1 protein expression, as previously reported (32).
Fig. 5.
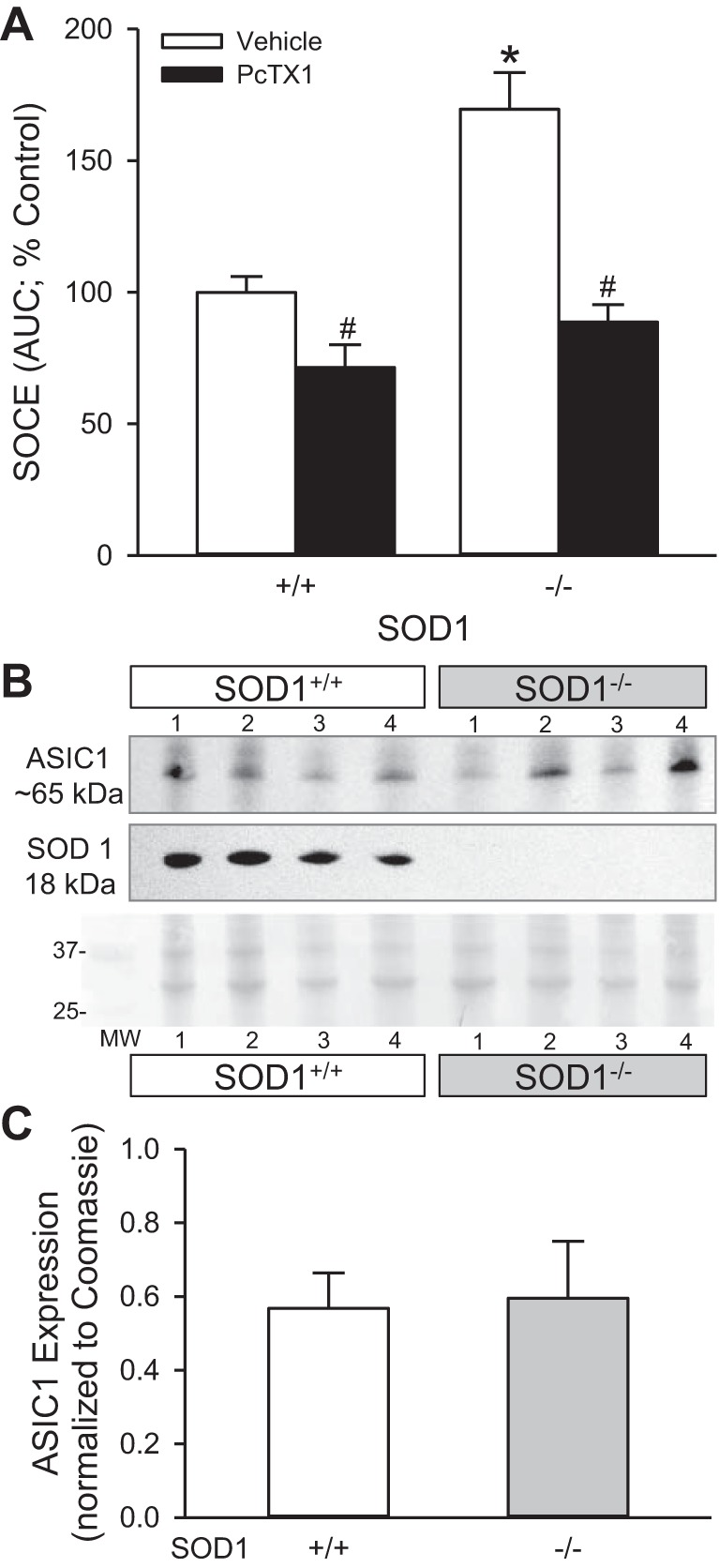
Acid-sensing ion channel 1 (ASIC1)-dependent store-operated Ca2+ entry (SOCE) is increased in pulmonary arteries from SOD1−/− mice. A: SOCE responses in isolated pressurized pulmonary arteries from SOD1+/+ and SOD1−/− mice in the presence or absence of psalmotoxin-1(PcTX1; 20 nM). Representative Western blots (B) and summary data (C) for ASIC1 (∼65 kDa) and SOD1 (18 kDa) protein expression in isolated pulmonary arteries from SOD1+/+ and SOD1−/− mice. ASIC1 expression was normalized to total protein using the corresponding Coomassie-stained blot. Values are means ± SE; n = 4–5 animals per group; *P < 0.05 vs. SOD1+/+; #P < 0.05 vs. respective vehicle.
CH Increases Pulmonary Arterial Antioxidant Capacity
If SOD is the rate-limiting step in the formation of H2O2, then addition of PEG-SOD should increase H2O2 levels to similar levels in pulmonary arteries from control and CH rats. PEG-SOD and the SOD mimetic, tiron, increased Amplex Red fluorescence in both groups above that observed in untreated tissue (Fig. 3, A, inset and Fig. 6A), and, therefore, SOTS-1 was not utilized in these experiments. However, H2O2 levels were still less in pulmonary arteries from CH rats (Fig. 6B), suggesting that antioxidant mechanisms other than SOD contribute to the overall H2O2 levels. Consistent with this possibility, preventing the catalysis of H2O2 with blockers of catalase (AT) and GPx (mercaptosuccinic acid) increased H2O2 levels similarly in pulmonary arteries from control and CH rats (Fig. 6B).
Fig. 6.
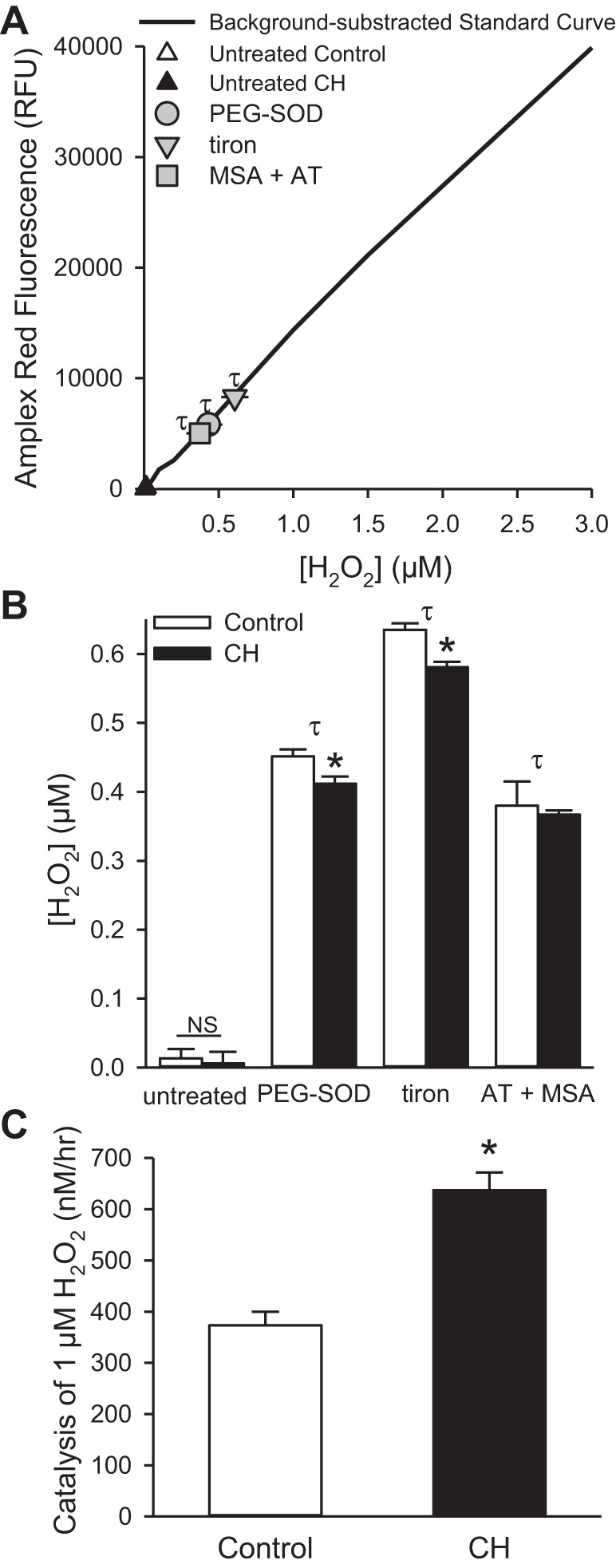
CH increases pulmonary arterial antioxidant capacity. A: Amplex Red RFU from control intrapulmonary arteries following treatment with PEG-SOD (50 U/ml), the SOD mimetic, tiron (10 mM), or inhibition of catalase and glutathione peroxidase (GPx) with 3-amino-1,2,4-triazole (AT; 5 mM) and mercaptosuccinic acid (MSA; 3 mM) are within the linear range of the background-subtracted standard curve. B: summary data of H2O2 levels in intrapulmonary arteries from control and CH rats following above treatments. C: rate of H2O2 catalysis (1 μM) by control and CH intrapulmonary arteries measured by Amplex Red fluorescence. Values are means ± SE; n = 6–8 animals per group; τP < 0.05 vs. untreated pulmonary arteries; *P < 0.05 vs. pulmonary arteries from control rats.
To specifically examine differences in the rate of H2O2 catalysis, we measured the rate of H2O2 decomposition following administration of 1 μM H2O2 to the arteries from each group (Fig. 6C). After 1 h, pulmonary arteries from CH rats had degraded significantly more H2O2 (637 ± 34 nM) compared with control tissue (373 ± 26 nM). Together, these data suggest that CH increases the rate of H2O2 catalysis in pulmonary arteries.
CH Increases GPx Expression and Activity
Next we determined the effect of CH on the expression and activity of the two main enzymes responsible for H2O2 degradation in the vasculature, catalase, and GPx-1. Although there was a tendency for catalase expression and activity to be decreased following CH, there was not a statistically significant difference between control and CH groups (Fig. 7). Conversely, GPx-1 expression and activity were augmented in pulmonary arteries from CH rats (Fig. 7).
Fig. 7.
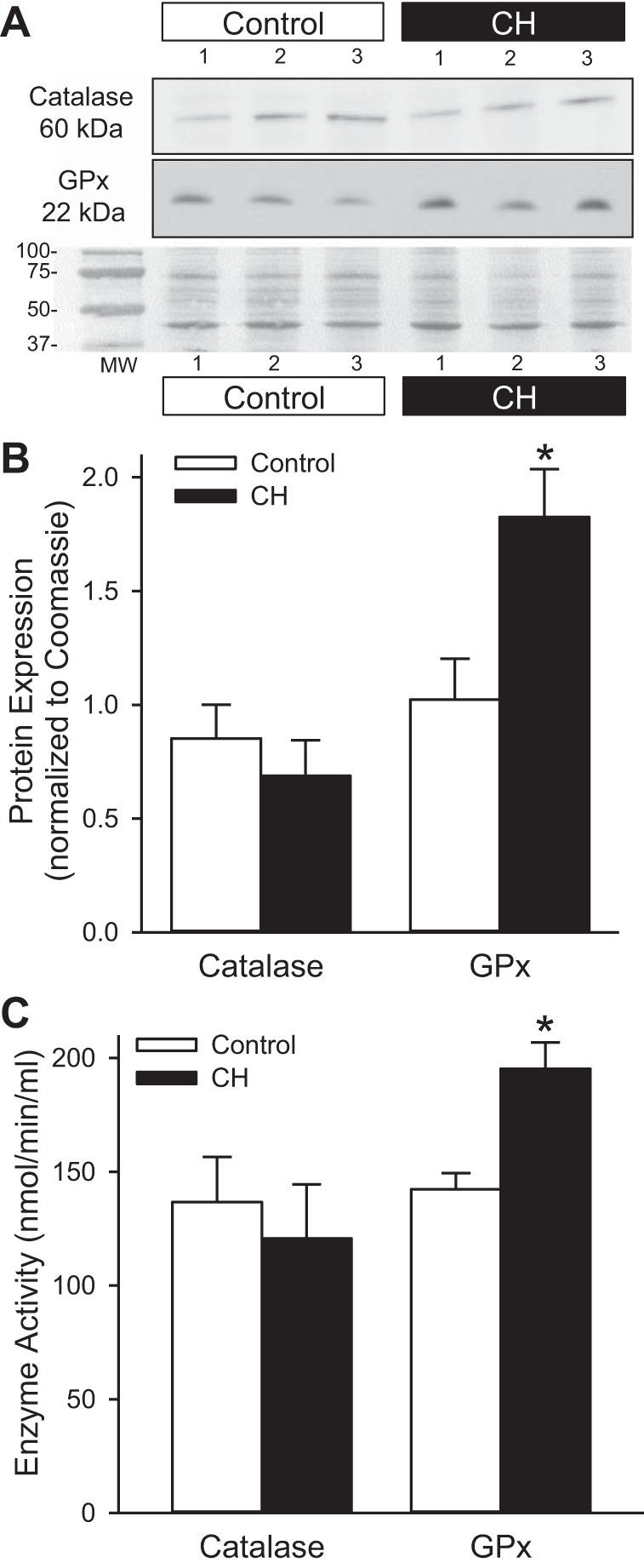
GPx expression and activity are increased following CH. Representative Western blots (A) and summary data showing protein expression (B) and enzyme activity (C) of catalase and GPx in isolated pulmonary arteries from control and CH rats. Protein expression was normalized to total protein using the corresponding Coomassie-stained blot. Values are means ± SE; n = 6 animals per group; *P < 0.05 vs. control.
H2O2 Inhibits ASIC1-dependent SOCE
We examined the PcTx1-sensitive component of SOCE to determine the influence of H2O2 on ASIC1. In time-control experiments, we found that SOCE was similar between the first and second response (Fig. 8B). Therefore, data are expressed as the PcTX1-sensitive component of SOCE, which is derived from subtracting the SOCE response in the presence of PcTX1 from that of vehicle (Fig. 8C). Consistent with previous data from our laboratory (23, 32), we observed an increase in ASIC1-mediated SOCE in PASMC from CH rats (Fig. 9). Decreasing levels of H2O2 with PEG-catalase (Fig. 9A) or the GPx mimetic, ebselen (Fig. 9B), increased SOCE in PASMC from control rats but had no further effect to increase SOCE in PASMC from CH rats. Addition of H2O2 largely attenuated SOCE in PASMC from both control and CH rats (Fig. 9A). Likewise, increasing H2O2 levels by blocking GPx with mercaptosuccinic acid diminished ASIC1-dependent SOCE (Fig. 9B). Together these data suggest that H2O2 negatively regulates ASIC1 activity.
Fig. 8.

ASIC1-dependent SOCE in PASMC. A: representative experiment showing SOCE and determination of area under curve (AUC). Representative traces and summary data showing time-control (B) or SOCE (C) responses in the presence of vehicle or PcTX1 separated by 30 min. SOCE responses are expressed as the PcTX1-sensitive component (open bar). Values are means ± SE; n = 3 experiments per group; *P < 0.05 vs. vehicle.
Fig. 9.

H2O2 inhibits ASIC1-dependent SOCE. PcTX1-sensitive SOCE in PASMC from control and CH rats in the presence of vehicle (physiological saline solution) (A), PEG-catalase (250 U/ml), or H2O2 (25 μM) and vehicle (DMSO) (B), ebselen (30 μM), or mercaptosuccinic acid (3 mM). Values are means ± SE; n = 4–10 experiments from at least 3 animals per group; *P < 0.05 vs. control; #P < 0.05 vs. respective vehicle treatment.
DISCUSSION
Previous studies from our laboratory show that the contribution of ASIC1 to the development of hypoxic pulmonary hypertension is independent of an increase in ASIC1 protein expression (32). Changes in ROS occur following CH that could potentially alter the activity of ASIC1 because ASIC1 is redox sensitive. The goal of the current study was to better understand the changes in ROS that occur following CH and how these changes influence ASIC1-dependent SOCE. The major findings from this study are that 1) CH increased PASMC O2·− and decreased pulmonary arterial H2O2 production; 2) CH attenuated Cu/Zn SOD expression and activity while increasing GPx expression and activity; 3) H2O2 inhibited ASIC1-dependent SOCE in PASMC from both control and CH animals; and 4) catalase and the GPx mimic, ebselen, augmented ASIC1-mediated SOCE in PASMC from control rats but had no further effect in PASMC from CH rats. These data suggest that, under control conditions, H2O2 inhibits ASIC1-dependent SOCE. Furthermore, H2O2 levels are decreased following CH as a result of diminished O2·− dismutase and increased H2O2 catalysis through GPx, leading to the augmented ASIC1-dependent SOCE (Fig. 10).
Fig. 10.

Summary diagram showing the effect of CH on H2O2 levels and ASIC1-dependent SOCE. A: H2O2 inhibits ASIC1-dependent SOCE in PASMC from control animals. B: H2O2 levels are decreased following CH as a result of diminished O2·− dismutation and increased H2O2 catalysis through GPx. This leads to the augmented ASIC1-dependent SOCE in PASMC following CH.
Endogenous O2·− and H2O2 are physiologically important second messengers that regulate a variety of downstream signaling pathways. However, an imbalance in ROS homeostasis has been implicated in the progression of various disease states, including pulmonary hypertension. Several lines of evidence, including studies from our laboratory, suggest that CH increases O2·− generation from multiple sources, including mitochondrial electron transport chain, xanthine oxidase, cytochrome P-450, nitric oxide synthase, and NADPH oxidase (34, 38, 43). Our laboratory further demonstrates an important contribution of O2·− to induce RhoA activation, myofilament Ca2+ sensitization, and enhanced vasoconstriction in hypertensive pulmonary arteries (7, 8, 24, 33). Consistent with our previous studies, which examined O2·− generation in whole artery preparations (7, 24, 33), in the current study, we found that O2·− is specifically elevated in PASMC from CH rats. Pretreatment of PASMCs with PEG-SOD decreased O2·− in both groups and prevented the CH-induced increase in O2·−. DHE is expected to selectively detect O2·− over H2O2, and this is demonstrated in PASMC from control animals. Interestingly, we found that PEG-catalase diminished O2·− in PASMC from CH animals. We also observed a tendency for PEG-catalase to decrease SOTS-1-induced O2·− (i.e., DHE fluorescence); however, this did not reach statistical significance (Fig. 2; P = 0.073). Although the reason(s) for this effect of PEG-catalase is unclear, it is possible that catalase is removing a H2O2-mediated inhibition of SOD (20, 21, 29, 56). Exogenous H2O2 has been shown to inactivate Cu/ZnSOD through oxidation of copper at the enzyme active site (20, 21). In theory, PEG-catalase would increase SOD activity. Under conditions such as CH and SOTS-1 where there is more O2·− substrate for SOD, the effect of PEG-catalase to enhance SOD activity resulting in increased O2·− dismutation may be more apparent compared with control conditions. Although we did not specifically measure the effect of PEG-catalase on SOD activity, important studies from Wedgwood et al. (56) have recently reported this in the fetal pulmonary circulation. Relative to fetal control lambs, DHE fluorescence was increased, and SOD3 activity was decreased in a lamb model of persistent pulmonary hypertension of the newborn (PPHN) (56). This was further exacerbated in fetal PPHN lambs ventilated with 100% O2. Catalase treatment decreased DHE fluorescence similar to levels observed in fetal control lambs and greatly augmented SOD3 activity in ventilated PPHN lambs (56). In addition, Wedgwood et al. (56) show that exogenous H2O2 increases and PEG-catalase decreases SOD3 mRNA expression. This correlation between H2O2 and SOD expression is consistent with our current findings, where CH resulted in a decrease in H2O2 levels and SOD1 and SOD3 expression.
In contrast to effects of CH to increase O2·− levels, we found that CH exposure resulted in a paradoxical decrease in H2O2 levels. Although O2·− is generally associated with contraction of pulmonary arteries, both contraction and relaxation have been observed in response to H2O2 (reviewed in Ref. 25). This varied response to H2O2 in the pulmonary circulation appears to be dependent on the level of vascular tone present and the concentration of H2O2 applied. At physiological concentrations, H2O2 is a vasodilatory and antiproliferative signaling molecule (9, 30). In response to acute hypoxia, basal production of H2O2 in the rat pulmonary circulation is diminished (3, 30). This prompted the general notion that, under normoxic conditions, H2O2 inhibits smooth muscle contraction, whereas under hypoxia H2O2 levels fall, resulting in hypoxic pulmonary vasoconstriction. A decrease in H2O2 levels has also been reported following long-term exposure to CH (6, 13, 40, 41), as well as in other experimental models of spontaneous pulmonary hypertension and in humans with pulmonary arterial hypertension (4, 6, 40). This decrease in H2O2 is thought to contribute to proproliferative and antiapoptotic effects that are, in part, mediated by hypoxia-inducible factor 1α (HIF-1α) and involve a decrease in Kv1.5 channel expression and elevated [Ca2+]i levels (23).
To achieve a detectable Amplex Red signal in the current study, it was necessary to stimulate H2O2 production with the addition of SOTS-1, PEG-SOD, tiron, or mercaptosuccinic acid. Determination of H2O2 level was made in dissected intrapulmonary arteries as opposed to whole lung tissue, which may have contributed to the low detection levels. This may be further complicated by the fact that Amplex Red only detects extracellular H2O2. To address some of these questions, we examined both the upstream involvement of SOD and downstream involvement of catalase and GPx in the regulation of H2O2 levels.
Superoxide is highly reactive and rapidly dismutates to H2O2; therefore, an additional means of regulating O2·− and H2O2 levels is through SOD. Indeed, a number of studies have implicated the downregulation of various SOD isoforms in the development of pulmonary hypertension (4, 6, 13, 36, 40). Although SOD can be regulated by a variety of transcription factors, HIF-1α and HIF-2α directly bind the hypoxia-responsive element of SOD2 and SOD1 promoter, resulting in negative regulation of gene expression (18, 27, 52). Likewise, a loss of SOD1, SOD2, and SOD3 leads to the hypoxic activation of HIF-1α, suggesting that stabilization of HIF-1α is redox sensitive and closely linked to SOD function and the oxidant/antioxidant equilibrium (19, 45, 46, 48). Fawn-hooded rats, which develop spontaneous pulmonary hypertension, have an epigenetic silencing of SOD2 expression/activity resulting in decreased H2O2 production (4, 6). Although we did not detect differences in SOD2 expression or activity, we did find that CH decreased SOD1 and SOD3 expression and activity, which is consistent with studies from 3- and 10-day-old CH piglets (13, 15). CH also decreases lung SOD3 protein expression and activity in mice (36), whereas overexpression of SOD3 attenuates both hypoxic- and monocrotaline-induced pulmonary hypertension (1, 26, 36).
Our laboratory has recently shown that SOD1−/− mice develop spontaneous pulmonary hypertension, and this response is associated with increases in pulmonary arterial O2·− and decreases in H2O2 levels (40). In the current study, we demonstrate that the loss of SOD1 and subsequent imbalance in O2·−/H2O2 mimics the effects of hypoxia and leads to enhanced ASIC1-dependent SOCE. Similar to our findings following CH (32), this occurs independently of a significant change in ASIC1 protein expression in pulmonary arteries from SOD−/− compared with SOD+/+ mice. Together, these data strongly support the concept that the downregulation of SOD1 and SOD3, and subsequent decrease in H2O2, following CH contributes to ASIC1 activation. Additionally, we found that the addition of either PEG-SOD or tiron improved, but did not normalize, H2O2 levels in pulmonary arteries from CH rats to those of controls. This suggested that CH may additionally augment the enzymatic decomposition of H2O2 by catalase and/or GPx. Consistent with this possibility, we found that expression and activity of GPx-1 was augmented in pulmonary arteries from CH rats. Other studies also indicate that CH exposure in rats results in increased lung GPx-1 (17, 31, 58). Because CH alters the state of various redox couples, it is difficult to determine the physiological impact of increased GPX-1 expression and activity on the overall cellular redox environment. This is further complicated by the fact that redox responses can differ markedly among subcellular compartments (55). Nonetheless, changes to the redox potential represent an important mechanism of ion channel regulation, including regulation of ASIC1a.
Although H2O2 has been shown to increase [Ca2+]i and constriction of pulmonary arteries, these responses were unaffected by removal of extracellular Ca2+ or by nonselective channel inhibitors, arguing against involvement of store- and receptor-operated Ca2+ channels (28, 39). Rather than activation, H2O2 seems to cause a profound inhibition of SOCE in vascular smooth muscle and endothelial cells (14, 16, 37, 47), an effect widely observed in a variety of nonvascular cells (49, 51). This is consistent with our current findings that H2O2 inhibited ASIC1-dependent SOCE in PASMC from both control and CH rats. Consistently, removing H2O2 through the addition of PEG-catalase or the GPx mimetic, ebselen, increased SOCE in PASMC from control animals. In contrast, these inhibitors were without effect on SOCE following CH, suggesting that inhibition of SOCE by endogenous H2O2 is absent in PASMC from CH animals. However, whether this is due to a direct effect of decreased H2O2 levels on ASIC1 or an indirect effect mediated by a shift in the cytosolic redox environment is currently unknown. Furthermore, whether CH induces an oxidized or reduced shift in the redox potential is unclear, as both have been reported (41, 55).
Reducing agents such as dithiothreitol and glutathione potentiate ASIC1 current, increase pH sensitivity, and decrease channel inactivation (2, 10, 11, 60). In cortical neurons, this leads to enhanced acid-induced membrane depolarization and Ca2+ influx (11). Site-directed mutagenesis identified involvement of extracellular cysteine and lysine residues in the modulation of ASICs by oxidizing and reducing agents, respectively (11). Other studies show that intracellular COOH-terminal cysteine residues are important in the intersubunit disulfide bond formation induced by H2O2. The formation of intersubunit disulfide bonds inhibits acid-evoked currents and reduces the amount of ASIC1a present on the cell surface (60). It is therefore likely that redox reagents affect both trafficking of ASIC1 to the membrane as well as altering of the kinetics of channel activation.
Oxidative stress reflects an imbalance in the production of free radicals and antioxidant defenses. In lungs, the main enzymatic antioxidants are SOD, catalase, GPx, and thioredoxin (38). In the current study, we show that CH decreases pulmonary arterial SOD1 and SOD3 and increases GPx-1 expression and activity. The resulting decrease in H2O2 production and increase in degradation lead to an overall reduction in pulmonary arterial H2O2 levels following CH. H2O2, either through direct mechanisms or indirectly through redox modification, inhibits ASIC1-dependent SOCE. Loss of endogenous H2O2 following CH leads to enhanced ASIC1-mediated SOCE (Fig. 10). Further studies are necessary to specifically address key questions regarding how the redox changes during CH correlate to ASIC1 activity and the mechanisms involved in this response.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-92598 (to N. L. Jernigan), HL-111084 (to N. L. Jernigan), HL-88151 (to L. V. Gonzalez Bosc), HL-88192 (to T. C. Resta), and HL-95640 (to B. R. Walker).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.R.P., L.M.H., T.R.Y., and N.L.J. performed experiments; D.R.P. and N.L.J. analyzed data; D.R.P. and N.L.J. prepared figures; D.R.P. and N.L.J. drafted manuscript; D.R.P., L.M.H., T.R.Y., T.C.R., L.V.G.B., B.R.W., and N.L.J. edited and revised manuscript; D.R.P., L.M.H., T.R.Y., T.C.R., L.V.G.B., B.R.W., and N.L.J. approved final version of manuscript; T.C.R. and N.L.J. conception and design of research; T.C.R., L.V.G.B., B.R.W., and N.L.J. interpreted results of experiments.
ACKNOWLEDGMENTS
The authors thank Carlos Nitta for technical assistance.
REFERENCES
- 1.Ahmed MN, Zhang Y, Codipilly C, Zaghloul N, Patel D, Wolin M, Miller EJ. Extracellular superoxide dismutase overexpression can reverse the course of hypoxia-induced pulmonary hypertension. Mol Med 18: 38–46, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrey F, Tsintsadze T, Volkova T, Lozovaya N, Krishtal O. Acid sensing ionic channels: Modulation by redox reagents. Biochim Biophys Acta 1745: 1–6, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Archer SL, Huang J, Henry T, Peterson D, Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res 73: 1100–1112, 1993 [DOI] [PubMed] [Google Scholar]
- 4.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thebaud B, Husain AN, Cipriani N, Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121: 2661–2671, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benarroch EE. Acid-sensing cation channels: structure, function, and pathophysiologic implications. Neurology 82: 628–635, 2014 [DOI] [PubMed] [Google Scholar]
- 6.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Broughton BRS, Jernigan NL, Norton CE, Walker BR, Resta TC. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol 298: L232–L242, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broughton BRS, Walker BR, Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 294: L797–L806, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Burke TM, Wolin MS. Hydrogen peroxide elicits pulmonary arterial relaxation and guanylate cyclase activation. Am J Physiol Heart Circ Physiol 252: H721–H732, 1987 [DOI] [PubMed] [Google Scholar]
- 10.Cho J, Askwith C. Potentiation of acid-sensing ion channels by sulfhydryl compounds. Am J Physiol Cell Physiol 292: C2161–C2174, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Chu XP, Close N, Saugstad JA, Xiong ZG. ASIC1a-specific modulation of acid-sensing ion channels in mouse cortical neurons by redox reagents. J Neurosci 26: 5329–5339, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chu X, Xiong Z. Physiological and pathological functions of acid-sensing ion channels in the central nervous system. Curr Drug Targets 13: 263–271, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297: L596–L607, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elliott SJ, Doan TN. Oxidant stress inhibits the store-dependent Ca(2+)-influx pathway of vascular endothelial cells. Biochem J 292: 385–393, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fike CD, Dikalova A, Slaughter JC, Kaplowitz MR, Zhang Y, Aschner JL. Reactive oxygen species-reducing strategies improve pulmonary arterial responses to nitric oxide in piglets with chronic hypoxia-induced pulmonary hypertension. Antioxid Redox Signal 18: 1727–1738, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Florea SM, Blatter LA. The effect of oxidative stress on Ca2+ release and capacitative Ca2+ entry in vascular endothelial cells. Cell Calcium 43: 405–415, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Frank L. Protection from O2 toxicity by preexposure to hypoxia: lung antioxidant enzyme role. J Appl Physiol Respir Environ Exercise Physiol 53: 475–482, 1982 [DOI] [PubMed] [Google Scholar]
- 18.Gao YH, Li CX, Shen SM, Li H, Chen GQ, Wei Q, Wang LS. Hypoxia-inducible factor 1α mediates the down-regulation of superoxide dismutase 2 in von Hippel-Lindau deficient renal clear cell carcinoma. Biochem Biophys Res Commun 435: 46–51, 2013 [DOI] [PubMed] [Google Scholar]
- 19.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol 91: 807–819, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Hink HU, Santanam N, Dikalov S, McCann L, Nguyen AD, Parthasarathy S, Harrison DG, Fukai T. Peroxidase properties of extracellular superoxide dismutase: role of uric acid in modulating in vivo activity. Arterioscler Thromb Vasc Biol 22: 1402–1408, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Hodgson EK, Fridovich I. The interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide: inactivation of the enzyme. Biochemistry 14: 5294–5299, 1975 [DOI] [PubMed] [Google Scholar]
- 22.Ingold KU, Paul T, Young MJ, Doiron L. Invention of the first azo compound to serve as a superoxide thermal source under physiological conditions: Concept, synthesis, and chemical properties. J Am Chem Soc 119: 12364–12365, 1997 [Google Scholar]
- 23.Jernigan NL, Herbert LM, Walker BR, Resta TC. Chronic hypoxia upregulates pulmonary arterial ASIC1: a novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am J Physiol Cell Physiol 302: C931–C940, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones RD, Morice AH. Hydrogen peroxide—an intracellular signal in the pulmonary circulation: involvement in hypoxic pulmonary vasoconstriction. Pharmacol Ther 88: 153–161, 2000 [DOI] [PubMed] [Google Scholar]
- 26.Kamezaki F, Tasaki H, Yamashita K, Tsutsui M, Koide S, Nakata S, Tanimoto A, Okazaki M, Sasaguri Y, Adachi T, Otsuji Y. Gene transfer of extracellular superoxide dismutase ameliorates pulmonary hypertension in rats. Am J Respir Crit Care Med 177: 219–226, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Kojima I, Tanaka T, Inagi R, Kato H, Yamashita T, Sakiyama A, Ohneda O, Takeda N, Sata M, Miyata T, Fujita T, Nangaku M. Protective Role of Hypoxia-Inducible Factor-2α against Ischemic Damage and Oxidative Stress in the Kidney. J Am Soc Nephrol 18: 1218–1226, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Lin MJ, Yang XR, Cao YN, Sham JSK. Hydrogen peroxide-induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 292: L1598–L1608, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Marklund SL. Properties of extracellular superoxide dismutase from human lung. Biochem J 220: 269–272, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, Gurtu R, Archer SL. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res 90: 1307–1315, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Nakanishi K, Tajima F, Nakamura A, Yagura S, Ookawara T, Yamashita H, Suzuki K, Taniguchi N, Ohno H. Effects of hypobaric hypoxia on antioxidant enzymes in rats. J Physiol 489: 869–876, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nitta CH, Osmond DA, Herbert LM, Beasley BF, Resta TC, Walker BR, Jernigan NL. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 306: H41–H52, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norton CE, Broughton BR, Jernigan NL, Walker BR, Resta TC. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxid Redox Signal 18: 1777–1788, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nozik-Grayck E, Stenmark KR. Role of reactive oxygen species in chronic hypoxia-induced pulmonary hypertension and vascular remodeling. Adv Exp Med Biol 618: 101–112, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Nozik-Grayck E, Suliman HB, Majka S, Albietz J, Van Rheen Z, Roush K, Stenmark KR. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 295: L422–L430, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nozik-Grayck E, Suliman HB, Majka S, Albietz J, Van Rheen Z, Roush K, Stenmark KR. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 295: L422–L430, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pande J, Dimmers G, Akolkar G, Skelley L, Samson SE, Grover AK. Store operated Ca2+ entry dependent contraction of coronary artery smooth muscle: Inhibition by peroxide pretreatment. Cell Calcium 51: 149–154, 2012 [DOI] [PubMed] [Google Scholar]
- 38.Perez-Vizcaino F, Cogolludo A, Moreno L. Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir Physiol Neurobiol 174: 212–220, 2010 [DOI] [PubMed] [Google Scholar]
- 39.Pourmahram GE, Snetkov VA, Shaifta Y, Drndarski S, Knock GA, Aaronson PI, Ward JPT. Constriction of pulmonary artery by peroxide: role of Ca2+ release and PKC. Free Radic Biol Med 45: 1468–1476, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Ramiro-Diaz JM, Nitta CH, Maston LD, Codianni S, Giermakowska W, Resta TC, Gonzalez Bosc LV. NFAT is required for spontaneous pulmonary hypertension in superoxide dismutase 1 knockout mice. Am J Physiol Lung Cell Mol Physiol 304: L613–L625, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reeve HL, Michelakis E, Nelson DP, Weir EK, Archer SL. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J Appl Physiol 90: 2249–2256, 2001 [DOI] [PubMed] [Google Scholar]
- 42.Rehman J, Archer S. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: The Warburg Model of pulmonary arterial hypertension. In: Membrane Receptors, Channels and Transporters in Pulmonary Circulation, edited by Yuan JXJ, Ward JPT. New York, NY: Humana, 2010, pp. 171–185 [DOI] [PubMed] [Google Scholar]
- 43.Resta T, Broughton BS, Jernigan N. Reactive oxygen species and rhoa signaling in vascular smooth muscle: Role in chronic hypoxia-induced pulmonary hypertension. In: Membrane Receptors, Channels and Transporters in Pulmonary Circulation, edited by Yuan JXJ, Ward JPT. New York, NY: Humana, 2010, pp. 355–373 [DOI] [PubMed] [Google Scholar]
- 44.Resta TC, Chicoine LG, Omdahl JL, Walker BR. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol Heart Circ Physiol 276: H699–H708, 1999 [DOI] [PubMed] [Google Scholar]
- 45.Sasabe E, Yang Z, Ohno S, Yamamoto T. Reactive oxygen species produced by the knockdown of manganese-superoxide dismutase up-regulate hypoxia-inducible factor-1α expression in oral squamous cell carcinoma cells. Free Radic Biol Med 48: 1321–1329, 2010 [DOI] [PubMed] [Google Scholar]
- 46.Sato K, Morimoto N, Kurata T, Mimoto T, Miyazaki K, Ikeda Y, Abe K. Impaired response of hypoxic sensor protein HIF-1α and its downstream proteins in the spinal motor neurons of ALS model mice. Brain Res 1473: 55–62, 2012 [DOI] [PubMed] [Google Scholar]
- 47.Schach C, Xu M, Platoshyn O, Keller SH, Yuan JXJ. Thiol oxidation causes pulmonary vasodilation by activating K+ channels and inhibiting store-operated Ca2+ channels. Am J Physiol Lung Cell Mol Physiol 292: L685–L698, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Suliman HB, Ali M, Piantadosi CA. Superoxide dismutase-3 promotes full expression of the EPO response to hypoxia. Blood 104: 43–50, 2004 [DOI] [PubMed] [Google Scholar]
- 49.Suzuki Y, Yoshimaru T, Inoue T, Ra C. Discrete generations of intracellular hydrogen peroxide and superoxide in antigen-stimulated mast cells: Reciprocal regulation of store-operated Ca2+ channel activity. Mol Immunol 46: 2200–2209, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Szasz T, Thompson JM, Watts SW. A comparison of reactive oxygen species metabolism in the rat aorta and vena cava: focus on xanthine oxidase. Am J Physiol Heart Circ Physiol 295: H1341–H1350, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tornquist K, Vainio PJ, Bjorklund S, Titievsky A, Dugue B, Tuominen RK. Hydrogen peroxide attenuates store-operated calcium entry and enhances calcium extrusion in thyroid FRTL-5 cells. Biochem J 351: 47–56, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tuller ER, Beavers CT, Lou JR, Ihnat MA, Benbrook DM, Ding WQ. Docosahexaenoic acid inhibits superoxide dismutase 1 gene transcription in human cancer cells: The involvement of peroxisome proliferator-activated receptor α and hypoxia-inducible factor-2α signaling. Mol Pharmacol 76: 588–595, 2009 [DOI] [PubMed] [Google Scholar]
- 53.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature 386: 173–177, 1997 [DOI] [PubMed] [Google Scholar]
- 54.Ward JPT, McMurtry IF. Mechanisms of hypoxic pulmonary vasoconstriction and their roles in pulmonary hypertension: new findings for an old problem. Curr Opin Pharmacol 9: 287–296, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D, Schumacker PT. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res 106: 526–535, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wedgwood S, Lakshminrusimha S, Fukai T, Russell JA, Schumacker PT, Steinhorn RH. Hydrogen peroxide regulates extracellular superoxide dismutase activity and expression in neonatal pulmonary hypertension. Antioxid Redox Signal 15: 1497–1506, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci 14: 461–471, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.White CW, Jackson JH, McMurtry IF, Repine JE. Hypoxia increases glutathione redox cycle and protects rat lungs against oxidants. J Appl Physiol 65: 2607–2616, 1988 [DOI] [PubMed] [Google Scholar]
- 59.Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci USA 101: 6752–6757, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zha X, Wang R, Collier D, Snyder P, Wemmie J, Welsh M. Oxidant regulated inter-subunit disulfide bond formation between ASIC1a subunits. Proc Natl Acad Sci USA 106: 3573–3578, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]