

Abstract
Background
Ataluren was developed to restore functional protein production in genetic disorders caused by nonsense mutations, which are the cause of cystic fibrosis (CF) in 10% of patients..
Methods
This randomized, double-blind, placebo-controlled study enrolled 238 patients ≥6 years with nmCF to receive oral ataluren 10 mg/kg in the morning, 10 mg/kg mid-day, and 20 mg/kg in the evening or matching placebo for 48 weeks. The primary endpoint was relative change in % predicted forced expiratory volume in one second (FEV1) at Week 48; the secondary endpoint was the rate of pulmonary exacerbations. This study is registered with ClinicalTrials.gov, number NCT00803205.
Findings
There was no statistically significant difference in relative change from baseline in % predicted FEV1between ataluren and placebo at Week 48(-2•5% vs -5•5%, p=0.1235). The rate of pulmonary exacerbations was not statistically different between treatment arms (rate ratio 0.77 (95% CI 0.57, 1.05), p=0.0992). However, post hoc analysis of the subgroup of patients not using chronic inhaled tobramycin showed a 5.7% difference in relative change from baseline in % predicted FEV1 between ataluren and placebo at Week 48 (-0.7% vs -6.4%, nominal p=0•008, adjusted for multiplicity p = 0•024) and 40% fewer exacerbations in ataluren-treated patients (OR 0.60 (95% CI 0•42, 0•86), nominal p=0•006, adjusted for multiplicity p = 0•018).
Interpretation
While there was no statistically significant improvement in lung function or exacerbation rate in the ITT population of cystic fibrosis patients with nonsense mutations treated with ataluren, treatment might be beneficial for nmCF patients not receiving chronic inhaled tobramycin.
Keywords: Cystic fibrosis, Nonsense mutation, Pulmonary exacerbation, Forced expiratory volume, Cystic fibrosis transmembrane conductance regulator
Introduction
Cystic fibrosis (CF) is a disabling and life-threatening autosomal recessive disorder resulting from mutations that cause dysfunction in the cystic fibrosis transmembrane conductance regulator (CFTR). Approximately 10% of patients have CF due to a Class I nonsense mutation in at least one allele of the CFTR gene.1Available therapies for treatment of lung manifestations of CF, such as inhaled antibiotics and dornasealfado not address the underlying defect. Ivacaftor, a recently approved potentiator for the Class III G551D CFTR mutation, does not target the defect associated with CF caused by nonsense mutations (nmCF),2 which are associated with severe CF lung disease.3–7
Ataluren (PTC124) (3-[5-(2-fluorophenyl)-[1,2,4]oxadiazol-3-yl]-benzoic acid) is an orally bioavailable, investigational agent that promotes ribosomal readthrough of premature termination codons,8-11 and production of full-length, functional CFTR11-12 and is being developed as a disease-modifying treatment for diseases due to nonsense mutations, including nmCF. Phase 2 studies in 77 patients (ages 6-57 years) with nmCF receiving oral ataluren for periods of 14 days through 12 weeks showed that ataluren was well tolerated12-15 and that ataluren treatment can generate production of full-length CFTR protein, localized in the apical nasal epithelium.12 Nasal potential difference (NPD) assessments exhibited a pattern of improved total chloride transport from baseline to end of treatment followed by a return to baseline values in the post treatment period in three of the four Phase 2 studies.13-15 These open-label studies had short-term treatment durations and were conducted in a small number of investigative sites with experience in assessing NPD. While no significant changes in % predictedFEV1 were observed in short 14 day studies,12,14,17other small, non-significant, positive effects were observed in a 12-week study, suggesting a time-dependent effect.15 This Phase 3 clinical trialwas designed to evaluate the efficacy and safety of ataluren in nmCF and is the largest randomized controlled trial ever conducted in nmCF patients.
Methods
Study Oversight
The protocol was reviewed and approved by the ethics committee or institutional review board of each participating institution. Written informed consent was obtained from patients or their custodians prior to patient screening. External oversight of the study was provided by a Study Steering Committee and an independent Data Monitoring Committee.
Study Design and Participants
This randomised, double-blind, placebo-controlled, phase 3 trial was performed between August 2009 and November 2011 at 36 sites in 11 countries in North America and Europe.
Inclusion criteria included age ≥6 years; abnormal NPD (a less electrically negative value than 5 mV for total chloride conductance [Δchloride-free+isoproterenol]); sweat chloride >40 mmol/L; documentation of the presence of a nonsense mutation in at least one allele of the CFTR gene; the ability to perform a valid, reproducible spirometry test that demonstrated an FEV1≥40% and ≤90% of predicted for age, gender, and height16,17; confirmed screening laboratory values within pre-specified central laboratory ranges; and willingness and ability to comply with all study procedures, scheduled visits, and restrictions.
Major exclusion criteria included known hypersensitivity to ataluren; any change in a chronic treatment or prophylaxis regimen for CF within four weeks of starting study treatment; systemic aminoglycoside antibiotics treatment within two weeks before the date of baseline NPD assessment; major complications of lung disease within eight weeks prior to start of study; history of Grade 3 or higher creatinine elevation due to aminoglycoside nephrotoxicity; and ongoing participation in any other clinical trial (see Supplementary Appendix for all inclusion and exclusion criteria).
Randomisation and masking
Eligible subjects were randomized in a 1:1 fashion to ataluren or placebo by means of interactive response technology, using a block size of 4 within the cells created by the interaction of the 3 stratification factors (age [<18 vs ≥18 years], chronic inhaled antibiotic use [yes vs no], and % predicted FEV1 [40 to <65% vs ≥65 to 90%]). The random allocation sequence was generated by the contract research organization. Patients, medical and ancillary staff, the study investigators, and the sponsor were masked to treatment assignment. Only designated personnel at the contract research organization had access to treatment assignments.
Procedures
The active study medication ataluren, a powder for oral suspension, was dosed based on milligrams per kilogram of body weight three times per day - 10 mg/kg in the morning, 10 mg/kg at mid-day, and 20 mg/kg in the evening for 48 weeks. Placebo was identical in appearance. The primary endpoint for this study was the relative change in % predicted FEV1 from baseline to Week 48 as assessed by spirometry. Spirometry was performed at screening, at randomization, and every eight weeks during the 48 week study duration. All sites used a study-specific spirometer in their assessment of FEV1, FVC,and FEF25-75, while adhering to American Thoracic Society/European Respiratory Society guidelines.18,19
The secondary endpoint was the rate of pulmonary exacerbations. Comprehensive respiratory event data forms were used throughout the study to record the occurrence of signs and symptoms, hospitalization, and the incidence of investigator-defined pulmonary exacerbation. Events meeting the modified Fuchs' criteria20 (at least 4 of 12 signs and symptoms with or without intravenous antibiotic treatment; Supplementary Appendix), were counted over the 48-week treatment period to determine the pulmonary exacerbation rate for each arm. The remainder of the endpoints presented were tertiary or exploratory based on the predetermined statistical analysis plan. NPD measurement and sweat chloride concentration, by pilocarpineiontophoresis, were assessed every 16 weeks.21,22 Safety was evaluated via adverse events reporting and laboratory assessments. Ataluren plasma concentrations were obtained prior to, and two hours after, the morning dose every 16 weeks.
In vitro studies
The hypothesis that aminoglycosides interfere with ataluren at the ribosomal level was explored in a functional cell-based translation assay. In this assay, the firefly luciferase gene23 containing a premature stop codon at position 190 is inserted into human embryonic kidney (HEK293) cells growing in a medium containing fetal bovine serum. Translational readthrough at the site of the nonsense mutation is directly correlated to the level of luciferase-mediated light production (chemoluminescence) produced in the cells.
Post-hoc, ataluren was also tested in combination with tobramycin to determine it'seffect on tobramycin's antibacterial activity when both compounds were present. P. aeruginosa bacteria were grown in rich media and used in a checkerboard titration experiment with both ataluren and tobramycin present at concentrations ranging from 0·24 to 125 μg/mL and 0·1 to 6·25 μg/mL, respectively.24 The minimum inhibitory concentration (MIC) of tobramycin was determined at all combinations.
Statistical Analysis
The sample size was calculated to detect a 6% difference between ataluren and placebo in mean relative change in % predicted FEV1 from baseline at Week 48, the primary endpoint, with power of >0.90 using a 2 sided t-test at a 0.05 significance level. The targeted treatment difference (6%) was in the range of that previously observed with approved CF therapies. Patients were stratified by age (<18 vs ≥18 years), chronic inhaled antibiotic use (yes vs no), and % predicted FEV1 (40 to <65% vs ≥65 to 90%). Efficacy analyses were performed on the intent-to-treat (ITT) population, defined as those patients who had at least 1 valid post-baseline spirometry measurement.
The predetermined statistical plan called for Mixed-model repeated-measures (MMRM) analysis to compare the difference in relative change in % predicted FEV1 between ataluren and placebo at 48 weeks as well as the average treatment effect across all post-baseline visits. The relative strengths of the interactions between treatment and the prespecified stratification factors for FEV1 were determined by a model that included baseline FEV1 and the other stratification factors. In the case that the interaction was statistically significant, results within the subgroup are presented. The analysis of pulmonary exacerbations was performed using the generalized linear model by the GENMOD procedure (SAS v 9·2) with a negative binomial distribution for the number of exacerbations to test the ratio of exacerbation rates. MMRM was used for all continuous tertiary endpoints (Supplementary Appendix).
A p-value is reported as nominal when not adjusted for multiplicity. For the post-hoc analysis of subgroups determined by type of concomitant inhaled antibiotic (colistin, aztreonam, or tobramycin), p-values were adjusted for multiplicity by a factor of 3. This study is registered with ClinicalTrials.gov, number NCT00803205.
Role of the funding source
The study sponsor oversaw trial management, data collection, statistical analyses, and the writing and review of the report. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication.
Results
238 patients (the as-treated population) were randomly assigned to the ataluren 10, 10, 20 mg/kg treatment arm, or to the placebo arm. Six patients did not have a valid post-baseline spirometry measurement, therefore the ITT population comprised 232 patients, 116 in each treatment arm (Supplementary Figure 1). Types of nonsense mutation were generally well-balanced between treatment groups, and the most commonly present in one or both alleles of the CFTR gene were W1282X (86 patients), G542X (83 patients), R1162X (22 patients), and R553X (18 patients). There were no clinically significant differences among genotype combinations. Treatment arms were generally well balanced by demographic characteristics (Table 1) and by stratification factors (see Supplementary Table 1). In the ITT population, 127 patients (54·7%)were treated chronically with inhaled antibiotics and usage was similar across treatment groups (Table 1). Other concomitant medications (eg, pancreatic enzyme preparations, mucolytics, selective beta-2-adrenoreceptor agonists) also were generally well balanced between treatment groups (see Supplementary Table 2). There were 20 patients in the ataluren arm and 14 patients in the placebo arm that withdrew from the study, and this difference in withdrawals between treatment groups was not statistically significant (p=0.36). 204 patients completed the study. A summary of concomitant medications is presented in Supplementary Table 2.
Table 1. Patient Demographics and Baseline Characteristics (ITT Population).
| Characteristic | Treatment Arm | |
|---|---|---|
|
| ||
| Ataluren | Placebo | |
|
| ||
| N=116 | N=116 | |
|
| ||
| Age, years | ||
| Mean (SD) | 22.8 (10.18) | 23.2 (9.32) |
| Median | 22.0 | 22.0 |
| Range | 6, 49 | 8, 53 |
|
| ||
| Sex, n (%) | ||
| Male | 60 (51.7%) | 58 (50.0%) |
| Female | 56 (48.3%) | 58 (50.0%) |
|
| ||
| Body weight, kg | ||
| Mean (SD) | 53.5 (13.94) | 56.0 (13.15) |
| Median | 54.4 | 57.2 |
| Range | 21, 105 | 24, 93 |
|
| ||
| % predicted FEV1 at baseline | ||
| Mean (SD) | 62.1 (13.62) | 60.2 (15.14) |
| Median | 63.4 | 59.0 |
| Range | 38.4, 90.3 | 36.2, 92.6 |
|
| ||
| Sweat chloride at baselinea | N=114 | N=111 |
| Mean (SD) | 100.1 (14.22) | 96.6 (15.93) |
| Median | 101.5 | 100.0 |
| Range | 22.5, 128.0 | 22.0, 117.5 |
|
| ||
| Inhaled antibiotic use at randomizationb | 64 (55.2%) | 63 (53.4%) |
|
| ||
| Aminoglycoside (tobramycin) | 44 (37.9%) | 42 (35.6%) |
|
| ||
| Colistin | 30 (25.9%) | 22 (18.6%) |
|
| ||
| Aztreonam | 10 (8.3%) | 8 (6.8%) |
The results of historical sweat chloride tests were used for eligibility assessment of 2 patients whose centrally analyzed baseline sweat chloride value was subsequently found to be <40 mEq/L.
A patient was considered to be using inhaled antibiotics at baseline even if the patient was in the “off” portion of an intermittent cycling regimen. Patients may have been using >1 antibiotic at baseline.
Note: There were no statistical differences between the 2 arms
Abbreviations: FEV1 = forced expiratory volume in 1 second, SD = standard deviation
Spirometry
At baseline, mean % predicted FEV1, the primary endpoint,was 62·1% for ataluren vs 60·2% for placebo. The mean relative change in % predicted FEV1 from baseline to Week 48 was -2·5% for ataluren vs -5·5% for placebo (Table 3). The difference between ataluren and placebo in mean relative change in % predicted FEV1 from baseline to Week 48 was 3·0% favoring ataluren (p=0·124). The average difference between ataluren and placebo in mean relative change in % predicted FEV1 across all post-baseline visits favored ataluren by 2·5% (p=0·048) (Figure 1). A test of the interaction between treatment and each pre-specified stratification factor demonstrated that only the use of chronic inhaled antibiotics was significant (p = 0·022, multiplicity-adjusted). Based on this finding, further analysis of this subgroup was performed (see Supplementary Appendix).
Table 3. Pulmonary Exacerbation Rate Using the Modified Fuchs Definition by Chronic Inhaled Antibiotic Use (ITT Population).
| Population | Ataluren Rate | Placebo Rate | Rate Ratio |
|---|---|---|---|
| Overall | N=116 | N=116 | |
| Ratea (95% CI) | 1.42 (1.06, 1.95) | 1.78 (1.28, 2.68) | 0.77 (0.57, 1.05) |
| Nominal p-value | -- | -- | 0.0992 |
| Inhaled antibiotics = no | N=52 | N=53 | -- |
| Ratea (95% CI) | 1.30 (0.76, 1.85) | 2.15 (1.51, 278) | 0.57 (0.37, 0.89) |
| Nominal p-value | -- | -- | 0.0134 |
| Inhaled antibiotics = yes | N=64 | N=63 | -- |
| Ratea (95% CI) | 1.52 (1.01, 2.02) | 1.46 (0.97, 1.96) | 1.01 (0.67, 1.54) |
| Nominal p-value | -- | -- | 0.952 |
| Inhaled tobramycin = no | N=72 | N=74 | |
| Ratea (95% CI) | 1.42 (0.92,1.93) | 2.18 (1.62,2.74) | 0.60 (0.42, 0.86) |
| Nominal p-value | 0.006 | ||
| Inhaled tobramycin = yes | N=44 | N=42 | |
| Ratea (95% CI) | 1.42 (0.89,1.95) | 1.06 (0.67,1.45) | 1.33 (0.79,2,25) |
| Nominal p-value | 0.297 |
Rates are from observed data over 48 weeks and rate ratio was estimated using a generalized linear model.
Abbreviations: CI = confidence interval, ITT = intent-to-treat
Figure 1. Mean Relative Change in % predicted FEV1 from Baseline to Week 48 (ITT Population).
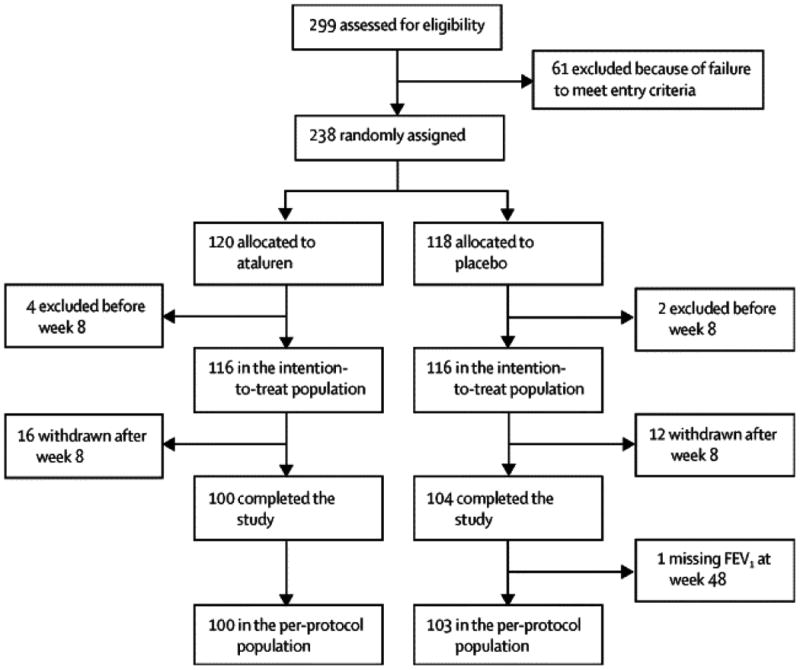
The plotted values represent observed data (±SEM). The dotted lines represent the average treatment effect across all post-baseline visits. The p-values were obtained from a mixed-model repeated measures (MMRM) analysis. Covariates were baseline % predicted FEV1, treatment, visit, treatment-by-visit interaction, baseline % predicted FEV1-by-visit interaction, and the stratification factors of baseline inhaled antibiotics (yes vs no), baseline age (<18 vs ≥18 years), and baseline % predicted FEV1 (40 to <65% vs ≥65 to 90%).
Abbreviations: FEV1 = forced expiratory volume in 1 second, ITT = intent-to-treat, MMRM = mixed-model repeated-measures
A posthoc analysis of the subgroup of patients receiving chronic inhaled tobramycin vs patients not receiving chronic inhaled tobramycin was performed. In patients not receiving chronic inhaled tobramycin, the difference in mean relative change from baseline in % predicted FEV1 at Week 48 was 5·7% favoring ataluren (95% CI = 1·5 to 10·1, nominal p=0·008, adjusted for multiplicity p = 0·024), with a mean change from baseline of -0·7% in the ataluren arm, and -6·4% in the placebo arm (Figure 2). In contrast, in patients who received chronic inhaled tobramycin, there was no significant difference in mean relative change from baseline in % predicted FEV1 at Week 48 between ataluren and placebo. The use of other inhaled antibiotics (eg, colistin and aztreonam) did not appear to modify the treatment effect of ataluren (see Supplementary Appendix Figure 4).
Figure 2. Week 48 Relative Change in % predicted FEV1 by Chronic Inhaled Antibiotic Use (ITT Population).
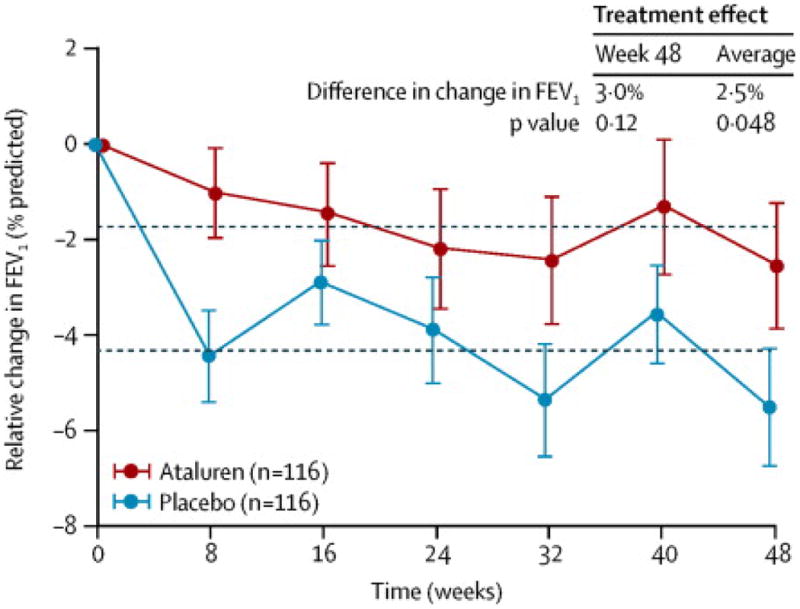
The plotted values represent observed data (±SEM). The plot on the left includes patients who were not taking tobramycin (either no antibiotics or antibiotics other than tobramycin) chronically at baseline. The plot on the right includes patients who were taking tobramycin (either alone or in combination with other antibiotics) chronically.
Abbreviations: %FEV1 = % predicted forced expiratory volume in 1 second, ITT = intent-to-treat
There was some missing data; this is permitted in the MMRM analysis. The percentage of missing values in % predicted FEV1 was not unusual; the missing values were 12% (104/840) in the ataluren treatment group and 9% (73/820) in the placebo group. Most of the missing values were due to not meeting the ATS (American Thoracic Society) criteria for valid spirometry measurement.
Pulmonary exacerbation rate
The mean (SD) pulmonary exacerbation rate over 48 weeks (the secondary endpoint), was 23% lower in the ataluren arm than placebo (Table 3): 1·42 (2·00) for ataluren vs 1·78 (2·15) for placebo, resulting in a rate ratio of 0·77 (95% CI 0·57, 1·05; p = 0·099).
In patients not receiving chronic inhaled antibiotics, the mean (SD) pulmonary exacerbation rate was 43% lower in the ataluren arm than placebo (Table 3):1·30 (1·97) for ataluren vs 2·15 (2·32) for placebo resulting in a rate ratio of 0·57 (95% CI 0·37, 0·89; nominal p=0·0134). In patients receiving chronic inhaled antibiotics, the pulmonary exacerbation rates were similar in both treatment arms.
In patients not receiving chronic inhaled tobramycin, the mean (SD) pulmonary exacerbation rate was 40% lower in the ataluren arm: 1·42 (2·15) for ataluren vs 2·18 (2·44) for placebo, resulting in a rate ratio of 0·60 (95% CI 0·42, 0·86; nominal p = 0·006, adjusted for multiplicity p = 0·018).
The use of other inhaled antibiotics did not appear to modify the treatment effect of ataluren. Pulmonary exacerbation rate was also assessed using other definitions (see Supplementary Table 7). Times to first and second pulmonary exacerbation were also assessed; differences favoring ataluren were seen in time to second exacerbation (see Supplementary Appendix).
Sweat Test and Nasal Potential Difference
The mean change in sweat chloride concentration from baseline to Week 48 was small in both ataluren (Δ = -1·3 mmol/L [95% CI =-3·1, 0·5]) and placebo (Δ = -0·6 mmol/L [95% CI = -2·7, 1·5]), resulting in a non-significant difference between treatment arms of 0·7 mmol/L; p=0·9919 (see Supplementary Table 8).
Total chloride transport (the change induced by zero chloride plus isoproteronol) was assessed by NPD. The mean change in total chloride transport from baseline to Week 48 was small in both ataluren (Δ = 0·31 mV [95% CI = -0·69, 1·32]) and placebo (Δ = 0·14 mV [95% CI = -0·99, 1·27]), resulting in a non-meaningful difference between treatment arms of 0·17 mV; p=0·8587 (see Supplementary Table 9).
A chloride transport response, defined as at least a -5 mV improvement in the change induced by zero chloride plus isoproteronol at Week 48 vs baseline, was observed in similar frequencies in both ataluren and placebo arm 13 (13·0%) patients in the ataluren arm, and 16 (15·4%) patients in the placebo arm). A hyperpolarized chloride transport value, defined as a change induced by zero chloride plus isoproteronol that is at least as electrically negative as -5 mV, was also observed in similar frequencies in both arms; ataluren arm = 6·0% and placebo = 5·8% (see Supplementary Table 10).
Other Endpoints
Evaluations of other endpoints of this study, ie, cough frequency, inflammation, computerized tomography, weight/BMI, and health-related quality of life (HRQL), showed results which were not statistically significant, and are included in the Supplementary Appendix.
Safety and compliance
In the as-treated population, defined as all patients who received at least one dose of blinded study treatment, 120 patients were exposed to ataluren 10, 10, 20 mg/kg (range, 1·7 to 55·3 weeks), and 118 patients were exposed to placebo (range, 0·7 to 55·9 weeks). The median compliance rate, based on study drug accountability, was ∼90% for ataluren compared to ∼85% for placebo.
Concomitant medications were generally well-balanced between treatment groups. A summary of all concomitant medications is presented in Supplementary Table 2.
Safety profiles were generally similar for ataluren and placebo, except for the occurrence of creatinine elevations (see Supplementary Table 14). Most treatment emergent adverse events were of mild (Grade 1) or moderate (Grade 2) severity, and no life-threatening adverse events were reported (Table 4). Most serious adverse events reported in this study were CF pulmonary exacerbations and were considered unrelated to ataluren treatment. Eight patients in the ataluren arm and three patients in the placebo arm discontinued treatment due to an adverse event (Table 4).
Table 4. Summary of Safety.
| Parameter, n (%) | Treatment Arm | |
|---|---|---|
| Ataluren N = 120 |
Placebo N = 118 |
|
| ≥1 Treatment emergent adverse event | 118 (98.3%) | 115 (97.5%) |
| ≥1 Treatment emergent serious adverse event | 45 (37.5%) | 48 (40.7%) |
| Adverse events by severity | ||
| Grade 1 (mild) | 18 (15.0%) | 20 (16.9%) |
| Grade 2 (moderate) | 81 (67.5%) | 65 (55.1%) |
| Grade 3 (severe) | 19 (15.8%) | 30 (25.4%) |
| Grade 4 (life-threatening) | 0 | 0 |
| Grade 4 (death) | 0 | 0 |
| Discontinued treatment due to adverse event | 8 (6.7%) | 3 (2.5%) |
| Treatment emergent adverse events leading to discontinued treatmenta | ||
| Congenital, familial and genetic disorders | 1 (0.8%) | 0 |
| Cystic fibrosis-related diabetes | 1 (0.8%) | 0 |
| Gastrointestinal disorders | 2 (1.7%) | 2 (1.7%) |
| Abdominal pain | 0 | 1 (0.8%) |
| Abdominal pain upper | 0 | 1 (0.8%) |
| Diarrhoea | 1 (0.8%) | 0 |
| Pancreatitis | 1 (0.8%) | 0 |
| General disorders and administration site conditions | 1 (0.8%) | 0 |
| Pyrexia | 1 (0.8%) | 0 |
| Infections and infestations | 1 (0.8%) | 0 |
| Urinary tract infection | 1 (0.8%) | 0 |
| Nervous system disorders | 0 | 1 (0.8%) |
| Headache | 0 | 1 (0.8%) |
| Renal and urinary disorders | 5 (4.2%) | 0 |
| Hypercreatininaemia | 2 (1.7%) | 0 |
| Nephrolithiasis | 1 (0.8%) | 0 |
| Renal failure | 2 (1.7%) | 0 |
| Respiratory, thoracic and mediastinal disorders | 1 (0.8%) | 1 (0.8%) |
| Pulmonary exacerbation | 1 (0.8%) | 0 |
| Haemoptysis | 0 | 1 (0.8%) |
| Skin and subcutaneous tissue disorders | 2 (1.7%) | 0 |
| Rash | 2 (1.7%) | 0 |
Adverse events leading to study drug discontinuation are displayed alphabetically by MedDRA System Organ Class and from highest to lowest incidence across both treatment arms within each System Organ Class. Patients may have had more than one adverse event.
Overall, there were 18 (15·0%) treatment-emergent events of acute kidney injury (ie, creatinine elevations) in the ataluren arm, vs 1 (0·01%) in the placebo arm. These events were alternatively reported as renal failure, acute renal failure, renal impairment, or hypercreatininemia. Cases of reversible Grade 3-4 creatinine elevations were observed in the ataluren arm, which were associated with the treatment of exacerbations with nephrotoxic systemic antibiotics (aminoglycosides and vancomycin) and, in some cases, dehydration. Therefore, the protocol was amended to prohibit the concomitant use of these antibiotics with ataluren, and to encourage patients to maintain adequate hydration. The changes were implemented ∼7 months into the 27-month study duration and successfully addressed this issue. No deaths occurred during the study. The frequency of all adverse events that occurred >10% by-patient is included in the Supplementary Appendix.
Effect of aminoglycosides on ataluren in vitro
The luciferase assay, performed post hoc, demonstrated that ataluren-induced readthrough of premature stop codons is diminished when cells are coincubated with ataluren and aminoglycosides (tobramycin or gentamicin), but not with colistin or aztreonam, inhaled antibiotics that do not interfere with ribosomal function (see Supplementary Appendix Figure 4). In a separate experiment, also performed post-hoc, it was demonstrated that there is no effect of ataluren on the antibacterial activity of tobramycin (see Supplementary Appendix).
Discussion
Class I (eg, nonsense mutations) and class II (eg, ΔF508) CFTR mutations usually result in complete absence of CFTR at the epithelial cell membrane, generally resulting in a severe disease phenotype. In contrast, CFTR class III (eg, G551D) mutations usually result in full-length CFTR that is appropriately trafficked to the epithelial cell membrane and retains some residual activity. These differences in the underlying CFTR defect may be expected to influence the nature of the clinical efficacy produced by drugs targeting different mutation classes.
Similarly, treatment that alleviates signs and symptoms (eg, inhaled antibiotics)addresses a complication of disease (eg, infection) whereas ataluren treatment is disease-modifying, targeting the underlying cause of disease (nonsense mutations), suggesting that the expectations for clinical efficacy may also be different.
The goal of this study was to assess ataluren's tolerability and activity in CF caused by at least one nonsense mutation using pulmonary function and pulmonary exacerbations as the primary and secondary endpoints, respectively.
Neither relative change in % predicted FEV1at Week 48 nor pulmonary exacerbation rate over 48 weeks were statistically different between ataluren and placebo arms in this trial. For % predicted FEV1, the average treatment effect across all post-baseline visits demonstrated a statistically significant result favoring ataluren vs placebo (2·5%) and a 3% difference favoring ataluren in mean relative change in % predicted FEV1 at Week 48, which was not statistically significant. In both analyses, ataluren treatment resulted in a smaller decrease in FEV1 compared to placebo-treated patients, who had a relative decline in FEV1 of -5·5% (or 3·1% absolute), reflecting the severity of disease in these nmCF patients.
Although an increase in FEV1 has generally been expected in interventional studies in CF, preservation of lung function is also a clinically important goal of CF treatment. For example, treatment of chronic P. aeruginosa infection with inhaled antibiotics is aimed at preservation of lung function for as long as possible.25
The pulmonary exacerbation rate was 23% lower over 48 weeks for ataluren vs placebo (p=0·099).
The three pre-specified stratification factors (baseline age [<18 years vs ≥18 years], use of inhaled antibiotics [yes vs no], and baseline % predicted FEV1 [<65% vs ≥65%]) balanced patient allocation across treatment arms, and were identified a priori as clinically relevant. Particularly, stratification by use of inhaled antibiotics was intended to evenly allocate patients who were receiving these drugs; this group typically comprises a sicker population of patients who have Pseudomonas aeruginosa infection.26 The most commonly used inhaled antibiotic, tobramycin, is an aminoglycoside with a bacterial ribosomal binding mechanism of action.27 Ataluren also acts through modulation of the ribosome to enable readthrough of nonsense mutations. It is likely that the mode of binding of tobramycin may interfere with ataluren's mechanism of action. In vitro testing demonstrating the interference of aminoglycoside antibiotics with ataluren activity supports this clinical finding (see Supplementary Appendix). In a post hoc analysis of patients not receiving chronic inhaled tobramycin there was a 5·7% treatment effect favoring ataluren in FEV1 at Week 48 (nominal p = 0·008; adjusted for multiplicity p = 0·024). Similarly, in patients who were not administered chronic inhaled tobramycin, the pulmonary exacerbation rate was 40% lower in the ataluren arm vs placebo (nominal p = 0·006; adjusted for multiplicity p = 0·018). The disparity observed in multiple endpoints between the subgroup of patients who were not prescribed chronic inhaled tobramycin and the subgroup of patients who were prescribed chronic inhaled tobramycin, supports the hypothesis that inhaled tobramycin may interfere with ataluren's mechanism of action. In a separate post-hoc in vitro experiment, it was demonstrated that there is no effect of ataluren on the antibacterial activity of tobramycin (see Supplementary Appendix).
The use of other inhaled antibiotics did not appear to modify the treatment effect of ataluren. These data support the development of a confirmatory trial targeting the subgroup of patients who do not use chronic inhaled aminoglycosides (panel).
An unexpected result of this study was the lack of difference between treatment groups in NPD, particularly in the context of the ataluren effect on NPD demonstrated in the Phase 2 program.12,14,15 This study was the first to assess NPD in a large, multicenter, long-term, placebo controlled clinical trial. Notably, a pre-specified analysis in this study showed a relatively high NPD “response” rate (ie, change ≥ 5.0 mV from baseline) in the placebo arm. Of placebo-dosed patients, 16 (15·4%) had an NPD response at Week 48. When even larger thresholds for defining NPD response are used (eg, change ≥-8.0 mV from baseline), the ataluren response rate was 6% and the placebo arm response rate was 7.7%, signifying that there were a large number of false positives. These results, and the complexity of performing NPD at many sites that did not have previous experience, may have contributed to the failure to reproduce previous results with this outcome measure.
Sweat chloride concentrations also did not change during this study. Whereas NPD results were positive in the prior Phase 2 studies, sweat chloride results were not. Both sweat chloride concentration and NPD have been utilized as pharmacodynamic endpoints in a number of clinical trials of CFTR restoration therapies, at times with discrepant results.28-30 Organ (eg, sweat gland vs lung) effects may vary due to differences in tissue drug availability, CFTR regulation in different cell types, or the responsiveness of mutant CFTR in different tissue compartments.31 This issue of organ specificity is noted in the 2010 EMA guideline on clinical development of medicinal products for the treatment of CF and may be relevant to the effects of ataluren.
In this study, there was no meaningful change in BMI or CFQ-R respiratory domain score in either the ataluren treatment arm or placebo over 48 weeks, and differences between arms were small and not significant. Neither BMI nor the CFQ-R respiratory score decreased over 48 weeks in the placebo group.
Ataluren was generally well tolerated over 48 weeks in this trial. Safety profiles were similar for ataluren and placebo, other than cases of acute kidney injury (ie, creatinine elevations), the most severe of which were associated with the combination of potentially nephrotoxic antibiotics with ataluren. Thus, the combination of ataluren and aminoglycosides has been prohibited, effectively managing those clinically significant creatinine elevations.
This was a well-designed, well-controlled study of ataluren in nmCF. The main limitation of the study was the choice of a patient population that includes patients who used chronic inhaled tobramycin, which is hypothesized to interfere with ataluren on the ribosomal level. Another limitation of the study was inability to find a biomarker that could be implemented on a global scale and could differentiate ataluren treatment from placebo.
Conclusions
Ataluren has been developed as a potential treatment of the underlying cause of CF in patients with nonsense mutations. In the overall population of this phase 3 study, ataluren showed no statistically significant difference compared to placebo for relative change in % predicted FEV1 at Week 48 and pulmonary exacerbation rate over 48 weeks. Concomitant administration of inhaled tobramycin interfered with readthrough of CFTR nonsense mutations by ataluren in vitro and in the clinic. In nmCF patients not administered chronic inhaled tobramycin, ataluren produced a notable treatment effect compared with placebo in the endpoints of % predicted FEV1and pulmonary exacerbation rate. Given the subgroup effect on spirometry and pulmonary exacerbation rate in patients not receiving chronic inhaled tobramycin, and a favorable safety profile, this study supports further testing of ataluren as a first-in-class, safe and effective treatment for nmCF patients not receiving chronic inhaled tobramycin. A confirmatory Phase 3 efficacy and safety trial of ataluren in nmCF patients not receiving chronic inhaled tobramycin (ACT CF trial) is ongoing.
Supplementary Material
Figure 3.
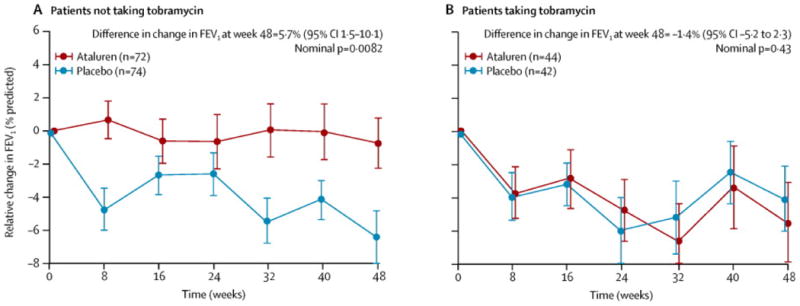
Mean relative change in percent-predicted FEV1 from baseline to week 48, by chronic inhaled antibiotic use in the intention-to-treat population (A) Patients who were not taking tobramycin (either no antibiotics or antibiotics other than tobramycin) chronically at baseline. (B) Patients who were taking tobramycin (either alone or in combination with other antibiotics) chronically. Datapoints show the mean change in FEV1 since baseline at each timepoint; error bars show SE. FEV1=forced expiratory volume in 1 s
Table 2. Summary of % predicted FEV1 at Baseline and Week 48 (ITT Population).
| Visit/Parameter | Treatment Arm (All) |
Treatment Arm (No chronic inhaled tobramycin) |
||
|---|---|---|---|---|
| Ataluren | Placebo | Ataluren | Placebo | |
| Baseline, N | 116 | 116 | 72 | 74 |
| Mean (SD) | 62.09 (13.62) | 60.23 (15.14) | 61.6 (13.11) | 60.5 (15.68) |
| Week 48, N | 100 | 103 | 62 | 63 |
| Mean (SD) | 60.37 (16.68) | 57.18 (16.70) | 61.2 (15.77) | 56.976 (16.84) |
| Δ from baseline to Week 48 | ||||
| Mean (SD) | -1.34 (8.50) | -3.10 (7.39) | -0.3 (7.53) | -3.7 (7.58) |
| P-value1 | 0.1170 | 0.0122 | ||
| Relative Δ from baseline to Week 48 | ||||
| Mean (SD) MMRM P-value2 |
-2.53 (13.25) | -5.50 (12.56) | -0.7 (11.93) | -6.4 (12.64) |
| 0.1235 | 0.00823 | |||
Abbreviations: FEV1 = forced expiratory volume in 1 second, MMRM = mixed model repeated measures, SD = standard deviation
P-value based on t-test comparison of ataluren and placebo.
P-value based on MMRM analysis for comparison of ataluren and placebo.
P-value not adjusted for multiplicity.
Systematic Review.
We searched PubMed up to April 9, 2014, using the search term (“Codon, Nonsense”[Mesh]) AND “Cystic Fibrosis”[Mesh]) AND “Clinical Trial” [Publication Type] to identify clinical trials in patients with nonsense mutation cystic fibrosis (nmCF). We identified 3 prior Phase 2 clinical trials of ataluren in nmCF and 1 prior Phase 2 clinical trial of gentamicin in nmCF. Unlike the current large, long-term, registration-directed trial, these previous trials were small, short-term, proof-of-concept studies. It was appropriate to conduct this Phase 3 trial based on the results of the sponsor's previous Phase 2 studies of nmCF, which showed that ataluren was well-tolerated and effective in restoration of CFTR function as shown by changes in chloride conductance measured by NPD and the restoration of CFTR to the cell surface as seen by immunofluorescence staining of nasal epithelial cells. A review of the therapies recommended by international groups and/or approved by regulatory agencies for treatment of cystic fibrosis (eg, tobramycin, dornase alpha, hypertonic saline) was performed in order to help target an appropriate treatment effect that would determine the sample size.
Interpretation.
In preclinical and Phase 2 clinical studies, ataluren was shown to restore in vitro and in vivo functional CFTR production in genetic disorders caused by nonsense mutations. Building on this earlier work, this Phase 3 double-blind placebo-controlled study was conducted; a post hoc analysis of data showed a statistically significant treatment effect in patients not using inhaled tobramycin on pulmonary function and respiratory exacerbations, suggesting that ataluren can enable readthrough of premature stop codons in patients with CF. This was supported by an in vitro study showing that tobramycin may interfere with ataluren's mechanism of action. Although the primary outcome was not achieved, the findings are encouraging since it demonstrates a positive outcome of a disease-modifying therapy of a corrector of a genetic defect that results in the absence of CFTR on the cell surface.
Acknowledgments
We thank the patients and families who committed their time and effort to this study. We thank the Principal Investigators, supporting investigators, clinical coordinators, the clinical evaluator trainers, clinical evaluators, and study coordinators and INC Research for statistical programming support. We also thank The CFFT-TDNCC and the European Cystic Fibrosis Society Clinical Trials Network, (ECFS-CTN) for their protocol review; the University of Alabama at Birmingham Center for Clinical and Translational Science for their technical assistance; and the patient advocacy organizations, particularly the Cystic Fibrosis Foundation, for the collaboration and support which made this trial possible. Michael W. O'Donnell, a medical writer employed by PTC Therapeutics, Inc., assisted in preparation of this manuscript for publication.
Funding: This study was sponsored by PTC Therapeutics, Inc. with grant support from the Cystic Fibrosis Foundation and the Food and Drug Administration's Office of Orphan Products Development and investigator grant support from the NIH.
Footnotes
Address for Reprints info@ptcbio.com
Contributors: EK, MWK, KB, FJA, JSE, GLE, JB, RJS, SWP, TA, and SMR contributed to study design, data collection, interpretation of the data, intellectual content of the manuscript, writing of the manuscript, and reviewing the final version of the manuscript. PM, IB, IF, AM, DBR, PAW, SAM, CK, SQ, ER, PLZ contributed to study design, data collection, interpretation of the data, and reviewing the final version of the manuscript. ISG and MW contributed to data collection, intellectual content of the manuscript, and reviewing the final version of the manuscript. EMW and AB designed and analyzed the in vitro combination experiments, contributed to the intellectual content of the manuscript, and reviewed the final version of the manuscript.
Conflicts of Interest: JB, GLE, EMW, AB, RJS, SWP, MWO, and TA are employees of PTC Therapeutics, Inc., the sponsor of this clinical trial, and hold financial interests in the company. MWK and SMR received compensation for consultant services from PTC Therapeutics, Inc. during this study.
EK, KB, ISG, and MW received compensation for travel expenses for meetings related to the study. The other authors do not currently hold a direct financial interest in the sponsor's company and they do not have a direct financial interest in the outcome of the study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boyle M, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. The Lancet Respiratory Med. 2013 Apr;1(2):158–163. doi: 10.1016/S2213-2600(12)70057-7. [DOI] [PubMed] [Google Scholar]
- 2.US Food and Drug Administration. Drug Approval Package. Kalydeco (ivacaftor 150 mg Tablets. FDA Web site. [Accessed February 06, 2013]; Updated January 31, 2012. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203188s000TOC.cfm.
- 3.Shoshani T, Augarten A, Gazit E, et al. Association of a nonsense mutation (W1282X), the most common mutation in the Ashkenazi Jewish cystic fibrosis patients in Israel, with presentation of severe disease. Am J Hum Genet. 1992 Jan;50(1):222–8. [PMC free article] [PubMed] [Google Scholar]
- 4.Cystic Fibrosis Genotype-Phenotype Consortium. Correlation between genotype and phenotype in patients with cystic fibrosis. N Engl J Med. 1993 Oct 28;329(18):1308–13. doi: 10.1056/NEJM199310283291804. [DOI] [PubMed] [Google Scholar]
- 5.Kerem E, Kerem B. Genotype-phenotype correlations in cystic fibrosis. PediatrPulmonol. 1996 Dec;22(6):387–95. doi: 10.1002/(SICI)1099-0496(199612)22:6<387::AID-PPUL7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 6.de Gracia J, Mata F, Alvarez A, et al. Genotype-phenotype correlation for pulmonary function in cystic fibrosis. Thorax. 2005 Jul;60(7):558–63. doi: 10.1136/thx.2004.031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKone EF, Goss CH, Aitken ML. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest. 2006 Nov;130(5):1441–7. doi: 10.1378/chest.130.5.1441. [DOI] [PubMed] [Google Scholar]
- 8.Peltz SW, Morsy M, Welch, Jacobson A. Ataluren as an Agent for Therapeutic Nonsense Suppression. Annu Rev Med. 2013;64:407–25. doi: 10.1146/annurev-med-120611-144851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007 May 3;447(7140):87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Hilarion S, Beghyn T, et al. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis. 2012 Aug 31;7:58. doi: 10.1186/1750-1172-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. ProcNatlAcadSci U S A. 2008 Feb 12;105(6):2064–9. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sermet-Gaudelus I, Boeck KD, Casimir GJ, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med. 2010 Nov 15;182(10):1262–72. doi: 10.1164/rccm.201001-0137OC. [DOI] [PubMed] [Google Scholar]
- 13.Clancy JP, Konstan MW, Rowe SM, et al. A Phase 2 study of PTC124 in cystic fibrosis patients harboring premature stop mutations. PediatrPulmonol. 2006;40(Suppl29):301. (abst #269) [Google Scholar]
- 14.Kerem E, Hirawat S, Armoni S, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008 Aug 30;372(9640):719–27. doi: 10.1016/S0140-6736(08)61168-X. [DOI] [PubMed] [Google Scholar]
- 15.Wilschanski M, Miller LL, Shoseyov D, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J. 2011 Jul;38(1):59–69. doi: 10.1183/09031936.00120910. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Dockery DW, Wypij D, Fay ME, Ferris BG., Jr Pulmonary function between 6 and 18 years of age. PediatrPulmonol. 1993 Feb;15(2):75–88. doi: 10.1002/ppul.1950150204. [DOI] [PubMed] [Google Scholar]
- 17.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J RespirCrit Care Med. 1999 Jan;159(1):179–87. doi: 10.1164/ajrccm.159.1.9712108. [DOI] [PubMed] [Google Scholar]
- 18.Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P, Jensen R, Johnson DC, MacIntyre N, McKay R, Navajas D, Pedersen OF, Pellegrino R, Viegi G, Wanger J ATS/ERS Task Force. Standardisation of spirometry. EurRespir J. 2005 Aug;26(2):319–38. doi: 10.1183/09031936.05.00034805. [DOI] [PubMed] [Google Scholar]
- 19.Miller MR, Crapo R, Hankinson J, et al. ATS/ERS Task Force. General considerations for lung function testing. EurRespir J. 2005 Jul;26(1):153–61. doi: 10.1183/09031936.05.00034505. [DOI] [PubMed] [Google Scholar]
- 20.Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994 Sep;331(10):637–42. doi: 10.1056/NEJM199409083311003. comment in N Engl J Med. 1994 Sep;331(10):672-3. [DOI] [PubMed] [Google Scholar]
- 21.Knowles M, Gatzy J, Boucher R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med. 1981 Dec 17;305(25):1489–95. doi: 10.1056/NEJM198112173052502. [DOI] [PubMed] [Google Scholar]
- 22.Knowles MR, Carson JL, Collier AM, Gatzy JT, Boucher RC. Measurements of nasal transepithelial electric potential differences in normal human subjects in vivo. Am Rev Respir Dis. 1981 Oct;124(4):484–90. doi: 10.1164/arrd.1981.124.4.484. [DOI] [PubMed] [Google Scholar]
- 23.Gould SJ, Subramani S. Firefly luciferase as a tool in molecular and cell biology. Anal Biochem. 1988 Nov 15;175(1):5–13. doi: 10.1016/0003-2697(88)90353-3. [DOI] [PubMed] [Google Scholar]
- 24.Brenbaum MC. A method for testing for synergy with any number of agents. J Infect Dis. 1978 Feb;137(2):122–30. doi: 10.1093/infdis/137.2.122. [DOI] [PubMed] [Google Scholar]
- 25.Frederiksen B, Koch C, Høiby N. Antibiotic treatment of initial colonization with Pseudomonas aeruginosa postpones chronic infection and prevents deterioration of pulmonary function in cystic fibrosis. Pediatr Pulmonol. 1997;23:330–335. doi: 10.1002/(sici)1099-0496(199705)23:5<330::aid-ppul4>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 26.Bush A, Alton EWFW, Davies JC, Griesenbach U, Jaffe A, editors. Prog Respir Res. Vol. 34. Bassel: Karger; 2006. Cystic Fibrosis in the 21st Century; pp. 180–186. [Google Scholar]
- 27.Neu HC. Tobramycin: an overview. J Infect Dis. 1976;134(Suppl):S3–19. doi: 10.1093/infdis/134.supplement_1.s3. [DOI] [PubMed] [Google Scholar]
- 28.Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J RespirCrit Care Med. 1998 Feb;157(2):484–90. doi: 10.1164/ajrccm.157.2.9706088. [DOI] [PubMed] [Google Scholar]
- 29.Clancy JP, Bebok Z, Ruiz F, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J RespirCrit Care Med. 2001 Jun;163(7):1683–92. doi: 10.1164/ajrccm.163.7.2004001. [DOI] [PubMed] [Google Scholar]
- 30.Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012 Jan;67(1):12–8. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clancy JP, Bebok Z, Ruiz F, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J RespirCrit Care Med. 2001 Jun;163(7):1683–92. doi: 10.1164/ajrccm.163.7.2004001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.