

Abstract
Purpose
To investigate the clinical and genetic features of late-onset FCD on Tangier, an island in the Chesapeake Bay with an isolated population of approximately 500 individuals.
Design
Observational, cross-sectional study
Methods
A total of 156 individuals born to inhabitants of Tangier Island volunteered to undergo ophthalmic evaluation. Medical history was ascertained prior to examination. All participants underwent anterior segment examination with slit-lamp biomicroscopy. Retroillumination photographs were acquired from affected individuals and the disease severity was compared with individuals from large families ascertained previously. Genomic DNA samples were investigated for the presence of the recently identified risk allele rs613872, an intronic variant of TCF4.
Results
Of the 148 examined individuals who were at least 30 years of age, 32 showed the classical symptoms of late-onset FCD (21.6%), providing a minimum prevalence of 11% among individuals over the age of 50 years. Severity was significantly lower compared to 51 cases from unlinked families, among individuals either 50 to 70 or above 70 years of age (p = 0.05 and 0.01, respectively). Retroillumination photography analyses were suggestive of mild severity when compared with the disease phenotype associated with FCD1 and FCD2-linked families. The rs613872 variant was associated with a higher affectation rate (p=0.01), while the wild-type allele was correlated with a higher proportion of subclinical disease (p=0.01).
Conclusions
In this study population in Tangier, late-onset FCD manifests clinically with a mild phenotype and increased prevalence. The rs613872 variant correlates with increased affectation and a clinical disease phenotype.
Introduction
Fuchs Corneal Dystrophy (FCD) is a progressive, hereditary degenerative condition of the posterior cornea associated with endothelial cell loss, thickening of Descemet membrane and focal excrescences termed guttae. The phenotype was first described by the Austrian ophthalmologist Ernst Fuchs in 1910.1 Hereditary transmission was suggested by Clegg in 19152 and subsequent studies confirmed a familial component to disease.3-5 FCD is considered to affect 4% of the United States population above the age of 40 years and is more prevalent among women.
Recent studies have identified four genetic loci, FCD1, FCD2, FCD3, and FCD4 on chromosome 13, 18, 5 and 9 respectively;6-9 as well as two causally associated genes, TCF89 and SLC4A11.10 Of these, the FCD2 locus appears to be the most common contributor, with approximately 40% of large families in our cohort mapping to this locus.11 An intronic TCF4 intronic variant located at chromosome map location 18q21, rs613872, was recently associated with increased odds of developing FCD,12 and independently confirmed by our group and colleagues.13, 14
The extent to which the genetic susceptibility to FCD may be modifiable by environmental risk factors has yet to be elucidated; height, weight, smoking status and ultraviolet light exposure have been postulated as potential factors but no strong, repeatable correlation has been found.15-17 Determination of risk factors requires appropriately powered studies of populations at risk for developing FCD.
Here we describe the study of FCD among an isolated population on Tangier, an island in Virginia with a population of over 500 related individuals18 and a known presence of disease after an inhabitant presented to our clinic for management of FCD.
Methods
Subjects
Following Descemet Stripping Endothelial Keratoplasty of a 50-year-old male with classic signs of late-onset FCD, we traveled to Tangier to examine eight first- and second-degree relatives of the patient. We acquired a comprehensive pedigree of the island population, and thus the family pedigree was expanded to include the population of Tangier. A total of 156 individuals born to inhabitants of the island volunteered to undergo ophthalmic evaluation. Blood samples of approximately 10–15 mL were collected from all willing participants, and genomic DNA was extracted from white blood cells (Gentra Puregene Blood Kit; Qiagen Inc., Valencia, CA).
To determine the number of current inhabitants, we utilized the most recent published government estimate of 535.18 We then used the most recent US Census data (2000 at time of study), which publishes figures stratified by age range, to develop an estimate of the percentage of individuals above 50.
Examination
Medical history, including medications and comorbidities, was asked of each study participant prior to examination. We inquired into height, weight and smoking status, factors which have been previously associated with FCD.17 All participants underwent anterior segment examination by an ophthalmologist using a Haag-Streit 900 slit-lamp biomicroscope.
To grade severity, we utilized a scale described previously by Krachmer and colleagues.4 This scale includes six levels of severity: negative, defined as up to 12 central guttae; Grade 1, greater than 12 central non-confluent guttae; Grade 2, 1 to 2mm of central confluent guttae; Grade 3, 2 to 5mm of central confluent guttae; Grade 4, more than 5mm of central confluent guttae, and Grade 5, the addition of corneal stromal or epithelial edema to Grade 4 findings. We defined positive affectation as a minimum of one eye with Grade 1 severity in this scale. In this early stage, localization of guttae within the central 5mm zone distinguishes lesions from Hassall-Henle bodies, small extrusions of Descemet membrane that may appear in the periphery with age. Confluency represents an increase in the surface density of guttae such that excrescences appear adjacent to one another, either with direct slit-beam illumination or with corneal retroillumination.
Disease severity was compared with 51 cases from large families ascertained previously and of undetermined genetic linkage, acquired through similar testing in other geographic locations. To quantify and further assess severity, we then utilized slit-lamp retroillumination photography as described previously11 to develop a profile of the age-severity relationship of FCD on the island and compare it to previously reported cohorts with FCD.11, 19
Single Nucleotide Polymorphism (SNP) Genotyping
We interrogated genomic DNA samples of all individuals for rs613872, an intronic TCF4 SNP, as described previously.13 Briefly, polymerase chain reaction (PCR) was performed in 5μl volumes containing 10 ng of genomic DNA, 2.5 μl ABI Taqman SNP genotyping master mix, and 0.125 μl of ABI Taqman genotyping assay mix. Reactions for this SNP were amplified independently in a 9700 thermocycler (Applied Biosystems, Foster City, CA). The cycling parameters consisted of 2 minute incubation at 50°C and denaturation at 95°C for 10 minutes followed by 40 cycles of 10 seconds at 95°C, and 1 minute elongation at 72°C with a final 10 minute extension at 72°C. Amplified products were analyzed for the enrichment of specific alleles in an ABI 7900HT Sequence Detection System (Applied Biosystems).
Exclusion Analyses
All of the known late-onset FCD loci were excluded using closely spaced short tandem repeat (STR) markers. Primer sequences for all the STR markers are as described previously. PCR was completed in GeneAmp 9700 PCR System (Applied Biosystems, Foster city, CA). Briefly, each reaction was carried out in a 5 μl mixture containing 40 ng genomic DNA, various combinations of 10 μM fluorescently-labeled primer pairs, 0.5 μl 10x PCR Buffer (Applied Biosystems), 0.5 μl 10mM dNTP mix, 2.5 mM MgCl2 and 0.2 U of Taq DNA polymerase (Applied Biosystems). Initial denaturation was carried out for 5 minutes at 95°C, followed by 10 cycles of 15 sec at 94°C, 15 sec at 55°C and 30 sec at 72°C and then 20 cycles of 15 sec at 89°C, 15 sec at 55°C and 30 sec at 72°C. The final extension was performed for 10 minutes at 72°C and followed by a final hold at 4°C. PCR products from each DNA sample were pooled and mixed with a loading cocktail containing HD-400 size standards (Applied Biosystems). The resulting PCR products were separated in an ABI 3100 DNA sequencer and analyzed by using GENESCAN 4.0 software packages (Applied Biosystems).
Results
A total of 156 individuals participated in this study, which included approximately half (50.4%) of the predicted island population over 50 years of age. Mean and median ages were 57, with range of 18 to 87 years old. Of 148 individuals at least 30 years of age, 32 were affected (21.6%). Independent of severity, the affectation rate did not significantly increase with age in the study cohort, with age-dependent affectation rates of 19.5% (30-49 years old), 22.9% (50 to 69 years old), and 21.6% (70 years or older). Therefore, a more modest estimate, developed by considering all untested individuals to be unaffected, predicts that at least 11% of all individuals over 50 on the island would be positive. Participants included 92 females and 64 males. Although our studies are de facto limited by the size of the Tangier population, males appeared to be less commonly affected (14.1%) than females (25.3%); this however did not reach statistical significance (p<0.07). We observed no difference in age between the two groups (p<0.47).
Severity
As evidenced by histological analysis of Descemet membrane provided by the proband through endothelial keratoplasty, guttate changes observed during examination were consistent with common late-onset FCD. Severity increased with age, with age-dependent mean Krachmer scores of 1.44 (30 to 50 years old), 2.04 (50 to 70 years old), and 2.50 (70 years or older). Severity in both older groups represented a significant increase relative to the 30-49-year-old group (p=0.03 and p=0.001, respectively); only one individual progressed to transplantation. Severity was mild and significantly decreased relative to 51 cases from unlinked families, in both the 50-69-year-old (p<0.05) and 70-and-older (p<0.02) categories. Assessment of retroillumination photography reveals an increase of 10% annually (p<0.001). Comparisons with previously reported eyes associated with the FCD1 and FCD2 locus confirm a milder severity which develops later in life (Figure 1). Specifically, at age 60, the maximum age in the FCD1 sample, the number of guttae in affected eyes associated with FCD1 is 39.6 times higher (p<0.001) and FCD2 is 13.5 times higher (p<0.001) than in eyes associated with Tangier. There was no difference between left and right eyes (p=0.27).
Figure 1.
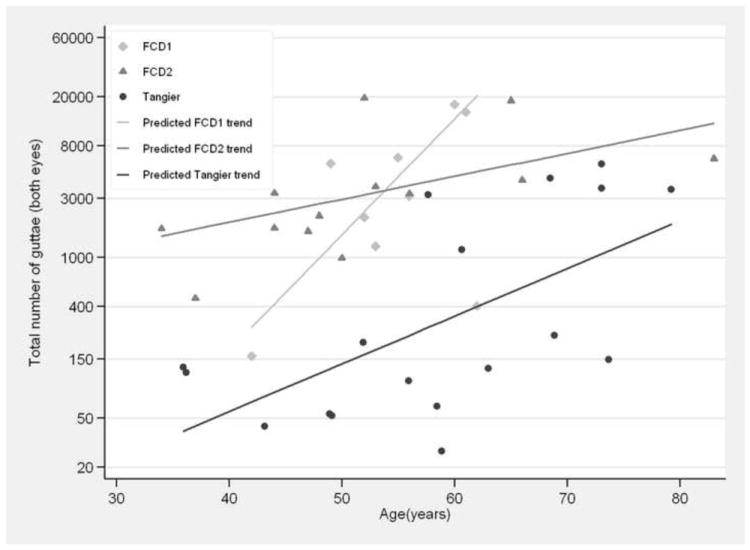
Age-severity relationship as measured by retroillumination photography among individuals affected with Fuchs Corneal Dystrophy. In Tangier, the number of guttae increase by approximately 10% each year (p<0.001). At age 60, the maximum age in the FCD1 sample, the number of guttae in affected eyes associated with FCD1 is 39.6 times higher (p<0.001) and FCD2 is 13.5 times higher (p<0.001) than in eyes associated with Tangier. Data points represent affected individuals for whom retroillumination photographs were acquired.
Risk factors
Next, we assessed our sample for previously identified factors associated with FCD. Average height was decreased significantly among patients with FCD by t-test (p=0.005). After stratifying for gender, this correlation approached but did not reach statistical significance among females (p=0.06) or males (p=0.37). Individuals with FCD also appeared to exhibit decreased weight (p=0.03); stratification by gender revealed no significant difference between affected and unaffected females (p=0.57), and a difference that fell short of significance among males (p=0.06). Approximately 28% of affected individuals demonstrated a history of regularly smoking tobacco compared to 24.8% of unaffected subjects; there was no correlation with FCD affectation by Fisher exact test (p=0.46). The most common medical comorbidities among the sample population were hypertension (35%), diabetes (16%), and dyslipidemia (13%), none of which correlated with disease status.
Genetics
Initially, we excluded all four known late-onset FCD loci (FCD1, FCD2, FCD3, FCD4) using closely spaced STR markers, as described in Methods. Next, we interrogated genomic DNA samples for the rs613872 SNP in TCF4, which revealed that the minor allele is present in Hardy-Weinberg equilibrium in this island population, in a relatively high frequency (0.37) nearing previously-reported cohorts of cases.13 Individuals homozygous for the minor allele comprised a minority (17%) of this isolated population compared to individuals heterozygous (40%) or homozygous (43%) for the wild-type allele. We observed the lowest affectation rate among individuals homozygous for the wild-type allele, of which 8.8% were affected. In contrast, subjects demonstrated a significantly higher affectation rate when heterozygous (24.5% affected, p=0.02) or homozygous (31.8% affected, p<0.02) for the minor allele. There was no interaction between rs613872 status and sex (p<0.64).
We also investigated whether presence of the minor allele would correlate with a difference in clinical severity. Among positive cases, defined by at least 12 central guttae in one eye, individuals demonstrated no significant increase in Krachmer grading if heterozygous (p=0.31) or homozygous (p=0.48) for the minor allele compared to wild-type individuals, consistent with previous reports.13 Since FCD on Tangier Island is manifested by a particularly mild phenotype, we also investigated whether subclinical, or mildest disease severity, may be modified in its presentation by the minor allele. We particularly inquired into individuals with documented guttae but less than 12 in number. Indeed, among individuals considered clinically negative, we observed that a significantly greater proportion of wild-type individuals demonstrated subclinical signs of FCD than those with the intronic variant (p=0.01; Figure 2).
Figure 2.
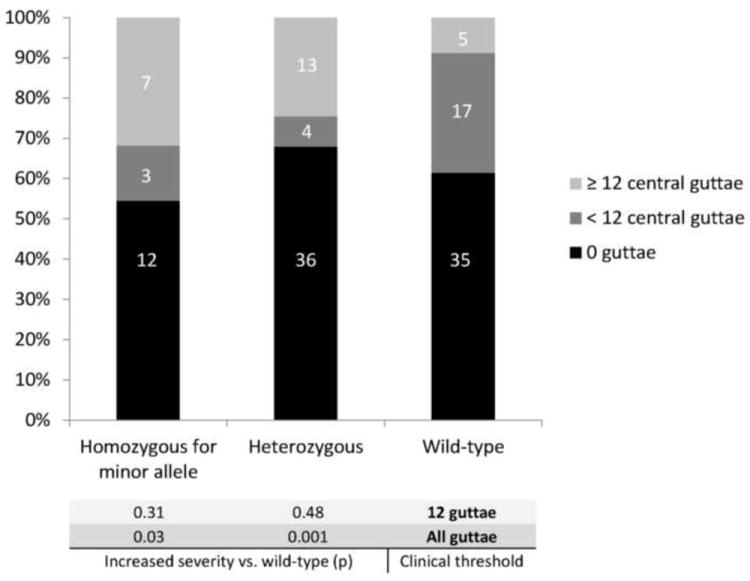
Interaction between Fuchs Corneal Dystrophy affectation, severity, and the rs613872 intronic variant in Tangier. Affectation rate (12 or more central guttae, green) was significantly higher among individuals either homozygous or heterozygous for the minor allele relative to individuals with the wild-type genotype (p=0.02). Subclinical disease (<12 central guttae, red) was significantly over-represented among clinically negative individuals with the wild-type genotype relative to those with at least one copy of the minor allele (p=0.01). Inclusion of all quantities of guttae in analysis eliminates the significant difference in affectation rate and results in lower severity among wild-type individuals, by Fisher exact test.
Inclusion of the subclinical individuals to have the mildest category of FCD severity does not result in a significantly increased prevalence of the disease phenotype among individuals homozygous (p<0.38) and heterozygous (p<0.31) for the minor allele. However, average severity is then significantly higher among homozygotes (p=0.03) and heterozygotes (p=0.001) relative to individuals with the wild-type allele, as measured by Fisher exact test.
Discussion
Tangier Island offers a unique study location due to its isolated geographical and genetic features which have been previously well-studied.20, 21 Inhabitants of the island have been associated previously with one large pedigree; this analysis of island inhabitants dating back to 1722 demonstrates an average inbreeding coefficient of 0.009.21 In this study, initial pursuit of a family with FCD and subsequent expansion resulted in an assessment of a little over 50% of individuals over 50 years of age on the island.
This study revealed an affectation rate among study subjects above 30 of approximately 21%, suggesting a prevalence of at least 11% of the population above 50 years of age. Previous studies suggest a prevalence in the United States of approximately 4 to 7%,22, 23 in Europe of 4.5 to 9%,15 and in Asia of 3.8 to 8.5%.16, 17, 24 Although the variation among studies in methods and thresholds utilized to define affectation, such as specular microscopy or slit-lamp examination, limits direct comparison, the data suggest an increased prevalence on the island. This finding is supported by the high frequency of the TCF4 variant allele noted in this isolated population.
In the Tangier study population, FCD demonstrates a mild phenotype that begins in the fourth or fifth decade of life and slowly develops in severity over time. This progression is evident through clinical comparison using slit-lamp biomicroscopy and analysis of retroillumination photography. Because of this mild clinical phenotype, we sought to assess the effect of theTCF4 susceptibility allele on severity.
Previously, Riazuddin et al.13 and Li et al.14 showed that the presence of the minor allele does not influence the severity of the disease phenotype in unrelated sporadic cases. Here, we replicate the increased affectation rate associated with the intronic variant, and find that in this isolated population, it appears to inversely correlate with subclinical disease. Although we are cautious at interpreting our data given the relatively small sample size, we speculate that the presence of the minor allele may be associated with a shift from subclinical to clinical disease, or that the wild-type allele may in fact represent a protective factor which prevents individuals from progressing to clinical FCD. Longitudinal study with enrollment of more individuals will better elucidate this phenomenon.
In the Tangier study population, height and weight correlate with disease with only marginal significance after stratification for gender. Previous reports describe a similar trend mildly associated with age and sex.15 Of the identified genetic lesions associated with disease, two are related to transcription control factors, a causative lesion in TCF8 and a SNP correlated at TCF4, which may play a role in development. As there may be a familial effect in height and weight distribution, further study is needed to ascertain the presence of such an effect.
In summary, we report a phenomenon of mild FCD with increased prevalence on Tangier Island and severity which appears to be modulated by presence of the rs613872 SNP.
Acknowledgments
Funding/Support: This study was supported in part by National Eye Institute Research Grant R01EY016835 (JDG, NK) and the A. Edward Maumenee Research Grant Award (AOE). Data analysis was supported by Wilmer Biostatistics Core Grant R01EY01765.
Conformity: The study protocol was prospectively approved by the Joint Committee on Clinical Investigation at The Johns Hopkins University School of Medicine and was conducted in accordance with the tenets of the Declaration of Helsinki. Informed written consent was obtained from all participants.
Other acknowledgements: We express our gratitude to the people of Tangier Island for their hospitality and to Cindy Parks for assistance with data collection and supplies.
Footnotes
Contributions of Authors: Design of the study (AOE, EJM, SAR, JDG); Conduct of the study (AOE, EJM, BWI, DE, JDG); Collection, management, analysis, and interpretation of the data (AOE, EJM, BWI, JW, SAR, JDG); Preparation of the manuscript (AOE, EJM, BWI, JW, SAR, NK, JDG); Review and approval of the manuscript (AOE, EJM, BWI, JW, DE, SAR, NK, JDG).
Financial Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Fuchs E. Dystrophia epithelialis corneae. Graefes Arch Clin Exp Ophthalmol. 1910;(76):478–508. [Google Scholar]
- 2.Clegg JG. Remarks on dystrophies of the cornea and glaucoma, with especial reference to a familial variety of the former. Transactions of the Ophthalmological Society of the United Kingdom. 1915;35:245–253. [Google Scholar]
- 3.Cross HE, Maumenee AE, Cantolino SJ. Inheritance of Fuchs’ endothelial dystrophy. Arch Ophthalmol. 1971;85(3):268–272. doi: 10.1001/archopht.1971.00990050270002. [DOI] [PubMed] [Google Scholar]
- 4.Krachmer JH, Purcell JJ, Jr, Young CW, Bucher KD. Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978;96(11):2036–2039. doi: 10.1001/archopht.1978.03910060424004. [DOI] [PubMed] [Google Scholar]
- 5.Rosenblum P, Stark WJ, Maumenee IH, Hirst LW, Maumenee AE. Hereditary Fuchs’ Dystrophy. Am J Ophthalmol. 1980;90(4):455–462. doi: 10.1016/s0002-9394(14)75011-1. [DOI] [PubMed] [Google Scholar]
- 6.Sundin OH, Jun AS, Broman KW, et al. Linkage of late-onset Fuchs corneal dystrophy to a novel locus at 13pTel-13q12.13. Invest Ophthalmol Vis Sci. 2006;47(1):140–145. doi: 10.1167/iovs.05-0578. [DOI] [PubMed] [Google Scholar]
- 7.Sundin OH, Broman KW, Chang HH, Vito EC, Stark WJ, Gottsch JD. A common locus for late-onset Fuchs corneal dystrophy maps to 18q21.2-q21.32. Invest Ophthalmol Vis Sci. 2006;47(9):3919–3926. doi: 10.1167/iovs.05-1619. [DOI] [PubMed] [Google Scholar]
- 8.Riazuddin SA, Eghrari AO, Al-Saif A, et al. Linkage of a mild late-onset phenotype of Fuchs Corneal Dystrophy to a novel locus at 5q33.1-q35.2. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-3764. [DOI] [PubMed] [Google Scholar]
- 9.Riazuddin SA, Zaghloul NA, Al-Saif A, et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet. 2010;86(1):45–53. doi: 10.1016/j.ajhg.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riazuddin SA, Vithana EN, Seet LF, et al. Missense mutations in the sodium borate cotransporter SLC4A11 cause late-onset Fuchs corneal dystrophy. Hum Mutat. 2010;31(11):1261–1268. doi: 10.1002/humu.21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGlumphy EJ, Yeo WS, Riazuddin SA, et al. Age-severity relationships in families linked to FCD2 with retroillumination photography. Invest Ophthalmol Vis Sci. 2010;51(12):6298–6302. doi: 10.1167/iovs.10-5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baratz KH, Tosakulwong N, Ryu E, et al. E2-2 protein and Fuchs’s corneal dystrophy. N Engl J Med. 2010;363(11):1016–1024. doi: 10.1056/NEJMoa1007064. [DOI] [PubMed] [Google Scholar]
- 13.Riazuddin SA, McGlumphy EJ, Yeo WS, Wang J, Katsanis N, Gottsch JD. Replication of the TCF4 Intronic Variant in Late-Onset Fuchs Corneal Dystrophy and Evidence of Independence from the FCD2 Locus. Invest Ophthalmol Vis Sci. 2011;52(5):2825–2829. doi: 10.1167/iovs.10-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li YJ, Minear MA, Rimmler J, et al. Replication of TCF4 through Association and Linkage Studies in Late-Onset Fuchs Endothelial Corneal Dystrophy. PLoS One. 2011;6(4):e18044. doi: 10.1371/journal.pone.0018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zoega GM, Fujisawa A, Sasaki H, et al. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology. 2006;113(4):565–569. doi: 10.1016/j.ophtha.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 16.Kitagawa K, Kojima M, Sasaki H, et al. Prevalence of primary cornea guttata and morphology of corneal endothelium in aging Japanese and Singaporean subjects. Ophthalmic Res. 2002;34(3):135–138. doi: 10.1159/000063656. [DOI] [PubMed] [Google Scholar]
- 17.Higa A, Sakai H, Sawaguchi S, et al. Prevalence of and risk factors for cornea guttata in a population-based study in a southwestern island of Japan: the Kumejima study. Arch Ophthalmol. 2011;129(3):332–336. doi: 10.1001/archophthalmol.2010.372. [DOI] [PubMed] [Google Scholar]
- 18.Fletcher KR. Tangier Island and the Way of the Watermen. Smithsonian.com. 4-1-2009 6-27-2011. [Google Scholar]
- 19.Meadows DN, Eghrari AO, Riazuddin SA, Emmert D, Katsanis N, Gottsch JD. Progression of Fuchs Corneal Dystrophy in a family linked to the FCD1 locus. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-3568. [DOI] [PubMed] [Google Scholar]
- 20.Mathias RA, Bickel CA, Beaty TH, et al. A study of contemporary levels and temporal trends in inbreeding in the Tangier Island, Virginia, population using pedigree data and isonymy. Am J Phys Anthropol. 2000;112(1):29–38. doi: 10.1002/(SICI)1096-8644(200005)112:1<29::AID-AJPA4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Mathias RA, Beaty TH, Bailey-Wilson JE, Bickel C, Stockton ML, Barnes KC. Inheritance of total serum IgE in the isolated Tangier Island population from Virginia: complexities associated with genealogical depth of pedigrees in segregation analyses. Hum Hered. 2005;59(4):228–238. doi: 10.1159/000087123. [DOI] [PubMed] [Google Scholar]
- 22.Goar EL. Dystrophy of the Corneal Endothelium (Cornea Guttata), with Report of a Histologic Examination. Trans Am Ophthalmol Soc. 1933;31:48–59. [PMC free article] [PubMed] [Google Scholar]
- 23.Lorenzetti DW, Uotila MH, Parikh N, Kaufman HE. Central cornea guttata. Incidence in the general population. Am J Ophthalmol. 1967;64(6):1155–1158. [PubMed] [Google Scholar]
- 24.Nagaki Y, Hayasaka S, Kitagawa K, Yamamoto S. Primary cornea guttata in Japanese patients with cataract: specular microscopic observations. Jpn J Ophthalmol. 1996;40(4):520–525. [PubMed] [Google Scholar]