

Abstract
Time series profiling is a powerful approach for obtaining information on protein expression dynamics and prevailing biochemical pathways. To date, such information could only be obtained at the mRNA level using mature and highly parallel technologies such as microarray gene expression profiling. The generation of time series data at the protein level has lagged due to the lack of robust and highly reproducible methodologies. Using a combination of SILAC strategy, SDS-PAGE and LC-MS/MS, we demonstrate successful monitoring of expression levels of the same set of proteins across different time points within the ER compartment of human primary fibroblast cells when exposed to ER stress inducers tunicamycin and thapsigargin. Data visualization was facilitated using GeneSpring GX analysis platform that was designed to process Affymetrix microarray data. This software also facilitated the generation of important parameters such as data normalization, calculation of statistical values to extract significant changes in protein expression, and the cross comparison of data sets.
Keywords: SILAC, Isotope Ratio, LC-MS/MS, Proteome profiling, Time series, ER stress, GRP78, Reticulocalbin
Introduction
Time series expression profiling has proven to be a powerful approach to monitor the dynamics of molecular events and build biochemical pathways in a variety of samples representing different biological states and experimental conditions.1,2 Large time series data sets are most commonly obtained at the mRNA level using mature and highly parallel technologies such as oligonucleotide and cDNA microarray gene expression profiling. Time series expression profiling at the protein level has been hindered by the slow process of generating substantive proteome profile data sets and by the lack of a robust and highly reproducible methodology. In the majority of experiments, stable isotope tagging strategies such as Isotope-Coded Affinity Tagging (ICAT),3 H 182O proteolytic labeling,4,5 or isobaric tag for relative and absolute quantitation (iTRAQ)6,7 have been used in combination with LC-MS/MS analysis to generate proteome profile data sets. Thus far, these techniques have been limited to pairwise comparisons, with the exception of the iTRAQ strategy where 4 or even 8 samples may be analyzed simultaneously in the near future.8,9 Moreover, proteome profiling using chemical tagging requires careful and complete derivatization of all the proteins or peptides in the samples in order to minimize false positive differential expressions introduced during experimental handling.
Alternative approaches such as label-free strategies in combination with LC-MS or LC-MS/MS are being tested and appear promising for generating large data sets. These are potentially suitable for data cross comparisons necessary for time series experiments.10,11 Nevertheless, these label-free strategies are still time-consuming, labor-intensive, and require replicate analysis for each sample. Additionally, the label-free approach requires standardization both at the instrumental level (e.g., reproducible LC separations and accurate mass spectrometer instruments) and at the data processing level (e.g., bioinformatics and statistical analysis).
While variations due to experimental handling still remain an issue when using chemical tagging in an ex-vivo system (e.g., tissue and body fluids), these variations have been greatly minimized in a cell culture system through the use of metabolic labeling such as stable isotope labeling by amino acid in cell culture (SILAC).12–14 SILAC consists of growing one set of cells in a medium where some of the amino acids, usually arginine (Arg) and/or lysine (Lys), are replaced with stable isotope-labeled ones, while the comparative sample is grown in unlabeled medium. Metabolic labeling by stable isotopes is the most systemic way to uniformly label all cellular proteins within the cell. In this method, variability due to sample preparation is circumvented as experimental pairs of labeled and unlabeled cells can be mixed before protein extraction and even before subcellular fractionation. Additionally, SILAC can allow accurate temporal proteome profiling15 and monitoring of protein phosphorylation to study signaling pathways.16–20
In this study, we implemented the SILAC strategy in combination with SDS-PAGE and LC-MS/MS to monitor temporal changes in the expression and dynamics of proteins within the endoplasmic reticulum (ER) compartment of human primary fibroblast cells exposed to ER stress inducers tunicamycin (Tun) and thapsigargin (Thp). To demonstrate the feasibility of temporal profiling using this method, we have chosen a model system using Tun and Thp, as these ER stress agents have been documented to induce temporal changes in a number of ER resident proteins within minutes to hours. Additionally, we implemented the use of these two ER stress induction agents that are known to act by two completely different mechanisms in order to demonstrate the sensitivity of this comparative proteomic profiling approach and identify novel proteomic differences between the two ER stress mechanisms. The methodologies applied to this system of normal primary human fibroblasts can be applied to further investigations of time series proteomic profiling utilizing cells obtained from disease patients versus controls.
Methods
Cell Culture
Human primary fibroblast cells of a 5 year old donor were obtained from the laboratory of Dr. Schiffmann (NINDS/NIH). The fibroblast cells were established from ex-plant cultures of punch skin biopsies in accordance with an NIH Institutional Review Board approved protocol. The cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) with GlutaMAX I, low glucose, 110 mg/L sodium pyruvate, pyridoxine HCl (Invitrogen Corporation, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen Corporation, Carlsbad, CA) and with 100 µg/mL penicillin and 100 µg/mL streptomycin (Invitrogen Corporation, Carlsbad, CA). To study the effect of Tun and/or Thp on the ER proteome, we implemented the SILAC strategy in combination with subcellular fractionation and proteome profiling using LC-MS/MS. Cells were seeded in 75 cm2 flasks and subcultured in custom-made DMEM medium (Atlanta Biologicals, Lawrenceville, GA) where Arg and Lys were replaced by 13C6-Arg (147.5 μg/mL), and 15N2, 13C6-Lys (91.25 μg/mL) (Cambridge Isotope Laboratories, Inc., Andover, MA). Usually, cellular proteins were fully labeled after incubation for at least 7 cell doublings (roughly four passages) in isotopically labeled custom-made DMEM. In parallel, the same amounts of cells were grown in unlabeled DMEM medium. The unlabeled cells were treated with tunicamycin (5 μg/mL) (Tun) or thapsigargin (final concentration 1μm) (Thp), while fully labeled cells were kept untreated. At time points 0, 15 min, 1, 6, 12 and 24 h, treated and nontreated cells were harvested and mixed at 1:1 ratio for subsequent subcellular fractionation.
Preparation of the ER Fraction
Figure 1 shows the overall experimental strategy used in this study. The labeled and unlabeled cell monolayers were washed three times with PBS solution. The cells were then scraped in 2 mL of ice-chilled TE Mix buffer containing 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 2.5 M sucrose (Sigma Aldrich, St. Louis, MO) and 1 complete mini protease inhibitor cocktail tablet (Roche Pharmaceuticals, Nutley, NJ) per 10 mL. Cells were collected by gentle centrifugation in preweighed polypropylene conical flasks for 5 min at 4000 rpm at 4 °C. After removing the supernatant, the cell pellets from unlabeled and labeled cells were mixed at a 1:1 ratio (w/w) and then resuspended in 1 mL of TE buffer mix. The cells were then homogenized by passing them 15 times through a 1 mL syringe with a 23 gauge needle. The suspension was centrifuged for 10 min at 4000g at 4 °C and the supernatant containing the microsomal fraction was transferred to a clean Ependorf tube and further centrifuged at 13 000g at 4 °C for 20 min to obtain the microsomal pellet. The microsomal pellet was then resuspended in protein extraction buffer containing urea (7 M), thiourea (2 M), CHAPS (2%, w/v) and DTT (50 mM). Protein concentration was determined in each fraction using Bio-Rad protein assay (Bio-Rad, Hercules, CA). Samples were stored at –80 °C until analysis.
Figure 1.

Overview of the experimental design used in this study. Control human primary fibroblasts were grown in medium where Lys and Arg were replaced by 15N2, 13C6-Lys and 13C6-Arg. The cells fully incorporated these amino acids after 6 to 7 cell doublings. To monitor the temporal effect of Tun or Thp on the ER proteome, labeled control cells were mixed at 1:1 ratio with Tun- or Thp-treated cells then processed for subcellular fractionation. Proteins were extracted from the ER fraction and further separated by SDS-PAGE. Thereafter, LC-MS/MS analysis was conducted on peptides generated by trypsin in-gel digestion of gel slices from SDS-PAGE. Protein ratios are determined from the peak areas of labeled and unlabeled peptide pairs.
Protein Separation and Mass Spectrometry Analysis
The ER fractions containing labeled and unlabeled proteins (60 μg of total proteins per sample) were further separated by SDS-PAGE and the gel stained with Bio-Safe Coomassie (Bio-Rad, Hercules, CA). Ge bands were sliced and digested with trypsin, and the resulting peptides were analyzed by LC-MS/MS using a LTQ mass spectrometer (Thermo Fisher Scientific, Waltham, MA) coupled online with a Dionex LC-Packing nano-HPLC system (Sunnyvale, CA). Each sample was injected via an autosampler and loaded onto a C18 trap column (5 μm, 300 μm i.d. × 5 mm, LC Packings) for 6 min at a flow rate of 10 μL/min, 100% A. The sample was subsequently separated by a C18 reverse-phase column (3.5 μm, 100 μm × 15 cm, Agilent, Santa Clara, CA) at a flow rate of 200 nL/min. The mobile phases consisted of water with 0.1% formic acid (A) and 90% acetonitrile with 0.1% formic acid (B). A 100 min linear gradient from 5 to 60% B was typically employed. Eluted peptides were introduced into the mass spectrometer via a 10 μm silica tip (New Objective, Inc., Ringoes, NJ) adapted to a nanoelectro-spray source (Thermo Fisher Scientific). The spray voltage was set at 1.7 kV and the heated capillary at 160 °C. The LTQ was operated in data-dependent mode in which one cycle of experiments consisted of one full-MS survey and subsequently three sequential pairs of intercalated zoom scans and MS/MS experiments. The targeted ion counts in the ion trap during full-MS, zoom scan, and MS/MS were 30 000, 3000, and 10 000, respectively. Peptides were fragmented in the linear ion trap using collision-induced dissociation with the collision gas (helium) pressure set at 1.3 mTorr and the normalized collision energy value set at 35%. The zoom scan events were of higher resolution and were used to determine the charge state of the ion as well as the ratio of labeled to unlabeled peptide pairs using the ZoomQuant software developed by Halligan, et al.22
Database Search
Protein identification was performed using BioWorks 3.1 software (Thermo Fisher Scientific). Each file was searched against a subset of the Swiss-Prot database containing human proteins only (UniProtKB/Swiss-Prot release 51.0, download November 6, 2006 with 14 987 human protein entries) and indexed with assumptions for fully enzymatic tryptic cleavage with two missed cleavages using the Sequest search engine set with the following criteria: signal threshold ≥1000, peptide mass tolerance ±1.5 Da, fragment ion mass tolerance ±0.35 Da and possible 16 Da shift for oxidized Met residue, 6 and 8 Da shifts for stable isotope labeled Arg and Lys, respectively. Full Sequest search results, prior to filtration, were loaded into ZoomQuant Software.22 XCorr filters of 2.5 for doubly charged and 3.5 for triply charged were applied. Singly charged peptides were omitted. Each peptide was manually validated by examining zoom scans to verify the labeled and unlabeled pairs. Identified peptides were required to have a matched peptide partner 6 Da greater for arginine terminating peptides, 8 Da greater for lysine containing peptides and other proper combinations for missed cleavage peptides. The combination of using the smaller, less redundant human only database combined with manual data analysis eliminated redundancy. Initial analysis of the ER fraction was performed by searching the same database using Bioworks 3.2 and filtering peptide identifications with the following acceptance criteria: DeltaCn (ΔCn) > 0.1, a variable threshold of Xcorr versus charge state (Xcorr = 1.9 for z = 1, Xcorr = 2.5 for z = 2, and Xcorr = 3.5 for z = 3), peptide probability based score with a p value < 0.001 and two or more unique peptides in at least one data set. Reverse database search using the same criteria gave false-positive rate of less than 2.6%, thus, increasing protein identification confidence (False Positive Rate = 2 × [Reverse Database Protein Hits/(Reverse + Forward Database Protein Hits)] × 100.21
Statistical Analysis
ZoomQuant software22 was used to determine protein ratios between 13C6-Arg/5N2, 13C6-Lys labeled and unlabeled cells by using the peak areas of labeled and unlabeled peptide pairs from zoom scan data. The data was analyzed using GeneSpring GX Analysis Platform (Agilent technologies, Inc., Santa Clara, CA). Peptide ratios were normalized to the median expression in each time point and treatment group. An average ratio with standard deviation was determined using the multiple peptides detected per protein. Only proteins with two or more identified peptides in at least one data set were retained for analysis. A two-sided p-value or z-score was generated for each data point to determine how closely the ratios of several peptides belonging to the same protein were close to each other. Proteins with at z-score < 0.005 were retained for further analysis. Fold changes of specific proteins were compared at the different time points between Thp- and Tun-treated cells.
Results
Characterization of Proteome Profiles in ER Fractions Prepared from Human Fibroblast Cells
At different time points, Thp- or Tun-treated cells were mixed at 1:1 ratio with nontreated cells and processed for subcellular fractionation, protein separation and mass spectrometry analysis (Figure 1). Briefly, a total of 12 ER fraction pairs corresponding to time 0, 15 min, 1, 6, 12, and 24 h following treatment with Tun or Thp were prepared and processed for SDS-PAGE. Each SDS-PAGE lane was excised into 14 bands resulting in a total of 84 gel bands per stress agent (168 gels bands combined) to be analyzed by LC-MS/MS.
We applied stringent filtrations and manual validation both at the protein identification level and the quantitation level to process our data (Figure 2). The proteome coverage in the ER fractions prepared from primary fibroblast cells was initially examined through a protein database search using the Sequest search engine against the Swiss-Prot human fasta database. Using stringent filtration (Δn ≥ 0.1; Xcorr ≥ 1.9, 2.5, and 3.5 for singly, doubly, and triply charged ions, respectively; peptide probability < 0.001; and retaining only proteins that were represented by two or more unique peptides in at least one data set), we confidently identified 596 proteins in the ER fraction from 3 combined pairwise comparisons (control versus control, Thp-treated versus control and Tun-treated versus control). Thereafter, these 596 proteins were classified based on their known subcellular localization and function using gene ontology (GO) information obtained from cross-referencing each protein's Swiss-Prot accession number to the GO localization information available on the NCBI protein database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=Protein). Approximately 62% of the identified proteins were found to be of ER origin (Figure 3) indicating that the ER fraction was effectively enriched. The identification of a large number of extracellular proteins, membrane proteins, ribosomal proteins and lysosomal proteins was expected since these proteins can be directly associated with the ER compartment. However, only 118 proteins among the 596 identified proteins were shared across the 3 different experiments (Supplemental Table 1). This poor overlap of 19.7% can be explained by the fact that the ER fractionation was not highly reproducible from one sample pair to the next, and this may have resulted in random organelle cross contamination during these multiple subcellular fractionations and also possibly due to poor reproducibility between LC-MS/MS runs. The use of SILAC strategy with sample mixing prior to subcellular fractionation alleviates the concern of poor percentage of protein overlap due to experimental handling. Furthermore, in the event of cross contamination during subcellular fractionation, the contaminant proteins are present equally in both samples. The differential distribution of protein abundances between sample pairs are therefore attributed to inherent variations in the experimental samples and conditions. Figure 4 displays the distribution profiles of labeled and unlabeled peptide pair ratios obtained from ER fractions prepared from 3 different paired samples after 24 h of stress induction. In the control sample (nontreated cells versus nontreated cells) (Figure 4A), the overall distribution of peptide ratios fits a normal Gaussian shape with values centered around 1 and standard deviation of ±0.12. The distribution of peptide ratios in Tun -treated (Figure 4B) or Thp-treated (Figure 4C) cells versus nontreated cells showed a broader Gaussian shape indicating large variations between treated and nontreated cells with average standard deviations of ±0.25 and 0.45, respectively. Narrower distribution was also observed on ER fractions prepared from three independent replicates of control versus control cells (data not shown) indicating the robustness and the accuracy of the technique.
Figure 2.

Chart depicting the processing and filtration steps used to process the LC-MS/MS data. Step 1: Raw LC-MS/MS data was searched against Swiss-Prot human database using Bioworks 3.1 with the indicated parameters. Step 2: Unfiltered identified peptides along with their zoom scans were uploaded into ZoomQuant, filtered based on XCorr and subjected to manual validation. Step 3: Peak area ratio calculations between labeled and unlabeled peptides were calculated using ZoomQuant. Step 4: The peptide list and quantitative ratios (treated/control) were uploaded to GeneSpring. Step 5: To adjust for errors when mixing labeled and unlabeled cells at 1:1 ratios, each data set was normalized to its median. Only proteins that were detected across different time points were retained for further analysis. Step 6: Performed paired comparison of time points 6, 12 and 24 h to time zero. Z-scores were calculated for each data point. Only average ratios with low standard deviation (z-score < 0.005) were retained.
Figure 3.
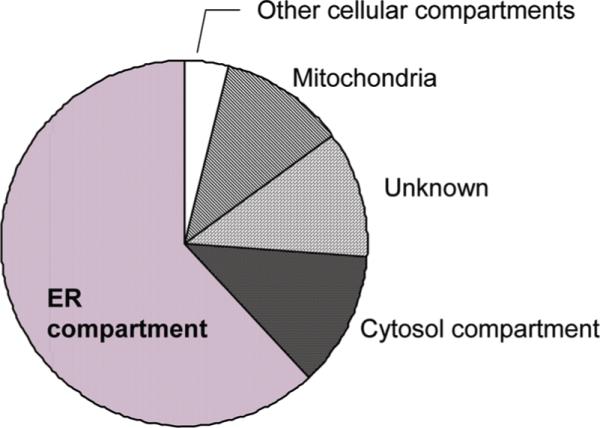
Cellular localizations of proteins identified in the crude ER fractionations prepared from human primary fibroblast cells. In total, 62% of the identified proteins were found to be of ER origin.
Figure 4.

Distribution profiles of protein abundances in the ER fraction of Thp- and Tun-treated fibroblast cells and nontreated cells. (A) Control versus control fibroblast cells: the overall distribution of peptide ratios fits a normal Gaussian shape with values centered around 1 and minimal standard deviation of ±0.12. (B) Tun versus Control fibroblast cells: a larger distribution of labeled and unlabeled peptide pair ratios indicates a greater differentiation of protein expression ratios between Tun-treated and nontreated cells. (C) Thp versus Control fibroblast cells: a larger distribution of labeled and unlabeled peptide pair ratios indicates a greater differentiation of protein expression ratios between Thp-treated and nontreated cells.
Monitoring Changes in the ER Proteome of Tun- and Thp-Treated Cells versus Nontreated Cells
Each of the ER fraction pairs of treated and nontreated cells corresponding to time 0, 15 min, 1, 6, 12, and 24 h following treatment with Tun or Thp were prepared and processed for LC-MS/MS analysis as described above (Figure 1). Proteins were identified as described above and protein ratios of Thp- or Tun-treated cells versus nontreated cells were determined from zoom scans of labeled and unlabeled peptide pairs using peak area and ZoomQuant software22,23 (Figure 2).
The list of labeled and unlabeled peptide pairs and corresponding expression ratios was directly uploaded into Gene-Spring software. The software, developed for microarray data analysis, readily adapted to this protein data set as it is engineered to recognize the accession number of a protein as it would a gene identifier. Additionally, the GeneSpring software calculates the number of peptides per protein as it would an array probe set count. Unlike with mircroarray expression profiling experiments in which time series data are often normalized to time 0 point, each data set in this experiment was normalized to its median to correct for mixing of labeled and unlabeled cells at 1:1 ratios. This normalization approach allowed a better assessment of differential protein expression both in treated versus nontreated cells as well as in control cells (e.g., nontreated versus nontreated cells). Subsequently, an average value including a z-score was calculated for each protein. The GeneSpring software was then used to graphically visualize the time series data of Thp and Tun experiments and to monitor the expression levels of identified proteins across the time points 0, 6, 12 and 24 h in both Thp- and Tun-treated cells (Figure 5). Since there was no significant differential protein expression at time point 15 min and 1 h, these data sets were removed from further analysis.
Figure 5.

Time series data as visualized in GeneSpring Software. The 118 proteins found in common between Tun and Thp over 0, 12, and 24 h of stress were visualized in GeneSpring. Protein expression patterns can easily be visualized to distinguish proteins behaving similarly or differently under the two stress mechanisms. The inset shows a magnified view of the temporal change in the abundance of GRP78 protein in both Tun- and Thp-treated cells.
The protein lists submitted into Genespring were further focused to only include proteins overlapping different time points (0, 6, 12 and 24 h) and different conditions (Thp and Tun). Nonoverlapping proteins, mostly originating from organelle cross contaminations during ER fractionation or irreproducible MS runs, were excluded. In this particular visualization, shown in Figure 5, the time series data set can be easily queried for both Tun and Thp experiments. Each interactive curve is clickable and will display the protein name with ratio and z-scores across the different time points (see inset for temporal change in the abundance of GRP78 protein in both Tun and Thp treated cells). Data can be also filtered based on fold changes, z-scores and select significant changes.
As shown in Figure 6, both Thp and Tun resulted in a significant temporal increase of the ER chaperone proteins and proteins involved in glycosylation and maturation of proteins transiting the ER. The most significant temporal changes were observed for the 78 kDa glucose-regulated protein (GRP78), also known as BiP, followed by protein disulfide-isomerase A4 (PDIA4), endoplasmin (ENPL), also known as GRP94, and calreticulin (CLAR). Glucosidase 2 subunit beta (GLU2B) was slightly more increased in Tun-treated cells than in Thp -treated cells. An additional common change exerted by Tun and Thp treatment is the significant temporal decrease in collagen isoforms (CO1A1, CO1A2 and CO6A1). While CO1A1 and CO1A2 isoforms began decreasing after 6 h of treatment with Thp or Tun, there was a slight delay in the decrease of CO6A1 isoform. Interestingly, reticulocalbin-3 (RCN-3) was significantly decreased following Thp treatment but did not change with Tun treatment even after 24 h treatment. Figure 7 summarizes all the proteins that were significantly altered in their expression after 24 h treatment with Thp or Tun. This histogram plot easily points out similarities and dissimilarities between the two ER stress inducer agents. Both Thp and Tun treatment resulted in an increase of ER chaperone proteins (GRP78, calreticulin CALR, endoplasmin ENPL, heat shock proteins and protein disulfide-isomerases, PDIA1, PDIA3, PDIA4, PDIA6), and glucosidases (GLU2B and GNAB), while proteins destined for secretion such as collagens (CO1A1, CO1A2, CO6A1, CO6A3) were decreased. Reticulocalbins RCN1 and RCN3 as well as calumenin (CALU) were significantly decreased after Thp treatment and remained unchanged after Tun treatment.
Figure 6.

Dynamic profiles of ER resident proteins in Thp- and Tun-treated fibroblast cells. Thp and Tun display a significant temporal increase of glycosylation proteins, proteins involved in maturation of proteins transiting the ER, and ER chaperones.
Figure 7.

Proteins significantly altered after 24 h of stress induction with Tun and Thp. Proteins displayed from left to right with Swiss-Prot accession number: Neutral alpha-glucosidase AB (Q14697), Protein disulfide-isomerase A1 (P07237), Endoplasmic reticulum protein ERp29 (P30040), Protein disulfide-isomerase A6 (Q15084), Glucosidase 2 subunit beta (P14314), Heat shock protein 90 (P08238), Protein disulfide-isomerase A3 (P30101), Pyruvate Kinase isozymes M1/M2 (P14618), Calreticulin (P27797), Endoplasmin (P14625), Protein disulfide-isomerase A4 (P13667), 78 kDa glucose-regulated protein (P11021), Reticulocalbin-3 (Q96D15), Collagen alpha-1(I) chain (P02452), Collagen alpha-1(VI) chain (P12109), Collagen alpha-2(I) chain (P08123), Collagen alpha-3(VI) chain (P12111), FK506-binding protein 10 (Q96AY3), Reticulocalbin-1 (Q15293), Collagen alpha-2(VI) chain (P12110), Calumenin (O43852), FK506- binding protein 9 (O95302).
Discussion
SILAC strategy in combination with SDS-PAGE and LC-MS/ MS has been implemented to generate a panel of differentially regulated proteins in the ER fraction of Tun- and Thp-treated human primary fibroblast cells. We demonstrate that a combination of these proteomic tools generates sensitive and accurate temporal proteomic profiling data. Moreover, we emphasize that the proteomic data interpretation can be standardized based on gene expression profile analysis techniques.
Current advances in proteomic profiling are hindered by a lack in complete reproducibility between LC-MS/MS experiments across several comparative samples. There is often a poor overlap in the proteins identified between replicate samples and even between replicate LC-MS run for a same sample. This is especially the case with low-abundance proteins. An additional impediment in comparative time series experiments is the generation of a large body of data and a lag in bioinformatic support for adequate data processing. We implemented proven gene expression analysis software to maximize the visualization of proteins shared between comparative samples. The GeneSpring software, designed to process and visualize microarray data, was used in this study to visualize the time series proteome profiling data and cross compare the temporal effects of Tun and Thp.
In our ER proteomic profile investigation, we demonstrate that both Tun and Thp exerted similar overall effects on the ER proteome profile. Tun induces ER stress by inhibiting N-glycosylation of newly synthesized proteins,24,25 where as Thp induces ER stress via Ca2+ discharge.26,27 Two overwhelming trends were observed with both agents, namely, an increase in expression over time of ER chaperone proteins (GRP78, CALR, and PDIs) and the concomitant decrease in the expression of proteins destined for secretion (e.g., collagen isoforms). This orchestrated response is classically characterized by the unfolded protein response (UPR) and is triggered when a cell senses an ER overload.28–31 UPR is initiated by ER transmembrane proteins (PERK, IRE1 and ATF6), which are activated by sensing the protein-folding status in the ER lumen. The activated PERK phosphorylates eIF2 alpha, which in turn inhibits eIF2B and synthesis of new proteins, while ATF6 and IRE1 induce transcription of specific ER chaperones and ER enzymes necessary to alleviate ER overload and ensure proper disposal of misfolded proteins. The marked temporal decrease observed for collagen isoforms could be explained by the fact that these proteins are the most ER trafficked proteins in fibroblast cells. Thus, the immediate decrease in the synthesis of this class of proteins will alleviate ER overload. The end result of the UPR can be either cell survival if the cell recovers from the stress or initiation of apoptosis (see review by Volchuk, et al. for more details).29
Although Thp causes a decrease of Ca2+ concentrations within the ER lumen, it was intriguing to identify a marked increase of Ca2+ binding chaperon proteins GRP78 and CLAR. This phenomenon has been previously reported and is believed to involve the control of Ca2+ homeostasis within the ER lumen and is necessary to maintain the proper function of ER chaperone proteins.32 Nevertheless, Thp, unlike Tun, caused a significant decrease of another class of Ca2+ binding protein (RCN and CALU) belonging to the CREC family (which is an acronym for Cab45), reticulocalbin, ERC-45 and calumenin.33
To our knowledge, this is the first identification of another class of ER proteins that was selectively affected by Thp treatment. RCN first characterized by Ozawa, et al.34 has been classified as a member of the CREC family.35 CREC members are lowaffinity Ca2+ binding proteins with six EF-hand motifs and are strictly localized into the ER lumen and secretory pathway.35
Although the function of these proteins is not well-known, it has been suggested that RCN may play an important role in normal cell behavior and cell life. In fact, homozygous deletion of the RCN gene results in embryonic lethality in mice.36 More recent studies have suggested that overexpression of RCN may play a role in tumor progression, invasiveness and resistance to chemotherapy.37 In this investigation, RCN-3 was the most affected protein, with a 7-fold down-regulation after 24 h treatment with Thp. Conversely, RCN-1, which shares 58% sequence homology with RCN-3 (Supplemental Figure 1), was only down-regulated by a factor of 1.65 ± 0.12. This selective decrease in RCN-3 suggests a specialized function of this protein in Thp-induced ER stress response. Furthermore, it has been demonstrated that RCN-3, but not RCN-1, interacts with and regulates the secretion of a serine endoprotease PACE4.38 Whether this interaction is specific to PACE4 or is more general to other secreted proteins remains to be further examined. Tun, which induces ER stress by blocking N-glycosylation, did not alter the expression of these RCN proteins, suggesting that the specific down-regulation of these proteins by Thp is most likely due to the decrease of Ca2+ in the ER lumen. However, further studies are still needed to verify this hypothesis and bring insight into the relationship between Ca2+ levels in the ER and expression of RCN-3.
Although, this study successfully generated a robust time response protein profiles of the ER stress series to Tun and Thp, it is probable that some important proteins were overlooked due to the continued obstacles faced in current proteomic analysis. Specifically, low-abundance proteins were identified with less fidelity between LC-MS/MS runs, and therefore could not be quantified across the entire time points. Additionally, although this investigation followed a large number of proteins over a sizable time series, further refined analysis and bioinformatic support are needed to conduct a more comprehensive high-throughput time series proteome profile. The development of new instruments that can provide better resolution, accuracy and throughput are likely to overcome some of these obstacles in the near future. This is typically true with the LTQ-orbitrap and triple quadrupole-trap instruments which are gaining popularity in the field of proteomic applications.39–43 A recent study using a combination of stable isotope labeling by iTRAQ and multiple reaction monitoring (MRM) clearly demonstrated the power of a triple quadrupole instrument to monitor the temporal phosphoryaltion of 222 tyrosine phosphorylated peptides following EGF signaling.42 Nevertheless, a priori knowledge of the peptide selected for monitoring is still required in this case.
Conclusion
Comparative time series proteomic profiling was conducted using SILAC strategy, SDS-PAGE, and LC-MS/MS to generate a panel of differentially regulated proteins specific to Tun- and Thp-modulated ER stress response. This method allowed the identification of specific ER stress expression changes as well as changes unique to Tun and Thp response. We found the use of SDS-PAGE followed by consistent band excision and ingel digestion provided stable and reproducible protein coverage over different time points and between different stress agents. Although the use of SILAC decreases protein variation due to mixing prior to experimental processing, variation in the number of proteins identified between LC-MS/MS runs within the same sample continues to be a hindrance. Reproducibility between LC-MS/MS experiments across several comparative samples is especially decreased for low-abundance proteins. An additional impediment in comparative time series experiments is the generation of a large body of data and a lag in bioinformatic support for parallel high-throughput data processing. These drawbacks strongly point toward a need for an automated platform to establish a true high-throughput proteome profiling. Nevertheless, the methods utilized in this study enabled the identification of both temporal changes of a robust set of proteins and attribute differential expression to different stress agents.
Supplementary Material
Acknowledgment
This work was supported in part by NIH grants: IDDRC 1P30HD40677 (Intellectual and Developmental Disabilities Research Centers), 5R24 HD050846 (IntegratedMolecularCoreforRehabilitationMedicine),K12HD001-399 (Children Health Research Center for Dr. Vanderver, the intramural program of the NINDS for Dr. Schiffmann), 5T32HD-046388 (Postdoctoral Training in Developmental Disabilities Research for Dr. Mintz) and by the Lynn and Doug Parsons foundation.
Abbreviations
- ICAT
Isotope-Coded Affinity Tagging
- iTRAQ
isobaric tag for relative and absolute quantitation
- ER
endoplasmic reticulum
- SILAC
stable isotope labeling by amino acid in cell culture
- Tun
tunicamycin
- Thp
thapsigargin
- DMEM
Dulbecco's Modified Eagle Medium
- GO
gene ontology
- GRP78
78 kDa glucose-regulated protein
- PDIA4
protein disulfide-isomerase A4
- ENPL
endoplasmin
- CLAR
calreticulin
- GLU2B
Glucosidase 2 subunit beta
- CO1A1, CO1A2, CO6A1
collagen isoforms
- RCN-3
reticulocalbin-3
- CALU
calumenin
- UPR
unfolded protein response
- PERK, IRE1, ATF6, eIF2 alpha
CREC family, acronym for Cab45, reticulocalbin, ERC-45 and calumenin
Footnotes
Supporting Information Available: Figure of the sequence alignment of RCN3 and RCN1; table listing the 118 proteins among the 596 identified proteins that were shared across the 3 different experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Zhao P, Iezzi S, Carver E, Dressman D, Gridley T, Sartorelli V, Hoffman EP. Slug is a novel downstream target of MyoD. Temporal profiling in muscle regeneration. J. Biol. Chem. 2002;277:30091–30101. doi: 10.1074/jbc.M202668200. [DOI] [PubMed] [Google Scholar]
- 2.Zhao P, Caretti G, Mitchell S, McKeehan WL, Boskey AL, Pachman LM, Sartorelli V, Hoffman EP. Fgfr4 is required for effective muscle regeneration in vivo. Delineation of a MyoDTead2-Fgfr4 transcriptional pathway. J. Biol. Chem. 2006;281:429–438. doi: 10.1074/jbc.M507440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 4.Fenselau C, Yao X. Proteolytic labeling with 18O for comparative proteomics studies: preparation of 18O-labeled peptides and the 18O/16O peptide mixture. Methods Mol. Biol. 2007;359:135–142. doi: 10.1007/978-1-59745-255-7_9. [DOI] [PubMed] [Google Scholar]
- 5.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal. Chem. 2001;73:2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 6.DeSouza L, Diehl G, Rodrigues MJ, Guo J, Romaschin AD, Colgan TJ, Siu KW. Search for cancer markers from endome-trial tissues using differentially labeled tags iTRAQ and cICAT with multidimensional liquid chromatography and tandem mass spec-trometry. J. Proteome Res. 2005;4:377–386. doi: 10.1021/pr049821j. [DOI] [PubMed] [Google Scholar]
- 7.Choe LH, Aggarwal K, Franck Z, Lee KH. A comparison of the consistency of proteome quantitation using two-dimensional electrophoresis and shotgun isobaric tagging in Escherichia coli cells. Electrophoresis. 2005;26:2437–2449. doi: 10.1002/elps.200410336. [DOI] [PubMed] [Google Scholar]
- 8.Cong YS, Fan E, Wang E. Simultaneous proteomic profiling of four different growth states of human fibroblasts, using amine-reactive isobaric tagging reagents and tandem mass spectrometry. Mech. Ageing Dev. 2006;127:332–343. doi: 10.1016/j.mad.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Wolf-Yadlin A, Ross PL, Pappin DJ, Rush J, Lauffenburger DA, White FM. Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol. Cell. Proteomics. 2005;4:1240–1250. doi: 10.1074/mcp.M500089-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Wiener MC, Sachs JR, Deyanova EG, Yates NA. Differential mass spectrometry: a label-free LC-MS method for finding significant differences in complex peptide and protein mixtures. Anal. Chem. 2004;76:6085–6096. doi: 10.1021/ac0493875. [DOI] [PubMed] [Google Scholar]
- 11.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- 12.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 13.Ong SE, Kratchmarova I, Mann M. Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC). J. Proteome Res. 2003;2:173–181. doi: 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- 14.Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol. Biol. 2007;359:37–52. doi: 10.1007/978-1-59745-255-7_3. [DOI] [PubMed] [Google Scholar]
- 15.Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE, Lamond AI, Mann M. Nucleolar proteome dynamics. Nature. 2005;433:77–83. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- 16.Ibarrola N, Kalume DE, Gronborg M, Iwahori A, Pandey A. A proteomic approach for quantitation of phosphorylation using stable isotope labeling in cell culture. Anal. Chem. 2003;75:6043–6049. doi: 10.1021/ac034931f. [DOI] [PubMed] [Google Scholar]
- 17.Amanchy R, Kalume DE, Iwahori A, Zhong J, Pandey A. Phosphoproteome analysis of HeLa cells using stable isotope labeling with amino acids in cell culture (SILAC). J. Proteome Res. 2005;4:1661–71. doi: 10.1021/pr050134h. [DOI] [PubMed] [Google Scholar]
- 18.Bose R, Molina H, Patterson AS, Bitok JK, Periaswamy B, Bader JS, Pandey A, Cole PA. Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9773–9778. doi: 10.1073/pnas.0603948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z, Pandey A, Hart GW. Dynamic Interplay between O-Linked N-Acetylglucosaminylation and Glycogen Synthase Kinase-3-dependent Phosphorylation. Mol. Cell. Proteomics. 2007;6:1365–1379. doi: 10.1074/mcp.M600453-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Bonenfant D, Towbin H, Coulot M, Schindler P, Mueller DR, van Oostrum J. Analysis of dynamic changes in post-translational modifications of human histones during cell cycle by mass spectrometry. Mol. Cell. Proteomics. 2007;6:1917–1932. doi: 10.1074/mcp.M700070-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 22.Halligan BD, Slyper RY, Twigger SN, Hicks W, Olivier M, Greene AS. ZoomQuant: an application for the quantitation of stable isotope labeled peptides. J. Am. Soc. Mass Spectrom. 2005;16:302–306. doi: 10.1016/j.jasms.2004.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.An E, Lu X, Flippin J, Devaney JM, Halligan B, Hoffman EP, Strunnikova N, Csaky K, Hathout Y. Secreted proteome profiling in human RPE cell cultures derived from donors with age related macular degeneration and age matched healthy donors. J. Proteome Res. 2006;5:2599–2610. doi: 10.1021/pr060121j. [DOI] [PubMed] [Google Scholar]
- 24.Heifetz A, Keenan RW, Elbein AD. Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry. 1979;18:2186–2192. doi: 10.1021/bi00578a008. [DOI] [PubMed] [Google Scholar]
- 25.Dorner AJ, Wasley LC, Raney P, Haugejorden S, Green M, Kaufman RJ. The stress response in Chinese hamster ovary cells. Regulation of ERp72 and protein disulfide isomerase expression and secretion. J. Biol. Chem. 1990;265:22029–22034. [PubMed] [Google Scholar]
- 26.Booth C, Koch GL. Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell. 1989;59:729–737. doi: 10.1016/0092-8674(89)90019-6. [DOI] [PubMed] [Google Scholar]
- 27.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell. Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 29.Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda) 2007;22:193–201. doi: 10.1152/physiol.00050.2006. [DOI] [PubMed] [Google Scholar]
- 30.Urade R. Cellular response to unfolded proteins in the endoplasmic reticulum of plants. FEBS J. 2007;274:1152–1171. doi: 10.1111/j.1742-4658.2007.05664.x. [DOI] [PubMed] [Google Scholar]
- 31.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lievremont JP, Rizzuto R, Hendershot L, Meldolesi J. BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J. Biol. Chem. 1997;272:30873–30879. doi: 10.1074/jbc.272.49.30873. [DOI] [PubMed] [Google Scholar]
- 33.Yabe D, Nakamura T, Kanazawa N, Tashiro K, Honjo T. Calumenin, a Ca2+-binding protein retained in the endoplasmic reticulum with a novel carboxyl-terminal sequence, HDEF. J. Biol. Chem. 1997;272:18232–18239. doi: 10.1074/jbc.272.29.18232. [DOI] [PubMed] [Google Scholar]
- 34.Ozawa M, Muramatsu T. Reticulocalbin, a novel endoplasmic reticulum resident Ca(2+)-binding protein with multiple EF-hand motifs and a carboxyl-terminal HDEL sequence. J. Biol. Chem. 1993;268:699–705. [PubMed] [Google Scholar]
- 35.Honore B, Vorum H. The CREC family, a novel family of multiple EF-hand, low-affinity Ca(2+)-binding proteins localised to the secretory pathway of mammalian cells. FEBS Lett. 2000;466:11–18. doi: 10.1016/s0014-5793(99)01780-9. [DOI] [PubMed] [Google Scholar]
- 36.Kent J, Lee M, Schedl A, Boyle S, Fantes J, Powell M, Rushmere N, Abbott C, van Heyningen V, Bickmore WA. The reticulocalbin gene maps to the WAGR region in human and to the Small eye Harwell deletion in mouse. Genomics. 1997;42:260–267. doi: 10.1006/geno.1997.4706. [DOI] [PubMed] [Google Scholar]
- 37.Fukuda T, Oyamada H, Isshiki T, Maeda M, Kusakabe T, Hozumi A, Yamaguchi T, Igarashi T, Hasegawa H, Seidoh T, Suzuki T. Distribution and variable expression of secretory pathway protein reticulocalbin in normal human organs and nonneoplastic pathological conditions. J. Histochem. Cytochem. 2007;55:335–345. doi: 10.1369/jhc.6A6943.2006. [DOI] [PubMed] [Google Scholar]
- 38.Tsuji A, Kikuchi Y, Sato Y, Koide S, Yuasa K, Nagahama M, Matsuda Y. A proteomic approach reveals transient association of reticulocalbin-3, a novel member of the CREC family, with the precursor of subtilisin-like proprotein convertase, PACE4. Biochem. J. 2006;396:51–59. doi: 10.1042/BJ20051524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swatkoski S, Gutierrez P, Ginter J, Petrov A, Dinman JD, Edwards N, Fenselau C. Integration of residue-specific acid cleavage into proteomic workflows. J. Proteome Res. 2007;11:4525–7. doi: 10.1021/pr0704682. [DOI] [PubMed] [Google Scholar]
- 40.Hanke S, Besir H, Oesterhelt D, Mann M. Absolute SILAC for accurate quantitation of proteins in complex mixtures down to the attomole level. J. Proteome Res. 2008;7:1118–1130. doi: 10.1021/pr7007175. [DOI] [PubMed] [Google Scholar]
- 41.Lu B, Motoyama A, Ruse C, Venable J, Iii JR. Improving protein identification sensitivity by combining MS and MS/MS information for shotgun proteomics using LTQ-Orbitrap high mass accuracy data. Anal. Chem. 2008;80:2018–2025. doi: 10.1021/ac701697w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. U.S.A. 2007;104:5860–5865. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yates JR, Cociorva D, Liao L, Zabrouskov V. Performance of a linear ion trap-Orbitrap hybrid for peptide analysis. Anal. Chem. 2006;78:493–500. doi: 10.1021/ac0514624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.