

Abstract
Nephronophthisis (NPHP) is a recessive disorder of the kidney that is the leading genetic cause of end-stage renal failure in children. Egypt is a country with a high rate of consanguineous marriages; yet, only a few studies have investigated the clinical and molecular characteristics of NPHP and related ciliopathies in the Egyptian population. We studied 20 children, from 17 independent families, fulfilling the clinical and the ultrasonographic criteria of NPHP. Analysis for a homozygous deletion of the NPHP1 gene was performed by polymerase chain reaction on the genomic DNA of all patients. Patients were best categorized as 75% juvenile NPHP, 5% infantile NPHP, and 20% Joubert syndrome-related disorders (JSRD). The mean age at diagnosis was 87.5 + 45.4 months, which was significantly late as compared with the age at onset of symptoms, 43.8 ± 29.7 months (P <0.01). Homozygous NPHP1 deletions were detected in six patients from five of 17 (29.4%) studied families. Our study demonstrates the clinical phenotype of NPHP and related disorders in Egyptian children. Also, we report that homozygous NPHP1 deletions account for 29.4% of NPHP in the studied families in this cohort, thereby confir-ming the diagnosis of type-1 NPHP. Moreover, our findings confirm that NPHP1 deletions can indeed be responsible for JSRD.
Introduction
Nephronophthisis (NPHP) is an autosomal-recessive cystic kidney disease that constitutes the most frequent genetic cause of end-stage renal disease (ESRD) in the first three decades of life.1-3 The NPHP–medullary cystic kidney disease (NPHP–MCKD) complex describes a distinct clinico-pathologic entity of inherited diseases that lead to chronic renal failure on the pathologic basis of a chronic sclerosing tubulointerstitial nephritis.4
Three clinical forms of NPHP have been distinguished based on the age at onset of ESRD: infantile,5 juvenile,6 and adolescent NPHP,7 which manifest with ESRD at the median ages of one, 13, and 19 years, respectively. NPHP can be associated with retinitis pigmentosa (Senior-Loken syndrome), liver fibrosis, cerebellar vermis aplasia [Joubert syndrome (JBTS)], and ocular motor apraxia type Cogan.8-11
Because of the mild nature of symptoms and the lack of edema, hypertension, or urinary tract infections, there is often a delay in the diagnosis of NPHP.12 The most useful imaging technique in NPHP is ultrasonography. Kidneys are of normal or moderately reduced size and show increased echogenicity, loss of cortico-medullary differentiation, and, in later stages, cyst formation at the cortico-medullary border.13 Renal histology reveals a characteristic triad of tubular basement membrane thickening and disruption, interstitial infiltration and fibrosis, and tubular atrophy and dilatation, with or without cyst formation.14
Molecular genetic analysis is the only diagnostic procedure by which the diagnosis of NPHP-1, NPHP-2, or NPHP-3 can be made with certainty. However, due to the presence of additional loci for NPHP, the lack of detection of mutations in the NPHP1 gene does not exclude the diagnosis of NPHP. If molecular genetic diagnostics do not detect a molecular defect, the diagnosis of NPHP can be based on the combined results of typical clinical history with polyuria, polydipsia and anemia; the classical appearance of the kidney on ultrasound and renal histology.14 The appropriate diagnosis of NPHP is important not only for anticipating progressive renal failure but also for the implications on genetic counseling.
Twelve genes have been implicated in NPHP: NPHP1,15,16 NPHP2/INVS,17 NPHP3,18 NPHP4,19,20 NPHP5/IQCB1,21 NPHP6/CEP290,22,23 NPHP7/GLIS2,24 NPHP8/RPGRIP1L,25-27 NPHP9/NEK8,28 TMEM67,29 TTC21B,30 and XPNPEP3.31 Recently, massively parallel re-sequencing mutation analysis of 18 different NPHP-associated ciliopathy genes in 120 ascertained patients led to the identification of 57 mutated alleles, comprising 43 different mutations in 30 unrelated patients.32
Homozygous deletions in the NPHP1 gene account for approximately 21% of all NPHP cases, whereas the other genes contribute to less than 3% each. Interestingly, through positional cloning, many of the causative mutations have been mapped to genes involved in centrosome and cilia function. This had contributed to a unifying theory that defines cystic kidney diseases as “ciliopathies” based on the finding that all proteins mutated in cystic kidney diseases of humans or animal models are expressed in the primary cilia or centrosomes of renal epithelial cells.33
Little is known about the clinical characterization of NPHP and associated ciliopathies in the region, let alone the genetic molecular data on children afflicted with these diseases. This study was conducted to characterize the clinical phenotypes of infants and children with NPHP, whether isolated (only renal affection) or in the context of a complex ciliopathy (with one or more extra-renal associations). Additionally, we investigated the prevalence of NPHP1 mutations among the study group.
Patients and Methods
Patients
Children with a clinical diagnosis of NPHP, referred to the Center of Pediatric Nephrology and Transplantation, Cairo University, over a period of one year (early 2008–early 2009) were enrolled in this study. The clinical features of NPHP include presentation in the first two decades of life with symptoms of polyuria and polydipsia and signs of growth retardation and anemia. Renal ultrasound evaluation in NPHP demonstrates increased echogenicity, small to normal kidney size, and loss of cortico-medullary differentiation or small cysts at the cortico-medullary junction. Kidney biopsy, considered characteristic of NPHP, includes tubular atrophy with tubular basement membrane disruption, interstitial cellular infiltrates with fibrosis, and microcyst development.
We categorized the studied patients according to their clinical phenotype. The majority of enrolled patients had isolated NPHP, but a subset of patients had extra-renal manifestations, which included retinal degenerative changes, ocular motor apraxia, cerebellar vermis aplasia, and hepatic fibrosis (Table 1). Individuals were examined for recessive NPHP1 mutation irrespective of the presence or absence of their extra-renal manifestations. Informed consent was obtained from the parents of affected children. This study was approved by the Institutional Review Board at Cairo University Children’s Hospital.
Table 1.
Summary of clinical characteristics of the studied subjects.
| Characteristics | No. of subjects | % |
|---|---|---|
| Gender | ||
| Female | 11 | 55 |
| Male | 9 | 45 |
| Consanguineous parents | 15 | 75 |
| Sibling death* | 8 | 40 |
| Polyuria and polydipsia | 19 | 95 |
| Anemia | 20 | 100 |
| Growth retardation | 20 | 100 |
| End-stage renal disease | 15 | 75 |
| Hypertension | 4 | 20 |
Presumptive diagnosis was nephronophthisis
History taking included two generation family pedigree, age at onset of symptoms (polyuria, polydipsia, and secondary enuresis), age at onset of ESRD and renal replacement therapy (RRT), if any, as well as any visual or neurological symptoms. Clinical assessment included physical growth (height and weight centiles) and blood pressure (BP) measurement. Anomalies or signs of extra-renal involvement were documented. All patients had full ophthalmo-logic evaluation to rule out ocular motor apraxia and retinal degeneration.
Laboratory investigations included hemoglobin level, urine analysis, blood urea nitrogen (BUN), serum creatinine, and calculation of the glomerular filtration rate (GFR) at the time of presentation using modified Schwartz formula.34,35
Imaging
Abdominal ultrasonography was done for proper assessment of renal size, echogenicity, cortico-medullary differentiation, and to detect the presence of renal cysts, if any.13 It was also done to rule out congenital hepatic fibrosis as an extra-renal association (General Electric, Vivid 3 Pro, SyncMaster 591S device with a 3.5–7 MHz probe). All cases were examined by the same operator to avoid inter-observer variability. Magnetic resonance imaging (MRI) of the brain was performed in the subset of patients with clinical extra-renal neurological involvement to demonstrate the distinctive “molar tooth sign.”
Histopathology
Renal biopsy specimens were obtained, following informed parental consent, from eight of 20 patients. Kidney biopsy was not done in the remaining 12/20 patients as a result of medical contraindications (small renal size and/or uncontrollable hypertension) or parental decline to consent the procedure.
Mutational analysis
Genomic DNA was extracted from blood samples collected in EDTA tubes using the QIAGEN Blood and Cell Culture DNA kit according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA). Polymerase chain reaction (PCR) for mutation analysis was performed in the Hildebrandt laboratory, Ann Arbor Michigan. Cost was defrayed from the laboratory’s research funds. All individuals were screened for homozygous NPHP1 deletions, the most common cause of NPHP. Analysis for a homozygous deletion of the NPHP1 gene was performed by a multiplex PCR approach on genomic DNA of patients described earlier.32 Three pairs of primers amplifying three different exons of the NPHP1 gene (exons 5, 7, and 20) were PCR amplified in a single PCR reaction together with two control primer pairs from another gene (LHX9) from chromosome 1. Primers against two exons of LHX9 were used as internal controls to test for the presence of sufficient DNA and PCR accuracy. The primer sequences (5′ > 3′) used for the analysis are as follows: NPHP1-Exon5-Forward, CACTCATAGCTGGTCTGTTCTTG; NPHP1- Exon5-Reverse, CAGGTGTACAGGCAGAGTTTTC; NPHP1-Exon7-Forward, TGTTTTTACTGGAGGGTTAGGTG; NPHP1-Exon7-Reverse, CAGGTGTACAGGCAGAGTTTTC; NPHP1-Exon20-Forward, AATGGCACCCTCCATCCTAC NPHP1-Exon20-Reverse, AATCGTGGAGGATCCATCTG; LHX9-Exon4-Forward, ATATGGCTCTGCCTTGCTTC; LHX9-Exon4-Reverse, TTGGGCAA-AACACACTCTTG; LHX9-Exon6-Forward, ACCCCTAAAAGCCAAGTTGC; and LHX9-Exon6-Reverse, CCTAATAGTGTCTTTGTCTTCACTGC. One PCR reaction was set up by mixing 8 μL water (PCR-grade, 10 μL HotStarTaq®Master Mix (Qiagen), 0.1 μL of each primer (10 uM each), and 1 μL DNA (100 ng/μL). The following touchdown PCR protocol was used:
Initial denaturation: 94°C for 15 min; 24 cycles with an annealing temperature decreasing 0.7°C per cycle, starting at 72°C for 30 s; denaturation at 94°C for 30 s, and extension at 72°C for one min; addition of 24 cycles using a fixed low-annealing temperature of 55°C for 30 s and denaturation at 94°C for 30 s and extension at 72°C for one min; final extension was at 72°C for 10 min. About 10 μL of the PCR reaction was electrophoretically separated on a 1.5% agarose gel for 90 min at 150 V. Lack of amplification products of all three NPHP1 exons, was considered as a homozygous deletion in NPHP1.
Data Analysis
Numerical data were expressed as median and range. P-value less than 0.05 was considered significant.
Results
Twenty children from 17 families with renal findings of NPHP were included in this study. Sex distribution among the affected patients showed a slight preponderance of females, with a ratio of 1.2:1 (11 females and nine males). Seventy-five percent (15/20) were the products of consanguineous marriages. It is notable that the percentage of affected siblings was strikingly high, 65% (13/20 patients) and, likewise, the percentage of sibling death due to NPHP, which accounted for 40% (8/20) of the patients. This finding, together with the high degree of consanguinity, strongly suggests an autosomal-recessive mode of inheritance (Table 1).
While the median age of onset of symptoms in patients was 48 months (range 3–108 months), the median age at diagnosis was significantly delayed at 108 months (range 5–168 months) (P <0.01).
Fifteen of 20 patients (75%) presented with signs of ESRD. All the patients suffered from anemia and growth retardation when they first came to medical attention. Nineteen of 20 patients (95%) had a history typical of NPHP, with symptoms of polydipsia, polyuria, and secondary enuresis (Table 1). Four of 20 patients (20%) were hypertensive, with elevated blood pressure above the 95th percentile for age, gender, and height.
Clinically, the study patients were best categorized as: 13/20 (65%) patients with isolated juvenile NPHP and 3/20 (15%) patients as infantile NPHP, while the remaining 4/20 (20%) patients had extra-renal associations and, hence, were clinically categorized as JSRD, namely cerebello–oculo–renal syndrome (CORS).
We found extra-renal symptoms in 20% (4/20) of the study patients with neurological (mental retardation, ataxia, and MTS) and ophthalmo-logic (retinal degenerative changes and OMA) manifestations as the most frequently reported (Table 2). These four patients were categorized as syndromic juvenile NPHP, and three of them had ESRD at the time of diagnosis (age 84–168 months), whereas the fourth was de-fined as chronic kidney disease (CKD) stage III at the age of 84 months.
Table 2.
Extra-renal manifestations in four (20%) of the study patients.
| Extra-renal manifestations | Patients (n = 20) | % |
|---|---|---|
| Mental retardation | 4 | 20 |
| Dysmorphic facies | 3 | 15 |
| Ataxia | 3 | 15 |
| Molar tooth sign | 3 | 15 |
| Oculomotor apraxia | 3 | 15 |
| Retinal dystrophy/retinitis pigmentosa | 4 | 20 |
| Congenital hepatic fibrosis | 1 | 5 |
| Congenital heart disease* | 1 | 5 |
Prominent forehead, broad nasal bridge and hypertelorism
Ventricular septal defect
Homozygous deletions in the NPHP1 gene were identified in six patients from five independent families out of the 17 studied families (29.4%). Five patients had isolated NPHP, whereas the sixth patient was among the JSRD group (Figure 1) with the unique MTS detected in his brain MRI images (Figure 2). The clinical and genetic characteristics of all the study patients are demonstrated in Table 3.
Figure 1.
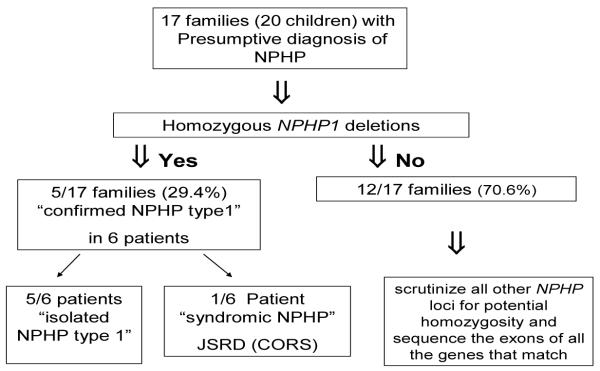
A lgorithm depicting characteristics of the study patients. NPHP: nephronophthisis, JSRD: Joubert syndrome related disorder, CORS: Cerebello oculo renal syndrome
Figure 2.
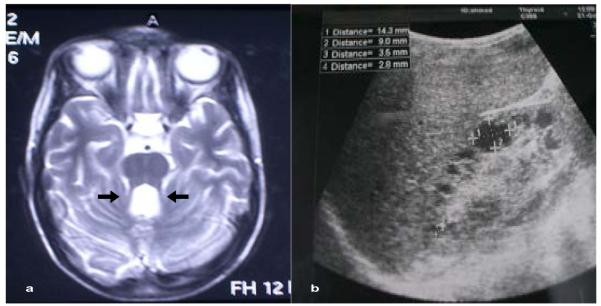
(a) Axial brain MRI scan of case no. 15 showing MTS with elongated superior cerebellar peduncles (arrows); (b) renal sonogram of patient no. 7 showing hyperechogenic kidney with multiple cortico-medullary and cortical cysts.
MRI: magnetic resonance imaging, MTS: molar tooth sign
Table 3.
Clinical and genetic characteristics of the study patients.
| Serial # |
Family # | Gender | Age (mo) |
Bone age (mo) |
Age at onset (mo) |
Initial presentation |
Extra-renal manifestations |
CKD stage at diagnosis |
Age at developing ESRD (mo) (RRT)a |
Cysts on ultrasound |
Histopathologic triad |
NPHP 1 homozygous deletion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| 1 | A1421 II1 | F | 168 | 108 | 36 | P&P, Anemia, GR | OMA, RP, MR, MTS, CHF, VSD |
5 168 | [RTx]b | + | NDd | No |
|
| ||||||||||||
| 2 | A1451 II4 | F | 48 | 15 | 24 | P&P, Anemia, GR | − | 4 | 60 [HD] | + | + | No |
|
| ||||||||||||
| 3 | A1937 II4 | F | 108 | 68 | 48 | P&P, Anemia, GR | − | 5 | 108 [HD] | + | + | No |
| 4 | A1937 II6 | F | 48 | 30 | 36 | Anemia, GR, episode of generalized edema |
− | 2 | − | 0 | ND | |
|
| ||||||||||||
| 5 | A1944 II2 | M | 9 | 2 | 5 | FTT, Dehydration P&P, Anemia |
− | 5 | 9 [PD] | + | ND | No |
|
| ||||||||||||
| 6 | A1967 II6 | F | 144 | 106 | 108 | P&P, Anemia, GR | − | 5 | 144 [HD] | + | + | No |
|
| ||||||||||||
| 7 | A2202 II1 | M | 42 | 30 | 12 | P&P, Anemia, GR | − | 5 | 32 [HD] | + | + |
Homozygous
deletion |
|
| ||||||||||||
| 8 | A2228 II3 | F | 132 | 86 | 48 | P&P, Anemia, GR | − | 4 | − | + | ND |
Homozygous
deletion |
|
| ||||||||||||
| 9 | A2229 II5 | F | 78 | 45 | 36 | P&P, Anemia, GR | − | 5 | 84 [HD] | 0 | ND | No |
|
| ||||||||||||
| 10 | A2245 II4 | F | 89 | 63 | 50 | P&P, Anemia, GR | − | 5 | 89 [HD] | + | ND | No |
|
| ||||||||||||
| 11 | A2324 II3 | M | 84 | 54 | 36 | P&P, Anemia, GR | MR, RD | 3 | − | 0 | + | No |
| 12 | A2324 II1 | M | 120 | 84 | 36 | P&P, Anemia, GR | OMA, RD, MR, MTS |
5 | 84 [HD] | + | ND | No |
|
| ||||||||||||
| 13 | A2325 II1 | M | 144 | 96 | 84 | P&P, Anemia, GR | − | 5 | 144[HD] | + | ND |
Homozygous
deletion |
| 14 | A2325 II2 | M | 120 | 72 | 48 | P&P, Anemia, GR | − | 4 | 130 [RTx] | 0 | + |
Homozygous
deletion |
|
| ||||||||||||
| 15 | A2371 III1 | M | 108 | 66 | 48 | P&P, Anemia, GR | Seizures, OMA, RP, MR, MTS |
5 | 108 [HD] | 0 | ND |
Homozygous
deletion |
|
| ||||||||||||
| 16 | A2413 II6 | F | 126 | ND | 48 | P&P, Anemia, GR | − | 5 | 126 [HD] | + | ND |
Homozygous
deletion |
|
| ||||||||||||
| 17 | A2412 II1 | F | 125 | ND | 50 | P&P, Anemia, GR | − | 5 | 123 [HD] | + | + | No |
|
| ||||||||||||
| 18 | A3059 II3 | F | 97 | 59 | 48 | P&P, Anemia, GR | − | 5 | 97 [HD] | 0 | ND | No |
|
| ||||||||||||
| 19 | A3060 II8 | M | 119 | ND | 60 | P&P, Anemia, GR | − | 5 | 107 [HD] | + | ND | No |
|
| ||||||||||||
| 20 | A3061 II1 | M | 5 | ND | 3 | P&P, Anemia FTT, Dehydration |
− | 5 | 5 [PD] | + | + | No |
RRT: renal replacement therapy,
RTx: renal transplantation, HD: hemodialysis, PD: peritoneal dialysis,
ND: not done. Histopathological triad: tubular basement membrane thickening and disruption, interstitial infiltration and fibrosis, and tubular atrophy and dilatation with or without cyst formation. P&P: polyuria and polydipsia, FTT: failure to thrive, GR: growth retardation, RP: retinitis pigmentosa, RD: retinal dystrophy, OMA: ocular-motor apraxia, MR: mental retardation, MTS: molar tooth sign, CHF: congenital hepatic fibrosis, VSD: ventricular septal defect, ESRD: end-stage renal disease
Discussion
In this report, we studied a cohort of 20 patients with NPHP. The common criterion in the study patients was the renal involvement. Of this cohort, six patients had positive homozygous deletion of NPHP1 gene; five children had isolated NPHP and one patient had JSRD or CORS.
Of the 16 patients with isolated NPHP, five patients had homozygous deletion of NPHP1 gene. Interestingly, four of these patients were clinically diagnosed as juvenile NPHP, whereas the fifth patient had the classical phenotype of infantile NPHP (NPHP type-2). This patient showed most of the clinical and histopathological features of NPHP type-2 previously reported in the literature,4,17,37 and had been reported recently as the first patient with the clinical phenotype of infantile NPHP, yet with documented homozygous deletion of the NPHP1 gene.38
Infantile NPHP, frequently caused by mutations in the NPHP2/inversin gene, differs from the other types of NPHP by the early age of onset of ESRD, usually less than five years in all reported cases, whereas the median age of ESRD in juvenile NPHP (NPHP type-1 or type-4) is about 13 years.17 Renal cortical microcysts is another criterion where detailed analysis of a murine model of NPHP2/inversin demonstrates cystic dilatation of Bowman’s capsule, proximal tubule, thick ascending limb, and collecting duct.39
Co-occurrence of extra-renal findings was observed in four of the study patients (20%), two of whom were siblings. The retinal degenerative changes and neurological involvement are the most frequently reported (Table 2). It is worth mentioning that there was a striking phenotypic heterogeneity between the two described siblings, although they both tested negative for NPHP1 homozygous deletion. The two siblings shared NPHP and retinal dystrophy; nevertheless, the younger brother lacked MTS, OMA, and ataxia described in his elder sibling.
In this subset of study patients categorized as JSRD, only one had homozygous deletion of NPHP1 gene. This patient had both neurologycal and ophthalmologic involvement as extra-renal manifestations. Notably, he had a more severe form of neurological involvement as compared with the other three JSRD patients who had no NPHP1 mutational defect. His mental capabilities were profoundly compromised with seizures and his ataxic manifestations were more pronounced as compared with the non-NPHP1 JSRD patients.
Marked intra-familial variability of associated extra-renal symptoms was observed by Caridi et al in familial cases of JSRDs with homozygous deletion of NPHP1 (nephrocystin).40 They reported neurological defects varying from subtle involvement of cerebellum with thickened peduncle to JSRD, showing the typical neurological signs of JBTS (hypotonia, ataxia, psychomotor delay, and oculomotor apraxia-type Cogan).
In contrast, Parisi et al reported that the subset of JBTS patients with an NPHP1 deletion have a form of JBTS at the mild end of the clinical spectrum for this disorder, as they were not severely mentally retarded and lacked the respiratory disturbance often seen in this disorder.41
The fourth patient described in this work as JSRD had more extra-renal manifestations than the other three JSRD children. She had the typical neurological signs with the distinct MTS, retinitis pigmentosa, congenital hepatic fibrosis, as well as ventricular septal defect that was surgically closed at the age of seven years. No NPHP1 mutation was detected in this patient.
Homozygous NPHP1 deletions, the most fre-quent NPHP1 mutation known, will be detec-ted with the multiplex approach used in the present study. However, in more than 6% of all NPHP1 cases, the underlying mutation has been found to be a heterozygous deletion com-bined with a single point mutation.36 As for the patients with no homozygous NPHP1 deletion, we will investigate for heterozygous deletions in a future study by applying the ligation-dependent probe amplification (MLPA) tech-nique. Further analysis of all other NPHP loci for potential homozygosity in consanguineous families by total genome search for linkage using Affimetrix SNP arrays is also consi-dered. Sequencing all exons of NPHP genes located within regions of significant homozy-gosity is crucial to identify the underlying ge-netic defect.
Acknowledgment
The authors thank the patients and their fami-lies for generously donating DNA samples and clinical information.
References
- 1.Hildebrandt F, Otto E. Cilia and centrosomes: A unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005;6:928–930. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- 2.Smith C, Graham J. Congenital medullary cysts of the kidneys with severe refractory anemia. Am J Dis Child. 1945;69:369–77. [Google Scholar]
- 3.Fanconi G, Hanhart E, von Albertini A, Uhlinger E, Dolivo G, Prader A. Familial, juvenile nephronophthisis (idiopathic paren-chymal contracted kidney) [in German] Helv Pediatr Acta. 1951;6:1–49. [PubMed] [Google Scholar]
- 4.Hildebrandt F. Juvenile nephronophthisis. In: Harmon WE, editor. Pediatric Nephrology. Williams & Wilkins; Baltimore: 2004. pp. 665–73. [Google Scholar]
- 5.Gagnadoux MF, Bacri JL, Broyer M, Habib R. Infantile chronic tubulo-interstitial nephritis with cortical microcysts: Variant of nephro-nophthisis or new disease entity. Pediatr Nephrol. 1989;3:50–5. doi: 10.1007/BF00859626. [DOI] [PubMed] [Google Scholar]
- 6.Hildebrandt F, Waldherr R, Kutt R, Brandis M. The nephronophthisis complex: Clinical and genetic aspects. Clin Investig. 1992;70:802–8. doi: 10.1007/BF00180751. [DOI] [PubMed] [Google Scholar]
- 7.Omran H, Fernandez C, Jung M, et al. Identi-fication of a new gene locus for adolescent nephronophthisis, on chromosome 3q22 in a large Venezuelan pedigree. Am J Hum Genet. 2000;66:118–27. doi: 10.1086/302705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Senior B, Friedmann AI, Braudo JL. Juvenile familial nephropathy with tapetoretinal dege-neration: A new oculorenal dystrophy. Am J Ophthalmol. 1961;52:625–33. doi: 10.1016/0002-9394(61)90147-7. [DOI] [PubMed] [Google Scholar]
- 9.Boichis H, Passwell J, David R, Miller H. Con-genital hepatic fibrosis and nephronophthisis. A family study. Q J Med. 1973;42:221–33. [PubMed] [Google Scholar]
- 10.Saraiva JM, Baraitser M. Joubert syndrome: A review. Am J Med Genet. 1992;43:726–31. doi: 10.1002/ajmg.1320430415. [DOI] [PubMed] [Google Scholar]
- 11.Saunier S, Morin G, Calado J, Benessay F, Silbermann F, Antignac C. Large deletions of the NPH1 region in Cogan syndrome (CS) associated with familial juvenile nephronoph-thisis (NPH) Am J Hum Genet. 1997;61:A346. [Google Scholar]
- 12.Hildebrandt F, Zhou W. Nephronophthisis-Associated Ciliopathies. J Am Soc Nephrol. 2007;18:1855–71. doi: 10.1681/ASN.2006121344. [DOI] [PubMed] [Google Scholar]
- 13.Blowey DL, Querfeld U, Geary D, Warady BA, Alon U. Ultrasound findings in juvenile nephronophthisis. Pediatr Nephrol. 1996;10:22–4. doi: 10.1007/BF00863431. [DOI] [PubMed] [Google Scholar]
- 14.Waldherr R, Lennert T, Weber HP, Fodisch HJ, Scharer K. The nephronophthisis complex. A clinicopathologic study in children. Virchows Arch A Pathol Anat Histol. 1982;394:235–54. doi: 10.1007/BF00430668. [DOI] [PubMed] [Google Scholar]
- 15.Hildebrandt F, Otto E, Rensing C, et al. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Gene. 1997;17:149–53. doi: 10.1038/ng1097-149. [DOI] [PubMed] [Google Scholar]
- 16.Saunier S, Calado J, Heilig R, et al. A novel gene that encodes a protein with a putative src homology 3 domain is a candidate gene for familial juvenile nephronophthisis. Hum Mol Genet. 1997;6:2317–23. doi: 10.1093/hmg/6.13.2317. [DOI] [PubMed] [Google Scholar]
- 17.Otto EA, Schermer B, Obara T, et al. Muta-tions in INVS encoding inversin cause nephro-nophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–20. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olbrich H, Fliegauf M, Hoefele J, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet. 2003;34:455–9. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- 19.Mollet G, Salomon R, Gribouval O, et al. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat Genet. 2002;32:300–5. doi: 10.1038/ng996. [DOI] [PubMed] [Google Scholar]
- 20.Otto E, Hoefele J, Ruf R, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, con-served in evolution. Am J Hum Genet. 2002;71:1167–71. doi: 10.1086/344395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otto E, Loeys B, Khanna H, et al. Nephro-cystin-5, a ciliary IQ domain protein, is mutated in Senior-Løken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37:282–8. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 22.Sayer JA, Otto EA, O’Toole JF, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–81. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 23.Valente EM, Silhavy JL, Brancati F, et al. Mutations in CEP290, which encodes a centro-somal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–5. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 24.Attanasio M, Uhlenhaut NH, Sousa VH, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet. 2007;39:1018–24. doi: 10.1038/ng2072. [DOI] [PubMed] [Google Scholar]
- 25.Delous M, Baala L, Salomon R, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–81. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 26.Wolf MT, Saunier S, O’Toole JF, et al. Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephro-nophthisis. Kidney Int. 2007;72:1520–6. doi: 10.1038/sj.ki.5002630. [DOI] [PubMed] [Google Scholar]
- 27.Roepman R, Bernoud-Hubac N, Schick DE, et al. The retinitis pigmentosa GTPase regulator (RPGR) interacts with novel transport-like proteins in the outer segments of rod photo-receptors. Hum Mol Genet. 2000;9:2095–105. doi: 10.1093/hmg/9.14.2095. [DOI] [PubMed] [Google Scholar]
- 28.Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol. 2008;19:587–92. doi: 10.1681/ASN.2007040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otto EA, Tory K, Attanasio M, et al. Hypo-morphic mutations in meckelin (MKS3/ TMEM67) cause nephronophthisis with liver fibrosis (NPHP11) J Med Genet. 2009;46:663e70. doi: 10.1136/jmg.2009.066613. [DOI] [PubMed] [Google Scholar]
- 30.Davis EE, Zhang Q, Liu Q, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43:189–96. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Toole JF, Liu Y, Davis EE, et al. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronoph-thisis-like nephropathy. J Clin Invest. 2010;120:791–802. doi: 10.1172/JCI40076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otto EA, Ramaswami G, Janssen S, et al. Mutation analysis of 18 nephronophthisis asso-ciated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet. 2011;48:88–92. doi: 10.1136/jmg.2010.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hildebrandt F, Attanasio M, Otto E. Nephro-nophthisis: Disease Mechanisms of a Ciliopathy. J Am Soc Nephrol. 2009;20:23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwartz GJ, Haycock GB, Edelmann CM, Spitzer A. A simple method estimate of glome-rular filtration rate in children derived from body length and plasma creatinine. Pediatrics. 1976;58:259–63. [PubMed] [Google Scholar]
- 35.van Rossum LK, Mathot RA, Cransberg K, Zietse R, Vulto AG. Estimation of the glome-rular filtration rate in children: Which algo-rithm should be used? Pediatr Nephro. 20:1769–75. doi: 10.1007/s00467-005-2001-y. 2005l. [DOI] [PubMed] [Google Scholar]
- 36.Otto EA, Helou J, Allen SJ, et al. Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum Mutat. 2008;29:418–26. doi: 10.1002/humu.20669. [DOI] [PubMed] [Google Scholar]
- 37.Simms RJ, Eley L, Sayer JA. Nephronoph-thisis. Eur J Hum Genet. 2009;17:406–16. doi: 10.1038/ejhg.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soliman NA, Hildebrandt F, Allen SJ, Otto EA, Nabhan MM, Badr AM. Homozygous NPHP1 deletions in Egyptian children with nephronophthisis including an infantile onset patient. Pediatr Nephrol. 2010;25:2193–4. doi: 10.1007/s00467-010-1539-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips CL, Miller KJ, Filson AJ, et al. Renal cysts of inv/inv mice resemble early infantile nephronophthisis. J Am Soc Nephrol. 2004;15:1744–55. doi: 10.1097/01.asn.0000131520.07008.b3. [DOI] [PubMed] [Google Scholar]
- 40.Caridi G, Dagnino M, Rossi A, et al. Nephro-nophthisis type 1 deletion syndrome with neurological symptoms: Prevalence and signi-ficance of the association. Kidney Int. 2006;70:1342–7. doi: 10.1038/sj.ki.5001768. [DOI] [PubMed] [Google Scholar]
- 41.Parisi MA, Bennett CL, Eckert ML, et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]