

Abstract
Low-density lipoproteins (LDLs, also known as ‘bad cholesterol’) are the major carriers of circulating cholesterol and the main causative risk factor of atherosclerosis. Plasma LDLs are 20- to 25-nm nanoparticles containing a core of cholesterol esters surrounded by a phospholipid monolayer and a single copy of apolipoprotein B (550 kDa). An early sign of atherosclerosis is the accumulation of LDL-derived lipid droplets in the arterial wall. According to the widely accepted ‘response-to-retention hypothesis’, LDL binding to the extracellular matrix proteoglycans in the arterial intima induces hydrolytic and oxidative modifications that promote LDL aggregation and fusion. This enhances LDL uptake by the arterial macrophages and triggers a cascade of pathogenic responses that culminate in the development of atherosclerotic lesions. Hence, LDL aggregation, fusion, and lipid droplet formation are important early steps in atherogenesis. In vitro, a variety of enzymatic and nonenzymatic modifications of LDL can induce these reactions and thereby provide useful models for their detailed analysis. Here, we summarize current knowledge of the in vivo and in vitro modifications of LDLs leading to their aggregation, fusion, and lipid droplet formation; outline the techniques used to study these reactions; and propose a molecular mechanism that underlies these pro-atherogenic processes. Such knowledge is essential in identifying endogenous and exogenous factors that can promote or prevent LDL aggregation and fusion in vivo and to help establish new potential therapeutic targets to decelerate or even block these pathogenic reactions.
Keywords: early events in atherosclerosis; hydrophobic interactions; lipoprotein hydrolysis; low-density lipoprotein (LDL) fusion kinetics; thermal, chemical, mechanical denaturation
Introduction
Atherosclerosis claims more than 600 lives annually in the USA and is the leading cause of death in the developed world (1). In atherosclerosis, cholesterol deposits accumulate in the arterial intima, leading to the gradual thinning of the blood vessel lumen, thus impeding blood flow and increasing the risk of coronary artery disease, heart attack, and stroke. Low-density lipoproteins (LDLs, also known as ‘bad cholesterol’) are the strongest causative risk factor for atherosclerosis (2). LDLs are the major plasma carriers of cholesterol in the form of cholesterol esters. An LDL particle (average diameter 22 nm) contains a hydrophobic core consisting of apolar lipids, mainly cholesterol esters and up to 20% triacylglycerol (3). This core is surrounded by an amphipathic surface containing a single copy of apolipoprotein B (apoB), a glycosylated 550-kDa protein, which is one of the largest known proteins, and a monolayer of polar lipids, mostly phosphatidylcholine (PC) and sphingomyelin (Figure 1). The physiological function of plasma LDLs is to deliver cholesterol to peripheral tissues via whole-particle endocytosis mediated by low-density lipoprotein receptor (LDLR) (4). LDL uptake by cells via LDLR is non-atherogenic because it down-regulates cholesterol biosynthesis (5). In the alternative pro-atherogenic pathway, LDLs are taken up by arterial macrophages via the scavenger receptors, leading to macrophage conversion into foam cells (6, 7).
Figure 1. LDL aggregation, fusion, and lipid droplet formation.

(Top) Cartoon representations of intact, aggregated, and fused LDLs in cross section. Lower panels show negative stain electron micrographs of intact, aggregated, and fused LDLs, and of LDL-derived lipid droplets (29). An intact LDL is a spheroidal particle (d ~ 22 nm) comprised of a core of apolar lipids, mainly cholesterol esters and triacylglycerides (gray), surrounded by a phospholipid monolayer (yellow) and one copy of apoB (blue and red arc). LDL aggregation probably involves changes in the conformation of the protein and lipid moieties (29) but not necessarily in the particle size. Fusion merges the contents of two or more LDL particles to form an enlarged lipoprotein. Rupture involves lipoprotein disintegration and release of the apolar core lipids that coalesce into droplets. Both fused and ruptured LDLs tend to aggregate. Enhanced uptake of aggregated and fused LDLs and lipid droplets by macrophages leads to formation of foam cells and initiates atherosclerosis.
According to the ‘response-to-retention hypothesis’ (8), atherogenesis is initiated upon LDL binding and retention by extracellular matrix components such as proteoglycans in the arterial wall. The retained lipoproteins undergo various modifications, including oxidation, lipolysis, and proteolysis by resident hydrolytic and oxidative enzymes. These modifications cause LDL fusion that further augments LDL retention in the arterial wall, triggering a cascade of inflammatory and apoptotic responses that contribute to atherogenesis.
The initial sign of atherogenesis is the appearance of cholesterol-rich extracellular lipid droplets up to 400 nm in size in the subendothelial space (9). Biochemical and morphological analysis of such droplets from human atherosclerotic lesions suggests that they are derived mainly from the entrapped LDLs (10, 11). Animal model studies strongly support this conclusion and show that accumulation of extracellular lipid droplets can be experimentally reproduced in rabbit arterial intima hours upon injection of large amounts of human LDL in circulation, as well as in isolated rabbit cardiac valves upon incubation with human LDL (12, 13).
Although the molecular mechanism of LDL retention and lipid droplet formation in the arterial subendothelium is not fully understood, it is increasingly clear from studies by the groups of Kovanen, Camejo and Hurt-Camejo, Sanchez-Quesada, Parasassi, and others that aggregation and fusion of modified LDLs prevent their exit from the arterial wall and contribute to atherogenesis (11, 14–20). Several lines of evidence support the presence of LDL aggregates in the arterial wall (21, 22) and their involvement in LDL retention by arterial proteoglycans during atherogenesis. For example, Frank and Fogelman (23) used freeze-etch electron microscopy (EM) to show that the aortic intima in Watanabe heritable hyperlipidemic and cholesterol-fed rabbits contained aggregated lipoproteins bound to subendothelial matrix. Steinbrecher and Lougheed (24) reported that LDL aggregates isolated from atherosclerotic lesions induced macrophage foam cell formation in a process independent of LDL uptake by scavenger receptors. In addition, aggregated LDLs have been reported to induce cholesterol accumulation in coronary vascular smooth muscle cells and turn them into foam cells, possibly by upregulating the level of LDLR-related protein (25). These and other studies convincingly showed that LDL aggregation, fusion, and coalescence into lipid droplets are important triggering events in early atherosclerosis (Figure 1).
In contrast to modified LDLs, native LDLs do not readily aggregate or fuse under physiological conditions, suggesting that lipoprotein modifications drive these transitions (26). The accepted view is that such major modifications in vivo include apoB proteolysis, LDL lipolysis, oxidation, and glycation. Many aspects of these reactions remain unclear, e.g., how do the apparently disparate chemical or physical modifications exert similar structural responses in LDL? Is there a synergy among numerous factors that influence LDL fusion? Which enzymatic or nonenzymatic modifications are particularly important in promoting or preventing LDL fusion in vivo? What are specific steps in LDL aggregation, fusion, and lipid droplet formation, and what therapeutic agents can block these pathogenic processes? These and other unanswered questions reflect the fact that atherosclerosis is a very complex chronic disease that can be influenced by an immense number of factors, many of which are not well understood.
In a complementary approach, in vitro studies can provide tractable experimental models to determine how individual factors, alone or in combination, influence LDL fusion. A number of different in vitro modifications can induce LDL aggregation, fusion, and coalescence into lipid droplets. These modifications include brief vortexing (27) or prolonged exposure to elevated temperatures (28) or acidic pH (29). Although such in vitro treatments do not necessarily mimic physiological conditions, they provide useful model systems to study the immensely complex process and elucidate its molecular mechanism. The results of such in vitro studies can provide sharper insights into the structural basis underlying LDL aggregation, fusion, and lipid droplet formation upon various biophysical and biochemical modifications, quantify the rate and the extent of these LDL reactions, and help design strategies aimed to decelerate or even block these pathogenic processes.
In this review, we summarize current knowledge of the in vivo and in vitro processes leading to LDL aggregation, fusion, and coalescence into lipid droplets; outline the techniques used to study them; and propose a molecular mechanism that underlies these pro-atherogenic processes. Whenever possible, we try to differentiate among lipoprotein aggregation, fusion, and coalescence into lipid droplets (Figure 1). However, many experimental studies do not make this distinction; in these instances, we use the term preferred by the authors or refer to it as ‘aggregation and fusion’.
Biochemical modifications
Lipolysis
Sphingomyelinase (SMase) hydrolyzes sphingomyelin to phosphocholine and ceramide. Secretory SMase is a zinc-dependent acidic metalloenzyme secreted by macrophages and smooth muscle cells that is found in the arterial intima (30). This enzyme hydrolyzes LDL sphingomyelin that accounts for 20%–25% of the phospholipids on LDL surface (31). Upon hydrolysis, water-soluble phosphocholine is released from the surface, whereas water-insoluble ceramide is retained in the core of the LDL. This leads to the increase in the apolar core lipids at the expense of the polar surface lipids, resulting in a hydrophobic mismatch between the core and surface, which is expected to cause lipoprotein fusion. In fact, ceramide accumulation leads to LDL fusion (described below). LDL fusion upon SMase reaction in vivo is supported by the observation that aggregated LDLs in atherosclerotic lesions are enriched in ceramide (32). Moreover, treatment of isolated LDLs with SMase can induce lipoprotein aggregation and fusion in vitro (18, 33). These observations suggest that secretory SMase can contribute to atherogenesis by mediating LDL fusion.
Phospholipase A2 (PLA2) superfamily enzymes hydrolyze sn-2 acyl bond in PC to generate free fatty acids (FFAs) and lyso-PC, which are important mediators of inflammation (34). Secretory nonpancreatic PLA2, which is secreted by endothelial cells and macrophages, is found in the arterial intima of atherosclerotic and healthy subjects and is associated with extracellular matrix and lipid droplets (35). Lipoprotein-associated PLA2, which is secreted by leucocytes, is associated with circulating lipoproteins and macrophages in atherosclerotic plaques (36). Importantly, type II secretory nonpancreatic PLA2 and lipoprotein-associated PLA2 preferentially hydrolyze oxidized PC in lipoproteins and serve as biomarkers of atherosclerosis (36).
Earlier studies reported that LDL lipolysis by PLA2 in the presence of serum albumin, which removes FFAs from LDLs, results in lipoprotein aggregation but not fusion (33). Later studies showed that if FFAs produced by PLA2 are not removed, lipoprotein coalescence into lipid droplets is greatly enhanced (37). Furthermore, lipolysis by secretory nonpancreatic PLA2 was reported to induce fusion of the proteoglycan-bound lipoproteins, thereby enhancing their retention in the arterial wall (38). Thus, multiple lines of evidence indicate that LDL hydrolysis by PLA2 family enzymes contributes to atherogenesis by inducing LDL aggregation, fusion, and retention by arterial proteoglycans. Notably, several studies reported that PLA2 is preferentially enriched in small, dense LDLs (14) and in electronegative LDLs (16); the latter probably reflects the negative charge on the FFAs accumulated in LDLs upon PLA2 hydrolysis. These findings suggest that PLA2 potentially contributes to the enhanced pro-atherogenic properties of small, dense LDLs and electronegative LDLs.
Phospholipase C (PLC) hydrolyzes PC to generate phosphocholine and diacylglycerol. Polar phosphocholine is released while apolar diacylglycerol is redistributed between the lipoprotein surface and the core. This lipid redistribution from the surface to the core generates hydrophobic mismatch that is expected to promote lipoprotein aggregation and fusion. In fact, LDL aggregation and fusion upon PLC hydrolysis, which was first observed in 1989 (39), became a standard technique to induce these LDL transitions in vitro. Notably, the authors also found that aggregated and fused LDLs were taken up much faster by macrophages as compared with normal LDLs, which helped establish the link among LDL aggregation, fusion, and atherogenesis (39). Recent report suggests that, similar to PLA2, PLC is preferentially associated with electronegative LDLs in plasma, which may potentially contribute to the enhanced pro-atherogenic properties of these particles (16, 40). Other studies showed that PLC-induced LDL aggregation and fusion can be prevented by the exchangeable (water-soluble) apolipoproteins that have high affinity for lipid surface (41). This observation supports the idea that lipoprotein fusion upon PLC hydrolysis results from the surface exposure of hydrophobic lipid moieties.
Cholesterol esterase hydrolyzes cholesterol esters, the most abundant lipids in LDL core. In contrast to LDLs, which contain mainly esterified cholesterol, biochemical analysis of lipid droplets isolated from atherosclerotic lesions detected mainly unesterified cholesterol (42). To understand the precursor-product relationship between LDLs and lesional lipid droplets, Kruth and colleagues hydrolyzed LDLs using cholesterol esterase (43). For hydrolysis to proceed, the enzyme needs to gain access to the lipoprotein core. Native LDLs did not provide such access and hence were not readily hydrolyzed by cholesterol esterase. However, core lipids became accessible to hydrolysis upon apoB proteolysis on LDL surface. Upon completion of hydrolysis, LDLs were converted to liposome-like structures that were chemically and morphologically similar to the extracellular lipid droplets in atherosclerotic lesions. This supports the precursor-product relationship between LDLs and extracellular lipid droplets (43).
Lipoprotein lipase exerts both enzymatic and nonenzymatic effects that contribute to lipoprotein remodeling in vivo. In its catalytically active dimeric form, lipoprotein lipase hydrolyzes triacylglycerol into diacylglycerol, monoacylglycerol, and FFAs. This reaction is key to the metabolism of triglyceride-rich lipoproteins such as very-low-density lipoproteins (VLDLs), which are metabolic precursors of LDLs (44). The enzymatic action of lipoprotein lipase on VLDL is an obligatory early step in VLDL maturation to LDL and is largely anti-atherogenic. Notably, lipid core hydrolysis depletes the core and expand the surface, generating excess surface material that dissociates from VLDL in the form of small particles that join the plasma pool of high-density lipoproteins (HDLs) (45). This contrasts with the hydrolysis by PLA2, PLC, or SMase, which depletes the surface lipids and promotes lipoprotein fusion. Therefore, in contrast to PLA2, PLC, or SMase, which induce lipoprotein fusion, enzymatic action of lipoprotein lipase is expected to promote lipoprotein fission rather than fusion.
Endothelial lipoprotein lipase that is anchored to the arterial endothelium via a flexible linker hydrolyzes VLDL triglycerides in vivo. As a structural anchor, the enzyme can bind lipoproteins and link them to subendothelial proteoglycans and various cell surface receptors, enhancing LDL retention in the subendothelial space (46). In contrast to the anti-atherogenic properties of its enzymatic function, the anchoring function of lipoprotein lipase on LDL, which enhances LDL retention in the arterial wall, is pro-atherogenic. Furthermore, LDL affinity for lipoprotein lipase was reported to increase upon LDL oxidation (47) probably because native LDLs preferentially bind to the monomeric catalytically inactive enzyme, whereas oxidized LDLs bind to the dimeric catalytically active form (48). Structural studies suggested that LDL binding to lipoprotein lipase is mediated entirely by the lipids and does not involve apoB (48). In vitro study showed that lipoprotein lipase can induce LDL aggregation at higher than equimolar ratios of the enzyme to LDL (49). This suggests that lipoprotein aggregation in these experiments was due to the nonenzymatic anchoring action of lipoprotein lipase.
Proteolysis
A single copy of apoB comprises over 95% of LDL protein content and covers more than 20% of LDL surface (50). This large multidomain protein of 4536 amino acids directs LDL metabolism and serves as a structural scaffold and an important functional ligand for LDL interactions with LDLR and with arterial proteoglycans. Therefore, even partial loss of apoB upon proteolysis can influence functional interactions of LDL and cause conformational changes in their protein and lipid moieties, leading to the reorganization of the entire particle. This can influence interactions between LDL particles and augment their aggregation, fusion, and lipid droplet formation. Therefore, apoB proteolysis is a potential mechanism for generating extracellular lipid droplets.
Kovanen and Kokkonen (51) observed that incubation with exocytosed rat mast cell granules can convert LDLs into lipid droplets whose morphology resembles that of the extracellular lipid droplets found in atherosclerotic lesions (10). Two neutral proteases, chymase and carboxypeptidase A, were responsible for apoB degradation and lipid droplet formation in these experiments. Tests of additional proteases that cleave apoB revealed two distinct effects. Plasmin, kallikrein, and thrombin, whose action on LDLs led to apoB fragmentation without release of proteolytic fragments, did not cause LDL fusion; in contrast, trypsin, α-chymotrypsin, and pronase, whose action led to apoB fragmentation followed by release of proteolytic fragments from LDL surface, triggered LDL fusion (52). The authors concluded that LDL fusion after proteolysis occurs only upon dissociation of proteolytic fragments from the lipoprotein surface (18).
Oxidation
Oxidative modification hypothesis of atherosclerosis originated 30 years ago from observations that oxidized LDLs are toxic to cultured cells (53–56) and are readily ingested via the scavenger receptors by macrophages, converting them into foam cells (6, 57, 58). The latter was attributed to oxidative modifications in apoB, which impair its interactions with LDLR and enhance LDL binding to macrophage scavenger receptors. Later studies showed that LDLs can be oxidized in circulation and in the arterial wall (59–61). The pathogenic properties of oxidized LDLs have been attributed to their ability to support foam cell formation as well as aid the recruitment of circulating monocytes to the arterial initima, induce platelet aggregation, and other pro-inflammatory and pro-thrombotic effects [reviewed in ref. (62)]. The molecular basis underlying these effects is difficult to establish because of the complexity of LDL oxidation, which involves an immense number of possible modifications to various lipid and protein moieties. The problem is further compounded by the heterogeneity of plasma LDLs and the products of their oxidation. These products depend upon the oxidants used, the extent of oxidation, the biochemical composition of intact LDLs that can vary from batch to batch (e.g., the amount of antioxidants such as carotenoids in LDL core), and many other factors that are beyond the scope of this review. As a result of this extreme biochemical complexity, the topic remains controversial, which is further compounded by the clinical trials that failed to show beneficial effects of antioxidants such as vitamin E in cardiovascular disease (63). Excellent reviews of this topic can be found in refs. (64–66). Here, we briefly outline the role of oxidation in the aggregation and fusion of LDLs, which is also not free from controversy.
One of the mechanisms responsible for the enhanced uptake by macrophages of oxidized LDLs is their increased tendency to aggregate (11). LDL aggregation and enhanced uptake by macrophages were consistently observed in cell culture upon action of various oxidative agents such as copper, a radical oxidant whose primary target is lipids, and hypochlorite, a nonradical oxidant whose primary target is proteins (67–70). In contrast to aggregation, evidence for LDL fusion upon oxidation is limited to a report that after days of LDL incubation with 5 μM Cu2+ at 37°C, extensive LDL oxidation led to complete lipoprotein disintegration (18, 70), which may or may not involve LDL fusion.
Studies from our laboratory were aimed to quantify the effects of oxidation on the extent of the heat-induced LDL fusion (71). Single-donor LDLs were isolated from human plasma and were modified to various degrees by several radical or nonradical oxidants including copper, hypochlorite, and myeloperoxidase, which produces hypochlorite and contributes to lipoprotein oxidation in vivo. In this system, LDL oxidation caused no significant changes in the particle size or morphology at ambient temperatures, as observed by nondenaturing polyacrylamide gel electrophoresis (PAGE) and negative stain EM. Surprisingly, the extent of the heat-induced LDL fusion and lipid droplet formation [monitored by circular dichroism (CD), turbidity, and EM] gradually decreased upon progressive oxidation by various agents (71). Biochemical analysis of LDLs at various stages of oxidation suggested that apoB fragmentation and cross-linking were partially responsible for this effect. To reconcile these observations with numerous in vivo and cell culture studies, we note that these studies provided a clear evidence for the aggregation but not necessarily fusion of oxidized LDLs. Moreover, different studies used different experimental systems that could contain additional factors such as PLA2 that acts synergistically with oxidation in vivo. In fact, our later studies showed that FFAs produced by PLA2 or other means are potent fusion-inducing agents in lipoproteins (37). This suggests that enhanced in vivo lipolysis of oxidized PC by PLA2 family enzymes generates FFAs whose accumulation greatly enhances LDL fusion.
In summary, oxidation produces numerous chemical modifications in the protein and lipid moieties, which often occur in parallel. Some of these modifications enhance LDL remodeling by other factors. For example, oxidized PCs are avidly hydrolyzed by PLA2, followed by removal of most lipolytic products by albumin. This leads to complex structural changes that can increase solvent exposure of the apolar groups on LDL surface and promote aggregation, fusion, and ultimate disintegration of LDLs (59, 72, 73).
Glycation
Glycation, which covalently links a sugar molecule to a protein or lipid moiety, is a ubiquitous LDL modification that contributes to atherogenesis. In contrast to enzymatic glycosylation, which occurs at specific sites, is subject to tight enzymatic control, and is often functionally important, nonenzymatic glycosylation (also known as glycation) is not well regulated and often results in impaired macromolecular function. In vivo LDL glycation is often linked to oxidation, and the combined effects, termed glycoxidation, are deleterious to LDL function (74, 75).
LDL glycation in vivo occurs both in diabetic and in nondiabetic patients (76) and principally affects lysines (Lys), which are abundant in apoB. Typically, in LDLs isolated from human plasma, 2%–17% of all Lys are glycated (77). As expected, LDLs isolated from plasma of diabetic patients contain higher proportion of glycated Lys as compared with nondiabetic controls (78). This increased Lys glycation in apoB potentially contributes to the link between diabetes and cardiovascular disease. Interestingly, small, dense LDLs, which are proposed to form a particularly pro-atherogenic subclass, are preferentially glycated in vitro and in vivo as compared with larger particles (76, 79). This difference in glycation, which may result from different apoB conformations on the large and small particles (50), potentially contributes to the enhanced pathogenic properties of small, dense LDLs.
LDL glycation is linked to several pro-atherogenic events, including increased LDL binding to proteoglycans, increased susceptibility to oxidation, and impaired binding to LDLR [ref. (80) and references therein]. The latter is probably due to the modifications in the Lys-rich LDLR-binding sites of apoB. As a result, LDL glycation promotes LDL clearance by macrophage scavenger receptors, leading to foam cell formation (75, 80). In addition, LDL glycation reportedly promotes LDL aggregation in vitro (18). Although the molecular mechanism responsible for aggregation of glycated LDLs is unknown, we speculate that changes in the surface charge distribution on apoB upon Lys glycation probably contribute to this effect.
Prolonged storage
Storage, or LDLs ‘aging’ in vitro, involves various hydrolytic and oxidative modifications to the protein and lipid moieties, which promote LDL aggregation and fusion. These changes can be decelerated, but not completely abolished, by storing LDLs at 4°C in the dark under anaerobic conditions in the presence of EDTA. An even safer way of LDL storage is flash-freezing with 20% sucrose as a cryoprotectant to prevent LDL fusion and rupture at low temperatures (unpublished data). Spontaneous changes that occur during LDL storage include lipid peroxidation and apoB fragmentation, which is attributed in part to its weak autoproteolytic activity. These deleterious changes can increase LDL susceptibility to other hydrolytic modifications. For example, minimal lipolytic activity of secretary PLA2 was observed when freshly isolated plasma LDLs were used as substrates; however, lipolytic activity increased up to 25-fold upon LDL storage at 6°C for 8 weeks or at 37°C for 15 h, which was probably due to PC oxidation (81). In another study, plasma incubation at 37°C produced a subpopulation of LDLs that were prone to aggregation, fusion, and lipid droplet formation as detected by dynamic light scattering and atomic force microscopy (82). Kinetics analysis suggested that particle aggregation in these experiments was driven by interactions between a limited number of specific surface sites on LDLs (82). This mechanism differed from massive aggregation and fusion observed upon other LDL modifications such as copper-induced oxidation. Our own studies using nondenaturing PAGE, size-exclusion chromatography (SEC), and negative stain EM showed that during storage of lipoproteins isolated from human plasma, a subpopulation of LDLs was converted into enlarged particles whose size (~40 nm) was consistent with LDL dimerization (29). In addition, we observed gradual formation of larger particles (≥100 nm) resulting from LDL fusion and coalescence into lipid droplets and their aggregates (29).
Other biochemical modifications
In vitro, LDLs can be chemically modified in numerous ways, some of which result in aggregation and fusion, which invariably enhance LDL uptake by macrophages and foam cell formation in cell culture studies (83). Such chemical modifications include acetylation and maleylation (26); acetoacetylation (84); carbamylation, malondialdehyde, or glutaraldehyde treatment (85, 86); desialylation (87); and treatment with certain flavonoids (88). Similar effects of these diverse modifications on lipoprotein morphology suggest that LDL aggregation and fusion provide a general structural response to a broad range of biochemical perturbations.
Proteins, lipids, small molecules, and polymers that promote or prevent LDL aggregation and fusion
Aggregation and fusion of lipoproteins are initiated by their surface contacts. Because the LDL surface is composed of a phospholipid monolayer and apoB, reagents that promote [FFA, polyethylene glycol (PEG)] or prevent (amphipathic molecules) fusion of phospholipid bilayers are expected to have similar effects on LDL fusion. In addition, changes in apoB conformation and in the core lipid composition can also contribute to LDL fusion. Below we discuss some agents, natural or engineered, that have been observed to promote or prevent LDL fusion. The latter are of particular interest because they may help develop novel therapeutics aimed to inhibit LDL fusion in vivo.
Ceramide
Ceramide is the product of sphingomyelin hydrolysis by SMase, an enzyme that promotes LDL aggregation and fusion in vivo as described above (18, 33). LDLs from atherosclerotic lesions contain 10–50 times more ceramide than plasma LDLs from healthy donors (11, 32). Furthermore, in lesional tissues, aggregated LDLs contain ceramide, whereas native nonaggregated LDLs do not (32). These data suggest that ceramide is directly involved in LDL aggregation and fusion in vivo and probably contributes to the development of atherosclerosis. Quantitative studies support this notion and show that increasing ceramide concentration increases the size of LDL aggregates (89). The physical basis for the ceramide-induced LDL aggregation and fusion appears straightforward, as conversion of polar PC into apolar ceramide molecules shifts the balance between the polar surface and the apolar core of LDLs and promotes solvent exposure of the apolar surface moieties. In summary, ceramide is a potent inducer of LDL fusion in vivo and in vitro.
Free fatty acids
FFAs are produced in vivo by PLA2-family enzymes that hydrolyze PC, by lipoprotein lipase that hydrolyses triaclylglycerides, and by hepatic lipase that hydrolyses both triacylglycerides and PC. Elevated levels of plasma FFAs are observed in inflammation (where excess FFAs are produced upon clearance of lipid membranes from the dying cells) and in metabolic syndrome and diabetes (which are characterized by hypertriglyceridemia) (90–92); all these conditions are associated with an increased risk of cardiovascular disease (93). Notably, FFAs are potent promoters of cell membrane fusion because they perturb molecular packing in phospholipid monolayers (94). This prompted us to test whether incorporation of additional FFAs into the PC monolayer on the lipoprotein surface promotes lipoprotein fusion. The results clearly showed that heat-induced aggregation, fusion, and lipid droplet formation are greatly enhanced upon FFA incorporation into LDLs or other lipoproteins; this effect could be achieved either via the enzymatic action of PLA2 or hepatic lipase on PC or via lipoprotein enrichment with exogenous oleic acid (37). Importantly, FFA removal by albumin reversed the effect and hampered lipoprotein fusion, indicating that FFAs play a direct role in LDL fusion. We concluded that similar to lipid bilayer fusion, lipoprotein fusion is greatly enhanced upon increasing the FFAs in the surface monolayer. In vivo, such an increase in lipoprotein-associated FFAs can result from impaired action of albumin in the acidic environment of deep atherosclerotic lesions as well as from elevated plasma FFAs in inflammation, metabolic syndrome, and diabetes (90–92, 95). Consequently, the ability of FFAs to promote LDL fusion may contribute to the well-established association of these diseases with atherosclerosis (93).
Albumin
Albumin is the most abundant protein in human plasma that acts as a carrier of FFA, lyso-PC, and other endogenous hydrophobic molecules as well as drugs. Khoo et al. (41) showed that albumin reduced LDL aggregation upon vortexing. Talbot et al. (96) showed that at physiological concentrations, albumin reduced flow-induced LDL aggregation. Notably, heat-denatured and fatty acid-stripped albumin was particularly effective in protecting LDLs from aggregation. This protective effect was attenuated upon progressive oxidation of LDLs (97). In our studies of the heat-induced lipoprotein fusion and lipid droplet formation, only FFA-free albumin showed a protective effect that could be fully attributed to FFA removal from LDLs (37). Taken together, these results suggest that albumin can serve as an important modulator of lipoprotein fusion in vivo and that the albumin’s ability to protect LDLs from fusion depends, at least in part, on its ability to effectively remove FFAs from LDLs.
Exchangeable apolipoproteins
In contrast to the nonexchangeable apoB that is permanently associated with its host particle, exchangeable apolipoproteins can transfer among the lipoproteins via the water-soluble globular form. These proteins are relatively small (6–32 kDa) as compared with apoB (550 kDa) and are comprised almost exclusively of amphipathic α-helices whose large apolar faces are optimized for lipid surface binding (98). One example is the major HDL protein, apolipoprotein A-I (apoA-I, 28 kDa). Plasma levels of HDL (also known as ‘good cholesterol’) and apoA-I correlate inversely with the risk of atherosclerosis (99, 100). This cardioprotective action of HDL is thought to result from their role in removing excess cell cholesterol via the reverse cholesterol transport as well as their antioxidant, antithrombolytic, and anti-inflammatory effects (101, 102). In addition, apoA-I helps prevent LDL aggregation in vitro and, potentially, in vivo. Studies of isolated lipoproteins have shown that LDL aggregation induced by vortexing or by PLC lipolysis is inhibited in the presence of HDL or apoA-I (41). This inhibitory effect persists in high salt, suggesting the importance of hydrophobic interactions between apoA-I and LDLs. The authors proposed that amphipathic α-helices in apoA-I can bind to the exposed hydrophobic moieties on LDL surface and thereby block the intermolecular interactions leading to LDL aggregation and fusion. Our unpublished studies showed that apoA-I and HDL help protect LDLs from the heat-induced fusion via a similar mechanism.
This protective effect is not limited to apoA-I but extends to other exchangeable apolipoproteins. One example is apolipoprotein E (apoE, 32 kDa) that circulates on VLDL and HDL and is important for the metabolism of the triglyceride-rich lipoproteins in plasma and for lipid transport in the brain (103). Similar to apoA-I, apoE can inhibit LDL aggregation and fusion upon lipolysis (41). Similarly, PLC-treated LDLs did not aggregate in the presence of apolipophorin, an exchangeable apolipoprotein of insect origin (104). In sum, LDL aggregation and fusion upon various biochemical (hydrolytic) or physical (thermal, mechanical) perturbations can be inhibited by the exchangeable apolipoproteins in vitro and, possibly, in vivo. This effect may contribute to the cardioprotective action of apoA-I, apoE, and related proteins.
Estradiol
Epidemiological studies show that premenopausal women are protected from cardiovascular disease, which is attributed to estrogens (105, 106). The anti-atherogenic action of estrogens is thought to result from their antioxidant effects on LDLs (107–109). Specifically, Parasassi and colleagues reported that 17-β-estradiol binds to apoB on LDLs, rendering the particle more compact and more resistant to copper-induced oxidative modifications (110). The authors hypothesized that by inhibiting LDL oxidation, estradiol can also inhibit LDL fusion. They tested this idea by determining the effects of 17-β-estradiol on electronegative LDLs, a pro-atherogenic subclass that was proposed to trigger aggregation of total LDLs. They reported that estradiol-stabilized LDLs were resistant to aggregation in the presence of electronegative LDLs and proposed that estradiol binding to specific sites on apoB was responsible for this effect (110).
Amphipathic polymers
The endogenous inhibitors of LDL aggregation and fusion, such as apoA-I, apoE, and estradiol, are amphipathic molecules whose protective effects apparently result from their ability to bind to the solvent-exposed hydrophobic moieties on LDL surface. Similarly, other amphipathic molecules that bind to LDL surface may potentially block LDL aggregation and fusion. Such molecules potentially provide new therapeutic agents for atherosclerosis.
To this end, pluronic block copolymers were tested for their ability to prevent LDL aggregation and fusion (111). These polymers are nanomaterials consisting of hydrophilic poly-(ethylene oxide) and hydrophobic poly-(propylene oxide) blocks arranged in A-B-A tri-block structure. These polymers can incorporate into cell membranes and translocate in the cells where they affect various cellular functions (112). A series of pluronic copolymers was tested for their ability to inhibit LDL fusion (111). LDL aggregation, which was induced by incubation at 37°C with constant stirring, was greatly diminished in a manner that was proportional to the hydrophobicity of the copolymer. This further supports the central role of hydrophobic interactions in LDL aggregation and fusion.
Physical perturbations
LDL aggregation and fusion can also result from physical perturbations, including mechanical or thermal stress, chemical denaturants such as guanidinium hydrochloride (Gdn HCl) that interacts with the protein moiety, solutes that promote fusion of lipid membranes and lipoproteins such as PEG (113, 114), reduction in pH, increase in solvent ionic strength, divalent metal ions, LDL crowding, etc. (29). Some of these factors and their effects on LDL fusion in vitro, in silico, and, potentially, in vivo are outlined below.
Mechanical stress
In 1988, the pioneering studies by Steinberg and colleagues showed that even brief 30-s vortexing of LDL solutions at room temperature caused irreversible aggregation, as indicated by increased turbidity and sedimentation coefficients (27). These aggregated LDLs were avidly ingested and degraded by macrophages, converting them into cholesterol ester-rich foam cells. Later studies using fluid stress model confirmed time-dependent LDL aggregation, which was monitored by light attenuation and sedimentation (96, 97). Notably, LDL aggregation was partially inhibited in total plasma, apparently due to the protective effects of other apolipoproteins that are also expected to inhibit LDL aggregation in circulation (described above).
Thermal and chemical denaturation
Protein unfolding upon heating or addition of denaturants such as Gdn HCl is a method of choice for measuring structural stability of globular proteins, water-soluble apolipoproteins, and lipoproteins in solution. Our studies during the last decade showed that thermal or chemical denaturation of all major lipoprotein classes, including LDLs, involves irreversible lipoprotein aggregation, fusion, and coalescence into lipid droplets (28, 29, 115, 116). This is not surprising because even partial protein denaturation should disrupt lipid-protein interactions, leading to dissociation of a portion of the protein from the lipoprotein surface; this is expected to increase solvent exposure of the hydrophobic ‘sticky patches’ that promote lipoprotein aggregation and fusion. In our studies, thermal or Gdn HCl-induced LDL aggregation, fusion, and lipid droplet formation have been detected by negative stain EM, nondenaturing PAGE, SEC, turbidity, CD spectroscopy (Figures 1 and 2), and calorimetric methods (28, 29). These studies showed that, similar to membrane fusion, lipoprotein fusion is thermodynamically irreversible and involves a high activation energy that is particularly high for LDLs (Ea= 100 kcal/mol as compared with ~50 kcal/mol measured for HDL and VLDL) (29, 115, 116). We postulated that this unusually high activation energy, together with the sigmoidal reaction kinetics, which is also unique to LDLs (Figure 2A), reflects the large size of apoB domains whose conformational changes prime LDLs for fusion (29). Further, the high activation energy of LDL fusion reflects the steep temperature dependence of the reaction rate; this limits the range of temperatures at which one can accurately measure this reaction rate. In addition, the rate of LDL fusion also depends strongly on solvent composition, pH, LDL concentration (29), and other factors described below. The combined effects of these factors limit the range of experimental conditions allowing quantitative kinetic analysis of LDL fusion. Nevertheless, in vitro kinetic analysis of LDL fusion provides a useful quantitative tool to determine how individual factors, alone or in combination, influence the rate of this pathogenic reaction.
Figure 2. Integrated approach for the analysis of morphological LDL transitions [modified from ref. (29)].

Single-donor human LDLs (0.5 mg/ml apoB, 20 mM Na phosphate, pH 7.5) were incubated at 85°C, and the time course of the increase in the particle size was monitored by turbidity (A). Aliquots taken at various stages, from 0 (intact LDLs) to 6 (fully denatured), were analyzed by (A) native PAGE, (B) SEC, and (C) negative-stain EM. SEC identified three peaks: I, intact size LDLs (~22 nm); II, enlarged lipoproteins (~40 nm); III, much larger particles (≥100 nm). Native PAGE resolved bands corresponding to peaks I and II, and EM of isolated peak II suggested LDL dimerization (A). Negative-stain EM showed progressive increase in the number of fused (violet arrows, stage 2) and ruptured LDLs (orange arrows, stages 5 and 6) and their aggregates. Similar changes in the particle size and morphology were observed upon lipolysis, sample aging, prolonged ultracentrifugation, or chemical denaturation of LDLs (18, 29).
Solvent ionic conditions
In atherosclerotic lesions, the extracellular pH varies from near-neutral to acidic, reaching as low as pH 5.5 in deep hypoxic areas (117, 118). LDL fusion at acidic pH can be augmented via two independent mechanisms. First, many hydrolytic and oxidative enzymes that modify LDLs in the arterial intima have optimal activity at acidic pH (118–121). Second, reduction in pH from near-neutral to acidic greatly enhances LDL fusion in vitro (29) and, probably, in vivo. This strong pH effect indicates the importance of electrostatic interactions in LDL fusion.
In addition to pH, salt ions also importantly influence LDL fusion. Our in vitro studies showed that increasing NaCl concentration from 0 to 150 mM greatly accelerates heat-induced LDL fusion (29), probably due to electrostatic screening of repulsive interactions between the particles or their specific sites. Moreover, LDLs spontaneously fuse and form lipid droplets at room temperature upon addition of low-millimolar concentrations of divalent metal ions such as Ca2+ or Mg2+, which is likely due to divalent metal binding by acidic groups in apoB (unpublished data). This effect probably underlies a fast laboratory method in which Mg-induced precipitation of total plasma LDLs is used to estimate the fraction of small, dense LDLs that are diagnostic markers of atherosclerosis (122, 123). Because small LDLs are more resistant to fusion than their larger counterparts, we proposed that small, dense LDLs remain in solution at Mg2+ concentrations that cause fusion and coalescence of larger LDLs into lipid droplets (29).
Lipoprotein crowding
Elevated concentration of plasma LDLs is the strongest causative risk factor of atherosclerosis (124, 125). The higher the concentration of LDLs in plasma, the higher the pro-atherogenic LDL uptake by arterial macrophages. In addition, LDL crowding at elevated concentrations may contribute to atherogenesis via two independent mechanisms. First, in the ‘lattice model’ proposed for LDL binding to LDLR, steric hindrance produced by the receptor-bound LDL decreases the binding of additional LDL particles to the adjacent receptors (126). Second, our experimental studies of isolated plasma LDL revealed that increasing LDL concentration in physiologically relevant range greatly accelerates heat-induced fusion and lipid droplet formation (Figure 3). Kinetic analysis of the reaction rate as a function of LDL concentration indicated a high-order reaction (Figure 3C) (29). This suggests that LDL crowding in the arterial subendothelium, together with the steric effects of the receptor-bound LDLs, potentially contributes to atherogenesis.
Figure 3. Effect of LDL concentration on aggregation, fusion, and lipid droplet formation.
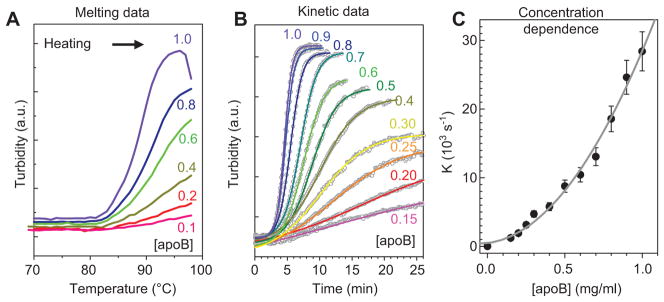
Single-donor human LDLs contained 0.1–1.0 mg/ml apoB (shown by numbers) in 20 mM Na phosphate, pH 7.5. The samples were (A) heated from 4°C to 98°C at a rate of 11°C/h to record the melting data or (B) incubated at 85°C to record the kinetic data. The increase in particle size upon LDL aggregation, fusion, and lipid droplet formation was monitored by turbidity at 320 nm. Kinetic data at each LDL concentration were approximated by a sigmoidal function, V(t) = V0 + (V1−V0){1+ exp[(t−t1/2)/k)}, where V0 and V1 are the initial and final values of turbidity, t1/2 is the midpoint, and k is the rate constant of the reaction. The plot of k versus apoB concentration is nonlinear (C); polynomial fitting suggests second-order reaction (gray line). These results show that increasing LDL concentration greatly increases the rate and the extent of LDL fusion and lipid droplet formation.
Detection of LDL aggregation, fusion, and lipid droplet formation
Conversion of intact LDLs (~22 nm) into lipid droplets (100–400 nm) is a complex process whose individual steps are difficult to discern experimentally, in part, due to particle heterogeneity. Some of these steps include (i) LDL aggregation, which may involve conformational changes in the protein and lipid but little or no changes in the particle size; (ii) LDL fusion, which produces enlarged lipoprotein-like particles that can undergo additional rounds of aggregation and fusion; (iii) lipoprotein disintegration (rupture) and release of the apolar core lipids, which coalesce into lipid droplets, often forming large aggregates (Figure 1). Dissecting this complex process is necessary to elucidate its underlying molecular basis and key determinants and to establish therapeutic targets to block its specific steps. Experimental techniques that are used to determine the lipoprotein size and morphology, differentiate between the aggregated and fused LDLs and lipid droplets, assess changes in the particle size distribution, and monitor these changes in real time are outlined below.
Methods to distinguish between lipoprotein aggregation and fusion
Transmission EM is a method of choice to visualize particle morphology and distinguish between the fused and aggregated lipoproteins and lipid droplets (Figures 1 and 2). For example, Guyton and colleagues used negative stain EM to detect aggregated and fused LDLs in human atherosclerotic lesions and to compare their size and morphology to those of similar particles formed upon LDL vortexing or aging in vitro [ref. (10) and references therein]. Another example is our negative stain EM analysis of LDL subfractions isolated by SEC from total LDLs upon various perturbations, which enabled us to identify LDL dimerization as a novel early step in aggregation and fusion (Figure 2) (29). As an alternative to EM, atomic force microscopy has been used to assess LDL aggregation and fusion (82). Although the resolution of the lipoprotein images attainable by negative stain EM (Figures 1 and 2) is superior to those obtained by atomic force microscopy, a potential drawback of negative stain preparation is that it can induce lipoprotein aggregation on the EM grids. This artifact can be eliminated by optimizing the staining technique (127). Still, negative-stain EM and atomic force microscopy are low-resolution techniques that can only resolve relatively large (>1 nm) structural features. Cryo-EM, which was used to determine LDL structure at up to 16-Å resolution (128, 129), can potentially provide a more detailed view of the aggregated and fused LDLs. The application of this technique to lipoproteins is restricted, in part, by sample heterogeneity. Furthermore, a general drawback of EM applications to heterogeneous samples is that the field views do not always represent the broader particle population. This necessitates the use of complementary techniques for accurate analysis of the particle size distribution in aggregated and fused LDLs, which are described below.
In addition to EM, proton nuclear magnetic resonance (NMR) has been used by Kovanen’s group to differentiate between LDL aggregation and fusion (130). The method is based upon the observation that 1H NMR resonances from the CH2 groups in lipoprotein lipids shift to higher frequencies upon increasing particle size; this effect apparently results from anisotropy in the magnetic susceptibility of the oriented molecules in the phospholipid monolayer of the lipoproteins surface (131). Although the interpretation of the data obtained by this indirect method involves approximations that may not strictly hold for lipoproteins, the method holds potential promise, as the results obtained in the studies of LDL aggregation and fusion using 1H NMR and EM were in good agreement (131).
Kovanen’s team developed another interesting approach to analyze fusion of LDLs in solution or bound to proteoglycans (132). Cholesterol esters labeled with fluorescence donor (pyrene) or acceptor (BODIPY) were incorporated into LDL core, and LDL fusion kinetics was monitored by fluorescence energy transfer (132). The advantage of the method is that it can differentiate between LDL aggregation and fusion and is applicable to lipoproteins both in fluid and in immobilized phase that mimics LDL binding to arterial matrix proteoglycans. The disadvantage is the necessity to label core lipids.
A promising label-free approach to directly visualize lipid assemblies is infrared imaging techniques based on coherent anti-Stokes Raman scattering (133). This novel approach utilizes strong infrared band from CH2 groups in lipid acyl chains, which eliminates the need for labeling. The method has been successfully applied for real-time imaging of cell organelles and lipid droplets (133). However, the diffraction limit in this and other infrared-based imaging techniques restricts their resolution to 100 nm. Therefore, optical advances will be needed before this novel technique can be used for imaging of lipoproteins (which range in size from about 10 to 100 nm) and their morphological transitions.
Methods to determine the size distribution in lipoproteins in solution
SEC and nondenaturing PAGE are the methods of choice to determine particle size distribution in lipoproteins in solution. Although these methods cannot differentiate between the aggregated and fused particles and are not suitable to discern large aggregated lipid droplets, they are extremely useful to monitor changes in the particle size during lipoprotein aggregation and fusion (Figure 2). In addition to providing an excellent analytical tool, SEC also provides a noninvasive preparative tool for isolating lipoproteins by size. The lipoproteins and their subfractions isolated by SEC can then be analyzed by other methods such as EM (Figure 2) (29).
As an alternative to SEC, preparative ultracentrifugation is the method of choice to isolate lipoproteins by size and density, whereas analytical ultracentrifugation is useful to determine particle size distribution. This method is more invasive than SEC and should be used with caution, as prolonged ultracentrifugation can mechanically perturb and remodel lipoproteins. For example, in our work, the total LDLs were isolated from human plasma by ultracentrifugation, followed by an additional round of ultracentrifugation to isolate large and small LDLs. This additional round led to particle size increase in a subset of LDLs, which was apparently due to LDL dimerization upon mechanical perturbation (unpublished data).
Because the size range of aggregated and fused LDLs and lipid droplets (100–400 nm) is commensurate with the wavelengths of UV-visible light, methods using UV-visible light scattering are useful in monitoring LDL aggregation and fusion. Dynamic light scattering is one of such methods that has been used to monitor lipoprotein size (82). In principle, measurements of dynamic light scattering can provide particle size distribution, especially in dilute solutions of spherical particles. In practice, the results are exquisitely sensitive to trace amounts of large particles such as dust, have limited accuracy in size analysis of nonuniformly shaped particles such as lipoprotein aggregates, and cannot differentiate between the aggregated and fused lipoproteins. Still, dynamic light scattering remains useful for monitoring changes in lipoprotein size.
Monitoring lipoprotein aggregation, fusion, and lipid droplet formation in real time
Static light scattering or turbidity measurements (i.e., attenuation in light intensity due to scattering) in the UV-visible range are useful in monitoring real-time changes in the lipoprotein size upon aggregation, fusion, and lipid droplet formation (41). We developed a method to record turbidity and right-angle light scattering in CD experiments (134) to monitor the time course of lipoprotein aggregation, fusion, and lipid droplet formation (Figures 2 and 3). The results are used for quantitative kinetic analysis to determine the Arrhenius activation energy (29, 116) or access the reaction order (Figure 3). Although such measurements alone cannot differentiate among aggregation, fusion, and lipid droplet formation, they can be combined with other methods such as SEC or EM to dissect these steps. Together with light scattering or turbidity, we also monitor near-UV CD that reports on lipoprotein rupture and release of core lipids that coalesce into droplets. We demonstrated that repacking of apolar lipids in this transition leads to a large negative induced CD peak centered circa 320 nm (Figure 4) (28, 116). Thus, this near-UV CD signal provides a convenient way to selectively monitor lipoprotein coalescence into lipid droplets.
Figure 4. LDL rupture and lipid droplet formation monitored by CD and turbidity.
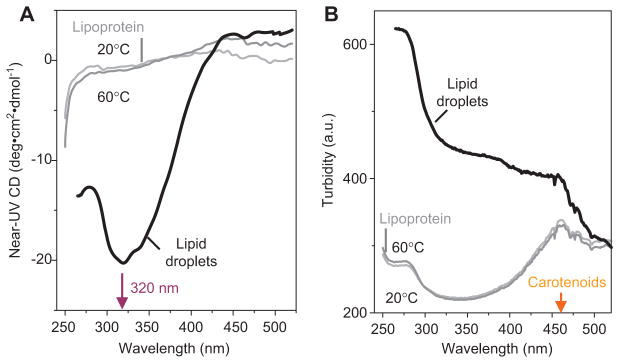
Near-UV-visible CD spectra (A) and turbidity (B) were recorded from human LDLs after incubation at 20°C, 60°C, or 90°C. EM revealed that LDL exposure to 90°C leads to irreversible rupture and release of apolar core lipids that coalesce into droplets. Lipid droplet formation induced a large negative CD signal centered at 320 nm (A), reflecting repacking of apolar moieties such as cholesterol esters, triacylglycerides, and carotenoids in LDL core. This induced CD was accompanied by increased turbidity reflecting increase in the particle size (B). In (B), the peak at 430–500 nm is characteristic of light absorption by carotenoids that are antioxidants in LDL core. We used CD at 320 nm to monitor LDL rupture and lipid droplet formation, together with turbidity to monitor increase in the particle size due to aggregation, fusion, and rupture of LDL [modified from ref. (28)].
Expert opinion
LDL aggregation, fusion, and lipid droplet formation are general structural responses to a wide variety of chemical and physical perturbations in the protein and lipid moieties. Numerous lines of evidence in vivo and in vitro directly link this process to atherogenesis. Therefore, elucidating the molecular mechanism involved in this pathogenic process and the factors that promote or prevent its specific steps may help establish new biomarkers and therapeutic targets for atherosclerosis. Analysis of lipolytic (PLA2, PLC, SMase) and proteolytic enzymes (trypsin, α-chymotrypsin, pronase) that promote LDL aggregation, fusion, and lipid droplet formation, as well as the amphipathic molecules (apoA-I, apoE, estradiol) and synthetic nanomaterials (pluronic copolymer) that hamper these reactions suggests that solvent exposure of hydrophobic moieties on LDL surface is a major driving force for particle aggregation and fusion (18, 33, 39, 41, 52, 110, 111). The strong effects of solvent ionic conditions (pH, monovalent and divalent cations) indicate that electrostatic interactions are also critically involved (29). Electrostatic effects probably also contribute to several important processes in vivo, such as LDL fusion and coalescence into lipid droplets at acidic pH in deep atherosclerotic lesions or during lysosomal degradation (117, 118, 122, 135) or to the effects of coronary artery calcium as a risk factor for atherosclerosis (136). Electrostatic effects in LDL aggregation, fusion, and lipid droplet formation also underlie laboratory methods such as LDL precipitation by magnesium salt, which is a useful tool to assess the patients’ risk of atherosclerosis (117, 118, 122, 135).
Outlook
Various chemical and structural changes in the protein and lipid moieties promote LDL aggregation, fusion, and lipid droplet formation. It remains unclear which of these changes or their combinations are particularly important during atherogenesis. Once these changes have been identified, they may provide viable therapeutic targets to hamper or even block this pathogenic process before it occurs and thereby complement the existing LDL-lowering drugs such as statins.
Highlights.
LDL aggregation, fusion, and coalesce into lipid droplets are directly linked to atherogenesis.
Various in vivo and in vitro modifications can trigger this pathogenic process, including phospholipid hydrolysis, apoB proteolysis, oxidative modifications, mechanical and thermal stress, etc.
Many of these modifications deplete the polar surface and/or expand the apolar core of LDL, creating hydrophobic packing defects on the LDL surface.
Such hydrophobic mismatch between the amphipathic surface and the apolar core is a major driving force for LDL aggregation, fusion, and lipid droplet formation.
Strong pH and salt dependence indicates that electrostatic effects are also key to this process.
Amphipathic molecules that bind to LDL surface and protect it from aggregation, fusion, and lipid droplet formation may provide potentially viable treatments for atherosclerosis.
Acknowledgments
We are grateful to Dr. Shobini Jayaraman for her help and extremely useful discussions as well as for sharing her unpublished data cited in this review. We thank our colleagues in the Department of Physiology and Biophysics at Boston University School of Medicine, particularly Drs. Haya Herscovitz, Yuhang Liu, and David Atkinson as well as Donald L. Gantz and Cheryl England for help with our LDL studies. This work was supported by the National Institutes of Health grants GM067260 and HL026355 and by institutional funds.
Abbreviations
- LDL
low-density lipoproteins
- LDLR
low-density lipoprotein receptor
- apo
apolipoprotein
- HDL
high-density lipoproteins
- VLDL
very low-density lipoproteins
- PC
phosphatidyl choline
- SMase
sphingomyelinase
- PLA2
phospholipase A2
- FFA
free fatty acids
- PLC
phospholipase C
- SEC
size-exclusion chromatography
- Gdn HCl
guanidinum hydrochloride
- PEG
polyethylene glycol
- EM
electron microscopy
- CD
circular dichroism
- NMR
nuclear magnetic resonance
- PAGE
polyacrylamide gel electrophoresis
Biographies
 Mengxiao Lu received her BS in Life Sciences from Peking University in 2006. She is currently a senior PhD student in Dr. Olga Gursky’s laboratory in the Department of Physiology and Biophysics of Boston University. She has been working on the structural stability of human low-density lipoproteins (LDLs). She has developed an experimental approach to perform quantitative kinetic analysis of LDL fusion and revealed a novel species in this process. The work was published in Journal of Lipid Research in 2012. The aim of her research is to understand the molecular mechanisms of LDL fusion and, ultimately, inhibit this pathological process at early stages.
Mengxiao Lu received her BS in Life Sciences from Peking University in 2006. She is currently a senior PhD student in Dr. Olga Gursky’s laboratory in the Department of Physiology and Biophysics of Boston University. She has been working on the structural stability of human low-density lipoproteins (LDLs). She has developed an experimental approach to perform quantitative kinetic analysis of LDL fusion and revealed a novel species in this process. The work was published in Journal of Lipid Research in 2012. The aim of her research is to understand the molecular mechanisms of LDL fusion and, ultimately, inhibit this pathological process at early stages.
 Olga Gursky obtained her BS and MS in Physics from Moscow State University (Russia) followed by a PhD from Brandeis University (USA). She is a Professor in the Department of Physiology and Biophysics at Boston University School of Medicine. During the last decade, studies in Gursky’s laboratory revealed that all lipoprotein classes are stabilized by kinetic barriers and showed that the major contribution to these barriers arises from protein dissociation and lipoprotein fusion and rupture. Similar energy barriers are proposed to modulate lipoprotein remodeling during metabolism. Ongoing research in Gursky’s laboratory addresses various aspects of structural stability and functional remodeling of high-, low-, and very-low-density lipoproteins and their protein constituents in health and disease.
Olga Gursky obtained her BS and MS in Physics from Moscow State University (Russia) followed by a PhD from Brandeis University (USA). She is a Professor in the Department of Physiology and Biophysics at Boston University School of Medicine. During the last decade, studies in Gursky’s laboratory revealed that all lipoprotein classes are stabilized by kinetic barriers and showed that the major contribution to these barriers arises from protein dissociation and lipoprotein fusion and rupture. Similar energy barriers are proposed to modulate lipoprotein remodeling during metabolism. Ongoing research in Gursky’s laboratory addresses various aspects of structural stability and functional remodeling of high-, low-, and very-low-density lipoproteins and their protein constituents in health and disease.
Contributor Information
Mengxiao Lu, Email: mxlu@bu.edu, Department of Physiology and Biophysics, Boston University School of Medicine, W321, 700 Albany Street, Boston, MA 02118, USA.
Olga Gursky, Department of Physiology and Biophysics, Boston University School of Medicine, W321, 700 Albany Street, Boston, MA 02118, USA.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castelli WP, Anderson K, Wilson PW, Levy D. Lipids and risk of coronary heart disease. The Framingham Study. Ann Epidemiol. 1992;2:23–8. doi: 10.1016/1047-2797(92)90033-m. [DOI] [PubMed] [Google Scholar]
- 3.Driscoll DM, Getz GS. Molecular and cell biology of lipoprotein biosynthesis. Methods Enzymol. 1986;128:41–70. doi: 10.1016/0076-6879(86)28062-3. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein JL, Brown MS, Anderson RG, Russell DW, Schneider WJ. Receptor-mediated endocytosis: concepts emerging from the LDL receptor system. Annu Rev Cell Biol. 1985;1:1–39. doi: 10.1146/annurev.cb.01.110185.000245. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–8. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henriksen T, Mahoney EM, Steinberg D. Enhanced macrophage degradation of low density lipoprotein previously incubated with cultured endothelial cells: recognition by receptors for acetylated low density lipoproteins. Proc Natl Acad Sci USA. 1981;78:6499–503. doi: 10.1073/pnas.78.10.6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP) Annu Rev Biochem. 1994;63:601–37. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- 8.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–61. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tirziu D, Dobrian A, Tasca C, Simionescu M, Simionescu N. Intimal thickenings of human aorta contain modified reassembled lipoproteins. Atherosclerosis. 1995;112:101–14. doi: 10.1016/0021-9150(94)05405-8. [DOI] [PubMed] [Google Scholar]
- 10.Guyton JR, Klemp KF, Black BL, Bocan TM. Extracellular lipid deposition in atherosclerosis. Eur Heart J. 1990;11:20–8. doi: 10.1093/eurheartj/11.suppl_e.20. [DOI] [PubMed] [Google Scholar]
- 11.Oorni K, Pentikainen MO, Ala-Korpela M, Kovanen PT. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: molecular mechanisms and effects on matrix interactions. J Lipid Res. 2000;41:1703–14. [PubMed] [Google Scholar]
- 12.Nievelstein PF, Fogelman AM, Mottino G, Frank JS. Lipid accumulation in rabbit aortic intima 2 hours after bolus infusion of low density lipoprotein. A deep-etch and immunolocalization study of ultrarapidly frozen tissue. Arterioscler Thromb. 1991;11:1795–805. doi: 10.1161/01.atv.11.6.1795. [DOI] [PubMed] [Google Scholar]
- 13.Nievelstein-Post P, Mottino G, Fogelman A, Frank J. An ultrastructural study of lipoprotein accumulation in cardiac valves of the rabbit. Arterioscler Thromb. 1994;14:1151–61. doi: 10.1161/01.atv.14.7.1151. [DOI] [PubMed] [Google Scholar]
- 14.Hurt-Camejo E, Camejo G, Sartipy P. Phospholipase A2 and small, dense low-density lipoprotein. Curr Opin Lipidol. 2000;11:465–71. doi: 10.1097/00041433-200010000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Guyton JR. The arterial wall and the atherosclerotic lesion. Curr Opin Lipidol. 1994;5:376–81. doi: 10.1097/00041433-199410000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Bancells C, Villegas S, Blanco FJ, Benitez S, Gallego I, Beloki L, Perez-Cuellar M, Ordonez-Llanos J, Sanchez-Quesada JL. Aggregated electronegative low density lipoprotein in human plasma shows a high tendency toward phospholipolysis and particle fusion. J Biol Chem. 2010;285:32425–35. doi: 10.1074/jbc.M110.139691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paananen K, Saarinen J, Annila A, Kovanen PT. Proteolysis and fusion of low density lipoprotein particles strengthen their binding to human aortic proteoglycans. J Biol Chem. 1995;270:12257–62. doi: 10.1074/jbc.270.20.12257. [DOI] [PubMed] [Google Scholar]
- 18.Pentikainen MO, Lehtonen EM, Kovanen PT. Aggregation and fusion of modified low density lipoprotein. J Lipid Res. 1996;37:2638–49. [PubMed] [Google Scholar]
- 19.Parasassi T, De Spirito M, Mei G, Brunelli R, Greco G, Lenzi L, Maulucci G, Nicolai E, Papi M, Arcovito G, Tosatto SC, Ursini F. Low density lipoprotein misfolding and amyloidogenesis. FASEB J. 2008;22:2350–6. doi: 10.1096/fj.07-097774. [DOI] [PubMed] [Google Scholar]
- 20.Camejo G, Hurt-Camejo E, Wiklund O, Bondjers G. Association of apo B lipoproteins with arterial proteoglycans: pathological significance and molecular basis. Atherosclerosis. 1998;139:205–22. doi: 10.1016/s0021-9150(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 21.Hoff HF, Morton RE. Lipoproteins containing apo B extracted from human aortas. Structure and function. Ann NY Acad Sci. 1985;454:183–94. doi: 10.1111/j.1749-6632.1985.tb11857.x. [DOI] [PubMed] [Google Scholar]
- 22.Aviram M, Maor I, Keidar S, Hayek T, Oiknine J, Bar-El Y, Adler Z, Kertzman V, Milo S. Lesioned low density lipoprotein in atherosclerotic apolipoprotein E-deficient transgenic mice and in humans is oxidized and aggregated. Biochem Biophys Res Commun. 1995;216:501–13. doi: 10.1006/bbrc.1995.2651. [DOI] [PubMed] [Google Scholar]
- 23.Frank JS, Fogelman AM. Ultrastructure of the intima in WHHL and cholesterol-fed rabbit aortas prepared by ultra-rapid freezing and freeze-etching. J Lipid Res. 1989;30:967–78. [PubMed] [Google Scholar]
- 24.Steinbrecher UP, Lougheed M. Scavenger receptor-independent stimulation of cholesterol esterification in macrophages by low density lipoprotein extracted from human aortic intima. Arterioscler Thromb. 1992;12:608–25. doi: 10.1161/01.atv.12.5.608. [DOI] [PubMed] [Google Scholar]
- 25.Llorente-Cortes V, Badimon L. LDL receptor-related protein and the vascular wall: implications for atherothrombosis. Arterioscler Thromb Vasc Biol. 2005;25:497–504. doi: 10.1161/01.ATV.0000154280.62072.fd. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein JL, Ho YK, Basu SK, Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci USA. 1979;76:333–7. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khoo JC, Miller E, McLoughlin P, Steinberg D. Enhanced macrophage uptake of low density lipoprotein after self-aggregation. Arteriosclerosis. 1988;8:348–58. doi: 10.1161/01.atv.8.4.348. [DOI] [PubMed] [Google Scholar]
- 28.Jayaraman S, Gantz D, Gursky O. Structural basis for thermal stability of human low-density lipoprotein. Biochemistry. 2005;44:3965–71. doi: 10.1021/bi047493v. [DOI] [PubMed] [Google Scholar]
- 29.Lu M, Gantz DL, Herscovitz H, Gursky O. Kinetic analysis of thermal stability of human low density lipoproteins: a model for LDL fusion in atherogenesis. J Lipid Res. 2012;53:2175–85. doi: 10.1194/jlr.M029629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marathe S, Kuriakose G, Williams KJ, Tabas I. Sphingomyelinase, an enzyme implicated in atherogenesis, is present in atherosclerotic lesions and binds to specific components of the subendothelial extracellular matrix. Arterioscler Thromb Vasc Biol. 1999;19:2648–58. doi: 10.1161/01.atv.19.11.2648. [DOI] [PubMed] [Google Scholar]
- 31.Nelson GJ, Freeman NK. The phospholipid and phospholipid fatty acid composition of human serum lipoprotein fractions. J Biol Chem. 1960;235:578–83. [PubMed] [Google Scholar]
- 32.Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J Clin Invest. 1996;98:1455–64. doi: 10.1172/JCI118934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oorni K, Hakala JK, Annila A, Ala-Korpela M, Kovanen PT. Sphingomyelinase induces aggregation and fusion, but phospholipase A2 only aggregation, of low density lipoprotein (LDL) particles. Two distinct mechanisms leading to increased binding strength of LDL to human aortic proteoglycans. J Biol Chem. 1998;273:29127–34. doi: 10.1074/jbc.273.44.29127. [DOI] [PubMed] [Google Scholar]
- 34.Corson MA. Emerging inflammatory markers for assessing coronary heart disease risk. Curr Cardiol Rep. 2009;11:452–9. doi: 10.1007/s11886-009-0065-1. [DOI] [PubMed] [Google Scholar]
- 35.Jonsson-Rylander AC, Lundin S, Rosengren B, Pettersson C, Hurt-Camejo E. Role of secretory phospholipases in atherogenesis. Curr Atheroscler Rep. 2008;10:252–9. doi: 10.1007/s11883-008-0039-6. [DOI] [PubMed] [Google Scholar]
- 36.Davis B, Koster G, Douet LJ, Scigelova M, Woffendin G, Ward JM, Smith A, Humphries J, Burnand KG, Macphee CH, Postle AD. Electrospray ionization mass spectrometry identifies substrates and products of lipoprotein-associated phospholipase A2 in oxidized human low density lipoprotein. J Biol Chem. 2008;283:6428–37. doi: 10.1074/jbc.M709970200. [DOI] [PubMed] [Google Scholar]
- 37.Jayaraman S, Gantz DL, Gursky O. Effects of phospholipase A(2) and its products on structural stability of human LDL: relevance to formation of LDL-derived lipid droplets. J Lipid Res. 2011;52:549–57. doi: 10.1194/jlr.M012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hakala JK, Oorni K, Pentikainen MO, Hurt-Camejo E, Kovanen PT. Lipolysis of LDL by human secretory phospholipase A(2) induces particle fusion and enhances the retention of LDL to human aortic proteoglycans. Arterioscler Thromb Vasc Biol. 2001;21:1053–8. doi: 10.1161/01.atv.21.6.1053. [DOI] [PubMed] [Google Scholar]
- 39.Suits AG, Chait A, Aviram M, Heinecke JW. Phagocytosis of aggregated lipoprotein by macrophages: low density lipoprotein receptor-dependent foam-cell formation. Proc Natl Acad Sci USA. 1989;86:2713–7. doi: 10.1073/pnas.86.8.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez-Quesada JL, Villegas S, Ordonez-Llanos J. Electronegative low-density lipoprotein. A link between apolipoprotein B misfolding, lipoprotein aggregation and proteoglycan binding. Curr Opin Lipidol. 2012;23:479–86. doi: 10.1097/MOL.0b013e328357c933. [DOI] [PubMed] [Google Scholar]
- 41.Khoo JC, Miller E, McLoughlin P, Steinberg D. Prevention of low density lipoprotein aggregation by high density lipoprotein or apolipoprotein A-I. J Lipid Res. 1990;31:645–52. [PubMed] [Google Scholar]
- 42.Kruth HS. Subendothelial accumulation of unesterified cholesterol. An early event in atherosclerotic lesion development. Atherosclerosis. 1985;57:337–41. doi: 10.1016/0021-9150(85)90045-0. [DOI] [PubMed] [Google Scholar]
- 43.Chao FF, Blanchette-Mackie EJ, Tertov VV, Skarlatos SI, Chen YJ, Kruth HS. Hydrolysis of cholesteryl ester in low density lipoprotein converts this lipoprotein to a liposome. J Biol Chem. 1992;267:4992–8. [PubMed] [Google Scholar]
- 44.Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: structure, function, regulation, and role in disease. J Mol Med (Berl) 2002;80:753–69. doi: 10.1007/s00109-002-0384-9. [DOI] [PubMed] [Google Scholar]
- 45.Musliner TA, Long MD, Forte TM, Nichols AV, Gong EL, Blanche PJ, Krauss RM. Dissociation of high density lipoprotein precursors from apolipoprotein B-containing lipoproteins in the presence of unesterified fatty acids and a source of apolipoprotein A-I. J Lipid Res. 1991;32:917–33. [PubMed] [Google Scholar]
- 46.Goldberg IJ. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J Lipid Res. 1996;37:693–707. [PubMed] [Google Scholar]
- 47.Auerbach BJ, Bisgaier CL, Wolle J, Saxena U. Oxidation of low density lipoproteins greatly enhances their association with lipoprotein lipase anchored to endothelial cell matrix. J Biol Chem. 1996;271:1329–35. doi: 10.1074/jbc.271.3.1329. [DOI] [PubMed] [Google Scholar]
- 48.Pentikainen MO, Oorni K, Kovanen PT. Lipoprotein lipase (LPL) strongly links native and oxidized low density lipoprotein particles to decorin-coated collagen. Roles for both dimeric and monomeric forms of LPL. J Biol Chem. 2000;275:5694–701. doi: 10.1074/jbc.275.8.5694. [DOI] [PubMed] [Google Scholar]
- 49.Walters MJ, Wrenn SP. Mechanistic roles of lipoprotein lipase and sphingomyelinase in low density lipoprotein aggregation. J Colloid Interface Sci. 2011;363:268–74. doi: 10.1016/j.jcis.2011.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McNamara JR, Small DM, Li Z, Schaefer EJ. Differences in LDL subspecies involve alterations in lipid composition and conformational changes in apolipoprotein B. J Lipid Res. 1996;37:1924–35. [PubMed] [Google Scholar]
- 51.Kovanen PT, Kokkonen JO. Modification of low density lipoproteins by secretory granules of rat serosal mast cells. J Biol Chem. 1991;266:4430–6. [PubMed] [Google Scholar]
- 52.Piha M, Lindstedt L, Kovanen PT. Fusion of proteolyzed low-density lipoprotein in the fluid phase: a novel mechanism generating atherogenic lipoprotein particles. Biochemistry. 1995;34:10120–9. doi: 10.1021/bi00032a004. [DOI] [PubMed] [Google Scholar]
- 53.Morel DW, Hessler JR, Chisolm GM. Low density lipoprotein cytotoxicity induced by free radical peroxidation of lipid. J Lipid Res. 1983;24:1070–6. [PubMed] [Google Scholar]
- 54.Henriksen T, Evensen SA, Carlander B. Injury to cultured endothelial cells induced by low density lipoproteins: protection by high density lipoproteins. Scand J Clin Lab Invest. 1979;39:369–75. doi: 10.3109/00365517909106121. [DOI] [PubMed] [Google Scholar]
- 55.Hessler JR, Robertson AL, Jr, Chisolm GM., 3rd LDL-induced cytotoxicity and its inhibition by HDL in human vascular smooth muscle and endothelial cells in culture. Atherosclerosis. 1979;32:213–29. doi: 10.1016/0021-9150(79)90166-7. [DOI] [PubMed] [Google Scholar]
- 56.Hessler JR, Morel DW, Lewis LJ, Chisolm GM. Lipoprotein oxidation and lipoprotein-induced cytotoxicity. Arteriosclerosis. 1983;3:215–22. doi: 10.1161/01.atv.3.3.215. [DOI] [PubMed] [Google Scholar]
- 57.Henriksen T, Mahoney EM, Steinberg D. Enhanced macrophage degradation of biologically modified low density lipoprotein. Arteriosclerosis. 1983;3:149–59. doi: 10.1161/01.atv.3.2.149. [DOI] [PubMed] [Google Scholar]
- 58.Steinbrecher UP, Parthasarathy S, Leake DS, Witztum JL, Steinberg D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc Natl Acad Sci USA. 1984;81:3883–7. doi: 10.1073/pnas.81.12.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jurgens G, Hoff HF, Chisolm GM, 3rd, Esterbauer H. Modification of human serum low density lipoprotein by oxidation – characterization and pathophysiological implications. Chem Phys Lipids. 1987;45:315–36. doi: 10.1016/0009-3084(87)90070-3. [DOI] [PubMed] [Google Scholar]
- 60.Heinecke JW. Mechanisms of oxidative damage of low density lipoprotein in human atherosclerosis. Curr Opin Lipidol. 1997;8:268–74. doi: 10.1097/00041433-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 61.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–24. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 62.Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 63.Kris-Etherton PM, Lichtenstein AH, Howard BV, Steinberg D, Witztum JL. Antioxidant vitamin supplements and cardiovascular disease. Circulation. 2004;110:637–41. doi: 10.1161/01.CIR.0000137822.39831.F1. [DOI] [PubMed] [Google Scholar]
- 64.Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50:S376–81. doi: 10.1194/jlr.R800087-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2311–6. doi: 10.1161/ATVBAHA.108.179697. [DOI] [PubMed] [Google Scholar]
- 66.Yoshida H, Kisugi R. Mechanisms of LDL oxidation. Clin Chim Acta. 2010;411:1875–82. doi: 10.1016/j.cca.2010.08.038. [DOI] [PubMed] [Google Scholar]
- 67.Hoff HF, O’Neil J. Lesion-derived low density lipoprotein and oxidized low density lipoprotein share a lability for aggregation, leading to enhanced macrophage degradation. Arterioscler Thromb. 1991;11:1209–22. doi: 10.1161/01.atv.11.5.1209. [DOI] [PubMed] [Google Scholar]
- 68.Hoff HF, Whitaker TE, O’Neil J. Oxidation of low density lipoprotein leads to particle aggregation and altered macrophage recognition. J Biol Chem. 1992;267:602–9. [PubMed] [Google Scholar]
- 69.Cynshi O, Takashima Y, Suzuki T, Kawabe Y, Ohba Y, Kodama T. Characterization of aggregated low density lipoproteins induced by copper-catalyzed oxidation. J Atheroscler Thromb. 1994;1:87–97. doi: 10.5551/jat1994.1.87. [DOI] [PubMed] [Google Scholar]
- 70.Dobrian A, Mora R, Simionescu M, Simionescu N. In vitro formation of oxidatively-modified and reassembled human low-density lipoproteins: antioxidant effect of albumin. Biochim Biophys Acta. 1993;1169:12–24. doi: 10.1016/0005-2760(93)90076-l. [DOI] [PubMed] [Google Scholar]
- 71.Jayaraman S, Gantz DL, Gursky O. Effects of oxidation on the structure and stability of human low-density lipoprotein. Biochemistry. 2007;46:5790–7. doi: 10.1021/bi700225a. [DOI] [PubMed] [Google Scholar]
- 72.Meyer DF, Nealis AS, Macphee CH, Groot PH, Suckling KE, Bruckdorfer KR, Perkins SJ. Time-course studies by synchrotron X-ray solution scattering of the structure of human low-density lipoprotein during Cu(2+)-induced oxidation in relation to changes in lipid composition. Biochem J. 1996;319:217–27. doi: 10.1042/bj3190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ettelaie C, Haris PI, James NJ, Wilbourn B, Adam JM, Bruckdorfer KR. Alterations in the structure of apolipoprotein B-100 determine the behaviour of LDL towards thromboplastin. Biochim Biophys Acta. 1997;1345:237–47. doi: 10.1016/s0005-2760(96)00185-3. [DOI] [PubMed] [Google Scholar]
- 74.Soran H, Durrington PN. Susceptibility of LDL and its subfractions to glycation. Curr Opin Lipidol. 2011;22:254–61. doi: 10.1097/MOL.0b013e328348a43f. [DOI] [PubMed] [Google Scholar]
- 75.Younis N, Sharma R, Soran H, Charlton-Menys V, Elseweidy M, Durrington PN. Glycation as an atherogenic modification of LDL. Curr Opin Lipidol. 2008;19:378–84. doi: 10.1097/MOL.0b013e328306a057. [DOI] [PubMed] [Google Scholar]
- 76.Younis N, Charlton-Menys V, Sharma R, Soran H, Durrington PN. Glycation of LDL in non-diabetic people: small dense LDL is preferentially glycated both in vivo and in vitro. Atherosclerosis. 2009;202:162–8. doi: 10.1016/j.atherosclerosis.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 77.Steinbrecher UP, Witztum JL. Glucosylation of low-density lipoproteins to an extent comparable to that seen in diabetes slows their catabolism. Diabetes. 1984;33:130–4. doi: 10.2337/diab.33.2.130. [DOI] [PubMed] [Google Scholar]
- 78.Schleicher E, Deufel T, Wieland OH. Non-enzymatic glycosylation of human serum lipoproteins. Elevated epsilon-lysine glycosylated low density lipoprotein in diabetic patients. FEBS Lett. 1981;129:1–4. doi: 10.1016/0014-5793(81)80741-7. [DOI] [PubMed] [Google Scholar]
- 79.Younis NN, Soran H, Sharma R, Charlton-Menys V, Greenstein A, Elseweidy MM, Durrington PN. Small-dense LDL and LDL glycation in metabolic syndrome and in statin-treated and non-statin-treated type 2 diabetes. Diab Vasc Dis Res. 2010;7:289–95. doi: 10.1177/1479164110383063. [DOI] [PubMed] [Google Scholar]
- 80.Tabas I. Nonoxidative modifications of lipoproteins in atherogenesis. Annu Rev Nutr. 1999;19:123–39. doi: 10.1146/annurev.nutr.19.1.123. [DOI] [PubMed] [Google Scholar]
- 81.Eckey R, Menschikowski M, Lattke P, Jaross W. Minimal oxidation and storage of low density lipoproteins result in an increased susceptibility to phospholipid hydrolysis by phospholipase A2. Atherosclerosis. 1997;132:165–76. doi: 10.1016/s0021-9150(97)00088-9. [DOI] [PubMed] [Google Scholar]
- 82.De Spirito M, Brunelli R, Mei G, Bertani FR, Ciasca G, Greco G, Papi M, Arcovito G, Ursini F, Parasassi T. Low density lipoprotein aged in plasma forms clusters resembling subendothelial droplets: aggregation via surface sites. Biophys J. 2006;90:4239–47. doi: 10.1529/biophysj.105.075788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tertov VV, Sobenin IA, Gabbasov ZA, Popov EG, Orekhov AN. Lipoprotein aggregation as an essential condition of intracellular lipid accumulation caused by modified low density lipoproteins. Biochem Biophys Res Commun. 1989;163:489–94. doi: 10.1016/0006-291x(89)92163-3. [DOI] [PubMed] [Google Scholar]
- 84.Mahley RW, Innerarity TL, Weisgraber KB, Oh SY. Altered metabolism (in vivo and in vitro) of plasma lipoproteins after selective chemical modification of lysine residues of the apoproteins. J Clin Invest. 1979;64:743–50. doi: 10.1172/JCI109518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fogelman AM, Shechter I, Seager J, Hokom M, Child JS, Edwards PA. Malondialdehyde alteration of low density lipoproteins leads to cholesteryl ester accumulation in human monocyte-macrophages. Proc Natl Acad Sci USA. 1980;77:2214–8. doi: 10.1073/pnas.77.4.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shechter I, Fogelman AM, Haberland ME, Seager J, Hokom M, Edwards PA. The metabolism of native and malondialdehyde-altered low density lipoproteins by human monocyte-macrophages. J Lipid Res. 1981;22:63–71. [PubMed] [Google Scholar]
- 87.Orekhov AN, Tertov VV, Mukhin DN, Mikhailenko IA. Modification of low density lipoprotein by desialylation causes lipid accumulation in cultured cells: discovery of desialylated lipoprotein with altered cellular metabolism in the blood of atherosclerotic patients. Biochem Biophys Res Commun. 1989;162:206–11. doi: 10.1016/0006-291x(89)91982-7. [DOI] [PubMed] [Google Scholar]
- 88.Rankin SM, de Whalley CV, Hoult JR, Jessup W, Wilkins GM, Collard J, Leake DS. The modification of low density lipoprotein by the flavonoids myricetin and gossypetin. Biochem Pharmacol. 1993;45:67–75. doi: 10.1016/0006-2952(93)90378-a. [DOI] [PubMed] [Google Scholar]
- 89.Walters MJ, Wrenn SP. Effect of sphingomyelinase-mediated generation of ceramide on aggregation of low-density lipoprotein. Langmuir. 2008;24:9642–7. doi: 10.1021/la800714w. [DOI] [PubMed] [Google Scholar]
- 90.Chisolm GM, Steinberg D. The oxidative modification hypothesis of atherogenesis: an overview. Free Radic Biol Med. 2000;28:1815–26. doi: 10.1016/s0891-5849(00)00344-0. [DOI] [PubMed] [Google Scholar]
- 91.de Winther MP, Hofker MH. Scavenging new insights into atherogenesis. J Clin Invest. 2000;105:1039–41. doi: 10.1172/JCI9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang WY, Schwartz E, Wang Y, Attrep J, Li Z, Reaven P. Elevated concentrations of nonesterified fatty acids increase monocyte expression of CD11b and adhesion to endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:514–9. doi: 10.1161/01.ATV.0000200226.53994.09. [DOI] [PubMed] [Google Scholar]
- 93.Chernomordik L. Non-bilayer lipids and biological fusion intermediates. Chem Phys Lipids. 1996;81:203–13. doi: 10.1016/0009-3084(96)02583-2. [DOI] [PubMed] [Google Scholar]
- 94.Kantor HL, Prestegard JH. Fusion of phosphatidylcholine bilayer vesicles: role of free fatty acid. Biochemistry. 1978;17:3592–7. doi: 10.1021/bi00610a027. [DOI] [PubMed] [Google Scholar]
- 95.Lahdesmaki K, Plihtari R, Soininen P, Hurt-Camejo E, Ala-Korpela M, Oorni K, Kovanen PT. Phospholipase A(2)-modified LDL particles retain the generated hydrolytic products and are more atherogenic at acidic pH. Atherosclerosis. 2009;207:352–9. doi: 10.1016/j.atherosclerosis.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 96.Talbot RM, del Rio JD, Weinberg PD. Effect of fluid mechanical stresses and plasma constituents on aggregation of LDL. J Lipid Res. 2003;44:837–45. doi: 10.1194/jlr.M200477-JLR200. [DOI] [PubMed] [Google Scholar]
- 97.Talbot RM, del Rio JD, Leake DS, Weinberg PD. Oxidation affects the flow-induced aggregation of low density lipoprotein and its inhibition by albumin. Biochim Biophys Acta. 2003;1634:24–9. doi: 10.1016/j.bbalip.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 98.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–66. [PubMed] [Google Scholar]
- 99.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–14. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 100.Boden WE. High-density lipoprotein cholesterol as an independent risk factor in cardiovascular disease: assessing the data from Framingham to the Veterans Affairs High – Density Lipoprotein Intervention Trial. Am J Cardiol. 2000;86:19L–22L. doi: 10.1016/s0002-9149(00)01464-8. [DOI] [PubMed] [Google Scholar]
- 101.Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211–28. [PubMed] [Google Scholar]
- 102.von Eckardstein A, Nofer JR, Assmann G. High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2001;21:13–27. doi: 10.1161/01.atv.21.1.13. [DOI] [PubMed] [Google Scholar]
- 103.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–30. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 104.Liu H, Scraba DG, Ryan RO. Prevention of phospholipase-C induced aggregation of low density lipoprotein by amphipathic apolipoproteins. FEBS Lett. 1993;316:27–33. doi: 10.1016/0014-5793(93)81730-n. [DOI] [PubMed] [Google Scholar]
- 105.Castelli WP. Epidemiology of coronary heart disease: the Framingham Study. Am J Med. 1984;76:4–12. doi: 10.1016/0002-9343(84)90952-5. [DOI] [PubMed] [Google Scholar]
- 106.Grodstein F, Stampfer M. The epidemiology of coronary heart disease and estrogen replacement in postmenopausal women. Prog Cardiovasc Dis. 1995;38:199–210. doi: 10.1016/s0033-0620(95)80012-3. [DOI] [PubMed] [Google Scholar]
- 107.Sack MN, Rader DJ, Cannon RO., 3rd Oestrogen and inhibition of oxidation of low-density lipoproteins in postmenopausal women. Lancet. 1994;343:269–70. doi: 10.1016/s0140-6736(94)91117-7. [DOI] [PubMed] [Google Scholar]
- 108.Shwaery GT, Vita JA, Keaney JF., Jr Antioxidant protection of LDL by physiological concentrations of 17 beta-estradiol. Requirement for estradiol modification. Circulation. 1997;95:1378–85. doi: 10.1161/01.cir.95.6.1378. [DOI] [PubMed] [Google Scholar]
- 109.Huber LA, Scheffler E, Poll T, Ziegler R, Dresel HA. 17 beta-estradiol inhibits LDL oxidation and cholesteryl ester formation in cultured macrophages. Free Radic Res Commun. 1990;8:167–73. doi: 10.3109/10715769009087990. [DOI] [PubMed] [Google Scholar]
- 110.Brunelli R, Mei G, Krasnowska EK, Pierucci F, Zichella L, Ursini F, Parasassi T. Estradiol enhances the resistance of LDL to oxidation by stabilizing apoB-100 conformation. Biochemistry. 2000;39:13897–903. doi: 10.1021/bi000341p. [DOI] [PubMed] [Google Scholar]
- 111.Melnichenko AA, Aksenov DV, Myasoedova VA, Panasenko OM, Yaroslavov AA, Sobenin IA, Bobryshev YV, Orekhov AN. Pluronic block copolymers inhibit low density lipoprotein self-association. Lipids. 2012;47:995–1000. doi: 10.1007/s11745-012-3699-5. [DOI] [PubMed] [Google Scholar]
- 112.Batrakova EV, Kabanov AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Control Release. 2008;130:98–106. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lentz BR. PEG as a tool to gain insight into membrane fusion. Eur Biophys J. 2007;36:315–26. doi: 10.1007/s00249-006-0097-z. [DOI] [PubMed] [Google Scholar]
- 114.Jayaraman S, Gantz DL, Gursky O. Poly(ethylene glycol)-induced fusion and destabilization of human plasma high-density lipoproteins. Biochemistry. 2004;43:5520–31. doi: 10.1021/bi036274r. [DOI] [PubMed] [Google Scholar]
- 115.Mehta R, Gantz DL, Gursky O. Human plasma high-density lipoproteins are stabilized by kinetic factors. J Mol Biol. 2003;328:183–92. doi: 10.1016/s0022-2836(03)00155-4. [DOI] [PubMed] [Google Scholar]
- 116.Guha M, England C, Herscovitz H, Gursky O. Thermal transitions in human very-low-density lipoprotein: fusion, rupture, and dissociation of HDL-like particles. Biochemistry. 2007;46:6043–9. doi: 10.1021/bi7001532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Naghavi M, John R, Naguib S, Siadaty MS, Grasu R, Kurian KC, van Winkle WB, Soller B, Litovsky S, Madjid M, Willerson JT, Casscells W. pH Heterogeneity of human and rabbit atherosclerotic plaques; a new insight into detection of vulnerable plaque. Atherosclerosis. 2002;164:27–35. doi: 10.1016/s0021-9150(02)00018-7. [DOI] [PubMed] [Google Scholar]
- 118.Oorni K, Kovanen PT. Enhanced extracellular lipid accumulation in acidic environments. Curr Opin Lipidol. 2006;17:534–40. doi: 10.1097/01.mol.0000245259.63505.c2. [DOI] [PubMed] [Google Scholar]
- 119.Dollery CM, Libby P. Atherosclerosis and proteinase activation. Cardiovasc Res. 2006;69:625–35. doi: 10.1016/j.cardiores.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 120.Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J Biol Chem. 1996;271:18431–6. doi: 10.1074/jbc.271.31.18431. [DOI] [PubMed] [Google Scholar]
- 121.Shamir R, Johnson WJ, Morlock-Fitzpatrick K, Zolfaghari R, Li L, Mas E, Lombardo D, Morel DW, Fisher EA. Pancreatic carboxyl ester lipase: a circulating enzyme that modifies normal and oxidized lipoproteins in vitro. J Clin Invest. 1996;97:1696–704. doi: 10.1172/JCI118596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hirano T, Ito Y, Saegusa H, Yoshino G. A novel and simple method for quantification of small, dense LDL. J Lipid Res. 2003;44:2193–201. doi: 10.1194/jlr.D300007-JLR200. [DOI] [PubMed] [Google Scholar]
- 123.Ai M, Otokozawa S, Asztalos BF, Ito Y, Nakajima K, White CC, Cupples LA, Wilson PW, Schaefer EJ. Small dense LDL cholesterol and coronary heart disease: results from the Framingham Offspring Study. Clin Chem. 2010;56:967–76. doi: 10.1373/clinchem.2009.137489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Manninen V, Tenkanen L, Koskinen P, Huttunen JK, Manttari M, Heinonen OP, Frick MH. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation. 1992;85:37–45. doi: 10.1161/01.cir.85.1.37. [DOI] [PubMed] [Google Scholar]
- 125.Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–47. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 126.Chappell DA, Fry GL, Waknitz MA, Berns JJ. Ligand size as a determinant for catabolism by the low density lipoprotein (LDL) receptor pathway. A lattice model for LDL binding. J Biol Chem. 1991;266:19296–302. [PubMed] [Google Scholar]
- 127.Zhang L, Song J, Cavigiolio G, Ishida BY, Zhang S, Kane JP, Weisgraber KH, Oda MN, Rye KA, Pownall HJ, Ren G. Morphology and structure of lipoproteins revealed by an optimized negative-staining protocol of electron microscopy. J Lipid Res. 2011;52:175–84. doi: 10.1194/jlr.D010959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu Y, Atkinson D. Enhancing the contrast of ApoB to locate the surface components in the 3D density map of human LDL. J Mol Biol. 2011;405:274–83. doi: 10.1016/j.jmb.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kumar V, Butcher SJ, Oorni K, Engelhardt P, Heikkonen J, Kaski K, Ala-Korpela M, Kovanen PT. Three-dimensional cryoEM reconstruction of native LDL particles to 16A resolution at physiological body temperature. PLoS One. 2011;6:e18841. doi: 10.1371/journal.pone.0018841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ala-Korpela M, Pentikainen MO, Korhonen A, Hevonoja T, Lounila J, Kovanen PT. Detection of low density lipoprotein particle fusion by proton nuclear magnetic resonance spectroscopy. J Lipid Res. 1998;39:1705–12. [PubMed] [Google Scholar]
- 131.Lounila J, Ala-Korpela M, Jokisaari J, Savolainen MJ, Kesaniemi YA. Effects of orientational order and particle size on the NMR line positions of lipoproteins. Phys Rev Lett. 1994;72:4049–52. doi: 10.1103/PhysRevLett.72.4049. [DOI] [PubMed] [Google Scholar]
- 132.Pentikainen MO, Lehtonen EM, Oorni K, Lusa S, Somerharju P, Jauhiainen M, Kovanen PT. Human arterial proteoglycans increase the rate of proteolytic fusion of low density lipoprotein particles. J Biol Chem. 1997;272:25283–8. doi: 10.1074/jbc.272.40.25283. [DOI] [PubMed] [Google Scholar]
- 133.Fu D, Lu FK, Zhang X, Freudiger C, Pernik DR, Holtom G, Xie XS. Quantitative chemical imaging with multiplex stimulated Raman scattering microscopy. J Am Chem Soc. 2012;134:3623–6. doi: 10.1021/ja210081h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Benjwal S, Verma S, Rohm KH, Gursky O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 2006;15:635–9. doi: 10.1110/ps.051917406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci USA. 1978;75:3327–31. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Greenland P, LaBree L, Azen SP, Doherty TM, Detrano RC. Coronary artery calcium score combined with Framingham score for risk prediction in asymptomatic individuals. J Am Med Assoc. 2004;291:210–5. doi: 10.1001/jama.291.2.210. [DOI] [PubMed] [Google Scholar]