

Abstract
Engineering enzymes with novel reaction modes promises to expand the applications of biocatalysis in chemical synthesis and will enhance our understanding of how enzymes acquire new functions. The insertion of nitrogen-containing functional groups into unactivated C–H bonds is not catalyzed by known enzymes but was recently demonstrated using engineered variants of cytochrome P450BM3 (CYP102A1) from Bacillus megaterium. Here, we extend this novel P450-catalyzed reaction to include intermolecular insertion of nitrogen into thioethers to form sulfimides. An examination of the reactivity of different P450BM3 variants toward a range of substrates demonstrates that electronic properties of the substrates are important in this novel enzyme-catalyzed reaction. Moreover, amino acid substitutions have a large effect on the rate and stereoselectivity of sulfimidation, demonstrating that the protein plays a key role in determining reactivity and selectivity. These results provide a stepping stone for engineering more complex nitrogen-atom-transfer reactions in P450 enzymes and developing a more comprehensive biocatalytic repertoire.
Introduction
Direct oxidation of unactivated C–H bonds and heteroatoms in small molecules is a valuable method for introducing complexity into industrially and medically important compounds.1 Modification of sulfur heteroatoms is particularly interesting given the potential for chirality in trivalent sulfur compounds and the efficacy of chiral sulfoxide therapeutics.2 Enzymes catalyze a wide range of reactions in nature, and oxygenases in particular are powerful catalysts of heteroatom oxidation and C–H bond activation.3 Enzymes capable of catalyzing sulfoxidation are well documented4 and have even found industrial application,5 underscoring the utility of biocatalysis in heteroatom functionalization.
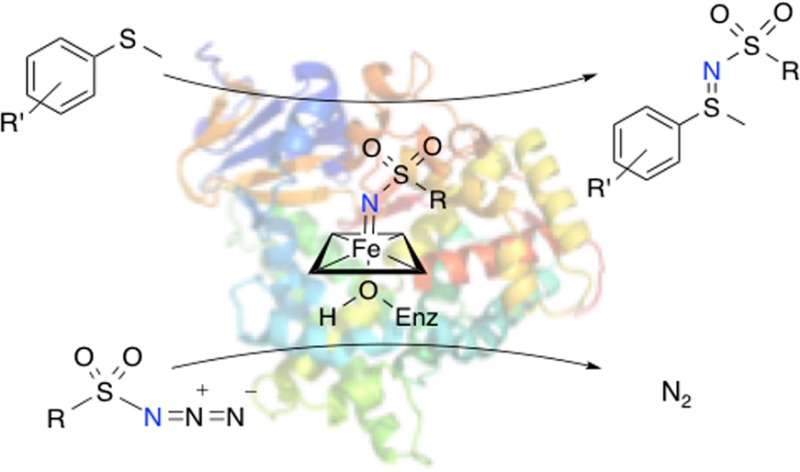
The nitrogen analogues of sulfoxides, known as sulfimides (Figure 1), are useful building blocks in chemical synthesis,6 ligands for metal catalysts,7 and are functional groups in agrochemicals.8 Subsequent oxidation to form sulfoximines can be achieved with good chirality transfer,9 and the resulting compounds are a promising source of novel derivatives of therapeutic small molecules.10 Furthermore, sulfides substituted at the sulfur position with allyl groups can undergo a 2,3-sigmatropic rearrangement upon sulfimidation, resulting in formation of a new C–N bond.11a Available methods for producing sulfimides are largely based on organometallic catalytic systems using iron or rhodium, with only one example of an iron-based asymmetric sulfimidation catalyst reported.9,11 These methodologies typically require iminoiodinane nitrene precursors and chiral ligand frameworks to achieve stereoselectivity. Transition-metal catalysts for nitrenoid transfer also require high catalyst loadings and anhydrous conditions and may also require high temperatures.12
Figure 1.

(A) P450-catalyzed sulfoxidation, shown proceeding through compound I. This reaction can also be mediated by compound 0 (hydroperoxy intermediate). (B) Serine-ligated P411-catalyzed sulfimidation (this work), believed to proceed through a nitrenoid intermediate formed from an azide with N2 as a byproduct.
Enzymes offer a “green” alternative to transition-metal catalysts: they are regio- and stereoselective, nontoxic, function in aqueous media and can be produced with ease in a suitable host organism. Whereas enzymatic sulfoxidation catalysts are well-known, enzymes that catalyze the analogous sulfimidation reaction have not been reported. Our laboratory has recently shown that enzymes of the iron porphyrin-containing cytochrome P450 family can catalyze carbenoid and nitrenoid insertion reactions with high activity and selectivity.13 Mutations to conserved residues T268 and C400 in P450BM3 from Bacillus megaterium were found to dramatically enhance reactivity for non-natural chemistry. The C400S mutation, which replaces the axial cysteine with serine, was particularly activating toward C–H amination and toward olefin cyclopropanation in whole cells.13b,13c The resulting shift in the ferrous-carbon monoxide (CO) Soret band from 450 to 411 nm prompted the “P411” description for P450BM3 variants having the C400S mutation (Figure 1). These “chemo-mimetic” enzyme systems offer several advantages over transition-metal complexes. Because they are genetically encoded, tuning of selectivity and reactivity can be achieved through directed evolution. Moreover, these novel enzyme catalysts operate under mild, aqueous conditions amenable to sustainable chemical synthesis.
Previously, we and Fasan have shown that variants of P450BM3 catalyzed the intramolecular C–H amination of arylsulfonylazides with high selectivities and hundreds of total turnovers.13c,22 To further develop enzymatic nitrene transfer, we wished to explore intermolecular reactions, given the clear synthetic applications in building complexity in a convergent manner. Additionally, we reasoned that an intermolecular nitrene-transfer reaction would allow a more detailed mechanistic characterization than was possible with the intramolecular reaction. Given that variants of P450BM3 are promising enantioselective sulfoxidation and C–H amination catalysts, we sought to determine whether P450 variants could catalyze the nitrogen analogue of sulfoxidation, the intermolecular insertion of nitrogen into organosulfur compounds. This study describes the first report of intermolecular nitrene-transfer catalyzed by an enzyme, in the context of imidation of organosulfur compounds.
Results and Discussion
In our previous work on intramolecular C–H amination, we were limited to aryl sulfonylazide substrates as nitrene precursors. Despite the success with this substrate class, we wished to assess the influence of the R-group on the nitrenoid transfer and thus tested a series of substrates displaying a range of stereoelectronic properties that have been shown to be effective nitrene precursors in other contexts (Figure S1, 1–6).11b,12
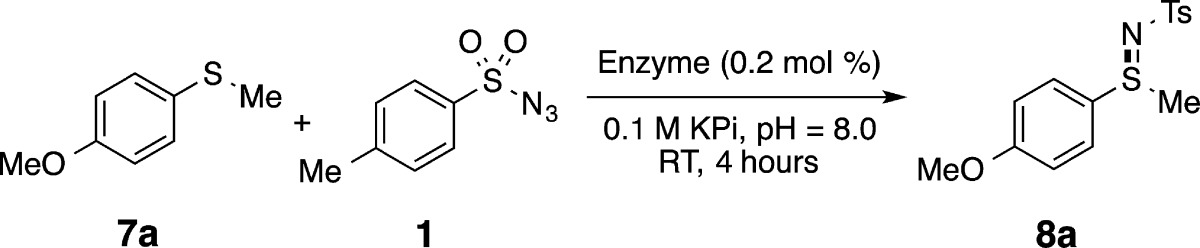
For the thioether acceptor substrate we chose thioanisole, which has been used in enzymatic sulfoxidation by cytochrome P450s and other oxygenase enzymes.4b,4d As a catalyst, we used the P411BM3-CIS T438S variant of cytochrome P450BM3, possessing the aforementioned C400S mutation. This enzyme, which contains 14 mutations relative to wild-type P450BM3 (Table S1), was previously shown to be a good catalyst in the activation of azides for intramolecular C–H insertion.13c Reaction conditions were similar to those reported previously for intramolecular C–H amination13c under anaerobic conditions with nicotine adenine dinucleotide phosphate (NADPH) supplied as a reductant.
Considering the small size of reactive oxygen species naturally produced by P450s, we anticipated that smaller azides, such as mesyl azide (4) would be less sterically demanding than aryl or arylsulfonyl azides and thus a more suitable partner for reaction with thioanisole. We were thus surprised to find that of all the azides initially examined, sulfimidation activity was only observed with tosyl azide (1) as the nitrene source (30 total turnovers, TTN, Figure S1). Control experiments confirmed that enzyme was necessary for sulfimide formation (Table S2). Free hemin showed no activity in this transformation. As we reported previously for intramolecular C–H amination, some of the azide was reduced to sulfonamide, in this case p-toluenesulfonamide (9).13c
In other non-natural P450 reactions reported to date, it was shown that amino acid substitutions could alter both the activity and stereoselectivity of the enzymes.13a,13c Thus, we tested how mutation of conserved residues C400 and T268 and other active-site residues affect sulfimidation activity (Table 1). For these experiments we used the more reactive sulfide 4-methoxythioanisole, for which we measured 300 TTN with P411BM3-CIS T438S (see below for more discussion of the effect of sulfide substituents on reactivity).
Table 1. Sulfimidation Activity and Selectivity of BM3 Variants Using Substrates and Reaction Conditions Showna.
| entry | enzyme | TTN | er |
|---|---|---|---|
| 1 | P411BM3-CIS T438S | 300 | 74:26 |
| 2 | P450BM3-CIS T438S | 7 | nd |
| 3 | P411BM3-CIS A268T T438S | 19 | nd |
| 4 | P411BM3-H2-5-F10 | 140 | 29:71 |
| 5 | P411BM3-H2-A-10 | 84 | 57:43 |
| 6 | P411BM3-H2-4-D4 | 32 | 70:30 |
| 7 | P450BM3 | 10 | nd |
| 8 | P411BM3 | 11 | nd |
| 9 | P450BM3-T268A | 19 | nd |
| 10 | P411BM3-T268A | 17 | nd |
| 11 | P411BM3-CIS I263A T438S | 320 | 18:82 |
Since activating mutations T268A and C400S were already present in P411BM3-CIS T438S, we tested the effects of reverting each mutation to the wild-type residue (Table 1, entries 1–3). Each revertant was much less active than the parent, supporting the benefit of having the C400S and T268A mutations for effective nitrene-transfer chemistry. Given the bulky nature of the aryl sulfonylazide nitrene sources and aryl thioethers, we next tested the C400S mutants of several P450BM3 variants that had been engineered via combinatorial alanine scanning to hydroxylate large substrates14 (Table 1, entries 4–6). While P411BM3-H2-5-F10 displayed comparably high levels of activity to P411BM3-CIS T438S (>100 TTN), the other mutants we tested from this library were less productive. We also tested the effects of introducing the activating mutations into wild-type P450BM3. Although these wild-type derivatives were highly active and stereoselective for intramolecular C–H amination,13c here we found that neither single mutant (T268A or C400S) nor the double mutant (T268A + C400S) were particularly active for intermolecular sulfimidation.
The turnover data presented above demonstrate the sequence dependence of sulfimidation productivity. The effects, however, could be due to changes in stability of the enzymes that lead to degradation over the course of the reaction. To address this possibility, we compared the initial rates of reaction using the most productive enzyme in terms of total turnover, P411BM3-CIS T438S, and the less productive P411BM3-H2-A-10 and P411BM3-H2-4-D4 enzymes (Figure S2). Differences in the total productivity (i.e., TTN) for each enzyme are mirrored in the initial rates of reaction (Table S3), suggesting that specific amino acids proximal to the heme influence binding and orientation of the substrates to effect catalytic rate enhancement.
The key role of active site architecture in guiding reaction trajectory is further supported by the effects of amino acid substitutions on the reaction stereochemistry. Indeed, we found enzymes capable of producing an excess of either sulfimide enantiomer: e.g., P411BM3-CIS T438S gave an er of 74:26, while expanded active site variant P411BM3-H2-5-F10 exhibited the opposite selectivity, giving 29:71 (Figure S3). Among the H2 mutants (which differ from P411BM3-CIS T438S by 3–5 amino acid substitutions, Table S1), H2-5-F10 was alone in containing the I263A mutation, suggesting this mutation is responsible for enantiomeric inversion observed in the P411BM3-H2-5-F10 variant. When the I263A mutation was placed in the P411BM3-CIS background, an even more pronounced inversion in selectivity was observed (er = 18:82 for the P411BM3CIS I263A T438S variant, compared to 74:26 for P411BM3CIS T438S). This enzymatic system not only induces asymmetry in sulfimide products but also provides tunability in which selectivity can be switched with just a few mutations.
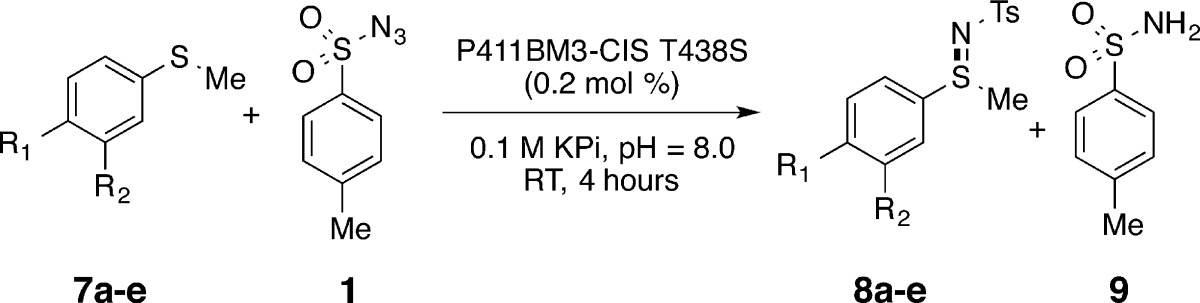
Previous studies of P450-catalyzed sulfoxidation15 as well as rhodium-catalyzed C–H amination16 suggest that the electronic properties of sulfide or alkyl acceptor substrates significantly impact reactivity. Thus, to better understand the mechanism of this new enzyme reaction, we sought to establish how thioether electronic properties affected enzyme-catalyzed sulfimidation. We chose a set of aryl sulfide substrates with substituents encompassing a range of electronic properties, from strongly donating to weakly withdrawing. As a first approximation of the effect of sulfide electronics, we examined the total number of turnovers catalyzed by P411BM3-CIS T438S in the reactions of different sulfides with tosyl azide (Table 2). In general, sulfides containing electron-donating substituents on the aryl sulfide ring were better substrates for sulfimidation. For example, the enzyme reaction containing 4-methoxythioanisole (7a) gave the highest levels of activity (300 TTN). In contrast, the electron-deficient p-aldehyde substrate (7e) gave only trace amounts of sulfimide product. Further, some azides that initially appeared entirely inactive gave small amounts of sulfimide products when reacted with 4-methoxythioanisole, underscoring the importance of sulfide electronics in this reaction (Table S4). The identity of substrates also exerted a modest influence on the enantioselectivity of sulfimidation. In particular, P411BM3-CIS T438S gave er values for substrates 8a-8d that ranged from 59:41 for 8c to 87:13 for 8d (Table S5, Figures S4–S6). While it is possible that some sulfides were poorer substrates due to the steric influence of the para substituent, the overall trend is strongly suggestive of electron induction to the aryl sulfide being a major contributor to activity. One notable aspect of these reactions is that significantly more sulfonamide (9) was produced when less reactive sulfides were used.
Table 2. Impact of Sulfide Substituents on Sulfimidation Activity with P411BM3-CIS T438S.
| entry | R1 in 7 | R2 in 7 | TTN 8 | TTN 9 |
|---|---|---|---|---|
| a | -OMe | -H | 300 | 270 |
| b | -Me | -H | 190 | 400 |
| c | -H | -Me | 100 | 390 |
| d | -H | -H | 30 | 500 |
| e | -CHO | -H | <1a | 510 |
Trace product observed by liquid chromatography-mass spectrometry (LC-MS).
Although the total turnover data suggest that sulfide electronics influence reactivity, this result could also be due to other factors, such as substrate-dependent enzyme inactivation. To assess the effect of sulfide substituents on reactivity more directly, we measured the initial rates of reaction of tosyl azide with the sulfides 7a–7d in Table 2. The initial rates correlated well with the total turnover data presented above, with p-OMe showing the highest rate of reaction (Figure S7). Given this correlation, we sought to obtain more mechanistic information by fitting the data to a Hammett plot that correlates the observed rates with each substituent’s Hammett parameter.17 We found a strong, linear relationship with a Hammett value of ρ = −4.0 (Figure 2), which suggests that during the rate-limiting step there is a buildup of positive charge on the sulfide that is stabilized by electron-donating substituents. This observation is consistent with Hammett values obtained for the oxidation of thioanisoles in P450-catalyzed sulfoxidation reactions, though the magnitude of ρ for sulfimidation is significantly greater than for sulfoxidation (−4.0 versus −0.2).15 One possible explanation for this difference is that the presumed nitrenoid intermediate of this reaction (Figure 1) is a weaker oxidant than compound I, making the nucleophilicity of sulfur an important contributor to the rate of nitrenoid transfer. The large difference in the magnitude of ρ could also indicates a change in mechanism relative to P450 sulfoxidation: whereas Watanabe and co-workers have proposed that sulfoxidation proceeds through a single electron-transfer process,15 sulfimidation may occur via direct nucleophilic attack of the thioether on the nitrenoid intermediate.
Figure 2.

Plot of reaction rate versus Hammett parameter of substituted aryl sulfides using the P411BM3-CIS T438S enzyme and tosyl azide as nitrogen source. Data points are labeled with aryl substituent and position (p- = para, m- = meta) and Hammett parameters obtained form Hansch et al.17
As noted above, we observed that a greater proportion of sulfonamide side product was formed when less reactive sulfides were used. The varying amounts of this side product prompted us to examine more closely how sulfonamide might be produced. We first tested the possibility that azide is reduced by some additive in the reactions (i.e., glucose oxidase, catalase, NADPH, etc.) by simply omitting the P450 enzyme from the reactions (Table S2). We found that no-enzyme controls yielded very little reduced sulfonamide product (more than 10-fold lower than with enzyme present). While these experiments showed that enzyme was likely involved in azide reduction, this still left several possibilities. Since P411BM3’s heme domain is fused to a reductase, we considered the possibility that azide reduction occurs via direct hydride transfer from the reductase, as has been observed for aldehyde reductions.18 We used carbon monoxide-inhibited reactions to investigate this possibility, since CO binding to the heme iron should have no effect on the reductase domain. In the presence of CO, we observed a significant decrease in the sulfonamide produced, suggesting that azide reduction occurs at the heme. Furthermore, only trace sulfonamide was observed when reactions were conducted in the presence of oxygen, further supporting the involvement of reduced heme in azide reduction. Since all the available evidence suggests that azide reduction and sulfimide formation both occur at the heme, the most parsimonious explanation is that both reactions stem from a common intermediate that can give rise to both sulfonamide and sulfimide products.
We propose a mechanism of sulfonamide and sulfimide formation that begins with the iron(III) heme gaining an electron from NADPH via the flavin cofactors of the reductase domain (Figure 3). Addition of azide substrate results in formation of a formal iron(IV) nitrenoid, which can either be reduced by subsequent electron transfer or “trapped” by sulfide to form sulfimide product. We hypothesize that a second electron transfer followed by protonation of the nitrenoid to generate sulfonamide restores the heme iron to its ferric state, and additional reductant is required to return to the catalytically active ferrous state (Figure 3, unproductive pathway).
Figure 3.

Proposed mechanisms of sulfimide (“productive”) and sulfonamide (“unproductive”) formation.
To test whether ferric heme is involved in the unproductive pathway, we monitored the change in the visible absorbance spectrum of the reduced holoenzyme P411BM3-CIS I263A T438S upon addition of NADPH followed by azide. The Ser-ligated P411 proteins exhibit different absorbance properties in the ferric and ferrous states compared with their Cys-ligated counterparts, such that the ferric, ferrous, and CO-ferrous Soret bands are shifted from 418, 408, and 450 nm to 405, 422, and 411 nm, respectively (Figure S8).13b When NADPH was added to a solution of enzyme under an anaerobic atmosphere, we observed reduction of the heme from the ferric to the ferrous state. When a degassed solution of azide was added to the ferrous protein, we observed an immediate shift back to the ferric state, with concomitant production of sulfonamide, verified by high-performance liquid chromatography (HPLC). This observation suggests that the unproductive azide reduction pathway occurs readily in the absence of sulfide and that, when provided only with azide, the catalyst rests in the ferric state.
To determine the resting state of the P411 catalyst in sulfimidation, we repeated the above experiment in the presence of both sulfide and azide. Addition of sulfide to a solution of enzyme and NADPH results in no change in the Q or Soret bands, with the iron heme remaining in the ferrous state. However, addition of azide to this solution causes the iron heme to shift to the ferric state. After 10 min, peaks corresponding to the ferrous heme begin to grow until the ferrous heme becomes the dominant species at 30 min (Figure S9). Both sulfonamide and sulfimide products are formed throughout the course of the reaction. Our observations are consistent with the competing reaction pathways outlined in Figure 3 and suggest that the catalyst rests as a mixture of ferric and ferrous hemes during sulfimidation. When the concentration of azide is high, as it is at the beginning of the reaction, the unproductive pathway is favored, and a ferric resting state dominates. As azide is consumed, both the ferric and ferrous resting states can be observed.
P450 monooxygenases are known to undergo an “oxidase uncoupling” side reaction in which compound I is reduced by two electrons to give water, which bears some similarity to the process of azide reduction we have observed here. One difference, however, is that only a single electron transfer is required to attain a reactive state in nitrene-transfer chemistry. This stands in contrast to P450 monooxygenase chemistry, where the generation of compound I from O2 requires the transfer of two electrons. Thus, one explanation for the relatively high proportion of reduced azide in these reactions is that the electron-transfer machinery in P450BM3 is evolved to carry out two-electron reductions.19 In the case of nitrene-transfer chemistry, reducing the ferric heme to the +2 state allows nitrenoid formation, but a second electron transfer would generate an unreactive iron(III) sulfonamide complex, as proposed by Fasan and coworkers for intramolecular C−H amination.22 Coupled with the fact that lower sulfide concentrations and less-reactive sulfides lead to increased azide reduction, these observations are consistent with the mechanism discussed above in which sulfimide formation competes with azide reduction. Since electron transfers from the reductase domain are quite rapid,20,13b only relatively reactive sulfides can successfully compete with reduction to form sulfimide.
The mechanistic picture described above suggests that achieving higher levels of sulfide occupancy in the active site should favor sulfimide formation and inhibit azide reduction. This could be achieved with tighter binding of the sulfide acceptor substrate or by increasing the concentration of sulfide relative to azide. We therefore tested whether excess sulfide or slow addition of azide would increase sulfimide formation relative to sulfonamide. Increasing the sulfide concentration decreased reduction of azide to sulfonamide and improved the ratio of sulfimide to sulfonamide, from 0.6 (with 0.5 equiv sulfide) to 1.8 (with 4 equiv sulfide) (Figure S10, Table S6). Slow azide addition slightly increased the TTN for sulfimide and decreased sulfonamide formation in a 2 h reaction (Figure S11). That higher concentrations of sulfide substrate improve sulfimide production suggest that protein engineering to improve the binding of sulfide acceptor substrates could also produce strong gains in the desired activity. Indeed, the specific activities of the enzyme catalysts reported here compare favorably with enantioselective iron-based catalysts, which routinely require catalyst loadings of 10 mol %.9b Furthermore, engineering the holoenzyme or reductase domain to favor one-electron transfers might improve the proportion of desired product relative to azide reduction, which could allow reaction with more challenging organic acceptor substrates.
Conclusions
This is the first report of intermolecular nitrene transfer catalyzed by an enzyme, allowing for a mechanistic analysis of this new enzyme activity. Similar to P450-catalyzed sulfoxidation, we find that the electronic properties of the sulfide substrates strongly influence reactivity, though the magnitude of the substituent effects is greater for nitrene transfer, possibly owing to the less oxidizing nature of the presumed nitrenoid intermediate as compared to compound I. The necessity of the C400S mutation for sulfimidation can be rationalized along similar lines: the less electron-donating axial serine ligand in P411 enzymes likely makes the nitrenoid species a more potent oxidant. The impact of sulfide substituents on sulfimide formation is also reflected in the generation of sulfonamide side product, suggesting the nitrenoid undergoes rapid reduction and can only productively insert into sufficiently reactive sulfides. Characterization of the redox state of the heme iron in the presence and absence of nitrene source and sulfide acceptor supports the proposal that nitrenoid “over-reduction” competes with productive sulfimide formation and that the former is a two-electron process resulting in regeneration of ferric heme. Another interesting aspect of this enzyme reaction is the strong preference for an aryl sulfonylazide nitrene source: although monooxygenation reactions use a small donor substrate (dioxygen), only trace sulfimidation was observed with small azides such as ethanesulfonyl azide (Table S4). The ability of the enzyme to accept larger aryl substrates may be beneficial for development of enantioselective intermolecular nitrene-transfer catalysts, as we have observed that a single mutation can dramatically affect the enantioselectivity of reaction. Intermolecular nitrene transfer in the form of sulfimidation can now be added to the impressive array of natural and non-natural chemistry catalyzed by cytochrome P450 enzymes.
Acknowledgments
The authors acknowledge the support of the Jacobs Institute for Molecular Engineering for Medicine at Caltech and the Department of the Navy, Office of Naval Research (grant N00014-11-1-0205). C.C.F. is supported by an NSF Graduate Research Fellowship (DGE-1144469). J.A.M., T.K.H., and Z.J.W. are supported by NIH Ruth L. Kirschstein National Research Service Awards (F32GM101792, F32GM108143, F32EB015846). We thank R. Kelly Zhang and Hans Renata for helpful comments on the manuscript, and Scott Virgil and the 3CS catalysis center for HPLC and LC-MS analysis.
Supporting Information Available
Text, tables, and figures describing methods and results for reaction set up and quantitation, determination of enzyme reaction rates, and enzyme mutant generation and composition. Chromatogram traces demonstrating dependence of enantioselectivity on enzyme sequence. Spectroscopic characterization of the P411BM3-CIS I263A T438S catalyst in the absence and presence of azide substrate as well as sulfide nitrene acceptor. Calibration curves for all products presented and LC-MS verification of product formation by enzyme catalysts. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Davies H. M. L.; Manning J. R. Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Paddon C. J.; Westfall P. J.; Pitera D. J.; Benjamin K.; Fisher K.; McPhee D.; Leavell M. D.; Tai A.; Main A.; Eng D.; Polichuk D. R.; Teoh K. H.; Reed D. W.; Treynor T.; Lenihan J.; Fleck M.; Bajad S.; Dang G.; Dengrove D.; Diola D.; Dorin G.; Ellens K. W.; Fickes S.; Galazzo J.; Gaucher S. P.; Geistlinger T.; Henry R.; Hepp M.; Horning T.; Iqbal T.; Jiang H.; Kizer L.; Lieu B.; Melis D.; Moss N.; Regentin R.; Secrest S.; Tsuruta H.; Vazquez R.; Westblade L. F.; Xu L.; Yu M.; Zhang Y.; Zhao L.; Lievense J.; Covello P. S.; Keasling J. D.; Reiling K. K.; Renninger N. S.; Newman J. D. Nature 2013, 496, 528–532. [DOI] [PubMed] [Google Scholar]
- a Carreno M. C. Chem. Rev. 1995, 95, 1717–1760. [Google Scholar]; b Fernandez I.; Khiar N. Chem. Rev. 2003, 103, 3651–3706. [DOI] [PubMed] [Google Scholar]
- a de Gonzalo G.; Pazmino D. E. T.; Ottolina G.; Fraaije M. W.; Carrea G. Tetrahedron: Asymmetry 2006, 17, 130–135. [Google Scholar]; b Feingersch R.; Shainsky J.; Wood T. K.; Fishman A. Appl. Environ. Microbiol. 2008, 74, 1555–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Colonna S.; Gaggero N.; Pasta P.; Ottolina G. Chem. Commun. 1996, 2303–2307. [Google Scholar]; d Allenmark S. G.; Andersson M. A. Tetrahedron: Asymmetry 1996, 7, 1089–1094. [Google Scholar]; e Colonna S.; Gaggero N.; Manfredi A.; Casella L.; Gullotti M.; Carrea G.; Pasta P. Biochemistry 1990, 29, 10465–10468. [DOI] [PubMed] [Google Scholar]; f Colonna S.; Gaggero N.; Carrea G.; Pasta P. J. Chem. Soc., Chem. Comm. 1992, 357–358. [Google Scholar]; g Dai L. H.; Klibanov A. M. Biotechnol. Bioeng. 2000, 70, 353–357. [DOI] [PubMed] [Google Scholar]; h Fruetel J.; Chang Y. T.; Collins J.; Loew G.; Demontellano P. R. O. J. Am. Chem. Soc. 1994, 116, 11643–11648. [Google Scholar]
- a Zhang J. D.; Li A. T.; Yang Y.; Xu J. H. Appl. Microbiol. Biotechnol. 2010, 85, 615–624. [DOI] [PubMed] [Google Scholar]; b Schallmey A.; den Besten G.; Teune I. G.; Kembaren R. F.; Janssen D. B. Appl. Microbiol. Biotechnol. 2011, 89, 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pordea A.; Creus M.; Panek J.; Duboc C.; Mathis D.; Novic M.; Ward T. R. J. Am. Chem. Soc. 2008, 130, 8085–8088. [DOI] [PubMed] [Google Scholar]; d O’Reilly E.; Corbett M.; Hussain S.; Kelly P. P.; Richardson D.; Flitsch S. L.; Turner N. J. Catal. Sci. Technol. 2013, 3, 1490–1492. [Google Scholar]; e Nikodinovic-Runic J.; Coulombel L.; Francuski D.; Sharma N. D.; Boyd D. R.; Ferrall R. M.; O’Connor K. E. Appl. Microbiol. Biotechnol. 2013, 97, 4849–4858. [DOI] [PubMed] [Google Scholar]
- a Bong Y. K.; Clay M.; Collier S.; Mijts B.; Vogel M.; Zhang X.; Zhu J.; Nazor J.; Smith D.; Song S. WO Patent 2,011,071,982, 2011.; b Ang E. L.; Clay M.; Behrouzian B.; Eberhard E.; Collier S.; Fu Fan J.; Smith D.; Song S.; Alvizar O.; Widegren M. WO Patent 2,012,078,800, 2012.; c Huisman G. W.; Collier S. J. Curr. Opin. Chem. Biol. 2013, 17, 284–292. [DOI] [PubMed] [Google Scholar]
- a Raghavan S.; Kumar C. N. Tetrahedron Lett. 2006, 47, 1585–1588. [Google Scholar]; b Padwa A.; Nara S.; Wang Q. Tetrahedron Lett. 2006, 47, 595–597. [Google Scholar]
- a Thakur V. V.; Kumar N.; Sudalai A. Tetrahedron Lett. 2004, 45, 2915–2918. [Google Scholar]; b Takada H.; Oda M.; Oyamada A.; Ohe K.; Uemura S. Chirality 2000, 12, 299–312. [DOI] [PubMed] [Google Scholar]
- a Gilchrist T. L.; Moody C. J. Chem. Rev. 1977, 77, 409–435. [Google Scholar]; b Park S. J.; Baars H.; Mersmann S.; Buschmann H.; Baron J. M.; Amann P. M.; Czaja K.; Hollert H.; Bluhm K.; Redelstein R.; Bolm C. ChemMedChem 2013, 8, 217–220. [DOI] [PubMed] [Google Scholar]; c Chen X. Y.; Buschmann H.; Bolm C. Synlett 2012, 23, 2808–2810. [Google Scholar]
- a Collet F.; Dodd R. H.; Dauban P. Org. Lett. 2008, 10, 5473–5476. [DOI] [PubMed] [Google Scholar]; b Wang J.; Frings M.; Bolm C. Angew. Chem., Int. Ed. 2013, 52, 8661–8665. [DOI] [PubMed] [Google Scholar]
- Lücking U. Angew. Chem., Int. Ed. 2013, 52, 9399–9408. [DOI] [PubMed] [Google Scholar]
- a Murakami M.; Uchida T.; Saito B.; Katsuki T. Chirality 2003, 15, 116–123. [DOI] [PubMed] [Google Scholar]; b Murakami M.; Uchida T.; Katsuki T. Tetrahedron Lett. 2001, 42, 7071–7074. [Google Scholar]; c Mancheno O. G.; Bolm C. Org. Lett. 2006, 8, 2349–2352. [DOI] [PubMed] [Google Scholar]; d Mancheno O. G.; Bolm C. Chem.—Eur. J. 2007, 13, 6674–6681. [DOI] [PubMed] [Google Scholar]; e Mancheno O. G.; Dallimore J.; Plant A.; Bolm C. Org. Lett. 2009, 11, 2429–2432. [DOI] [PubMed] [Google Scholar]
- a Cramer S. A.; Jenkins D. M. J. Am. Chem. Soc. 2011, 133, 19342–19345. [DOI] [PubMed] [Google Scholar]; b Ruppel J. V.; Jones J. E.; Huff C. A.; Kamble R. M.; Chen Y.; Zhang X. P. Org. Lett. 2008, 10, 1995–1998. [DOI] [PubMed] [Google Scholar]; c Fiori K. W.; Du Bois J. J. Am. Chem. Soc. 2007, 129, 562–568. [DOI] [PubMed] [Google Scholar]
- a Coelho P. S.; Brustad E. M.; Kannan A.; Arnold F. H. Science 2013, 339, 307–310. [DOI] [PubMed] [Google Scholar]; b Coelho P. S.; Wang Z. J.; Ener M. E.; Baril S. A.; Kannan A.; Arnold F. H.; Brustad E. M. Nat. Chem. Biol. 2013, 9, 485–487. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McIntosh J. A.; Coelho P. S.; Farwell C. C.; Wang Z. J.; Lewis J. C.; Brown T. R.; Arnold F. H. Angew. Chem., Int. Ed. 2013, 52, 9309–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang Z. J.; Peck N. E.; Renata H.; Arnold F. H. Chem. Sci. 2014, 5, 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Singh R.; Bordeaux M.; Fasan R. ACS Catal. 2014, 4, 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bordeaux M.; Singh R.; Fasan R. Bioorg. Med. Chem. 2014 doi: 10.1016/j.bmc.2014.05.015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. C.; Mantovani S. M.; Fu Y.; Snow C. D.; Komor R. S.; Wong C.-H.; Arnold F. H. ChemBioChem 2010, 11, 2502–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Watanabe Y.; Iyanagi T.; Oae S. Tetrahedron Lett. 1982, 23, 533–536. [Google Scholar]; b Watanabe Y.; Iyanagi T.; Oae S. Tetrahedron Lett. 1980, 21, 3685–3688. [Google Scholar]
- Collet F.; Lescot C.; Liang C. G.; Dauban P. Dalton Trans. 2010, 39, 10401–10413. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- Kaspera R. d.; Sahele T.; Lakatos K.; Totah R. A. Biochem. Biophys. Res. Commun. 2012, 418, 464–468. [DOI] [PubMed] [Google Scholar]
- McIntosh J. A.; Farwell C. C.; Arnold F. H. Curr. Opin. Chem. Biol. 2014, 19, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daff S. N.; Chapman S. K.; Turner K. L.; Holt R. A.; Govindaraj S.; Poulos T. K.; Munro A. W. Biochemistry 1997, 36, 13816–13823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.