

Abstract

We synthesized 3-aroyl-1-arylpyrrole (ARAP) derivatives as potential anticancer agents having different substituents at the pendant 1-phenyl ring. Both the 1-phenyl ring and 3-(3,4,5-trimethoxyphenyl)carbonyl moieties were mandatory to achieve potent inhibition of tubulin polymerization, binding of colchicine to tubulin, and cancer cell growth. ARAP 22 showed strong inhibition of the P-glycoprotein-overexpressing NCI-ADR-RES and Messa/Dx5MDR cell lines. Compounds 22 and 27 suppressed in vitro the Hedgehog signaling pathway, strongly reducing luciferase activity in SAG treated NIH3T3 Shh-Light II cells, and inhibited the growth of medulloblastoma D283 cells at nanomolar concentrations. ARAPs 22 and 27 represent a new potent class of tubulin polymerization and cancer cell growth inhibitors with the potential to inhibit the Hedgehog signaling pathway.
INTRODUCTION
Cancer is presently a major cause of death worldwide. The proportion of people suffering from cancer is estimated to continue rising, largely because of the aging of the population in most countries.1 Antitumor agents in clinical use generally show cytostatic or cytotoxic activity through interference with mechanisms responsible for cell division. Despite enormous efforts, cancer remains one of the most difficult diseases to treat, as most patients obtain only a longer survival or no benefit at all from current cancer treatments.
Microtubules (MTs) are formed from α,β-tubulin heterodimers and play a fundamental role in numerous cell functions. MTs undergo a dynamic equilibrium, and preventing proper MT function by either inhibiting tubulin polymerization or blocking MT disassembly invariably causes cell death. Colchicine (1),2 combretastatin A-4 (CSA4, 2)3 (Chart 1), vincristine (VCR), and vinblastine (VBL) prevent MT assembly by inhibiting tubulin polymerization. Taxoids and epothilones instead target a lumenal site on the β-subunit.4 These drugs enter the lumen through a binding site5 located at a pore on the MT surface formed by different tubulin heterodimers. At high concentrations, paclitaxel (PTX) stimulates MT polymerization and stabilization, whereas at lower concentrations it inhibits MT dynamics with little effect on the proportion of tubulin in polymer.6
Chart 1.

General Structures of Compounds 1–4
Interfering with MT formation or function has been a productive strategy for the development of highly successful antitumor drug classes.7 The classical antimitotic drugs are still one of the best approaches for cancer treatment. However, some problems related to drug resistance and to secondary toxicity still remain unsolved.9 Newer tubulin targeting agents have shown limited efficacy in clinical trials.8 Thus, the quest for better anticancer therapies based on alternative or synergistic anticancer drugs remains mandatory.
We have developed arylthioindole and aroylindole antimitotic agents as potent inhibitors of tubulin polymerization and of cancer cell growth.10 These compounds bind to the colchicine site on β-tubulin and inhibit the binding of [3H]colchicine to tubulin.10 Several compounds of this class were more potent than 1, 2, VBL, and PTX and have potential as novel therapeutic agents to treat cancer.10
Splitting of fused rings by following a disjunctive approach often yields new biologically active chemical entities that show the same mechanism of action, weaker cytotoxicity, and improved pharmacokinetic properties.11 This strategy allowed us to explore the potential of 3-aroyl-1-arylpyrrole (ARAP) derivatives as inhibitors of tubulin polymerization. In recent years, other groups have reported the discovery of tubulin binding agents sharing structural similarities to ARAPs.12 In light of these encouraging results, we designed a prototypic molecule, 4, by disjunction of the indole ring of 3 into 1-phenylpyrrole (Chart 1).
Because computer-aided drug design was useful in the development of ATIs,10 4 was first evaluated by docking studies (Figure 1). In the proposed binding mode, the phenylpyrrole moiety of 4 lay far from the indole of 3. Its binding pocket extended deep into the β-tubulin so that its binding mode was similar to those of other colchicine site agents, for example, ABT-751 (5),13 TN16 (6),13 G2N (7),14 and K2N (8)14 (Chart 2). Nevertheless, the position of the trimethoxyphenyl (TMP) group of 3 and 4 was virtually identical. These results encouraged us to investigate further this new structure scaffold.
Figure 1.

Binding mode of 4 (white) and parent compound 3 (cyan). The tubulin heterodimer is shown in strand format with pink for the α- and blue for the β-subunit. Residues within 3.5 Å of the inhibitors are also shown. Residues (C241 and T179) involved in polar contacts are represented as the thicker sticks.
Chart 2.

Structures of Reference Compounds 5–8
The biological evaluation confirmed our initial idea because 4 proved to be a potent tubulin polymerization inhibitor with an IC50 of 1.5 μM. These results prompted hit optimization studies of 4 by the synthesis of new ARAPs 9–55 (Table 1) and some correlated isomeric compounds 56–62 (Table 1S, Supporting Information). Our data show that ARAPs are a new class of potent tubulin polymerization and cancer cell growth inhibitors that have potential as novel anticancer agents.
Table 1.
Inhibition of Tubulin Polymerization and the Binding of Colchicine to Tubulin and Inhibition of Growth of MCF-7 Human Breast Carcinoma Cells by 3-Aroylpyrroles 4, 9–55, and Reference Compounds 1–3a

| |||||
|---|---|---|---|---|---|
| compd | R1 | R2 | Tubulin Assemblya IC50 ± SD (μM) | MCF-7b,c IC50 ± SD (nM) | Colchicine Bindingd (% ± SD) |
| 4 |

|

|
1.5 ± 0.2 | 700 ± 200 | 80 ± 2 |
| 9 | H |

|
>40 | ndf | nd |
| 10 | Me |

|
>40 | nd | nd |
| 11 |
|

|
>20e | ndf | nd |
| 12 |

|

|
8.8 ± 0.1 | 4500 ± 700 | nd |
| 13 |

|

|
2.2 ± 0.2 | 600 ± 0 | 58 ± 4 |
| 14 |

|

|
2.3 ± 0.02 | 200 ± 100 | 73 ± 2 |
| 15 |

|

|
1.0 ± 0.08 | 1000 ± 300 | 77 ± 2 |
| 16 |

|

|
2.1 ± 0.2 | 1300 ± 400 | 55 ± 5 |
| 17 |

|

|
1.4 ± 0.2 | 190 ± 40 | 78 ± 2 |
| 18 |

|

|
>20e | nd | nd |
| 19 |

|

|
8.9 ± 1 | 230 ± 60 | nd |
| 20 |

|

|
>20e | nd | nd |
| 21 |
|

|
6.8 ± 0.8 | nd | nd |
| 22 |

|

|
1.4 ± 0.2 | 15 ± 5 | 86 ± 2 |
| 23 |

|

|
1.3 ± 0.8 | 15 ± 1 | 81 ± 1 |
| 24 |

|

|
1.0 ± 0.04 | 30 ± 10 | 92 ± 0.7 |
| 25 |

|

|
2.5 ± 0.04 | 20 ± 0 | 53 ± 1 |
| 26 |

|

|
1.5 ± 0.2 | 1000 ± 0 | 60 ± 4 |
| 27h |

|

|
1.2 ± 0.1 | 20 ± 0 | 87 ± 0.8 |
| 28 |

|

|
0.86 ± 0.06 | 60 ± 30 | 84 ± 0.5 |
| 29 |

|

|
2.5 ± 0.07 | 43 ± 20 | 27 ± 4 |
| 30 |

|

|
2.5 ± 0.05 | 16 ± 6 | 23 ± 4 |
| 31 |

|

|
>20e | nd | nd |
| 32 |

|

|
13 ± 1 | nd | nd |
| 33 |

|

|
0.95 ± 0.02 | 50 ± 0 | 79 ± 1 |
| 34 |

|

|
0.90 ± 0.04 | 29 ± 10 | 95 ± 0 |
| 35 |

|

|
2.7 ± 0.5 | 3500 ± 700 | 50 ± 3 |
| 36 |

|

|
1.8 ± 0.2 | 600 ± 0 | 66 ± 3 |
| 37 |
|

|
1.2 ± 0.2 | 250 ± 70 | 76 ± 5 |
| 38 |

|

|
6.1 ± 1 | nd | nd |
| 39 |

|

|
2.5 ± 0.2 | 250 ± 50 | 17 ± 0.01 |
| 40 |

|

|
>20g | nd | nd |
| 41 |

|

|
>20g | nd | nd |
| 42 |

|

|
1.7 ± 0.03 | 530 ± 100 | 67 ± 0.6 |
| 43 |

|

|
1.2 ± 0.1 | 400 ± 100 | 86 ± 0.4 |
| 44 |

|

|
1.7 ± 0.1 | 450 ± 200 | 59 ± 1 |
| 45 |

|

|
15 ± 0.07 | 830 ± 200 | nd |
| 46 |

|

|
1.3 ± 0.01 | 470 ± 200 | 89 ± 0.2 |
| 47 |

|

|
1.3 ± 0.04 | 4500 ± 700 | 79 ± 3 |
| 48 |

|

|
2.1 ± 0.2 | 100 ± 0 | 56 ± 0.1 |
| 49 |

|

|
1.7 ± 0.2 | 1700 ± 1000 | 48 ± 6 |
| 50 |

|

|
>20g | nd | nd |
| 51 |

|

|
>40 | nd | nd |
| 52 |

|

|
>20e | nd | nd |
| 53 |

|

|
>20e | nd | nd |
| 54 |
|

|
>40 | nd | nd |
| 55 |

|
|
16 ± 0.6 | nd | nd |
| 1 | – | – | 3.2 ± 0.4 | 5 ± 1 | nd |
| 2 | – | – | 1.0 ± 0.1 | 13 ± 3 | 98 ± 0.6 |
| 3i | – | – | 3.5 ± 0.07 | 150 ± 50 | 26 ± 0.3 |
Inhibition of tubulin polymerization. Tubulin was at 10 μM in the assembly assay.
Inhibition of growth of MCF-7 human breast carcinoma cells.
Compounds that inhibited tubulin assembly with IC50 ≤ 5 μM were tested in the cellular and colchicine binding assays.
Inhibition of [3H]colchicine binding. Tubulin was at 1 μM. Both [3H]colchicine and inhibitor were at 5 μM.
Partial inibition at 20 μM.
No data.
Little or no activity at 20 μM.
Hydrochloride.
Lit.10a
CHEMISTRY
Compounds 4, 10–20, 32–41, 45, 47–55, and 56–60 were prepared by microwave (MW)-assisted reaction of the appropriate 1-substituted pyrrole with 3,4,5-trimethoxybenzoyl chloride in the presence of anhydrous aluminum chloride in 1,2-dichloroethane at 110 °C (150 W) for 2 min (Scheme 1a). Treatment of benzaldehyde or its 3,4,5-trimethoxy derivative with an acetophenone and sodium hydroxide in ethanol at 25 °C for 12 h gave the trans-chalcones 82 or 83, which cyclized to pyrroles 61 and 62 with p-toluenesulfonylmethyl isocyanide (TosMIC) in the presence of sodium hydride at 25°C for 15 min in dimethyl sulfoxide (DMSO)/diethyl ether. Sodium hydroxide hydrolysis of 1-phenylsulfonylpyrrole 55 by heating in methanol at reflux for 3 h furnished (1H-pyrrol-3-yl)-(3,4,5-trimethoxyphenyl)methanone 9 (Scheme 1b). Tin-(II) chloride dihydrate reduction of compounds 18–20 or 45 for 3 h in boiling ethyl acetate afforded amino derivatives, 21–23 or 46, respectively (Scheme 1c). Isopropoxyphenyl pyrroles 38–40 were converted into the corresponding hydroxyphenyl derivatives 2–44 by heating at reflux in chloroform with methanesulfonic acid for 2.5 h (Scheme 1c).
Scheme 1. Synthesis of Pyrroles 4, 9–23, and 32–62a.

aReagents and reaction conditions (R1 and R2, see Table 1): (a) appropriate benzoyl chloride, AlCl3, 1,2-dichloroethane, closed vessel, 110 °C, 150 W, 2 min, yield 25–80%; (b) appropriate acetophenone, NaOH, EtOH, 25 °C, 12 h, 41–67%; (c) NaH, TosMIC, DMSO/Et2O, 25 °C, 15 min, 38–75%; (d) 2 N NaOH, MeOH, reflux, 3 h, 85%; (e) SnCl2·2H2O, AcOEt, reflux, 3 h, 42–65%; (f) MeSO3H, CHCl3, reflux, 2.5 h, Ar, 28–51%.
Alkylation of 22 with dimethyl sulfate in acetone at 25 °C for 16 h in the presence of sodium carbonate gave 3-methyl (24) and 3-dimethylamino (26) derivatives that were separated by column chromatography (Scheme 2). Sodium cyanoborohydride reductive amination of 22 with acetone in methanol, tetrahydrofuran, and aqueous hydrochloric acid at 25 °C for 12 h furnished 25. Treatment of 22 with 1,4-dibromobutane or 1,5-dibromopentane in water in the presence of sodium carbonate at 110 °C (100 W) for 20 min yielded the corresponding 3-(pyrrolidin-1-yl) (30) or 3-(piperidin-1-yl) (31) derivatives. The guanidino (27) and methanesulfonamido (28) derivatives were prepared by treating 22 with cyanamide and 3.3 N hydrochloric acid in ethanol at 50 °C for 48 h or with methansulfonyl chloride in the presence of triethylamine (TEA) in tetrahydrofuran at 25 °C for 2 h, respectively. Pyrrole derivative 29 was obtained according to the Clauson–Kaas reaction15 by treating the appropriate aniline with 2,5-dimethoxytetrahydrofuran in glacial acetic acid at 80 °C for 2 h.
Scheme 2. Synthesis of Pyrroles 24–31a.

aReagents and reaction conditions: (a) Me2SO4, Na2CO3, anhydrous acetone, 25 °C, 16 h, Ar, 31%; (b) acetone, NaCNBH3, MeOH/THF, 6 N HCl, 25 °C, 12 h, 57%; (c) 1,4-dibromobutane, K2CO3, H2O, closed vessel, 110 °C, 100 W, 20 min, 35%; (d) 1,5-dibromopentane, K2CO3, H2O, closed vessel, 110 °C, 100 W, 20 min, 57%; (e) cyanamide, EtOH, 3.3 N HCl, 50 °C, 48 h, 45%; (f) MeSO2Cl, TEA, anhydrous THF, 25 °C, 2 h, 49%; (g) 2,5-(MeO)2–THF, AcOH, 80°C, 2 h, 82%.
Pyrrole derivatives 63–78 were obtained from an appropriate aniline and 2,5-dimethoxytetrahydrofuran under the aforementioned conditions of the Clauson–Kaas reaction (Scheme 3a). Reaction of methyl 3-hydroxy-4-methoxybenzoate with 2-iodopropane in DMF in the presence of potassium carbonate at 50 °C for 3 h furnished derivative 79. Lithium hydroxide hydrolysis of 79 in aqueous tetrahydrofuran at 25 °C for 12 h afforded the acid 80, which was converted to the corresponding acid chloride 81 by treatment with thionyl chloride at reflux temperature for 1.5 h (Scheme 3b).
Scheme 3. Synthesis of Intermediates 63–81a.

aReagents and reaction conditions: (a) (63 R″ = 2-Cl; 64 R″ = 3-Cl; 65 R″ = 3-NO2; 66 R″ = 2-Me; 67 R″ = 3-Me; 68 R″ = 4-Me; 69 R″ = 2-OMe; 70 R″ = 3-OMe; 71 R″ = 2-OCHMe2; 72 R″ = 3-OCHMe2; 73 R″ = 4-OCHMe2; 74 R″ = 3-OCH2Ph; 75 R″ = 4-OMe, 3-NO2; 76 R″ = 3-OCHMe2, 4-OMe; 77 R″ = 3,4-OMe2; 78 R″ = 3,4,5-OMe3) 2,5-OMe2THF, AcOH, 80 °C, 2 h, 34–72%; (b) 2-iodopropane, K2CO3, anhydrous DMF, 50 °C, 3 h, Ar stream, 76%; (c) LiOH·H2O, THF/H2O, 25°C, 12 h, 77%; (d) SOCl2, reflux, 1.5 h, Ar stream, used as a crude product.
RESULTS AND DISCUSSION
Inhibition of Tubulin Polymerization, the Binding of Colchicine to Tubulin and MCF-7 Breast Cancer Cell Growth
We synthesized ARAP compounds 4 and 9–55 to obtain structure–activity relationship (SAR) information regarding position and substituents on both the aroyl and aryl aromatic moieties. The effects of these compounds on tubulin polymerization in vitro, the binding of [3H]colchicine to tubulin, and the growth of MCF-7 human breast cancer cells are shown in Table 1. Sixteen new ARAPs (4, 15, 17, 22–24, 26, 27, 36, 37, 42–44, 46, 47, and 49) inhibited tubulin polymerization, with IC50 values in the 1.0–2.0 μM concentration range. Compounds 15, 24, 28, 33, and 34 yielded IC50 values ≤ 1.0 μM as compared with colchicine (1) (IC50 = 3.2 μM) and CSA4 (2) (IC50 = 1.0 μM). Compounds lacking the pendant 1-phenyl ring (9, 10) or the 3,4,5-trimethoxy group (11) did not inhibit tubulin assembly, emphasizing the essential role of these moieties for the antitubulin activity of ARAPs. Introduction of either a chlorine or fluorine atom on the 1-phenyl ring yielded potent tubulin polymerization inhibitors (compounds 12–17). However, regardless of the position of the halogen atom, these compounds were moderate to weak inhibitors of the growth of human MCF-7 nonmetastatic breast cancer epithelial cells. Among the nitrophenyl derivatives, only 1-(3-nitrophenyl)-pyrrole 19 showed significant inhibition of tubulin polymerization with an IC50 of 8.9 μM.
Reduction of the nitro group of 19 provided the corresponding amino derivative 22, and this compound potently inhibited tubulin assembly (IC50 = 1.4 μM). More importantly, compound 22 inhibited MCF-7 cell growth with an IC50 of 15 nM, 10-fold lower than the value obtained for reference compound 3, comparable with the value for 2 and only 3-fold higher than that of 1. For SAR studies regarding the 1-(3-aminophenyl)pyrrole 22, we synthesized ARAP derivatives 24–31. With the exception of 27, these derivatives potently inhibited tubulin polymerization with IC50 values ≤2.5 μM, with compound 28 (IC50 = 0.86 μM) being the most potent tubulin assembly inhibitor among all the ARAPs. As inhibitors of MCF-7 cell growth, 24, 25, and 27–30 yielded IC50 values from 16 (30) to 60 nM (28), and three compounds (25, 27, and 30) were comparable to 22.
Introduction of a methyl on the 1-phenyl group led to potent tubulin polymerization (33, IC50 = 0.95 μM; 34, IC50 = 0.90 μM) and cancer cell growth inhibitors (33, IC50 = 50 nM; 34, IC50 = 29 nM). Replacement of the methyl group with a methoxy also provided potent inhibitors (35–37), at least of tubulin polymerization (IC50s = 2.7, 1.8, and 1.2 μM, respectively, for 35, 36, and 37). Compounds 35–37 were, however, only moderate to weak inhibitors of MCF-7 cell growth. The hydroxyphenyl derivatives 42–44 strongly inhibited the polymerization of tubulin, with IC50 values in the low micromolar concentration range. Considering the typical substitution pattern of 2 and its amino derivatives, we synthesized two potent tubulin assembly inhibitors (both with IC50s of 1.3 μM), 1-(3-amino-4-methoxyphenyl)-(46) and 1-(3-hydroxy-4-methoxyphenyl)pyrroles (47). Compounds 42–49 were weak inhibitors of MCF-7 cell growth. Compounds 50–55 and, similarly, the isomeric 2-aroyl-1-arylpyrroles (56–60) and 3-aroyl-4-arylpyrroles (61, 62) all had little or no effect on tubulin polymerization (Table 1S, Supporting Information) and were not studied further.
In general, inhibition of colchicine binding was in accord with inhibition of tubulin binding (Figure 2). The compounds that inhibited assembly with IC50 values ≤1.0 μM inhibited colchicine binding (with tubulin at 1 μM and both colchicine and inhibitor at 5 μM) by 77–95%. Assembly inhibitors in the 1.0–1.5 μM range inhibited colchicine binding by 48–89%, those that inhibited assembly with IC50s in the 1.6–2.0 μM range inhibited colchicine binding by 48–67%, and assembly inhibitors with IC50s in the 2.1–2.7 μM range inhibited colchicine binding by 17–58% (CSA4: IC50 = 1.0 μM, colchicine binding inhibition = 98%).3 While the correlations between the three assays in Table 3 are reasonable, they are not perfect. For example, the best inhibitor of assembly was compound 28 (IC50, 0.86 μM), the best inhibitor of colchicine binding was compound 34 (95% inhibition), and the best inhibitors of MCF-7 cell growth were compounds 22 and 23 (IC50s, 15 nM). Nevertheless, these compounds, among others, had excellent activity in all three assays.
Figure 2.

Correlation between tubulin assembly (IC50 values, μM) and inhibition of colchicine binding (% values). Data of ARAP compounds 4 and 9–55 (open circles). CSA4 as reference compound is represented by a filled circle.
Table 3.
Inhibition of Growth of the OVCAR-8 and NCI/ ADR-RES and Messa and Messa/Dx Cell Line Pairs by Compounds 4, 22, 27, and 35–37 and Reference Compounds 1, 2, VRB, VBL, and PTXa
| compd | IC50 ± SD (nM)
|
|||
|---|---|---|---|---|
| OVCAR-8 | NCI/ADR-RES | Messab | Messa/Dx5b | |
| 4 | 45 ± 20 | 20 ± 0 | 56 ± 3 | 76 ± 7 |
| 22 | 7.0 ± 1 | 1.0 ± 0 | 1.8 ± 0.2 | 4.6 ± 0.3 |
| 27c | ndd | nd | 14 ± 4 | 16 ± 0.02 |
| 35 | 450 ± 70 | 200 ± 0 | 515 ± 29 | 653 ± 143 |
| 36 | 250 ± 70 | 90 ± 10 | 262 ± 74 | 471 ± 58 |
| 37 | 60 ± 20 | 17 ± 6 | 63 ± 7 | 87 ± 21 |
| 1 | 30 ± 1 | 420 ± 100 | 11 ± 6 | 329 ± 166 |
| 2 | 1.3 ± 0.6 | 1.3 ± 0.6 | 2.7 ± 2 | 2.6 ± 1 |
| VRB | 300 ± 0 | 5000 ± 1000 | ndd | nd |
| VBL | 15 ± 7 | 200 ± 0 | 3 ± 2 | 144 ± 61 |
| PTX | 5.0 ± 2 | 3300 ± 1000 | 4 ± 1 | 1764 ± 477 |
Inhibition of growth of the indicated cell lines.
Growth inhibition data of compounds 11, 12, 14, 18, 19, 27, 29, 38, and 51 are shown in Table 2S, Supporting Information.
Hydrochloride.
No data.
ARAPs 4, 22, 35–37, 47, and 49 were evaluated for growth inhibition of HeLa (human cervical carcinoma), HT29 (human colon adenocarcinoma), and A549 (human lung carcinoma) cells in comparison with 1, 2, VBL, and PTX (Table 2). With the exception of 47, and ignoring the high value obtained with 2 in the A549 cells, the compounds were uniformly inhibitory against the panel. When evaluated as inhibitor of HCT15 cells, the drug resistant cell line of HCT116 (human colon carcinoma cells), compound 4 yielded an IC50 of 39 ± 4 nM (data not shown in Table 2). The most potent ARAP 22 was of similar potency to 1 and VBL as inhibitor of the growth of HeLa, HT-29, and A549 cells.
Table 2.
Growth Inhibition of HeLa, HT-29, A549, and HCT116 Cell Lines by Compounds 4, 22, 35–37, 47, and 49 and Reference Compounds 1, 2, VBL, and PTXa
| compd | IC50 ± SD (nM)
|
|||
|---|---|---|---|---|
| HeLA | HT-29 | A549 | HCT116 | |
| 4 | 100 ± 18 | 90 ± 10 | 200 ± 50 | 77 ± 4 |
| 22 | 30 ± 1 | 60 ± 1 | 10 ± 4 | ndb |
| 35 | 900 ± 70 | 1000 ± 1400 | 1000 ± 300 | nd |
| 36 | 1000 ± 110 | 800 ± 20 | 3000 ± 500 | nd |
| 37 | 150 ± 1 | 200 ± 20 | 80 ± 7 | nd |
| 47 | 200 ± 10 | >10000 | >10000 | nd |
| 49 | 800 ± 033 | 800 ± 1 | 1000 ± 20 | nd |
| 1c | 28 ± 9 | 18 ± 4 | 20 ± 8 | 20 ± 2 |
| 2c | 20 ± 5 | 130 ± 12 | >10000 | 5 ± 0.4 |
| VBLc | 10 ± 0.6 | 30 ± 0.8 | 20 ± 2 | 3 ± 2 |
| PTXc | 5 ± 1 | 8 ± 1.5 | 7 ± 2 | 4 ± 0.4 |
Growth inhibition of the indicated cell lines (MTT method); incubation time was 48 h.
no data.
Lit.10b
Compounds 4, 22, 27, and 35–37 were evaluated as inhibitors of the ovarian carcinoma cell lines OVCAR-8 and its cognate P-glycoprotein (Pgp) overexpressing line NCI/ ADR-RES and of the human uterine sarcoma cell line Messa and its cognate MDR line Messa/Dx5, using 1, 2, vinorelbine (VRB), VBL, and PTX as reference compounds (Table 3). With the exception of 2, the reference agents were quite weak inhibitors of the MDR cell lines. ARAP 22 showed strong inhibition of MDR cells and was at the same level as 2.
Molecular Modeling Studies
To better understand the ARAPs binding mode, a series of molecular docking simulations were carried out. Among the available tubulin crystal structures with colchicine site inhibitors,13 3HKD16 was selected because of the structural similarity between the cocrystallized ligand and 4. The results obtained for compound 4 showed that the phenyl group was stabilized by a series of hydrophobic/aromatic contacts in a pocket formed mainly by the Y52, F169, Y202, V238, and L251 side chains; the pyrrole core was stabilized by L255, V238, and I378. Furthermore, we observed a weak H-bond interaction between the Y202 OH group and the π-aromatic cloud of the pyrrole. Interestingly, from the docking results, it appears that the ketone bridge is not involved in any interactions. Finally, the TMP moiety was in contact with residues C241, L245, L255, M258, A316, and A317 (Figure 1S, Supporting Information).
Induction of Mitotic Arrest and Cell Death in HeLa Cell Cultures
The ability of the ARAP molecules 4 and 22 to arrest mitotic progression was assessed in HeLa cells. HeLa cell cultures were treated for 24 h with 4 or 22, both used at 20 and 100 nM; VBL (20 nM) or 2 (20 nM) were used as reference compounds. Treated and untreated samples were first incubated with propidium iodide (PI) to analyze their genomic content in flow cytometry assays. We found that both 4 and 22 effectively arrested cell cycle progression when used at 100 nM, with the majority of cells accumulating with 4C DNA content, hence in the G2 or M phases (Figure 3A). At the 20 nM concentration, 22 proved to be as effective as VBL and 2, whereas 4 had only a minimal effect (data not shown). Light microscopy revealed that the majority of cells in treated cultures displayed a typical mitotic, rounded-up phenotype (Figure 3B), consistent with the cells being blocked in mitosis.
Figure 3.

Cell cycle arrest in HeLa cells after treatment with either 4 or 22. (A) Flow cytometric analysis of the cell cycle phase distribution in HeLa cell cultures after the indicated treatments. The histograms show mean values, and bars standard deviations, of cells with 2C (G1 phase), 4C (G2/M phases), or intermediate (S phase) DNA content detected by PI incorporation in 3 independent experiments. (B) Typical views from live, unfixed HeLa cell cultures under light microscopy (10× objective): 4 and 22, as well as VLB, induced an accumulation of rounded, detached cells, as occurs in mitotic arrest (compare to the adherent cells in the control (DMSO-treated) culture).
The effects of ARAPs on mitotic cells were observed by immunofluorescence (IF) methods (Figure 4). Both ARAPs 4 and 22 inhibited normal spindle formation, such that cells progressed normally into mitosis through nuclear envelope breakdown (depicted by the absence of lamin B1 staining), but treated cells failed to organize a proper mitotic apparatus and arrested with condensed chromosomes in a prometaphase-like state (Figure 4A). Noteworthy, 100 nM 4 and 20 nM 22 had similar effects, yielding evidently defective MTs that failed to form a normal mitotic spindle (Figure 4A, panel a): short MTs were formed, often arranged in aberrantly shaped, tripolar (Figure 4A, panel b) or multipolar (Figure 4A, panel c) structures. The abnormal MT structures failed to cause sustained mitotic arrest in some cells (mitotic “slippage”); such cells progressed toward aberrant cell division, eventually producing multi- and micronucleated daughter cells (Figure 4B). Only unstructured tubulin foci were seen with 100 nM 22, while MT elongation failed completely; under these conditions, the occurrence of mitotic slippage dropped significantly, in the same range as that observed with 2 (data not shown).
Figure 4.

Inhibition of normal spindle formation by 4 and 22. (A) Mitotic cells were identified by the lack of lamin B1 staining (red channel) and the organization of condensed chromosomes (blue channel), similar to those seen in a normal prophase (depicted in panel a). Representative examples from treated cultures are shown (panels b–d). (B) Frequency of mitotic figures (M) and micro-nucleated cells (MN), representing the products of mitotic slippage in HeLa cultures treated as indicated. At least 500 cells per condition were scored in two independent experiments; mean and SD values are shown. (C) Frequency of annexin V-reactive HeLa cells under the indicated conditions. Histograms represent mean values and bars standard deviations from three independent flow cytometry assays.
To determine whether ARAP-dependent MT damage induced cell death in treated cultures, we used fluorescently conjugated annexin V, which reveals the loss of organization of the cell membrane occurring in early cell death stages. Cell death was activated in cell populations treated by both ARAPs in proportions similar to those observed with VBL (Figure 4C). In summary, both ARAPs prevented mitotic MT organization into normal spindles and blocked mitotic progression in HeLa cells with concomitant activation of cell death. Compound 22 proved to be more effective than 4 and was comparable to VBL. With 100 nM 22, a reduction of mitotic slippage was observed with no effect on mitotic arrest or cell death induction.
The cell cycle inhibitory effects of 4 in HeLa cells, albeit less pronounced than those of 22, were significant and dose-dependent (Figure 3A). This differential response might have reflected a requirement for caspase-3 activity, which influences the cell response to MT-targeting drugs17 yet is defective in the MCF-7 cell line. Therefore, we compared the effects of 4 in MCF-7 and in a MCF-7-derived cell line stably expressing an exogenous caspase-3 gene (MCF-7/cas 3).18 However, by flow cytometry no differences between the caspase-3-reconstituted and the native MCF7 cell lines were detected in either cell cycle profile or in annexin V reactivity after treatment with 100 nM 4 (data not shown). We carried out a single-cell analysis after IF staining of α-tubulin, to stain mitotic MTs, and of active caspase-3 using a specific antibody to the processed form of the enzyme, to evaluate cell death. The IF analysis confirmed that 100 nM 4 induced mitotic arrest and cell death in HeLa cells but neither in MCF-7 nor in MCF-7/cas-3 parallel cultures treated under the same conditions. These data account for the very high IC50 value detected in the cell viability assays and rule out the possibility that caspase-3 would be the limiting factor in this response, suggesting that MCF-7 cells are intrinsically resistant to MT inhibition by 4.
Inhibition of PC-3, RD, and HepG2 Cancer Cell Growth
We wished to determine whether the effect of ARAPs on cell cycle distribution was observed more generally. Therefore, compounds 22 and 27 were evaluated as inhibitors of the growth of the rhabdomyosarcoma cell line RD, the human prostate cancer cell line PC-3, and the human liver hepatocellular carcinoma cell line HepG2 with VCR, VBL, and PTX as reference compounds (Table 4). ARAP 22 and 27 showed strong inhibition of these three cell lines as compared with PTX. Moreover, as an inhibitor of HepG2 cell growth, 27 was more effective than VCR and VBL.
Table 4.
Growth Inhibition of PC-3, RD, and HepG2 Cell Lines by Compounds 22 and 27 and Reference Compounds VBL, VCR, and PTXa
| compd | IC50 ± SD (nM)
|
||
|---|---|---|---|
| PC-3 | RD | HepG2 | |
| 22 | 9.8 ± 5.5 | 95.3 ± 1.7 | 274.5 ± 4.8 |
| 27 | 3.9 ± 1.9 | 803.2 ± 1.9 | 49.5 ± 1.8 |
| VBL | ndb | 52.5 ± 2.5 | 80.7 ± 2.4 |
| VCR | ndb | nd | 177 ± 1.6 |
| PTX | 3990 ± 1.9 | 14200 ± 1.6 | 2037 ± 2 |
Growth inhibition of the indicated cell lines (MTT method); incubation time was 48 h.
No data.
Effects on Cell Cycle Progression in the PC-3, RD, and HepG2 Cell Lines
ARAP molecules 22 and 27 were evaluated in PC-3, RD, and HepG2 cells, and accumulation of G2/M cells was observed in all of these cell lines (Figure 5). PC-3, RD, and HEPG2 cell cultures were treated for 24 h with increasing concentrations (500, 1000, and 2000 nM) of 22, 27, and the reference compounds PTX, VBL, and VCR. Treated cells and vehicle controls (0.1% DMSO) were incubated with PI to analyze their DNA content in flow cytometry assays.
Figure 5.

Cell cycle analysis of PC-3 (A), RD (B), and HepG2 (C) cells treated with 0.1% DMSO or 500, 1000, or 2000 nM PTX, VBL, VCR, 22, and 27 for 24 h. Representative cell cycle profiles derived from flow cytometric analysis of cell populations following treatment with DMSO or 2000 nM 22 and 27 are shown in the upper part of each panel. Histograms show the percent of cells with G0/G1, S, and G2/M DNA content expressed as mean values ± SD calculated from three independent experiments.
We found that both 22 and 27 effectively arrested cell cycle progression at the lowest concentration in the three cell lines. Nevertheless, there were differences in cell cycle distribution. As shown in Figure 5A,B, 22 and 27 induced an accumulation in G2 or M phase in PC-3 and RD cells that differed little from the patterns observed with the reference compounds. In contrast, in HepG2 cells, 22 and 27 caused a stronger effect on cell cycle progression as compared with PTX (Figure 5C). The significant increase in the proportion of cells in the G2/M phase was also reflected in abundant rounded cells observed under the light microscope in all cases. This indicates that ARAPs 22 and 27 exerted a selective action specifically at mitosis
Sequential treatment of PC-3, RD, and HepG2, consisting of 24 h exposure to 22, 27, or the reference compounds, followed by incubation in drug-free medium for 24 h, revealed a differential behavior in the cell lines analyzed. As shown in Figure 6A,B, except for RD cells following the 500 nM treatment, ARAPs induced an irreversible cell cycle arrest. In contrast, a substantial recovery of cell cycle progression, following drug washout, at all doses and with all compounds evaluated, was observed with the HepG2 cell line (Figure 6C). This indicates that a 24 h treatment with 22 or 27, or even the reference compounds, might be relatively ineffective in the HepG2 cell line.
Figure 6.

Cell cycle analysis of PC-3 (A), RD (B), and HepG2 (C) cells treated with 0.1% DMSO or 500, 1000, or 2000 nM PTX, VBL, VCR, 22, and 27 for 24 h and harvested after a further 24 h recovery in drug-free medium. Histograms show the percent of cells with G0/G1, S, and G2/M DNA content expressed as mean values ± SD calculated from three independent experiments.
Compound Effects on Cell Viability in the PC-3, RD, and HepG2 Cell Lines
To reveal whether ARAPs-dependent cell cycle arrest was associated with cell death, treated cells were incubated with fluorescently conjugated annexin V and PI. Comparable levels of cell death were triggered in cell populations treated with 22, 27, or with PTX, VBL, or VCR at all doses examined (Figure 7). A dose–response trend in cell death was observed only in the PC-3 cell line following 48 h of exposure to 27 and in the HepG2 cell line following treatment with PTX. Although a similar dose-dependent effect in RD and HepG2 cells was not observed with either ARAP, a stronger impact of both ARAPs on cell viability, in comparison with PTX, did occur.
Figure 7.

Cell death flow cytometric analysis of PC-3 (A), RD (B), and HepG2 (C) cells treated with 0.1% DMSO or 500, 1000, or 2000 nM PTX, VBL, VCR, 22, or 27 for 48 h. Flow cytometric profiles of cell populations following treatment with DMSO or 2000 nM 22 or 27 are shown in the upper part of each panel (annexin: apoptosis (annexin V-FITC staining) and in late apoptosis (annexin V-FITC and PI staining) expressed as mean values ± SD calculated from three independent experiments.
Survey of a Larger Number of ARAPs for Specific Antitubulin Activity, as Measured by Mitotic Index (MI) Studies in K562 Human Leukemia Cells
The studies above showed that compounds 4, 22, and 27 were clearly antitubulin agents, causing mitotic arrest, cellular microtubule disruption, and/or apoptosis. These are well-defined characteristics of a wide variety of antitubulin compounds. Nevertheless, we cannot readily exclude other targets for the other ARAPs. We therefore decided to do a MI study with additional compounds, comparing them with 22 and 27 as well as the extensively studied 2. We selected the human leukemia K562 line for this analysis, and the data are summarized in Table 5. We chose an additional six strong inhibitors of tubulin assembly (compounds 24, 25, 28, 29, 30, and 34) with IC50s against the MCF-7 cells ranging from 16 to 60 nM. In the K562 cells, the IC50s of these compounds ranged from 14 to 950 nM. We planned to obtain MIs at 10 times the IC50 value, but we were technically limited to a maximum concentration of 4 μM (due to toxicity of DMSO for the cells). In all cases elevated MIs were obtained, ranging from 39 to 73%, with the lowest value obtained with compound 30, where the concentration was only 4.2 times the IC50 value. Untreated cells yielded a MI of 3%. A high MI in drug-treated cells is a hallmark of antitubulin agents, and this finding makes it unlikely that an additional intracellular target plays a significant role in the cytotoxicity observed with these ARAPs.
Table 5.
Mitotic Index Values of ARAPs 22, 24, 25, 27–30, and 34
| compd | IC50 ± SD (nM)
|
mitotic index ± SD
|
|---|---|---|
| K562 | (concentrationa) | |
| 22 | 20 ± 6 | 60 ± 9 (at 10× IC50) |
| 24 | 30 ± 0 | 61 ± 1 (at 10× IC50) |
| 25 | 500 ± 100 | 69 ± 4 (at 4.0 nMb) |
| 30 | 950 ± 100 | 39 ± 6 (at 4.0 nMc) |
| 27 | 25 ± 7 | 71 ± 8 (at 10× IC50) |
| 28 | 19 ± 8 | 61 ± 1 (at 10× IC50) |
| 29 | 90 ± 10 | 47 ± 1 (at 10× IC50) |
| 34 | 30 ± 0 | 61 ± 1 (at 10× IC50) |
| 2 | nd | 65 ± 5 (at 0.1 nM) |
| controld | 3 ± 1 |
Highest concentration possible in mitotic index study was 4.0 μM.
IC50 500 ± 100 nM.
IC50 950 ± 100 nM.
Untreated cells.
Hedgehog Inhibiting Activity
In recent years, inhibitors of the Hedgehog (Hh) molecular signaling pathway have emerged as a new compound class with chemotherapeutic potential. A variety of small molecules targeting different components of the Hh pathway, i.e., Smoothened (Smo), Sonic hedgehog protein (Shh), and Gli1, have been identified.19 A number of inhibitors of the Smo receptor, the positive signaling transducer in the Hh signaling pathway, are presently in clinical trials.
We speculated that ARAPs may represent constraint analogues of MRT (i.e., 84 (MRT-10) and 85 (MRT-14)) Smo antagonists.20,21 (Chart 3). To validate this hypothesis, we carried out a series of in silico simulations (molecular docking and pharmacophore matching) on the Smo receptor to compare the structural features of ARAPs with 84 and 85. The docking results for both 4 and 85 showed that the TMP could be stabilized by hydrophobic contact with F484, M301, L221, W480, and P513.
Chart 3.

MRT–ARAP Structure Relationships
Furthermore, an H-bond contact between the 4-methoxy group of the two different compounds with N219 was also present (Figure 8). The carbonyl bridge also establishes an H-bond with the R400 residue. The pyrrole ring of compound 4 occupies the same space as the urea moiety of 85, although the pyrrole is mainly stabilized by Y394 and V386. Finally, the phenyl ring of both structures is in close contact with I389, M330, W281, and E517.
Figure 8.

Binding mode of 85 (cyan) and ARAP 4 (purple). Smo is shown as ribbon (green), residues within 3.5 Å are shown as gray lines, while residues involved in H-bonds are shown as gray sticks.
Our modeling studies propose a potential binding mode of ARAP compounds on the Smo receptor. Yet the proposed docking poses are in accordance with those of MRT derivatives20 as reference for the modeling studies. The pharmaco-phore matching showed clearly that 4 of the 6 features were satisfied by 4, namely the TMP group, fits into the F2:Acc and F3:Hyd features, the carbonyl matches the F4:Acc features, and the phenyl rings overlap with the F4:Hyd feature (Figure 2S, Supporting Information). The convergent results observed for the docking and pharmacophore studies suggested that the newly designed ARAPs could mimic the biological activity of the MRT compounds. Compounds 22 and 27 showed the highest score in the ARAP series, so they were selected to characterize the inhibitory properties of ARAP compounds on the Hh signaling pathway.
We examined the effects of 22 and 27 in NIH3T3 Shh-Light II (Shh-LII) cells stably incorporating an Hh-responsive (Gli-RE) reporter, in which induction of the pathway occurs following treatment with the Smo agonist SAG. This in vitro test, widely used to for characterizing Hh inhibitors, revealed that compounds 22 and 27 strongly reduced luciferase activity in cells treated with SAG in a dose-dependent manner (Figure 9) and showed the ability of both compounds to suppress the signaling pathway. In these assays, compound 22 yielded an IC50 of 676 nM and 27 an IC50 of 682 nM. We excluded the possibility that inhibition of Hh signaling in this assay was mediated by cytotoxicity because the investigated compounds did not decrease the control Renilla luciferase activity.
Figure 9.

Inhibition of endogenous Hh signaling in Shh-L II cells by 22 and 27. Dose–response curve of the indicated compounds in SAG-treated cells in comparison with untreated NIH3T3 Shh-Light II cells. Treatment time was 48 h, and normalization was against Renilla luciferase. Data from three independent experiments. Error bars indicate SD *P, 0.05 vs CTR.
Hh inhibitors have shown benefits in the treatment of Hh-dependent cancers, such as medulloblastomas.19 However, new Hh inhibitors are desired to overcome the problem of drug resistant Smo mutations arising during treatment. ARAPs 22 and 27 inhibited the growth of medulloblastoma D283 cells with IC50 values of 580 ± 150 and 675 ± 325 nM, respectively. These findings suggest that ARAPs can be developed as potential Hh-dependent anticancer agents.
Pharmacokinetic Studies
Intravenous pharmacokinetics and oral bioavailability of 22 was performed in a mouse model. Compound 22 showed a high clearance and a very high volume of distribution in the mouse after intravenous (IV) treatment at 5 mg/kg (Table 6, top panel). The average time spent between infusion and elimination (mean residence time, MRT) was about 31 min, whereas the elimination half-time (T1/2) was 74 min. After per os (PO) administration, the profile of 22 showed a very rapid absorption (Tmax = 5 min, time to reach Cmax), long MRT = 145 min, and oral bioavailability of 49% (Table 6, bottom panel). In Supporting Information are reported the profiles of distribution in plasma of 22 after IV infusion and PO administration (Figure 3S, Supporting Information) and a comparison of the kinetic profiles (Figure 4S, Supporting Information).
Table 6.
Pharmacokinetic Parameters for ARAP 22
| IV infusion | value |
|---|---|
| Cmax IV (ng/mL) | 3404 |
| Co (ng/mL) | 4152 |
| T1/2 (min) | 74 |
| Tlast (min) | 480 |
| Clast (ng/mL) | 2.3 |
| MRT (min) | 31 |
| Cl (mL/min/kg) | 80.8 |
| Vdss (L/kg) | 2.7 |
| AUC 0–480 IV (min·ng/mL) | 61660 |
| AUCinf IV (min·ng/mL) | 61906 |
|
| |
| PO administration | value |
|
| |
| Cmax PO (ng/mL) | 1767 |
| Tmax PO (min) | 5 |
| Tlast (min) | 480 |
| Clast (min) | 34 |
| T1/2 (min) | 179 |
| MRT (min) | 145 |
| AUC0–480 PO (min·ng/mL) | 79441 |
| AUCinf PO (min·ng/mL) | 88183 |
| Mean Fpo last (%) | 49 |
Caco-2 Cell Permeability
The intestinal permeability of compound 22 was evaluated in the human Caco-2 model in comparison with caffeine (high permeability) and cimetidine (low permeability, Pgp substrate). The apparent permeability (Papp) of 22 from the A (apical) to B (basolateral) side together with B to A was measured in order to predict the absorption from the lumen of the gut and potential efflux phenomena. Compound 22 showed high permeability in both the A → B (Papp = 123.2 nm/s) and B → A (Papp = 107.3 nm/s) directions (Table 7). Compound 22 did not show efflux phenomena (i.e., ratio of Papp(B → A) to Papp(A → B) of <2).
Table 7.
Caco-2 Cell Permeability and CYP450 Isoform Inhibition of Compound 22
| compd |
Papp (nm/s)a
|
CYP450 isoform (% inhibition at 1 μM)
|
|||||
|---|---|---|---|---|---|---|---|
| PA→B | PB→A | CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | |
| 22 | 123.2 | 107.3 | 66.8 | 72.6 | 28.9 | <5.0 | 12.2 |
Papp (nm/s): >50, high; 10–50, medium; <10, low; caffeine PA–B reference control, 206 ± 35 nm/s; cimetidine PA–B and PB–A reference control, 1.1 ± 0.1 and 21.3 ± 3.4 nm/s, respectively.
CYP450 Inhibition
The inhibition of the enzymatic activity of human cloned P450 isoforms CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 by 22 was measured using specific substrates for each isoform that produced a fluorescent metabolite upon CYP metabolism. Compound 22 was shown to inhibit weakly the isoforms CYP2C9, CYP2D6, and CYP3A4, with % of inhibition of 28.9, 5.0, and 12.2, respectively, whereas the isoforms CYP1A2 and CYP2C19 were inhibited by 66.8% and 72.6%, respectively (Table 5).
CONCLUSIONS
We synthesized 55 new pyrrole derivatives as potential anti-cancer agents, with the compounds having different substituents on the pendant 1-phenyl ring. Both the 1-phenyl ring and the 3-(3,4,5-trimethoxyphenyl)carbonyl moieties were mandatory to achieve potent inhibition of tubulin polymerization and cancer cell growth. Several new 3-aroyl-1-arylpyrroles (ARAPs) inhibited tubulin polymerization, with IC50 values in the 1.0–2.0 μM concentration range, and five compounds yielded IC50 values ≤ 1.0 μM. Twelve ARAPs inhibited the growth of human MCF-7 cells with IC50 values ≤50 nM. ARAP 22 inhibited MCF-7 cell growth with an IC50 of 15 nM and was similar to 1 and VBL as an inhibitor of the growth of HeLa, HT-29, and A549 cells. Compound 22 or 27 strongly inhibited PC-3, RD, and HepG2 cell growth as compared with PTX as reference compound. Furthermore, ARAP 27 proved to be a more effective inhibitor of proliferation in PC-3 and HepG2 cells in comparison with the compounds used as reference agents. A 24 h treatment with either 22 or 27 induced a robust accumulation of cells in the G2/M phase of the cell cycle at 500 nM, and 27 also showed a dose-dependent effect on PC-3 cell cycle progression. Besides the arrest of cell cycle progression, a strong induction of cell death was also detected in HeLa, PC-3, RD, and HepG2 cells exposed to 22 or 27 for 48 h. ARAP 22 showed strong inhibition of the Pgp-overexpressing NCI-ADR-RES and Messa/Dx5MDR cell lines. Against these MDR cell lines, 22 had activity similar to that of 2, while the reference agents 1, VRB, VBL, and PTX showed very limited inhibition. Compounds 4 and 22 effectively arrested cell cycle progression at 100 nM, with the majority of cells accumulating in the G2/M phase in HeLa, PC-3, RD, and HepG2 cells. At 20 nM, 22 was as effective as VBL or 2. In addition, eight ARAPs caused a marked increase in the mitotic index in K562 leukemia cells, a hallmark of antitubulin agents. Compounds 22 and 27 suppressed in vitro the Hh signaling pathway, strongly reducing luciferase activity in SAG treated NIH3T3 Shh-Light II cells, and both compounds inhibited the growth of medulloblastoma D283 cells at nanomolar concentrations. Available data suggest that the anticancer activity of ARAP derivatives is due to strong inhibition of tubulin polymerization (IC50 <5 μM: 4, 9–13, 18–26, 29–35, and 38–45) and of the Hedgehog signaling pathway (18 and 23). However, it should be noted that the overall observed cellular responses may be due as well to “off-target effects” not necessarily involving the above targets.
ARAP 22 showed high Caco-2 cell permeability, inhibited weakly the human cloned P450 isoforms CYP2C9, CYP2D6, and CYP3A4 and showed significant inhibition of CYP1A2 and CYP2C19. The isoform CYP1A2 is thought to be procarcinogenic, therefore, its inhibition may lead to a cancer-chemopreventive effect. In pharmacokinetics studies, ARAP 22 showed a long half-life, a rapid clearance and therefore a large volume of distribution, and a good oral bioavailability in the mouse. This is still a suboptimal pharmacokinetic profile, however, considering its very attractive cell growth inhibition profile, ARAP 22 represents an important lead compound for further in vivo proof-of-concept studies.
In short, ARAPs are a new potent class of tubulin polymerization and cancer cell growth inhibitors that have also the potential to inhibit strongly the Hh signaling pathway. Compounds 22 and 27 represent novel lead compounds of the ARAP class that pave the way for the development of new promising anticancer agents, including possible activity against Hh-dependent cancers.
EXPERIMENTAL SECTION
Chemistry
MW-assisted reactions were performed on a CEM Discover SP single mode reactor, controlling the reaction parameters and instrument settings with PC-running CEM Synergy 1.49 software. Closed vessel experiments were carried out in capped MW-dedicated vials (10 mL) with cylindrical stirring bar (length 8 mm, diameter 3 mm). Open vessel experiments were carried out in 100 mL round-bottom flasks equipped with a Dimroth reflux condenser and a cylindrical stirring bar (length 20 mm, diameter 6 mm). Stirring, temperature, irradiation power, maximum pressure (Pmax), PowerMAX (simultaneous cooling-while-heating), ActiVent (simultaneous venting-while-heating), and ramp and hold times were set as indicated. Temperature of the reaction was monitored by an external fiber optic temperature sensor. After completion of the reaction, the mixture was cooled to 25 °C via air-jet cooling. Melting points (mp) were determined on a Stuart Scientific SMP1 apparatus and are uncorrected. Infrared spectra (IR) were run on a PerkinElmer SpectrumOne FT-ATR spectrophotometer. Band position and absorption ranges are given in cm−1. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker 400 MHz FT spectrometer in the indicated solvent and corresponding fid files processed by MestreLab Research S.L. MestreReNova 6.2.1–769 software. Chemical shifts are expressed in δ units (ppm) from tetramethylsilane. Column chromatography was performed on columns packed with alumina from Merck (70–230 mesh) or silica gel from Macherey-Nagel (70–230 mesh). Aluminum oxide thin layer chromatography (TLC) cards from Fluka (aluminum oxide precoated aluminum cards with fluorescent indicator visualizable at 254 nm), and silica gel TLC cards from Macherey-Nagel (silica gel precoated aluminum cards with fluorescent indicator visualizable at 254 nm) were used for TLC. Developed plates were visualized with a Spectroline ENF 260C/FE UV apparatus. Organic solutions were dried over anhydrous Na2SO4. Evaporation of the solvents was carried out on a Büchi Rotavapor R-210 equipped with a Büchi V-850 vacuum controller and a Büchi V-700 or V-710 vacuum pump. All reagents and solvents are commercially available and were used as purchased, without further purification. Elemental analyses of the compounds were found to be within ±0.4% of the theoretical values. The purity of tested compounds was found to be >95% by high pressure liquid chromatography (HPLC) analysis. The HPLC system used (Dionex UltiMate 3000, Thermo Fisher Scientific Inc.) consisted of a SR-3000 solvent rack, a LPG-3400SD quaternary analytical pump, a TCC-3000SD column compartment, a DAD-3000 diode array detector, and an analytical manual injection valve with a 20 μL loop. Samples were dissolved in acetonitrile at 10 mg/mL. HPLC analysis was performed by using an Acclaim 120 C18 reversed-phase column (5 μm, 4.6 mm × 250 mm, Thermo Fisher Scientific Inc.) at 30 ± 1 °C with an isocratic gradient (acetonitrile:water = 90:10), flow rate of 1.0 mL/min and signal detector at 254 and 365 nm. Chromatographic data were acquired and processed by Chromeleon 6.80 software (Thermo Fisher Scientific Inc.). As an example, the HPLC analysis of compound 4 is shown in the Supporting Information.
General Procedure for the Preparation of Compounds 4, 10–20, 32–41, 45, and 47–60
Example: (1-Phenyl-1H-pyrrol-3-yl)-(3,4,5-trimethoxyphenyl)methanone (4) and (1-Phenyl-1H-pyrrol-2-yl)(3,4,5-trimethoxyphenyl)methanone (56)
A mixture of anhydrous AlCl3 (0.17 g, 0.0013 mol), 1-phenyl-1H-pyrrole (0.19 g, 0.0013 mol), and 3,4,5-trimethoxybenzoyl chloride (0.30 g, 0.0013 mol) in anhydrous 1,2-dichloroethane (2.0 mL) was placed into the MW cavity (closed vessel mode, Pmax = 250 psi). A starting MW irradiation of 150 W was used, the temperature being ramped from 25 to 110 °C while stirring vigorously. Once 110 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min. After cooling, the mixture was diluted with water, made acidic with 1 N HCl, and extracted with chloroform. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate: n-hexane = 1:1 as eluent) to give 56 (0.18 g, 42%), mp 120–125 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.92 (s, 6H), 3.95b(s, 3H), 6.37–6.39 (m, 1H), 6.96–6.98 (m, 1H), 7.16–7.17 (m, 1H), 7.22 (s, 2H), 7.33–7.35 (m, 2H), 7.39–7.47 ppm (m, 3H). IR: 1637 cm−1. Anal. (C20H19NO4 (337.37)) C, H, N. Further elution with the same eluent furnished 4 (0.19 g, 43%), mp 88–90 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.94 (s, 6H), 3.96 (s, 3H), 6.89–6.90 (m, 1H), 7.14–7.16 (m, 1H), 7.19 (s, 2H), 7.36–7.40 (m, 1H), 7.44–7.46 (m, 2H), 7.49–7.51 (m, 2H), 7.67–7.69 ppm (m, 1H). IR: ν 1621 cm−1. Anal. (C20H19NO4 (337.37)) C, H, N.
(1-Methyl-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (10)
Obtained as 4 from 1-methyl-1H-pyrrole. Yield 16%, mp 130–135 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.30 (s, 3H), 3.93 (s, 6H), 3.94 (s, 3H), 6.66–6.68 (m, 1H), 6.69–6.71 (m, 1H), 7.13 (s, 2H), 7.25–7.27 ppm (m, 1H). IR: ν 1610 cm−1. Anal. (C15H17NO4 (275.30)) C, H, N.
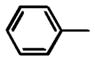
Phenyl(1-phenyl-1H-pyrrol-3-yl)methanone (11)
Obtained as 4 from 1-phenyl-H-pyrrole. Yield 29%. 1H NMR (CDCl3): δ 6.89–6.91 (m, 1H), 7.11 7.13 (m, 1H), 7.32–7.36 (m, 1H), 7.41–7.49 (m, 6H), 7.51–7.53 (m, 1H), 7.61–7.64 (m, 1H), 7.89–7.93 ppm (m, 2H). IR: ν 1630 cm−1.22
(1-(2-Chlorophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (12)
Obtained as 4 from 63. Yield 10%, mp 115–117 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.93 (s, 6H), 3.94 (s, 3H), 6.90–6.91 (m, 1H), 6.94–6.95 (m, 1H), 7.21 (s, 2H), 7.39–7.41 (m, 3H), 7.49–7.51 (m, 1H), 7.56–7.59 ppm (m, 1H). IR: ν 1633 cm−1. Anal. (C20H18ClNO4 (371.81)) C, H, Cl, N.
(1-(3-Chlorophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (13)
Obtained as 4 from 64. Yield 16%, mp 110–115 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.94 (s, 6H), 3.95 (s, 3H), 6.88–6.89 (m, 1H), 7.12 (dd, J = 2.3 and 3.1 Hz, 1H), 7.18 (s, 2H), 7.33–7.36 (m, 2H), 7.41–7.46 (m, 2H), 7.65–7.66 ppm (m, 1H). IR: ν 1616 cm−1. Anal. (C20H18ClNO4 (371.81)) C, H, Cl, N.
(1-(4-Chlorophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (14)
Obtained as 4 from 1-(4-chlorophenyl)-1H-pyrrole. Yield 14%, mp 125–128 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.94 (s, 6H), 3.95 (S, 3H), 6.8–6.88 (m, 1H), 7.00 (t, J = 2.7 Hz, 1H), 7.20 (s, 2H), 7.37–7.40 (m, 2H), 7.45–7.49 (m, 2H), 7.64 ppm (t, J = 2.1 Hz, 1H). IR: ν 1636 cm−1. Anal. (C20H18ClNO4 (371.81)) C, H, Cl, N.
(1-(2-Fluorophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (15)
Obtained as 4 from 1-(2-fluorophenyl)-1H-pyrrole. Yield 54%, mp 119–121 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.86 (s, 6H), 6.80–6.81 (m, 1H), 7.14 (s, 2H), 7.32–7.39 (m, 2H), 7.46–7.51 (m, 2H), 7.70 (t, J = 7.8 Hz, 1H), 7.84–7.85 ppm (m, 1H). IR: ν 1633 cm−1. Anal. (C20H18FNO4 (355.36)) C, H, F, N.
(1-(3-Fluorophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (16)
Obtained as 4 from 1-(3-fluorophenyl)-1H-pyrrole. Yield 70%, mp 107–114 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.77 (s, 3H), 3.87 (s, 6H), 6.80–6.82 (m, 1H), 7.14 (s, 2H), 7.16–7.21 (m, 1H), 7.51–7.57 (m, 1H), 7.61–7.63 (m, 2H), 7.71–7.75 (m, 1H), 8.13–8.15 ppm (m, 1H). IR: ν 1706 cm−1. Anal. (C20H18FNO4 (355.36)) C, H, F, N.
(1-(4-Fluorophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (17)
Obtained as 4 from 1-(4-fluorophenyl)-1H-pyrrole. Yield 54%, mp 140–144 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.86 (s, 6H), 6.78–6.79 (m, 1H), 7.13 (s, 2H), 7.36 (t, J = 2.6 Hz, 2H), 7.50–7.52 (m, 1H), 7.76–7.79 (m, 2H), 8.00–8.02 ppm (m, 1H). IR: ν 1634 cm−1. Anal. (C20H18FNO4 (355.36)) C, H, F, N.
(1-(2-Nitrophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (18)
Obtained as 4 from 1-(2-nitrophenyl)-1H-pyrrole. Yield 40%, mp 150–155 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.75 (s, 3H), 3.86 (s, 6H), 6.78–6.79 (m, 1H), 7.09 (s, 2H), 7.20–7.21 (m, 1H), 7.70–7.79 (m, 3H), 7.86–7.91 (m, 1H), 8.17 ppm (dd, J = 1.4 and 8.2 Hz, 1H). IR: ν 1635 cm−1. Anal. (C20H18N2O6 (382.37)) C, H, N.
(1-(3-Nitrophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (19)
Obtained as 4 from 65. Yield 46%, mp 145–150 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.78 (s, 3H), 3.87 (s, 6H), 6.84–6.85 (m, 1H), 7.16 (s, 2H), 7.72–7.73 (m, 1H), 7.80 (t, J = 8.2 Hz, 1H), 8.17–8.26 (m, 3H), 8.53–8.54 ppm (m, 1H). IR: ν 1640 cm−1. Anal. (C20H18N2O6 (382.37)) C, H, N.
(1-(4-Nitrophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (20)
Obtained as 4 from 1-(4-nitrophenyl)-1H-pyrrole. Yield 68%, mp 175–185 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.78 (s, 3H), 3.88 (s, 6H), 6.87–6.88 (m, 1H), 7.15 (s, 2H), 7.76–7.77 (m, 1H), 8.07 (d, J = 9.2 Hz, 2H), 8.25–8.26 (m, 1H), 8.34 ppm (d, J = 9.2 Hz, 2H). IR: ν 1633 cm−1. Anal. (C20H18N2O6 (382.37)) C, H, N.
(1-(2-Tolyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (32)
Obtained as 4 from 66. Yield 71%, mp 120–125 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 2.24 (s, 3H), 3.75 (s, 3H), 3.82 (s, 6H), 6.74–6.76 (m, 1H), 7.12–7.14 (m, 3H), 7.35–7.43 (m, 4H), 7.64–7.66 ppm (m, 1H). IR: ν 1632 cm−1. Anal. (C21H21NO4 (351.40)) C, H, N.
(1-(3-Tolyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (33)
Obtained as 4 from 67. Yield 80%, mp 105–109 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 1.28 (s, 3H), 130 (s, 3H), 3.77 (s, 3H), 3.86 (s, 6H), 4.73–7.81 (m, 1H), 6.77 (s, 1H), 6.89 (d, J = 7.3 Hz, 1H), 7.14 (s, 2H), 7.23–7.25 (m, 2H), 7.34–7.38 (m, 1H), 7.55 (s, 1H), 8.06 ppm (s, 1H). IR: ν 1712 cm−1. Anal. (C23H25NO5 (395.45)) C, H, N.
(1-(4-Tolyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (34)
Obtained as 4 from 68. Yield 62%, mp 100–105 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 2.35 (s, 3H), 3.76 (s, 3H), 3.86 (s, 6H), 6.77–6.78 (m, 1H), 7.13 (s, 2H), 7.30 (d, J = 8.2 Hz, 2H), 7.50 (t, J = 2.8 Hz, 1H), 7.60 (d, J = 8.4 Hz, 2H), 7.97–7.98 ppm (m, 1H). IR: ν 1632 cm−1. Anal. (C21H21NO4 (351.40)) C, H, N.
(1-(2-Methoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (35)
Obtained as 4 from 69. Yield 25%, mp 120–123 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.89 (s, 3H), 3.94 (S, 9H), 6.85–6.87 (m, 1H), 7.01–7.02 (dd, J = 2.1 and 3.0 Hz, 1H), 7.06–7.09 (m, 2H), 7.20 (s, 2H), 7.33–7.39 (m, 2H), 7.58 ppm (t, J = 2.0 Hz, 1H). IR: ν 1633 cm−1. Anal. (C21H21NO5 (367.40)) C, H, N.
(1-(3-Methoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (36)
Obtained as 4 from 70. Yield 10%. 1H NMR (DMSO-d6): δ 3.77 (s, 3H), 3.84 (s, 3H), 3.86 (s, 6H), 6.78–6.79 (m, 1H), 6.91–6.93 (m, 1H), 7.14 (s, 2H), 7.27–7.29 (m, 2H), 7.38–7.42 (m, 1H), 7.55–7.56 (m, 1H), 8.07 ppm (t, J = 2.1 Hz, 1H). IR: ν 1633 cm−1. Anal. (C21H21NO5 (367.40)) C, H, N.
(1-(4-Methoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (37)
Obtained as 4 from 1-(4-methoxyphenyl)-1H-pyrrole. Yield 58%, mp 140–145 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.81 (s, 3H), 3.86 (s, 6H), 6.76–6.77 (m, 1H), 7.03–7.07 (m, 2H), 7.13 (s, 2H), 7.42–7.43 (m, 1H), 7.61–7.65 (m, 2H), 7.92–793 ppm (m, 1H). IR: ν 1632 cm−1. Anal. (C21H21NO5 (367.40)) C, H, N.
(1-(2-Isopropoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (38)
Obtained as 4 from 71. Yield 42%, mp 130–133 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 0.98 (s, 3H), 0.99 (s, 3H), 3.77 (s, 3H), 3.82 (s, 6H), 4.40–4.46 (m, 1H), 6.36 (t, J = 3.2 Hz, 1H), 6.83–6.85 (m, 1H), 7.11 (s, 2H), 7.21–7.27 (m, 2H), 7.37 (t, J = 7.7 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.70–7.072 ppm (m, 1H). IR: ν 1618 cm−1. Anal. (C23H25NO5 (395.45)) C, H, N.
(1-(3-Isopropoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (39)
Obtained as 4 from 72. Yield 35% as a slurry. 1H NMR (DMSO-d6): δ 1.28 (s, 3H), 130 (s, 3H), 3.77 (s, 3H), 3.86 (s, 6H), 4.73–7.81 (m, 1H), 6.77 (s, 1H), 6.89 d, J = 7.3 Hz, 1H), 7.14 (s, 2H), 7.23–7.25 (m, 2H), 7.34–7.38 (m, 1H), 7.55 (s, 1H), 8.06 ppm (s, 1H). IR: ν 1634 cm−1. Anal. (C23H25NO5 (395.45)) C, H, N.
(1-(4-Isopropoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (40)
Obtained as 4 from 73. Yield 63%, mp 99–100 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 1.28 (s, 3H), 1.29 (s, 3H), 3.76 (s, 3H), 3.86 (s, 6H), 4.63–4.69 (m, 1H), 6.74–6.76 (m, 1H), 7.02 (d, J = 8.9 Hz, 2H), 7.13 (s, 2H), 7.42–7.43 (m, 1H), 7.59 (d, J = 8.9 Hz, 2H), 7.90–7.92 ppm (m, 1H). IR: ν 1629 cm−1. Anal. (C23H25NO5 (395.45)) C, H, N.
(1-(3-(Benzyloxy)phenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (41)
Obtained as 4 from 74. Yield 12% as a slurry. 1H NMR (DMSO-d6): δ 3.77 (s, 3H), 3.83 (s, 6H), 5.22 (s, 2H), 6.78–6.79 (m, 1H), 7.00–7.02 (m, 1H), 7.14 (s, 2H), 7.31–7.36 (m, 2H), 7.39–743 (m, 4H), 7.48 (t, J = 7.2 Hz, 2H), 7.55 (t, J = 2.6 Hz, 1H), 8.03–8.05 ppm (m, 1H). IR: ν 1633 cm−1. Anal. (C27H25NO5 (443.49)) C, H, N.
(1-(4-Methox y-3-ni tro phenyl)-1H-pyr rol-3-yl)(3,4,5-trimethoxyphenyl)methanone (45)
Obtained as 4 from 75. Yield 30%, mp 100–110 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.77 (s, 3H), 3.86 (s, 6H), 3.97 (s, 3H), 6.80–6.81 (m, 1H), 7.13 (s, 2H), 7.48 (d, J = 9.2 Hz, 1H), 7.57–7.58 (m, 1H), 8.05 (dd, J = 2.9 and 9.1 Hz, 1H), 8.10 (t, J = 1.9 Hz, 1H), 8.29 ppm (d, J = 2.9 Hz, 1H). IR: ν 1629 cm−1. Anal. (C21H20N2O7 (412.39)) C, H, N.
(1-(3-Hydroxy-4-methoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (47)
Obtained as 4 from 76. Yield 25%, mp 125–140 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.91 (s, 6H), 3.92 (s, 3H), 3.94 (s, 3H), 5.80 (broad s, disappeared on treatment with D2O, 1H), 6.82–6.83 (m, 1H), 6.89–6.91 (m, 2H), 7.00–7.03 (m, 2H), 7.15 (s, 2H), 7.55–7.56 ppm (m, 1H). IR: ν 1612, 2936, 3133 cm−1. Anal. (C21H21NO6 (383.39)) C, H, N.
(1-(3,4-Dimethoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (48)
Obtained as 4 from 77. Yield 46%, mp 110–120 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.80 (s, 3H), 3.85 (s, 3H), 3.86 (s, 6H), 6.75–6.77 (m, 1H), 7.04 (t, J = 8.8 Hz, 1H), 7.14 (s, 2H), 7.19 (dd, J = 2.6 and 8.6 Hz, 1H), 7.26 (d, J = 2.6 Hz, 1H), 7.46–7.47 (m, 1H), 8.00–8.01 ppm (m, 1H). IR: ν 1625 cm−1. Anal. (C22H23NO6 (397.42)) C, H, N.
(1-(3-Isopropoxy-4-methoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (49)
Obtained as 4 from 76. Yield 33%, mp 95–100 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 1.36 (s, 3H), 1.41 (s, 3H), 3.88 (s, 3H), 3.92 (s, 6H), 3.93 (s, 3H), 4.55–4.61 (m, 1H), 6.81–6.83 (m, 1H), 6.92–6.98 (m, 3H), 7.01–7.03 (m, 1H), 7.17 (s, 2H), 7.57–7.58 ppm (m, 1H). IR: ν 1625 cm−1. Anal. (C24H27NO6 (425.47)) C, H, N.
Phenyl(1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl)methanone (50)
Obtained as 4 from 78. Yield 26%, mp 125–130 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.90 (s, 3H), 3.93 (s, 6H), 6.64 (s, 2H), 6.86–6.88 (m, 1H), 7.06–7.08 (m, 1H), 7.49–7.53 (m, 2H), 7.57–7.61 (m, 2H), 7.90–7.93 ppm (m, 2H). IR: ν 1638 cm−1. Anal. (C20H19NO4 (337.37)) C, H, N.
(3,4,5-Trimethoxyphenyl)(1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl)methanone (51)
Obtained as 4 from 78. Yield 23%, mp 95 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.90 (s, 3H), 3.93 (s, 6H), 3.94 (s, 3H), 3.96 (s, 6H), 6.64 (s, 2H), 6.84–6.86 (m, 1H), 7.06–7.08 (m, 1H), 7.19 (s, 2H), 7.64–7.66 ppm (m, 1H). IR: ν 1615 cm−1. Anal. (C23H25NO7 (337.37)) C, H, N.
(3-Isopropoxy-4-methoxyphenyl)(1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl)methanone (52)
Obtained as 4 from 78 and 81. Yield 25%, mp 55–50 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.89 (s, 3H), 3.90 (s, 6H), 3.97 (s, 3H), 5.75 (broad s, disappeared on treatment with D2O, 1H), 6.64 (s, 2H), 6.84–6.85 (m, 1H), 6.95 (d, J = 8.2 Hz, 1H), 7.04–7.06 (m, 1H), 7.51–7.55 (m, 2H), 7.60–7.62 ppm (m, 1H). IR: ν 1600, 2933, 3133 cm−1. Anal. (C21H21NO6 (383.39)) C, H, N.
(3-Isopropoxy-4-methoxyphenyl)(1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl)methanone (53)
Obtained as 4 from 78 and 81. Yield 45%, mp 140–145 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 1.42 (s, 3H), 1.43 (s, 3H), 3.89 (s, 3H), 3.91 (s, 6H), 3.95 (s, 3H), 4.63–4.70 (m, 1H), 6.64 (s, 2H), 6.83–6.84 (m, 1H), 6.96 (d, J = 8.4 Hz, 1H), 7.05–7.07 (m, 1H), 7.53–7.62 ppm (m, 3H). IR: ν 1627 cm−1. Anal. (C24H27NO6 (425.47)) C, H, N.
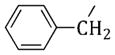
(1-Benzyl-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (54)
Obtained as 4 from 1-benzyl-1H-pyrrole. Yield 17% as an oil. 1H NMR (CDCl3): δ 3.88 (s, 6H), 3.93 (s, 3H), 5.12 (s, 2H), 6.74–6.76 (m, 2H), 7.11 (s, 2H), 7.20–7.23 (m, 2H), 7.31–7.41 ppm (m, 4H). IR: ν 1628 cm−1. Anal. (C21H21NO4 (351.40)) C, H, N.
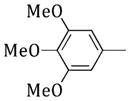
(1-(Phenylsulfonyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (55)
Obtained as 4 from 1-(phenylsulfonyl)-1H-pyrrole. Yield 64% as a slurry. 1H NMR (DMSO-d6): δ 3.75 (s, 3H), 3.82 (s, 6H), 6.78–6.80 (m, 1H), 7.04 (s, 2H), 7.53–7.55 (m, 1H), 7.66–7.70 (m, 2H), 7.78–7.82 (m, 1H), 7.97–7.99 (m, 1H), 8.13–8.15 ppm (m, 2H). IR: ν 1637 cm−1. Anal. (C20H19NO6S (401.43)) C, H, N, S.
(1-(3-Hydroxy-4-methoxyphenyl)-1H-pyrrol-2-yl)(3,4,5-trimethoxyphenyl)methanone (57)
Obtained as 56 from 76. Yield 14%, mp 145–150 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.90 (s, 6H), 3.92 (s, 3H), 3.96 (s, 3H), 5.83 (broad s, disappeared on treatment with D2O, 1H), 6.32–6.33 (m, 1H), 6.81–6.92 (m, 4H), 7.09–7.11 (m, 1H), 7.20 ppm (s, 2H). IR: ν 1634, 2939, 3402 cm−1. Anal. (C21H21NO6 (383.39)) C, H, N.
Phenyl(1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-2-yl)methanone (58)
Obtained as 56 from 78. Yield 10% as an oil. 1H NMR (CDCl3): δ 3.83 (s, 6H), 3.87 (s, 3H), 6.32–6.33 (m, 1H), 6.54 (s, 2H), 6.88 (dd, J = 1.7 and 4.0 Hz, 1H), 7.10–7.11 (m, 1H), 7.44–7.48 (m, 2H), 7.55–7.57 (m, 1H), 7.86–7.89 ppm (m, 2H). IR: ν 1630 cm−1. Anal. (C20H19NO4 (337.37)) C, H, N.
(3,4,5-Trimethoxyphenyl)(1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-2-yl)methanone (59)
Obtained as 56 from 78. Yield 13%, mp 130–135 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.86 (s, 6H), 3.90 (s, 3H), 3.92 (s, 6H), 3.95 (s, 3H), 6.35–3.38 (m, 1H), 6.56 (s, 2H), 6.95–6.97 (m, 1H), 7.13–7.15 (m, 1H), 7.20 ppm (s, 2H). IR: ν 1634 cm−1. Anal. (C23H25NO7 (337.37)) C, H, N.
(3-Hydroxy-4-methoxyphenyl)(1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-2-yl)methanone (60)
Obtained as 16 from 78 and 81. Yield 25%, mp 140–147 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.84 (s, 6H), 3.89 (s, 3H), 3.99 (S, 3H), 5.90 (broad s, disappeared on treatment with D2O, 1H), 6.33–6.34 (m, 1H), 6.54 (s, 2H), 6.90–6.94 (m, 2H), 7.10–7.11 (m, 1H), 7.50–7.53 ppm (m, 2H). IR: ν 1623, 2921, 3436 cm−1. Anal. (C21H21NO6 (383.39)) C, H, N.
(1H-Pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (9)
A solution of 55 (0.24 g, 0.6 mmol), 2 N NaOH (1.8 mL), and methanol (5.3 mL) was heated at reflux for 3 h. After cooling, the mixture was made acidic with 1 N HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl aceate:n-hexane = 3:2 as eluent) to give 9 (0.13 g, 85%), mp 155–160 °C (from ethanol/n-hexane) 1H NMR (CDCl3): δ 3.91 (s, 6H), 3.94 (s, 3H), 6.78–6.79 (m, 1H), 6.87–6.88 (m, 1H), 7.15 (s, 2H), 7.42–7.43 (m, 1H), 8.88 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1603, 3204 cm−1. Anal. (C14H15NO4 (261.27)) C, H, N.
(4-Phenyl-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (61)
A solution of 82 (0.50 g, 0.0017 mol) and TosMIC (0.33 g, 0.0017 mol) in anhydrous DMSO/Et2O (1:2, 15.0 mL) was added dropwise to a well-stirred suspension of NaH (0.33 g, 0.0076 mol; 55% in mineral oil) in anhydrous Et2O under an Ar stream. The reaction mixture was stirred at 25 °C for 15 min, diluted with water, and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:1 as eluent) to furnish 61 (0.43 g, 75%), mp 190 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.81 (s, 6H), 3.88 (s, 3H), 6.93 (m, 1H), 7.10 (s, 2H), 7.16–7.20 (m, 1H), 7.23–7.25 (m, 2H), 7.29–7.31 (m, 1H), 7.33–7.36 (m, 2H), 8.83 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1605, 3247 cm−1. Anal. (C20H19NO4 (337.37)) C, H, N.
Phenyl(4-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl)methanone (62)
Synthesized as 61 from 83. Yield 38%, mp 180 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.81 (s, 6H), 3.84 (s, 3H), 6.64 (s, 2H), 6.93–6.94 (m, 1H), 7.27–7.28 (m, 1H), 7.37 (t, J = 7.3 Hz, 2H), 7.48 (t, J = 8.0 Hz, 1H), 7.81–7.83 (m, 2H), 8.81 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1617, 3285 cm−1. Anal. (C20H19NO4 (337.37)) C, H, N.
General Procedure for the Preparation of Compounds 21–23 and 46
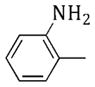
Example: (1-(2-Aminophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (21)
A mixture of 18 (0.2 g, 0.52 mmol) and SnCl2·2H2O (0.58 g, 0.0026 mol) in ethyl acetate was heated at reflux for 3 h. After cooling, the reaction mixture was made basic with a saturated aqueous solution of NaHCO3 and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:1 as eluent) to give 21 (0.12 g, 65%), mp 110–115 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.75 (s, 3H), 3.85 (s, 6H), 5.11 (broad s, disappeared on treatment with D2O, 2H), 6.63–6.65 (m, 1H), 6.76–6.77 (m, 1H), 6.87 (d, J = 7.04 Hz, 1H), 7.06 (t, J = 2.7 Hz, 1H), 7.10–7.12 (m, 2H), 7.17 (s, 2H), 7.55 ppm (t, J = 1.8 Hz, 1H). IR: ν 1615, 2990, 3126, 3324, 3445 cm−1. Anal. (C20H20N2O4 (352.38)) C, H, N.
(1-(3-Aminophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (22)
Synthesized as 21 from 19. Yield 60%, mp 160 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.86 (s, 6H), 5.36 (broad s, disappeared on treatment with D2O, 2H), 6.53–6.56 (m, 1H), 6.75–6.80 (m, 3H), 7.10–7.14 (m, 3H), 7.36–7.37 (m, 1H), 7.79 ppm (t, J = 1.9 Hz, 1H). IR: ν 1615, 2939, 3363 cm−1. Anal. (C20H20N2O4 (352.38)) C, H, N.
(1-(4-Aminophenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (23)
Synthesized as 21 from 20. Yield 58%, mp 55–60 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.85 (s, 6H), 5.26 (broad s, disappeared on treatment with D2O, 2H), 6.64 (d, J = 8.6 Hz, 2H), 6.70–6.72 (m, 1H), 7.11 (s, 2H), 7.29–7.32 (m, 3H), 7.75–7.77 ppm (m, 1H). IR: ν 1639, 3367, 3461 cm−1. Anal. (C20H20N2O4 (352.38)) C, H, N.
(1-(3-Amino-4-methoxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (46)
Synthesized as 21 from 45. Yield 42%, mp 165–170 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.80 (s, 3H), 3.85 (s, 6H), 5.01 (broad s, disappeared on treatment with D2O, 2H), 6.73–6.79 (m, 2H), 6.85–6.88 (m, 1H), 7.11 (s, 2H), 7.29 (dd, J = 2.2 and 3.0 Hz, 1H), 7.29 (dd, J = 2.2 and 3.0 Hz, 1H), 7.72 ppm (t, J = 1.9 Hz, 1H). IR: ν 1617, 2959, 3360, 3458 cm−1. Anal. (C21H22N2O5 (382.41)) C, H, N.
General Procedure for the Preparation of Compounds 42–44
Example: (1-(2-Hydroxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (42)
A mixture of 38 (0.17 g, 0.43 mmol) and methanesulfonic acid (1.33 g, 0.9 mL; 0.014 mol) in chloroform (9.0 mL) was refluxed for 2.5 h under an Ar stream. After cooling, the mixture was poured into ice/water and extracted with chloroform. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:1 as eluent) to afford 42 (0.04 g, 35%), mp 160 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.86 (s, 6H), 66.71–6.73 (m, 1H), 6.93 (t, J = 7.4 Hz, 1H), 7.06 (d, J = 8.2 Hz, 1H), 7.14 (s, 2H), 7.20–7.23 (m, 2H), 7.39 (d, J = 7.7 Hz, 1H), 7.76–7.78 (m, 1H), 9.94 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1610, 3240 cm−1. Anal. (C20H19NO5 (353.37)) C, H, N.
(1-(3-Hydroxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (43)
Synthesized as 42 from 39. Yield 51%, mp 160–164 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.77 (s, 3H), 3.86 (s, 6H), 6.75–6.78 (m, 2H), 7.01–7.03 (m, 1H), 7.08–7.13 (m, 3H), 7.28 (t, J = 8.1 Hz, 1H), 7.43–7.46 (m, 1H), 7.91 (s, 1H), 9.88 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1596, 2619, 2937, 2988, 3136 cm−1. Anal. (C20H19NO5 (353.37)) C, H, N.
(1-(4-Hydroxyphenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)-methanone (44)
Synthesized as 42 from 40. Yield 28%, mp 200–204 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.86 (s, 6H), 6.73–6.75 (m, 1H), 6.85 (d, J = 8.8 Hz, 2H), 7.12 (s, 2H), 7.36 (t, J = 2.4 Hz, 1H), 7.48 (d, J = 8.8 Hz, 2H), 7.82–7.84 (m, 1H), 9.73 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1610, 3240 cm−1. Anal. (C20H19NO5 (353.37)) C, H, N.
(1-(3-(Methylamino)phenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (24) and (1-(3-(Dimethylamino)-phenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (26)
A mixture of 22 (0.15 g, 0.43 mmol), dimethyl sulfate (0.054 g, 0.04 mL, 0.43 mmol), and Na2CO3 (0.12 g, 0.0011 mol) in anhydrous acetone (5.0 mL) was stirred at 25 °C for 16 h under an Ar stream. After dilution with water, the mixture was extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:1 as eluent) to furnish 26 (0.05 g, 31%) as a slurry. 1H NMR (CDCl3): δ 3.01 (s, 6H), 3.92 (s, 6H), 3.94 (s, 3H), 6.68–6.70 (m, 2H), 6.72–6.75 (m, 1H), 6.84–6.86 (m, 1H), 7.11–7.12 (m, 1H), 7.18 (s, 2H), 7.27–7.32 (m, 1H), 7.67 ppm (t, J = 1.8 Hz, 1H). IR: ν 1633 cm−1. Anal. (C22H24N2O4 (380.44)) C, H, N. Further elution with the same eluent furnished 24 (0.05 g, 31%) as a slurry. 1H NMR (DMSO-d6): δ 2.70 (s, 3H), 3.74 (s, 3H), 6.84 (s, 6H), 5.93 (broad s, disappeared after treatment with D2O, 1H), 6.51 (d, J = 8.6 Hz, 1H), 6.70–6.79 (m, 3H), 7.12 (s, 2H), 7.14–7.18 (m, 1H), 7.42 (s, 1H), 7.89 ppm (s, 1H). IR: ν 1610, 3937, 3396 cm−1. Anal. (C21H22N2O4 (366.41)) C, H, N.
(1-(3-(Isopropylamino)phenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (25)
NaCNBH3 (0.053 g, 0.85 mmol) was added to an ice-cooled mixture of 22 (0.25 g, 0.7 mmol) and acetone (0.041 g, 0.052 mL, 0.7 mmol) in methanol/THF (1:1, 8.9 mL) containing 6 N HCl/methanol (1:1, 0.12 mL). The reaction mixture was stirred at 25 °C for 12 h, made basic with a saturated aqueous solution of K2CO3, and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:1 as eluent) to furnish 25 (0.16 g, 57%) as a slurry. 1H NMR (CDCl3): δ 1.24 (s, 3H), 1.25 (s, 3H), 3.67–3.70 (m, 2H; one proton disappeared after treatment with D2O), 3.92 (s, 6H), 3.93 (s, 3H), 6.52–6.58 (m, 2H), 6.67–6.71 (m, 1H), 6.83–6.84 (m, 1H), 7.08–7.10 (m, 1H), 7.17 (s, 2H), 7.25 (t, J = 8.0 Hz, 1H), 7.63 ppm (t, J = 1.8 Hz, 1H). IR: ν 1610, 2965, 3368 cm−1. Anal. (C23H26N2O4 (394.46)) C, H, N.
1-(3-(3-(3,4,5-Trimethoxybenzoyl)-1H-pyrrol-1-yl)phenyl)-guanidine Hydrochloride (27)
Cyanamide (0.18 g, 0.0044 mol) was added in small portions with stirring to a solution of 22 (0.25 g, 0.71 mmol) in ethanol (5.0 mL) containing 3.3 N HCl (0.29 mL). The reaction mixture was heated at 50 °C for 48 h and cooled at 0 °C. The resulting suspension was filtered and the solid collected to give 27 (0.14 g, 45%), mp 196–200 °C (from ethanol). 1H NMR (DMSO-d6/D2O): δ 3.74 (s, 3H), 3.83 (s, 6H), 6.79 (s, 1H), 7.05–7.10 (m, 3H), 7.33–7.45 (m, 4H), 7.91 ppm (s, 1H). IR: ν 1642, 2590, 2841, 3200 cm−1. Anal. (C21H22N2O4·HCl (430.08)) C, H, N.
N-(3-(3-(3,4,5-Trimethoxybenzoyl)-1H-pyrrol-1-yl)phenyl)-methanesulfonamide (28)
To a solution of 22 (0.050 g, 0.14 mmol) and triethylamine (0.016 g, 0.02 mL, 0.16 mmol) in anhydrous THF (5.0 mL) was added dropwise a solution of methansulfonyl chloride (0.018 g, 0.01 mL, 0.16 mmol) in the same solvent (5.0 mL). The reaction mixture was stirred at 25 °C for 2 h, diluted with water, and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, chloroform:ethanol = 97:3 as eluent) to furnish 28 (0.03 g, 49%) as an oil. 1H NMR (CDCl3): δ 3.00 (s, 3H), 3.93 (s, 6H), 3.95 (s, 3H), 6.86–688 (m, 1H), 7.11 (t, J = 2.7 Hz, 1H), 7.19 (s, 2H), 7.21–7.28 (m, 3H), 7.42–7.49 (m, 2H; one proton disappeared after treatment with D2O), 7.69 ppm (t, J = 1.8 Hz, 1H). IR: ν 1606, 2853, 2924, 3237 cm−1. Anal. (C21H22N2O5 (382.41)) C, H, N.
General Procedure for the Preparation of compounds 29, 63–78
Example: (1-(3-(1H-Pyrrol-1-yl)phenyl)-1H-pyrrol-3-yl)-(3,4,5-trimethoxyphenyl)methanone (29)
A mixture of 22 (0.25 g, 0.7 mmol) and 2,5-dimethoxytetrahydrofuran (0.092 g, 0.09 mL, 0.7 mmol) in glacial acetic acid (0.19 mL) was heated at 80 °C for 2 h. After cooling, the solvent was evaporated, and the residue was made basic with a saturated aqueous solution of NaHCO3 and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:2 as eluent) to furnish 29 (0.23 g, 82%) as an oil. 1H NMR (DMSO-d6): δ 3.77 (s, 3H), 3.83 (s, 6H), 6.29–6.31 (m, 2H), 6.80–6.82 (m, 1H), 7.16 (s, 2H), 7.56–7.60 (m, 5H), 7.66–7.68 (m, 1H), 7.89–7.91 (m, 1H), 8.22–8.24 ppm (m, 1H). IR: ν 1632 cm−1. Anal. (C24H22N2O4 (402.44)) C, H, N.
1-(2-Chlorophenyl)-1H-pyrrole (63)
Synthesized as 29 from 2-chloroaniline. Yield 72% as an oil.23
1-(3-Chlorophenyl)-1H-pyrrole (64)
Synthesized as 29 from 3-chloroaniline. Yield 72%, mp 50–51 °C (from petroleum ether). Lit.24 50.5–51.5 °C.
1-(3-Nitrophenyl)-1H-pyrrole (65)
Synthesized as 29 from 3-nitroaniline. Yield 72%, mp 72–73 °C (from petroleum ether). Lit.24 75–76 °C.
1-(2-Tolyl)-1H-pyrrole (66)
Synthesized as 29 from o-toluidine. Yield 46% as an oil. Lit.24
1-(3-Tolyl)-1H-pyrrole (67)
Synthesized as 29 from m-toluidine. Yield 54% as an oil. Lit.24
1-(4-Tolyl)-1H-pyrrole (68)
Synthesized as 29 from p-toluidine. Yield 55%, mp 80–81 °C (from ethanol). Lit.24 82.5–83.5 °C.
1-(2-Methoxyphenyl)-1H-pyrrole (69)
Synthesized as 29 from 2-methoxyaniline. Yield 40% as an oil. Lit.25
1-(3-Methoxyphenyl)-1H-pyrrole (70)
Synthesized as 29 from 3-methoxyaniline. Yield 48% as an oil. Lit.24
1-(2-Isopropoxyphenyl)-1H-pyrrole (71)
Synthesized as 29 from 2-isopropoxyaniline. Yield 50% as an oil. 1H NMR (DMSO-d6): δ 1.22 (s, 3H), 1.24 (s, 3H), 5.52–4.58 (m, 1H), 6.17–6.19 (m, 2H), 7.00–7.07 (m, 3H), 7.19 (d, J = 7.4 Hz, 1H), 7.25–7.33 ppm (m, 2H).
1-(3-Isopropoxyphenyl)-1H-pyrrole (72)
Synthesized as 29 from 3-isopropoxyaniline. Yield 38% as an oil. 1H NMR (DMSO-d6): δ 1.63 (s, 3H), 1.65 (s, 3H), 5.04–5.10 (m, 1H), 6.58–6.60 (m, 2H), 7.14 (dd, J = 2.0 and 8.3 Hz, 1H), 7.43–7.46 (m, 2H), 7.66–7.72 ppm (m, 3H).
1-(4-Isopropoxyphenyl)-1H-pyrrole (73)
Synthesized as 29 from 4-isopropoxyaniline. Yield 53% as an oil. 1H NMR (DMSO-d6): δ 1.27 (s, 3H); 1.28 (s, 3H), 4.57–4.66 (m, 1H), 6.22 (t, J = 2.1 Hz, 2H), 6.97–7.00 (m, 2H), 7.22 (t, J = 2.1 Hz, 2H), 7.43–7.46 ppm (m, 2H).
1-(3-(Benzyloxy)phenyl)-1H-pyrrole (74)
Synthesized as 29 from 3-benzyloxyaniline. Yield 34%, mp 40 °C (from toluene). 1H NMR (DMSO-d6): δ 5.19 (s, 2H), 6.26 (t, J = 2.2 Hz, 2H), 6.90 (dd, J = 2.2 and 8.2 Hz, 1H), 7.14–7.16 (m, 1H), 7.22 (t, J = 2.2 Hz, 1H), 7.33–7.43 (m, 6H), 7.47–7.49 ppm (m, 2H).
1-(4-Methoxy-3-nitrophenyl)-1H-pyrrole (75)
Synthesized as 29 from 4-methoxy-3-nitroaniline. Yield 73%, mp 80 °C (from toluene). 1H NMR (DMSO-d6): δ 3.96 (s, 3H), 6.28 (t, J = 2.2 Hz, 2H), 7.40 (t, J = 2.2 Hz, 2H), 7.44 (d, J = 9.2 Hz, 1H), 7.89 (dd, J = 2.89 and 9.1 Hz, 1H), 8.10 ppm (d, J = 2.9 Hz, 1H).
1-(3-Isopropoxy-4-methoxyphenyl)-1H-pyrrole (76)
Synthesized as 29 from 3-isopropoxy-4-methoxyaniline. Yield 75%, mp 35–37 °C (from petroleum ether). 1H NMR (CDCl3): δ 1.40 (s, 3H), 1.42 (s, 3H), 3.90 (s, 3H), 4.55–4.63 (m, 1H), 6.33–6.36 (m, 2H), 6.91–7.01 (m, 3H), 7.27–7.28 ppm (m, 2H).
1-(3,4-Dimethoxyphenyl)-1H-pyrrole (77)
Synthesized as 29 from 3,4-dimethoxyaniline. Yield 50%, mp 60 °C (from petroleum ether). Lit.26
1-(3-Isopropoxy-4-methoxyphenyl)-1H-pyrrole (78)
Synthesized as 29 from 1-(3,4,5-trimethoxyphenyl)-1H-pyrrole. Yield 55%, mp 89–91 °C (from toluene). 1H NMR (DMSO-d6): δ 3.64 (s, 3H), 3.84 (s, 6H), 6.23 (s, 2H), 6.80 (s, 2H), 7.35 ppm (s, 2H).
(1-(3-(Pyrrolidin-1-yl)phenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (30)
A mixture of 22 (0.10 g, 0.28 mmol), 1,4-dibromobutane (0.065 g, 0.03 mL, 0.3 mmol), and K2CO3 (0.042 g, 0.3 mmol) in water (1.0 mL) was placed into the MW cavity (closed vessel mode, Pmax = 250 psi). A starting MW irradiation of 100 W was used, the temperature being ramped from 25 to 110 °C while stirring rapidly. Once 110 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 20 min, diluted with water, and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:1 as eluent) to afford 30 (0.04 g, 35%) as a slurry. 1H NMR (CDCl3): δ 2.03–2.06 (m, 4H), 3.31–3.34 (m, 4H), 3.93 (s, 6H), 3.94 (s, 3H), 6.52–6.54 (m, 2H), 6.66–6.68 (m, 1H), 6.83–6.85 (m, 1H), 7.11–7.13 (m, 1H), 7.18 (s, 2H), 7.26–7.30 (m, 1H), 7.67 ppm (t, J = 2.0 Hz, 1H). IR: ν 1631 cm−1. Anal. (C24H26N2O4 (406.47)) C, H, N.
(1-(3-(Piperidin-1-yl)phenyl)-1H-pyrrol-3-yl)(3,4,5-trimethoxyphenyl)methanone (31)
Synthesized as 30 from 1,5-dibromopentane. Yield 57% as a slurry. 1H NMR (CDCl3): δ 1.59–1.65 (m, 2H), 1.69–1.75 (m, 4H), 3.23–3.25 (m, 4H), 3.93 (s, 6H), 3.94 (s, 3H), 6.82–6.85 (m, 2H), 6.88–6.92 (m, 2H), 7.09–7.10 (m, 1H), 7.17 (s, 2H), 7.31 (t, J = 8.0 Hz, 1H), 7.65 ppm (t, J = 2.0 Hz, 1H). IR: ν 1627 cm−1. Anal. (C25H28N2O4 (420.50)) C, H, N.
Methyl 3-Isopropoxy-4-methoxybenzoate (79)
A mixture of methyl 3-hydroxy-4-methoxybenzoate (0.5 g, 0.0027 mol), anhydrous K2CO3, and 2-iodopropane (1.04 g, 0.61 mL, 0,0061 mol) in anhydrous DMF was heated at 50 °C for 3 h under an Ar stream. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:1) to furnish 79 (0.47 g, 76%), mp 50–55 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 1.38 (s, 3H), 1.39 (s, 3H), 3.88 (s, 3H), 3.91 (s, 3H), 4.56–4.64 (m, 1H), 6.88 (d, J = 8.5 Hz, 1H), 7.56 (d, J = 2.0 Hz, 1H), 7.65 ppm (dd, J = 2.0 and 8.4 Hz, 1H). IR: ν 1710 cm−1.
3-Isopropoxy-4-methoxybenzoic Acid (80)
A mixture of 79 (1.57 g, 0.0070 mol) and LiOH·H2O in THF/H2O (1:1, 70 mL) was stirred at 25 °C for 12 h. The reaction mixture was made acidic with 1 N HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by column chromatography (silica gel, ethyl acetate:n-hexane = 1:1 as eluent) to furnish 80 (1.14 g, 77%), mp 130–135 °C (from ethanol). Lit.27
3-Isopropoxy-4-methoxybenzoyl Chloride (81)
A mixture of 80 (1.0, 0.0052 mol) and SOCl2 (0.74 g, 0.46 mL, 0.0062 mol) was heated at reflux for 1.5 h under an Ar stream. Evaporation of the excess of SOCl2 left crude 77, which was used without further purification.
General Procedure for the Preparation of Compounds 82 and 83
Example: 3-Phenyl-1-(3,4,5-trimethoxyphenyl)-trans-prop-2-en-1-one (82)
A mixture of 1-(3,4,5-trimethoxyphenyl)ethanone (1.0 g, 0.0048 mol), benzaldehyde (0.51 g, 0.49 mL, 0.0048 mol), and NaOH (0.19 g, 0.0048 mol) in ethanol (19.2 mL) was stirred at 25 °C for 24 h. After filtering, the resulting solid was collected to provide 82 (0.58 g, 41%), mp 78–80 °C (from ethanol/n-hexane). Lit.28
1-Phenyl-3-(3,4,5-trimethoxyphenyl)-trans-prop-2-en-1-one (83)
Synthesized as 82 from 3,4,5-trimethoxybenzaldehyde and aceto-phenone. Yield 67%, mp 130–135 °C (from ethanol/n-hexane), Lit.29 135–136 °C.
Molecular Modeling
All molecular modeling studies were performed on a MacPro dual 2.66 GHz Xeon running Ubuntu 12.04 LTS. The tubulin structure was downloaded from the PDB code 3HKD.13 Hydrogen atoms were added to the protein, using Molecular Operating Environment (MOE) 2010,30 and the structures were energy minimized, keeping all the heavy atoms fixed until a rmsd gradient of 0.05 kcal mol−1 Å−1 was reached. Ligand structures were built with MOE and minimized using the MM-FF94x force field until a rmsd gradient of 0.05 kcal mol−1 Å−1 was reached. The Smo structure (PDB code 4JKV)31 was prepared in the same manner. The docking simulations were performed using Glide,32 PLANTS,33 and Auto-dock4.0.34 The pharmacophore model was created by the Pharmacophore query editor of MOE. The images presented here were created with Pymol.35
Biology
Tubulin Assembly
The reaction mixtures contained 0.8 M monosodium glutamate (pH 6.6 with HCl in a 2 M stock solution), 10 μM tubulin, and varying concentrations of drug. Following a 15 min preincubation at 30 °C, samples were chilled on ice, GTP to 0.4 mM was added, and turbidity development was followed at 350 nm in a temperature controlled recording spectrophotometer for 20 min at 30 °C. Extent of reaction was measured. Full experimental details were previously reported.36
[3H]Colchicine Binding Assay
The reaction mixtures contained 1.0 μM tubulin, 5.0 μM [3H]colchicine, and 5.0 μM inhibitor and were incubated 10 min at 37 °C. Complete details were described previously.37
Cell Cultures
Cell lines were obtained from the American Tissue Culture Collection (ATCC), unless otherwise specified. Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37 °C with 5% CO2. In all experiments, 3 × 105 cells were plated in 9 cm2 dishes and treated with a solution of test compound in DMSO (0.1% final concentration) at the indicated concentrations. HeLa, HT-29, and A549 cells were grown at 37 °C in DMEM containing 10 mM glucose supplemented with 10% FBS, 100 units/mL of penicillin and streptomycin, and 2 mM glutamine. At the onset of each experiment, cells were placed in fresh medium and cultured in the presence of test compound at 0.01–25 μM. HCT116, HCT15, Messa, and Messa/Dx5 were seeded into 96-well plates (Corning Inc., Costar) at a density of 2 × 103 cells/well in a volume of 50 μL of the appropriate tissue culture medium. Test compounds were added at different concentrations for the indicated incubation time at 37 °C in the presence of 5% CO2. The MCF-7/cas-3 cell line reconstituted with caspase-3 was a kind gift from Dr. Christopher J. Froelich (North Shore University, Health Systems Research Institute, Evanston, IL, USA). PC-3 cells were grown in DMEM containing 4.5 g/L of glucose, supplemented with 10% FBS, 100 units/mL of penicillin and streptomycin, and 2 mM glutamine. RD and HepG2 cells were grown in DMEM containing 1 g/L of glucose supplemented with 10% FBS, 100 units/mL of penicillin and streptomycin, and 2 mM glutamine and pyruvate.
Cell Viability Assays
The methodology for the evaluation of the growth of human MCF-7 breast carcinoma, OVCAR-8, and NCI/ ADR-RES cells, obtained from the National Cancer Institute drug screening laboratory, was previously described, except that cells were grown for 96 h for IC50 determinations.38 The drug screening laboratory also supplied the K562 cells, which were grown in suspension culture in the same medium as the other cell lines. They were grown in 5 mL of medium for 20 h. For IC50 determinations, cell number was the parameter quantitated, using a Beckman Coulter model Z-1 Coulter particle counter. For MI studies, cells were harvested by centrifugation (2000 rpm for 2 min for all centrifugations). The cells were washed in 10 mL of PBS and harvested by centrifugation. Each pellet was suspended in 0.5 mL of ice-cold half-strength PBS for 13 min (hypotonic solution to cause cells to swell to make nuclear morphology easier to evaluate), following which 15 mL of 0.5% ethanol–1.5% acetic acid was added to each tube to fix the cells. After at least 30 min, the cells were harvested by centrifugation. Each pellet was suspended in 0.5 mL of 75% ethanol–25% acetic acid. One drop of each suspension was placed on separate microscope slides. The slides were air died, stained with Giemsa for 10 min, washed with water, air-dried again, and examined under a light microscope. At least 100 cells were counted for each data point.
Cell viability of Hela, HT-29, and A549 cells was determined using the MTT colorimetric assay, whereby the mitochondrial dehydrogenase in viable cells transforms the yellow MTT reagent into a soluble blue formazan dye. Cells were seeded into 96-well plates to a density of 7 × 103/100 μL well. After 24 h of growth to allow attachment of cells to the wells, test compounds were added at 0.01–25 μM. After 48 h of growth and removal of the culture medium, 100 μL/well medium containing 1 mg/mL MTT was added. Cell cultures were further incubated at 37 °C for 2 h in the dark. The solutions were then gently aspirated from each well, and the formazan crystals within the cells were dissolved in DMSO (100 μL). Optical densities were read at 550 nm using a Multiskan Spectrum Thermo Electron Corporation reader. The results were expressed as % relative to vehicle-treated control (0.5% DMSO was added to untreated cells), and the IC50 values were determined by linear and polynomial regression analysis. Experiments were performed in triplicate.
Growth inhibition of HCT116, HCT15, Messa, and Messa/Dx5 tumor cell lines was evaluated using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI). The cells in exponential growth were incubated for 72 h at different concentrations of the inhibitors. Then an equivalent of the CellTiter-Glo reagent was added, and the solution was mixed for 2 min in order to induce cell lysis. Luminescence was recorded after an additional time of 10 min. IC50 values were calculated using nonlinear regression analysis (GraphPad Prism statistics software).
Cell viability of PC-3, RD, and HepG2 cells was determined using the MTT colorimetric assay. PC-3 and RD cells were seeded into 24-well plates to a density of 95 × 103/100 μL well. HepG2 cells were seeded into 24-well plates to a density of 120 × 103/100 μL well. After 24 h of growth to allow attachment of cells to the wells, test compounds were added at 0.01–25 μM. After 48 h of growth and removal of the culture medium, 500 μL/well of PBS containing 500 μM MTT was added. Cell cultures were further incubated at 37 °C for 2 h in the dark. The solutions were then gently aspirated from each well, and the formazan crystals within the cells were dissolved in propan-2-ol and 0.04 N HCl (200 μL). Optical densities were red at 550 nm using a Multiskan Spectrum Thermo Electron Corporation reader. The results were expressed as % relative to vehicle-treated control (0.1% DMSO), and IC50 values were calculated by nonlinear regression analysis (GraphPad Prism statistics software). Experiments were performed in triplicate.
Immunofluorescence and Light Microscopy
Cells were seeded in culture dishes containing sterile coverslips coated with poly-L-lysine. At the end of the indicated treatments, the cells were fixed in 3.7% paraformaldehyde, permeabilized in 0.1% Triton X-100 in PBS saline, and processed for IF using primary antibodies to lamin B1 (ab16048, Abcam, Cambridge, UK), active caspase-3 (ab13847, Abcam), or α-tubulin (clone B5.1.2, Sigma-Aldrich). Secondary antibodies were either conjugated to FITC (Jackson Immunoresearch Laboratories) or to rhodamine (Santa Cruz Biotechnology). The DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI). Samples were analyzed under a Nikon Eclipse 90i microscope equipped with a Qicam Fast 1394 CCD camera, and images were acquired using NIS-Elements AR 3.2 (Nikon). Unfixed cell cultures were observed under an inverted Nikon TE300 microscope with a 10× objective, and images were acquired using the ACT-1 software and a DMX1200 CCD (resolution 1280 × 1024 pixels).
Flow Cytometric Analysis
Cell cycle distribution was analyzed after incubation with PI (Sigma-Aldrich), whereas apoptosis was analyzed with annexin V-FITC (Immunological Sciences, IK-11120 or Becton Dickinson and Co., Milan, Italy) staining alone or with annexin V-FITC in combination with PI. Cell samples were analyzed in a Coulter Epics XL cytofluorimeter (Beckman Coulter) equipped with EXPO 32 ADC software. At least 105 cells per sample were acquired and analyzed using BD CellQuest Pro Software.
Gli-Dependent Luciferase Reporter Assay
The luciferase assay was performed in Shh-Light II (Shh-L II) cells, stably incorporating a Gli-responsive luciferase reporter and the pRL-TK Renilla (normalization control) for 48 h with SAG (200 nM) and the studied compounds. Luciferase and renilla activity were assayed with a dual-luciferase assay system according to the manufacturer’s instruction (Promega, Madison, WI, USA). Results are expressed as luciferase/renilla ratios and represent the mean ± SD of three experiments, each performed in triplicate. ARAP 18 and 23 did not modify the renilla activity.
Pharmacokinetic studies
CD-1 mice (27 + 27), male, were used for the pharmacokinetic studies. The IV bolus was injected through the caudal vein at a dose of 5 mg/kg; the PO gavage was at a dose of 15 mg/kg. In a Sirocco filter plate (Waters), 100 μL of plasma were added to 300 μL of MeOH spiked with 10 μL of IS (DDP-10228 1 μg/mL). The plate was shaken for 15 min and filtered under vacuum (15 mmHg) for 5 min. The samples were analyzed by LC/MS/MS: Premiere XE; eluent, water (solution A), acetonitrile with 0.1% HCOOH (solution B); gradient from 2% B to 80% B up to 0.8 min, then 100% B up to 1.3 min, then 0.5 min 100% B; flow rate 0.5 mL/ min; column Acquity BEH C18, 2.1 mm × 50 mm, 1.7 μm injection; volume 5 μL; temp column 50 °C. ESI positive, extractor 4 V; capillary 3.20 kV; temp source 115 °C; temp desolv 450 °C. The experiments were carried out in agreement with Italian Law (DL 116/92).
Caco-2 Cell Permeability
Caco-2 cells (ECACC) were cultured in DMEM, 10% FBS, 1% NEAA, 10 mM Hepes buffer, 50 U penicillin, and 50 μg/mL streptomycin and split at confluence by trypsinization. The cells (200000 cells/well) were seeded on Millicell 24-well cell culture plates. After 24 h at 37 °C in a humidified and 5% CO2 containing atmosphere, the medium was exchanged for enterocyte differentiation medium with additives (Becton Dickinson), which allows Caco-2 cells to establish within 3 days a differentiated enterocyte monolayer. TEER, measured with a Millicell-ERS (Millipore, Corp), must be >1000 Ω. Transport across the Caco-2 monolayer was determined by adding a 10 μM solution of test compound in DMEM (1% final concentration of DMSO) to the side from which permeability was to be determined. The A → B transport across Caco-2 monolayer cells was determined by adding the test compound to the apical side at pH 6.5. After 2 h, the basolateral side solution at pH 7.4 and the apical and starting solutions were analyzed by LC–MS/MS. In the B → A experiment, the test compound was added to the basolateral side and collected on the apical side. Monolayer integrity was assessed with a lucifer yellow assay at the end of each experiment. The apparent permeability (Papp) was calculated as J/C0 ratio, where J is the flux (dX/dt per A) and C0 is the donor concentration (μM) at t = 0; dX/dt is the change in mass (X, nmol) per time (t, s), and A is the filter surface area (cm2).
CYP450 Inhibition
Cytochrome P450 inhibition experiments were carried out according to the manufacturer’s instructions (BD Biosciences, Franklin Lakes, NJ, U.S.). Compound 22 was dissolved in a 96-well plate at a 10 μM final concentration in potassium phosphate buffer (pH 7.4) containing an NADPH regenerating system. For all enzyme/substrate pairs, the final cofactor concentrations were 1.3 mM NADP+, 3.3 mM glucose 6-phosphate, and 0.4 U/mL glucose 6-phosphate dehydrogenase. The reaction was initiated by the addition of specific isoenzymes (Supersomes, Gentest) and substrates at 37 °C. Furafylline (for CYP1A2, 100 mM), sulfaphenazole (for CYP2C9, 10 mM), tranylcypromine (for CYP2C19, 500 μM), quinidine (for CYP2D6, 0.5 mM), and ketoconazole (for CYP3A4, 1.66 mM) were employed as control inhibitors in one-third serial dilution. Incubations were carried out for 15 min (0.5 pmol of CYP1A2, 5 mM 3-cyano-7-ethoxycoumarin), 30 min (0.5 pmol of CYP2C19, 25 mM 3-cyano-7-ethoxycoumarin, 1 pmol of CYP3A4, 50 mM 7-benzyloxy-4-(trifluoromethyl)coumarin; 1.5 pmol of CYP2D6, 1.5 mM 3-[2-(N,N-diethyl-N-methylamino)-ethyl]-7-methoxy-4-methylcoumarin), or 45 min (1 pmol of CYP2C9, 75 mM 7-methoxy-4-(trifluoromethyl)-coumarin). The reaction was then quenched by adding 75 mL of a mixture containing 80% MeCN and 20% Tris base (0.5 m), and plates were read on a fluorimeter at the appropriate emission/excitation wavelengths. The percentage inhibition was calculated relative to enzyme samples without inhibitors.
Supplementary Material
Acknowledgments
This research was supported by grants PRIN 2010–2011 (2010W7YRLZ_001 and 2010W7YRLZ_001006), PRIN 2012–2013 (2012C5YJSK002), AIRC IG 14535, AIRC IG14723, Bando Futuro in Ricerca 2010 (grant no. RBFR10ZJQT), and Progetti di Ricerca di Università, Sapienza Università di Roma (grant no. C26H135FL5). We are grateful to Giulia Guarguaglini and Italia Anna Asteriti for microscopy analysis. We also thank Professor Claudia Piccoli and to Dr. Matteo Landriscina, University of Foggia, Italy, for providing cell lines.
ABBREVIATIONS USED
- MT
microtubule
- CSA4
combretastatin A-4
- VLB
vinblastine
- VCR
vincristine
- VRB
vinorelbine
- PTX
paclitaxel
- ARAP
3-aroyl-1-arylpyrrole
- TMP
trimethoxyphenyl
- MW
microwave
- DMSO
dimethyl sulfoxide
- TosMIC
p-toluenesulfonylmethyl isocyanide
- SAR
structure—activity relationship
- TPI
tubulin polymerization inhibition
- MI
mitotic index
- DOX
doxorubucin
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- DAPI
4′,6-diamidino-2-phenylindole
- PI
propidium iodide
- DMEM
Dulbecco’s Modified Eagle Medium
- FBS
fetal bovine serum
- PBS
phosphate-buffered saline
- IF
immunofluorescence
Footnotes
The authors declare no competing financial interest.
Binding mode of compound 4. Inhibition of tubulin polymerization and growth of MCF-7 cells by 56–62. Inhibition of growth of the Messa and Messa/Dx5 cells by 12–14, 19, 23, 25, 29, 30, 33, 34, and 45–49. Pharmacophore model for the Smo receptor. Kinetic profile of 22 in plasma after IV infusion or PO adiministration in the mouse. Comparison of the kinetic profiles of 22 after IV infusion or PO administration in mouse plasma. Purity HPLC analysis of compound 4. Elemental analysis of compounds 4 and 9–62. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cancer; Fact Sheet No. 297. World Health Organization; Geneva: Jan, 2013. http://www.who.int/mediacentre/factsheets/fs297/en/ [Google Scholar]
- 2.(a) Bhattacharyya B, Panda D, Gupta S, Banerjee M. Anti-mitotic acticity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev. 2008;28:155–183. doi: 10.1002/med.20097. [DOI] [PubMed] [Google Scholar]; (b) Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 3.Lin CM, Ho HH, Pettit GR, Hamel E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding to colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 4.(a) Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]; (b) Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. The binding mode of epothilone A on alpha,beta-tubulin by electron crystallography. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- 5.Buey RM, Calvo E, Barasoain I, Pineda O, Edler MC, Matesanz R, Cerezo G, Vanderwal CD, Day BW, Sorensen EJ, Lopez JA, Andreu JM, Hamel E, Diaz JF. Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nature Chem Biol. 2007;3:117–125. doi: 10.1038/nchembio853. [DOI] [PubMed] [Google Scholar]
- 6.Beckers T, Mahboobi S. Natural, semisynthetic and synthetic microtubule inhibitors for cancer therapy. Drugs Future. 2003;28:767–785. [Google Scholar]
- 7.(a) Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nature Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]; (b) Teicher BA. Newer cytotoxic agents: attacking cancer broadly. Clin Cancer Res. 2008;14:1610–1617. doi: 10.1158/1078-0432.CCR-07-2249. [DOI] [PubMed] [Google Scholar]
- 8.Marzo I, Naval J. Antimitotic drugs in cancer chemotherapy. Promises and pitfall. Biochem Pharmacol. 2013;86:703–710. doi: 10.1016/j.bcp.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt M, Bastians H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Update. 2007;10:162–181. doi: 10.1016/j.drup.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 10.(a) La Regina G, Sarkar T, Bai R, Edler MC, Saletti R, Coluccia A, Piscitelli F, Minelli L, Gatti V, Mazzoccoli C, Palermo V, Mazzoni C, Falcone C, Scovassi AI, Giansanti V, Campiglia P, Porta A, Maresca B, Hamel E, Brancale A, Novellino E, Silvestri R. New arylthioindoles and related bioisosteres at the sulfur bridging group. 4. Synthesis, tubulin polymerization, cell growth inhibition, and molecular modeling studies. J Med Chem. 2009;52:7512–7527. doi: 10.1021/jm900016t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) La Regina G, Bai R, Rensen WM, Di Cesare E, Coluccia A, Piscitelli F, Famiglini V, Reggio A, Nalli M, Pelliccia S, Da Pozzo E, Costa B, Granata I, Porta A, Maresca B, Soriani A, Iannitto ML, Santoni A, Li J, Miranda Cona M, Chen F, Ni Y, Brancale A, Dondio G, Vultaggio S, Varasi M, Mercurio C, Martini C, Hamel E, Lavia L, Novellino E, Silvestri R. Towards highly potent cancer agents by modulating the C-2 group of the arylthioindole class of tubulin polymerization inhibitors. J Med Chem. 2013;56:123–149. doi: 10.1021/jm3013097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morice C, Wermuth CG. The Practice of Medicinal Chemistry. 3. Chapter 16 Elsevier Ltd; Burlington, VT: 2008. Ring Transformations. [Google Scholar]
- 12.Chen J, Wang Z, Li CM, Lu Y, Vaddady PK, Meibohm B, Dalton JT, Miller DD, Li W. Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicines binding site in tubulin as potential anticancer agents. J Med Chem. 2010;53:7414–7427. doi: 10.1021/jm100884b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dorleans A, Gigant B, Ravelli RBG, Mailliet P, Mikol V, Knossow M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc Natl Acad Sci U S A. 2009;106:13775–13779. doi: 10.1073/pnas.0904223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barbier P, Dorleans A, Devred F, Sanz L, Allegro D, Alfonso C, Knossow M, Peyrot V, Andreu JM. Stathmin and interfacial microtubule inhibitors recognize a naturally curved conformation of tubulin dimers. J Biol Chem. 2010;285:31672–31681. doi: 10.1074/jbc.M110.141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clauson-Kaas N, Tyle Z. Preparation of cis and trans 2,5-dimethoxy-2-(acetamidomethyl)-2,5-dihydrofuran, cis and trans 2,5-dimethoxy-2-(acetamidomethyl)-tetrahydrofuran and 1-phenyl-2-(acetamidomethyl)-pyrrole. Acta Chem Scand. 1952;6:667–670. [Google Scholar]
- 16.Massarotti A, Coluccia A, Silvestri R, Sorba G, Brancale A. The tubulin colchicine domain: a molecular modeling perspective. ChemMedChem. 2012;7:33–42. doi: 10.1002/cmdc.201100361. [DOI] [PubMed] [Google Scholar]
- 17.Rensen WM, Roscioli E, Tedeschi A, Mangiacasale R, Ciciarello M, Di Gioia SA, Lavia P. RanBP1 downregulation sensitizes cancer cells to taxol in a caspase-3-dependent manner. Oncogene. 2009;28:1748–1758. doi: 10.1038/onc.2009.24. [DOI] [PubMed] [Google Scholar]
- 18.Yang XH, Sladek TL, Liu X, Butler BR, Froelich CJ, Thor AD. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Res. 2001;61:348–354. [PubMed] [Google Scholar]
- 19.Peukert S, Miller-Moslin K. Small-molecule inhibitors of the hedgehog signaling pathway as cancer therapeutics. ChemMedChem. 2010;5:500–512. doi: 10.1002/cmdc.201000011. [DOI] [PubMed] [Google Scholar]
- 20.Manetti F, Faure H, Roudaut H, Gorojankina T, Traiffort E, Schoenfelder A, Mann A, Solinas A, Taddei M, Ruat M. Virtual screening-based discovery and mechanistic characterization of the acylthiourea MRT-10 family as Smoothened antagonists. Mol Pharmacol. 2010;78:658–665. doi: 10.1124/mol.110.065102. [DOI] [PubMed] [Google Scholar]
- 21.Solinas A, Faure H, Roudaut H, Traiffort E, Schoenfelder A, Mann M, Manetti F, Taddei M, Ruat M. Acylthiourea, acylurea, and acylguanidine derivatives with potent Hedgehog inhibiting activity. J Med Chem. 2012;55:1559–1571. doi: 10.1021/jm2013369. [DOI] [PubMed] [Google Scholar]
- 22.Massa S, Di Santo R, Artico M, Costi R, Apuzzo G, Simonetti G, Artico M. Novel in vitro highly active antifungal agents with pyrrole and imidazole moieties. Med Chem Res. 1992;2:148–153. [Google Scholar]
- 23.Biswanath D, Digambar BS, Boddu SK, Jayprakash NK. Novel approach for the synthesis of N-substituted pyrroles starting directly from nitro compounds in water. Synth Commun. 2012;42:548–553. [Google Scholar]
- 24.Chen W, Wang J. Synthesis of pyrrole derivatives from diallylamines by one-pot tandem ring-closing metathesis and metal-catalyzed oxidative dehydrogenation. Organometallics. 2013;32:1958–1963. [Google Scholar]
- 25.Prakash Reddy V, Vijay Kumar A, Rama Rao K. New strategy for the synthesis of N-aryl pyrroles: Cu-catalyzed C–N cross-coupling reaction of trans-4-hydroxy-l-proline with aryl halides. Tetrahedron Lett. 2011;52:777–780. [Google Scholar]
- 26.Ramesh K, Murthy SN, Nageswar YVD. Synthesis of N-substituted pyrroles under catalyst- and solvent-free conditions. Synth Commun. 2012;42:2471–2477. [Google Scholar]
- 27.Pregel MJ, Hirth BH, Kane JL, Qiao S, Gregory J, Cuff L. Preparation of tetrahydroisoquinoline derivatives for treating diseases mediated by protein trafficking or chloride channel activity. 20050176761 A1. US Patent US. 2005 Aug 11;
- 28.Hermange P, Gogsig TM, Lindhardt AT, Taaning RH, Skrydstrup T. Carbonylative Heck reactions using CO generated ex situ in a two-chamber system. Org Lett. 2011;9:2444–2447. doi: 10.1021/ol200686h. [DOI] [PubMed] [Google Scholar]
- 29.Ducki S, Rennison D, Woo M, Kendall A, Fournier Dit Chabert J, McGown AT, Lawrence NJ. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: Synthesis and biological evaluation of antivascular activity. Bioorg Med Chem. 2009;22:7698–7710. doi: 10.1016/j.bmc.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 30.Molecular Operating Environment (MOE 2010.10) Chemical Computing Group, Inc; Montreal, Quebec, Canada: 2010. http://www.chemcomp.com. [Google Scholar]
- 31.Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013;497:338–343. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glide, version 5.5. Schrödinger, LLC; New York: 2009. [Google Scholar]
- 33.Korb O, Stützle T, Exner TE. Empirical scoring functions for advanced protein–ligand docking with plants. J Chem Inf Model. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 34.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.PyMol Molecular Graphics System. www.pymol.org.
- 36.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 37.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure–activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol Pharmacol. 1998;35:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 38.Ruan S, Okcu MF, Pong RC, Andreeff M, Levin V, Hsieh JT, Zhang W. Attenuation of WAF1/Cip1 expression by an antisense adenovirus expression vector sensitizes glioblastoma cells to apoptosis induced by chemotherapeutic agents 1,3-bis(2-chloroethyl)-1-nitrosourea and cisplatin. Clin Cancer Res. 1999;5:197–202. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.