

Abstract
Networks of protein interactions execute many different intracellular pathways. Small molecules either synthesized within the cell or obtained from the external environment mediate many of these protein-protein interactions. The study of these small molecule–mediated protein-protein interactions is important in understanding abnormal signal transduction pathways in a variety of disorders, as well as in optimizing the process of drug development and validation. In this study, we evaluated the rapamycin-mediated interaction of the human proteins FK506-binding protein (FKBP12) rapamycin-binding domain (FRB) and FKBP12 by constructing a fusion of these proteins with a split-Renilla luciferase or a split enhanced green fluorescent protein (split-EGFP) such that complementation of the reporter fragments occurs in the presence of rapamycin. Different linker peptides in the fusion protein were evaluated for the efficient maintenance of complemented reporter activity. This system was studied in both cell culture and xenografts in living animals. We found that peptide linkers with two or four EAAAR repeat showed higher protein-protein interaction–mediated signal with lower background signal compared with having no linker or linkers with amino acid sequences GGGGSGGGGS, ACGSLSCGSF, and ACGSLSCGS-FACGSLSCGSF. A 9 ± 2-fold increase in signal intensity both in cell culture and in living mice was seen compared with a system that expresses both reporter fragments and the interacting proteins separately. In this fusion system, rapamycin induced heterodimerization of the FRB and FKBP12 moieties occurred rapidly even at very lower concentrations (0.00001 nmol/L) of rapamycin. For a similar fusion system employing split-EGFP, flow cytometry analysis showed significant level of rapamycin-induced complementation.
Introduction
Because proteins associated with pathologic cellular processes are often engaged in abnormal protein-protein interactions, studying the complex process of protein-protein crosstalk is important for identifying the biochemical abnormalities in diseased states. A large amount of modern biological research is concerned with the how, when, and where of protein-protein interaction. The need for simpler approaches to study these interactions, particularly on a large scale, has escalated recently with the nearly complete list of players now available due to the completion of the human genome project. To understand these ubiquitous protein interactions, several techniques have been employed, including the inducible yeast two hybrid system (IY2H; ref. 1), split-ubiquitin system (2), β-galactosidase complementation (3), and the fluorescence resonance energy transfer system (4, 5). A review of various strategies may be found elsewhere (6, 7).
Complementation strategies for studying protein-protein interactions generally involve the fusion of split reporter protein fragments to the protein of interest in such a way that neither of the fragments retains significant activity by themselves. However, when the proteins of interest interact, the two inactive reporter protein fragments complement with each other such that activity is regained, providing a readout signal for indirectly following the protein-protein interaction. This protein fragment–assisted complementation strategy has been used with a variety of reporter proteins, including dihydrofolate reductase, β-Lactamase, green fluorescent protein (GFP), firefly luciferase and Renilla luciferase, and has been found to be a useful technique for studying protein-protein interactions in both bacteria and mammalian cells (8–12). Recent studies from our lab on protein fragment complementation using split firefly luciferase or split-Renilla luciferase (split-RLuc) have shown their use for studying both protein-protein interactions and small molecule–mediated protein-protein interaction non-invasively in small animals (13, 14). Others also have used similar strategy for studying the same in living animals (15, 16). The study of intracellular and extracellular dimerization kinetics of the protein-protein interaction of FK506-binding protein (FKBP12) rapamycin-binding domain (FRB) and FKBP12 mediated by the small molecule rapamycin and the associated Renilla luciferase complementation signals have been found to be relatively weak (11). For the purposes of increasing the sensitivity of the complementation system, it would be useful to have accelerated interaction kinetics between the protein partners. The protein-protein interaction through reporter fragment complementations in animals are low and this may be either due to (i) the recruitment of the interacting proteins from different cellular compartments, (ii) the imbalanced half-life of the interacting proteins and the reporter fragments, (iii) the unequal molar ratio of the interacting proteins with reporter fragments, and (iv) the relatively low intracellular concentration of the small drug of interest that mediates heterodimerization.
Because of all these assumed limitations, we reasoned that engineering a single fusion protein containing all the different components of the system may help to enhance the sensitivity (Fig. 1A). This increased sensitivity may occur at the expense of no longer being able to study protein pairs that depend in part on differential compartmentalization for mediating their interaction. To show this experimentally, we used our previously reported rapamycin-mediated FRB-FKBP12 interaction system (11) as a starting point for the construction of the single fusion protein system that was used in the current study. To avoid erroneous self-complementation, we also studied a series of peptide linkers to keep the reporter protein fragments separated until heterodimerization of FRB and FKB12 occurs. The performance of this fusion protein was compared with our previously developed nonfusion approach, both in cell culture and in the context of noninvasive imaging in living animals. To examine the generalizability of this single fusion protein strategy and to show its use in high-throughput screening of small molecules by flow cytometry, we also developed a fusion protein complementation system based on the split enhanced GFP (split-EGFP). The methods developed in this study will potentially increase the sensitivity of studying protein-protein interactions in living animals and also will increase the sensitivity in screening small molecules that modulate protein-protein interactions in cell culture.
Figure 1.
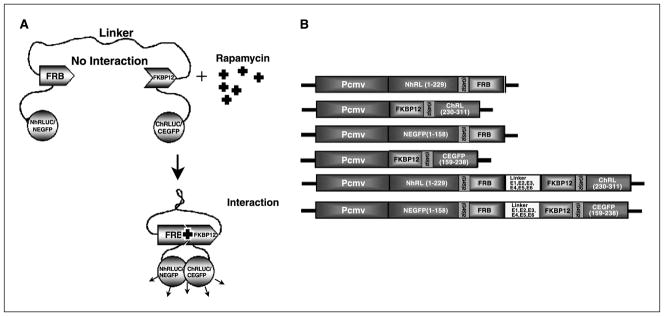
A, schematic diagram of a fusion protein–based approach for studying protein-protein interactions using a rapamycin-mediated complementation strategy with either split-RLuc or split-EGFP protein fragments. In this strategy, the NH2-terminal and COOH-terminal fragments of a reporter protein (RLuc or EGFP) are each attached to one of two interacting proteins (FRB and FKBP12) through a short peptide linker (GGGGSGGGGS). The two interacting proteins (FRB and FKBP12) are in turn attached with a different peptide linker that has the property of thermodynamically favoring a conformation such that the split reporter moieties of the fusion protein are kept away from each other. Ideally, this thermodynamic barrier is low enough that when given a suitable signal for protein-protein interaction to occur (rapamycin in this example), the two interacting proteins cause conformational changes such that the split reporter moieties can interact and complement themselves. B, schematic representation of different plasmid constructs made and used in this study. Shown are the components of the genes (N-rluc, NH2-terminal fragment of Renilla luciferase; C-rluc, COOH-terminal fragment of Renilla luciferase; N-egfp, NH2-terminal fragment of EGFP; C-egfp, COOH-terminal portion of EGFP; FRB and FKBP12 are the rapamycin-binding proteins). CMV is the cytomegalovirus early promoter/enhancer elements. (G4S)2 is the amino acid sequence for the linkers between the reporter fragments and the interacting proteins.
Materials and Methods
Chemicals, enzymes, and reagents
Restriction and modification enzymes and ligase were from New England Biolabs (Beverly, MA). TripleMaster Taq DNA polymerase was from Brinkmann Eppendorf (Hamburg, Germany). The plasmid pCMV-hRL from Promega (Madison, WI) containing a human codon optimized version of Renilla luciferase, was used as the template for the amplification of reporter gene fragments. The rapamycin-dimerizing proteins FRB and FKBP12 were obtained from the constructs pcDNA-Nrluc-FRB and pcDNA-FKBP12-Crluc from our previous study (13). Rapamycin was from Sigma (St. Louis, MO). LipofectAMINE transfection reagent was from Invitrogen (Carlsbad, CA). The plasmid extraction kit and DNA gel extraction kit were from Qiagen (Valencia, CA). Coelenterazine was from Nanolight (Pinetop, AZ). Bacterial culture media were from BD Diagnostic Systems (Sparks, MD). All cell culture media, fetal bovine serum (FBS), the antibiotics streptomycin, and penicillin, were from Invitrogen. Oligonucleotides were synthesized by the Stanford Protein and Nucleic Acid facility. Primary anti-Renilla luciferase antibody from Chemicon (Temecula, CA) and secondary goat anti-mouse/horseradish peroxidase (HRP) conjugate from Bio-Rad (Hercules, CA) were used. The nitrocellulose membrane for Western blot analysis from Schleicher and Schuell (Keene, NH) was used. Enhanced chemiluminescence Western blot detection reagents was from Cell Signaling Technology, Inc. (Beverly, MA).
Construction of plasmids
Nrluc-FRB fragment without stop codon was amplified from pcDNA-Nrluc-FRB (13) using the forward primer flanking NH2-terminal portion of rluc synthesized with NheI restriction site and a reverse primer flanking COOH-terminal portion of FRB fragment synthesized without stop codon and with EcoRI restriction enzyme site was digested by NheI and EcoRI restriction enzymes. The digested fragment was inserted in to pcDNA vector backbone digested with corresponding enzymes and constructed pcDNA-Nrluc-FRB. Similarly, PCR was used to generate a FKBP12-Crluc fragment from pcDNA-FKBP12-Crluc (13) with an NH2-terminal primer with EcoRI site flanking NH2-terminal portion of FKBP12 and the reverse primer designed with XhoI restriction enzyme site flanking COOH-terminal portion of Crluc fragment with stop codon (13) was digested with EcoRI and XhoI restriction enzymes and inserted to pcDNA-Nrluc-FRB digested with the same restriction enzymes and constructed pcDNA-Nrluc-FRB-FKBP12-Crluc. Sense and antisense oligonucleotides containing EcoRI sites were designed for generating the different linkers. The annealed digested oligonucleotides were ligated with the EcoRI-digested, dephosphorylated pcDNA-Nrluc-FRB-FKBP12-Crluc to generate pcDNA-Nrluc-FRB-L-FKBP12-Crluc. The selected clones were confirmed by PCR and nucleic acid sequencing to check the orientation, and the clones with correct orientation were used for further studies.
The pcDNA-Nrluc-FRB-FKBP12-Crluc plasmid was used to construct a vector with split-EGFP, entitled pcDNA-Negfp-FRB-FKBP12-Cegfp, by replacing the Nrluc and Crluc portions with PCR-amplified fragments of Negfp (1-158 amino acids, aa) and Cegfp (159-238 aa; ref. 12), respectively. The individual vectors pcDNA-Negfp-FRB and pcDNA-FKBP12-Cegfp were constructed from pcDNA-Negfp-FRB-FKBP12-Cegfp by amplifying the fragments Negfp-FRB and FKBP12-Cegfp such that each contained an NH2-terminal Nhe I site and a COOH-terminal stop codon and Xho I site, appropriately digested and ligated into the pcDNA backbone (Fig. 1B). All constructs were verified by sequencing.
Cell culture
Human 293T embryonic kidney cancer cells (American Type Culture Collection, Manassas, VA) were grown in MEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Chinese hamster ovary (CHO) cells were grown in DMEM F12-Ham supplemented with 10% FBS and 1% penicillin/streptomycin. Rat C6 glioma cells were maintained in glucose-deficient DMEM supplemented with 0.01% histidinol, 10% FBS, and 1% penicillin/streptomycin/glutamate.
Cell transfection and Renilla luciferase assay
Transfections were done on 24-hour-old cultures of 293T and CHO cells (~80% confluent). For transfections, 250 ng per well of DNA was used in 12-well culture plates. LipofectAMINE amounts were as recommended by the manufacturer. For cell culture heterodimerization experiments, 40 nmol/L rapamycin was added to each well immediately after transfection. Cells were assayed after a 24-hour incubation at 37°C and in 5% CO2. Renilla luciferase activity was assayed as previously published (17). In brief, cells were lysed in 200 μL of 1× passive lysis buffer (Promega), shaken for 15 minutes at room temperature, and centrifuged for 5 minutes at 10,000 rpm, 4°C. Twenty microliters of the supernatant were assayed by adding 100 μL of 0.05 mol/L sodium phosphate buffer (pH 7.0) and 1 μL of coelenterazine (1 μg/μL dissolved in anhydrous ethanol) followed by light measurement in a luminometer (Turner Designs, 20/20, Sunnyvale, CA) for 10 seconds. Bio-Rad protein assay reagent was used for measuring the protein concentrations in the cell lysates. Renilla luciferase activity was represented as relative light units (RLU) per microgram of protein per minute.
Extra cellular dimerization kinetics analysis of proteins FRB and FKBP12 in fusion and nonfusion split reporter protein systems
To study the dimerization kinetics of FRB and FKBP12 at the extra cellular level, 293T cells were either co-transfected with two-vector system (pcDNA-Nrluc-FRB + pcDNA-FKBP12-Crluc), or singly transfected with one of the fusion protein vectors (pcDNA-Nrluc-FRB-FKBP12-Crluc, pcDNA-Nrluc-FRB-E2-FKBP12-Crluc and pcDNA-Nrluc-FRB-E4-FKBP12-Crluc). The cells were lysed 24 hours post transfection in 200 μL of passive lysis buffer, and total cellular protein concentrations were measured. 10 μg of total cellular protein in 100 μL sodium phosphate buffer were incubated at room temperature with 40 nmol/L rapamycin. At several time points (0, 1, 5, 15, 30 and 60 minutes) the Renilla luciferase activity was assessed as mentioned above.
Rapamycin concentration-dependent extracellular dimerization analysis of proteins FRB and FKBP12 in fusion and nonfusion protein systems
To study the rapamycin concentration–dependent extracellular dimerization kinetics of FRB and FKBP12 in fusion and nonfusion protein system, the 293T cells were singly transfected with fusion vectors (pcDNA-Nrluc-FRB-E4-FKBP12-Crluc) or cotransfected with a two-vector system (pcDNA-Nrluc-FRB + pcDNA-FKBP12-Crluc). Again, cells were lysed 24 hours post-transfection and total cellular protein concentrations were measured. Equal concentrations of total cellular protein were incubated at room temperature for 60 minutes in the presence of varying concentrations of rapamycin (0.00004, 0.0004, 0.004, 0.04, 0.4, 4.0, 40, and 400 nmol/L) and measured Renilla luciferase activity.
Western blot analysis
Protein from cells that had been transiently transfected with the different fusion vector constructs and the vector constructs express proteins separately and grown in the presence or absence or rapamycin were used for Western blot analysis with a monoclonal antibody against NH2-terminal portion of Renilla luciferase. For this, 10 μg of protein were run on a 4% to 12% gradient SDS polyacrylamide gel. The protein was then transferred to nitrocellulose membrane using a Hoefer TE 70 semidry electroblot apparatus (Amersham Bioscience, Piscataway, NJ) and blocked with 5% nonfat milk powder in TBST for 1 hour. The membrane was then incubated with the anti-Renilla primary antibody overnight at 4°C with shaking. The washed membrane was then incubated with the HRP-conjugated anti-goat secondary antibody for 1 hour at room temperature, washed thrice with TBST, and exposed to film for 5 minutes. As an internal loading control, the same membrane was washed and incubated with anti-tubulin antibody.
Fluorescent microscopy
For microscopic analysis using the split-EGFP vectors, cells were either cotransfected with two-vector system (pcDNA-Negfp-FRB + pcDNA-FKBP12-Cegfp), or singly transfected with fusion vector (pcDNA-Negfp-FRB-FKBP12-Cegfp, pcDNA-Negfp-FRB-E2-FKBP12-Cegfp, pcDNA-Negfp-FRB-E3-FKBP12-Cegfp, and pcDNA-Negfp-FRB-E4-FKBP12-Cegfp) and incubated in the presence or absence of rapamycin. Cells were viewed and photographed at 24, 48, and 72 hours post-transfection using a fluorescent microscope, with three random fields analyzed for each sample at each time point.
Flow cytometry analysis
Using a FACSCalibur (Becton Dickinson, San Jose, CA), flow cytometry analysis was done in triplicate on the samples containing the abovementioned split-EGFP combinations for the 72-hour time point. The fluorescent intensities of the cells were quantified to analyze for the protein-protein interaction–mediated complementation of the split GFP.
Stable cell lines expressing fusion and nonfusion constructs
Stable 293T cells expressing either the fusion construct pcDNA-Nrluc-FRB-E4-FKBP12-Crluc or coexpressing the nonfusion constructs pcDNA-Nrluc-FRB and pcDNA-FKBP12-Crluc were generated by transfection or cotransfection with the appropriate plasmids and selection with Puromycin. Stably transfected clones were confirmed by luciferase activity following exposure to rapamycin.
Optical imaging in living mice
All animal handling was done in accordance with Stanford University Animal Research Committee guidelines. For imaging in nude mice (nu/nu), one million 293T cells stably transfected with pcDNA-Nrluc-FRB-E4-FKBP12-Crluc or 4.5 million 293T cells cotransfected with pcDNA-Nrluc-FRB + pcDNA-FKBP12-Crluc were s.c. implanted on the low dorsal side of the animal. The unequal numbers of cells used were based on achieving approximately equivalent total luciferase signal after exposure to 20 nmol/L rapamycin in cell culture. Six control and six rapamycin treatment animals were used for the study. All 12 animals were imaged immediately after cell implantation by injecting coelenterazine (20 μg dissolved in ethanol further diluted in 100 μL PBS) via the tail vein. After obtaining these baseline images, the six experimental animals received i.p. injections of 25 μg rapamycin in carrier DMSO, with the six control animals receiving i.p. injections of the carrier. The animals were imaged again at 24, 48, and 72 hours with repeat injections of coelenterazine via the tail vein, with the animals receiving i.p. reinjections of 25 μg rapamycin or carrier following each imaging session.
Mice were anesthetized by i.p. injection of ~40 μL of a ketamine and xylazine (4:1) solution. All mice were imaged using a cooled CCD camera (Xenogen IVIS, Xenogen Corp., Alameda, CA). The animals were placed prone in the imaging system, a low-level illumination reference image was taken, and then bioluminescent photons emitted from the subject were collected over 5 minutes. Images were analyzed using Igor Image Analysis Software (Wavemetrics, Seattle, WA). To quantify the measured light, regions of interest were drawn over the area of the implanted cells and the maximum photons/s/cm2/steradian (sr) were obtained as validated previously (17).
Results
The highest signal to background ratio was achieved for the fusion protein–based split-Renilla luciferase complementation system when linker peptides containing amino acids of EAAAR repeats were chosen
To develop an efficient fusion protein–based split-RLuc complementation system for studying small molecule–mediated protein-protein interactions, six different linker peptides of different lengths were screened in a rapamycin-inducible system. Three of the six linker peptides showed a significant complementation signal and were selected for further study. The complemented Renilla luciferase signal from these three peptide linkers was 9 ± 2-fold more than from the system expressing the proteins separately using two different vectors. When comparing the background signal with the signal achieved after the addition of rapamycin, we found that the peptide with the repeats of EAAAR amino acid sequences [(EAAAR)2 and (EAAAR)4] showed a very low background activity while giving a high complementation signal. The proteins expressed by these constructs showed a 12 ± 3-fold signal versus background ratio (4,500 ± 200 versus 300 ± 75 RLU/μg protein/min; Fig. 2).
Figure 2.

The luminometer assay results of 293T cells 24 hours post-transfection with either one of the split-RLuc reporter protein–based fusion protein vectors or cotransfected with the two-vector system. The two-vector system (Nrluc-FRB/FKBP12-Crluc) showed significant (P < 0.001) Renilla luciferase signal upon exposing the cells to 40 nmol/L rapamycin for a period of 24 hours, whereas the equivalent cells without rapamycin showed signal that is similar to mock-transfected cells. The fusion protein system without a linker between the interacting proteins (Nrluc-FRB-FKBP12-Crluc) shows equal amount of signals from cells with and without rapamycin exposure. The cells transfected with the fusion protein vector containing linkers (Nrluc-FRB-E2-FKBP12-Crluc, Nrluc-FRB-E3-FKBP12-Crluc, and Nrluc-FRB-E4-FKBP12-Crluc) showed significant (P < 0.01) increase in luciferase signal upon exposure to rapamycin. The fusion protein system without rapamycin showed signal that is significantly (P < 0.05) higher than mock-transfected cells. Bars, SE for triplicate determinations.
The fusion protein–based approach shows a significantly higher level of complementation-associated luciferase signal compared with the system that separately expresses proteins with reporter fragments
To compare the fusion protein–based protein-protein interaction system with the system expressing proteins separately from two vectors, 293T cells transfected with pcDNA-Nrluc-FRB-E4-FKBP12-Crluc or cotransfected with pcDNA-Nrluc-FRB + pcDNA-FKBP12-Crluc were assayed 24 hours post-transfection. Significant level of signal (P < 0.01) from both the systems were seen when the cells were incubated in the presence of rapamycin. In the absence of rapamycin, the fusion protein system showed significantly higher (P < 0.05) background signal compared with either the mock-transfected cells or the cells transfected with the two-vector system. The level of signal achieved in the presence of rapamycin from the fusion protein system is 9 ± 2-fold (P < 0.001) greater than the two-vector system that separately expresses the proteins (Fig. 2).
Extracellular dimerization kinetics of the FRB/FKBP-based split-Renilla luciferase fusion system shows significant levels of signal for much lower concentrations of rapamycin compared with the two-vector system
To study the rapamycin concentration–dependent extracellular dimerization kinetics of the FRB/FKBP12-based split-RLuc systems, 293T cells transfected with pcDNA-Nrluc-FRB-E4-FKBP12-Crluc or cotransfected with pcDNA-Nrluc-FRB + pcDNA-FKBP12-Crluc were lysed after a 24-hour transfection. Equal amounts of total cellular protein from the lysates were exposed to different concentrations of rapamycin (0.00004-400 nmol/L) and assayed following 60 minutes of incubation at room temperature. After incubation, significant levels of signal (P < 0.001) were seen for the fusion protein system even at very low concentration of rapamycin (0.00004, 0.0004, 0.004, 0.04, and 0.4 nmol/L). The two-vector system that separately expresses the proteins shows signal that is significantly less than the fusion system even at relatively high concentrations (400 nmol/L) of rapamycin but is significantly (P < 0.05) higher than the mock-transfected cells (Fig. 3A). To rule out effects of differing levels of protein expression, the cell lysates were studied by Western blot analysis. This showed comparable quantities of expressed protein from both the fusion and two-vector systems with (40 nmol/L) and without rapamycin (Fig. 3B).
Figure 3.

A, luminometer assay results for the lysates of 293T cells exposed to different concentrations of rapamycin that had been transfected with the vectors of split-RLuc reporter protein–based fusion protein approach containing different linkers (E0, E2, and E4) or cotransfected with the two-vector system. The result shows significant signal (P < 0.01) from the fusion protein system at all the concentrations studied. The fusion protein system with the linker E4 between the interacting proteins FRB and FKBP12 showed significantly higher (P < 0.05) signal than the cells lysates from cells transfected either with the two-vector system, or the fusion protein system containing either the linker E2 or without any linker. B, Western blot analysis of protein samples extracted from the cells transfected with the two-vector system [S (R− and R+)], and the fusion vector system with different linkers (E0, E2, and E4), both with and without rapamycin conditions (R− and R+).
Extracellular dimerization of FRB/FKBP-based split-Renilla luciferase fusion system shows faster reaction kinetics compared with the system that separately expresses the proteins
To compare the extracellular dimerization kinetics of the two-vector and fusion systems, 293T cells transfected with pcDNA-Nrluc-FRB-E4-FKBP12-Crluc or cotransfected with pcDNA-Nrluc-FRB + pcDNA-FKBP12-Crluc were lysed 24 hours post-transfection. Equal amounts of total cellular protein from the lysates were exposed to 20 nmol/L final concentrations of rapamycin; incubated at room temperature; and assayed at 1, 5, 15, 30, and 60 minutes. The cell lysates from the fusion protein system showed significant levels of signal (P < 0.001) immediately following exposure to rapamycin. The cell lysates from the two-vector system showed significantly less (P < 0.01) signal than the fusion system but significantly higher signal than the mock-transfected cells after 15 minutes of exposures (Fig. 4). These experiments were done on the same cell lysates as the previous subsection, so that protein amounts were comparable with those in Fig. 3B.
Figure 4.

Luminometer assay results for lysates of 293T cells at different time points following exposure to rapamycin. The cells had either been transfected with one of the split-RLuc-based fusion protein vectors, or cotransfected with the two-vector system. The results show rapid dimerization of FRB and FKBP12 in the fusion protein system immediately after exposure to rapamycin, whereas the two-vector strategy shows signal that is little above the background signal after 60 minutes of exposure to rapamycin. Bars, SE for triplicate determinations.
Rapamycin-mediated heterodimerization of FRB and FKBP12 with the split enhanced green fluorescent protein shows similar results compared with the split Renilla luciferase system in transiently transfected 293T cells
To develop a system usable for high-throughput screening by flow cytometry, we replaced the split Renilla luciferase fragments with NH2-terminal (aa 1-158) and COOH-terminal (aa 159-238) portions of EGFP in both the fusion and two-vector–based systems. Both of these systems were studied in transiently transfected 293T cells. Cells 24, 48, and 72 hours post-transfection were photographed in three randomly selected fields. The result showed complementation as evidenced by green fluorescence (Fig. 5). The cells were also quantitatively analyzed by flow cytometry. Cytometry results were tabulated by splitting each cell into one of three windows based on their intensity (low, medium, and high). Of 25,000 cells transiently transfected with an intact EGFP vector, 52 ± 2% were fluorescent, with 8.8 ± 1% in the high intensity window. Following exposure to rapamycin for 72 hours, the fusion and nonfusion systems showed equal numbers of fluorescent cells (26 ± 1%) with the cells expressing the fusion protein system exhibiting 2-fold higher levels of high intensity fluorescence cells compared with the two-vector system. Without rapamycin, neither system showed significant numbers of high intensity cells (Fig. 6).
Figure 5.

The fluorescent microscope photograph of 293T cells taken 72 hours post-transfection with constructs using the heterodimerizing proteins FRB and FKBP12 incubated in the presence or absence of rapamycin. The cells were either transfected with one of the fusion-based split-EGFP protein vectors containing the different linkers (E0, E2, E3, and E4), or cotransfected with the two-vector system. The result shows significant fluorescent signal upon exposure to rapamycin for the fusion systems containing linkers, whereas the fusion protein system with no linker (E0) shows no signal from both the cells with and without rapamycin.
Figure 6.

Flow cytometry of 293T cells transfected with one of the fusion-based split-EGFP protein vectors containing the different linkers (E3 and E4), or cotransfected with the two-vector system (SR). The results were also compared with the cells transfected with full-length EGFP. In both systems, significant green fluorescence intensity shifting was only seen for cells that were exposed to rapamycin. The cells transfected with the fusion system with linker E3 showed very few (3 ± 2%) fluorescent cells in the high intensity region selected for the analysis even without exposure to rapamycin.
Comparison of rapamycin-mediated split Renilla luciferase complementation for fusion and two-vector systems shows significant level of signal improvement by the fusion system for imaging in living mice
To compare the rapamycin-mediated split-RLuc fusion and two-vector complementation systems for imaging in living animals, two million 293T cells either stably transfected with pcDNA-Nrluc-FRB-E4-FKBP12-Crluc and 4.5 million cells stably cotransfected with pcDNA-Nrluc-FRB + pcDNA-FKBP12-Crluc, or mock transfected were implanted s.c. at sites B, C, and A, respectively. Immediately after, these tumors were imaged by injecting 25 μg of coelenterazine via the tail vein. Half of these animals were injected i.p. with 25 μg of rapamycin, whereas the other half received sham injections of DMSO. All of these animals were repeatedly imaged at 24-hour intervals using additional injections of coelenterazine. Immediately after implantation, significant signal was seen only on the site implanted with the cells containing the fusion complementation system (B), whereas the site containing cells expressing the two-vector system showed signal not significantly different (P < 0.2) from the mock-transfected cells. At the 24-hour time point, significant signal was seen in the animals that had received rapamycin at the implantation site of the cells containing the fusion complementation system (P < 0.001). The implantation site with cells expressing the two-vector system exhibited signal that is 3 ± 2-fold above mock-transfected cells even after receiving rapamycin. In the animals that did not receive rapamycin, none of the implantation sites were significantly different from background. The signal from the fusion system in animals that received rapamycin was maintained at all time points (24, 48, 72, and 96 hours) studied (P < 0.001). After 20 days, the well-grown tumors from the cells containing the fusion system and in the presence of rapamycin show 20-fold greater signal (P < 0.01) compared with tumors containing the same system and not exposed to rapamycin, as well as tumors containing the two-vector system that had been exposed to rapamycin (Fig. 7).
Figure 7.
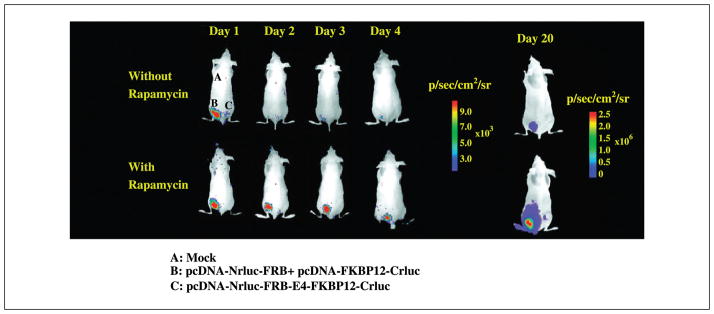
Optical imaging of living mice carrying two million s.c. injected 293T cells stably transfected with pcDNA-Nrluc-FRB-E4-FKBP12-Crluc (site B), 4.5 million cells cotransfected with pcDNA-Nrluc-FRB + pcDNA-FKBP12-Crluc (site C), and mock-transfected cells (site A). The animals were imaged immediately (time 0), and at 24, 48, 96 hours, and 20 days after injecting repeated doses of 25 μg rapamycin (i.p.) or DMSO. A significant increase in complemented Renilla luciferase signal was observed only from the group that had received repeated doses of rapamycin and only in the site implanted with cells stably transfected with the fusion protein–based Renilla luciferase complementation system.
Discussion
In this study, we developed a regulated protein interaction monitoring system for the rapamycin-mediated interaction of the proteins FRB and FKBP12 by fusing these proteins along with split-RLuc fragments into a single reporter construct. This system, along with a similar system employing split-EGFP fragments, was tested in a variety of in vitro and in vivo conditions. Our results show that the fusion protein system generates nearly 10-fold higher complemented signal intensity than the system that expresses proteins separately with split-RLuc system. The developed fusion vector system for studying drug modulated protein-protein interaction showed similar sensitivity when using the strategy for cells in culture and cell implants in living animals. The two-vector system expressing proteins separately showed good sensitivity in cell culture but was not as robust in animal models (13).
The drug-mediated protein-protein interactions could be relatively efficient in cell culture, even when exposing the cells to very low concentrations of drugs. But similar investigation in a living animal system would critically be dependent on many factors, including the availability of an efficient and sensitive (high output) reporter system and a highly sensitive imaging modality to help achieve a detectable signal (18). In this study, we found that expressing all the components of the system as a single fusion protein leads to significant improvements in the complementation-associated Renilla luciferase sensitivity compared with using two distinct vectors. Although the fusion system has a higher background signal before exposure to rapamycin in cells, the signal is not high enough for imaging in animals where lower signal levels are not detectable. Although the fusion strategy does not mimic all the cellular conditions of the native protein-protein interactions, such as expression in the correct cellular compartments, the major criteria to convince here are that the two proteins must eventually colocalize to a same cellular compartment for interaction by assuming that the compartmentalization is not part of modulating the interaction to begin with. In the current study, as few as two million implanted cells containing the new fusion system resulted in a 10-fold greater signal intensity than in our previous study for the same amount of rapamycin. At 20 days, the tumors containing both the fusion and two-vector systems showed signals, but the fusion system is significantly higher (20 ± 4-fold).
To date, several techniques have been developed for studying protein-protein interactions in cells, each with their own advantages and limitations (18). The formation of homotetramers by intracistronic complementation in mutants of β-galactosidase (10) has previously been used for studying protein-protein interactions. Fragments or mutants of reporter proteins with larger molecular size may be sterically hindered during the complementation process (19). Although there are many techniques available for studying protein-protein interactions in cell lines, very few (e.g., IY2H and the split firefly luciferase complementation system; refs. 14, 20) have been successfully extended for use in living animals. The IY2H system has limited applications, as it requires the interaction of the two proteins studied to occur in the nucleus. Protein-protein interactions mediated by the small molecule rapamycin have previously been studied in cell culture by using β-galactosidase complementation (3). In a previous study, we succeeded in developing a split-RLuc protein fragment–assisted complementation system (11) and employed it for noninvasively imaging of the rapamycin-mediated heterodimerization of FRB and FKBP12 in living mice (13). Recently, this same rapamycin-mediated FRB-FKBP12 interaction has been studied in living animals by using a split firefly luciferase complementation system (16). We also showed that this split-RLuc system could be titrated with rapamycin in vivo in living animals leading to detectable heterodimerization of FRB and FKBP12. The heterodimerization system with split-RLuc showed greater levels of sensitivity to rapamycin in cell culture, but the sensitivity lowered several fold when employed in living mice. Therefore, we attempted in this study to develop a system with greater sensitivity for detecting lower levels of drug-mediated protein-protein interactions in vivo.
The enzyme RLuc, a 36-kDa monomeric bioluminescent reporter protein, is currently the smallest optical reporter protein identified for studying protein-protein interactions through a protein fragment–assisted complementation strategy. This reporter protein, when rationally split at particular sites (11), functions efficiently in both cell culture and in living animals, as shown with several different protein partners studied to date (13, 14, 21, 22). One limitation associated with the use of Renilla luciferase is its relatively rapid reaction kinetics, requiring measurements immediately after injection of its substrate (17). Although split firefly luciferase may have some advantages for small animal imaging compared with split-RLuc, the smaller size of Renilla luciferase favors it in many fusion-based strategies. Further work will be needed to compare the relative advantages of firefly versus Renilla luciferase in split reporter strategies. Although split-EGFP is smaller than Renilla luciferase, absolute quantitation based on fluorescent intensity in cells is lacking. The imaging in animals with fluorescent approaches is less efficient mainly due to the autofluorescence and poor tissue penetration of the external excitation source of light. Future improvements in the ability to use split red fluorescent reporters and software tools for correction of autofluorescence may help lead to more sensitive fluorescent split-reporter strategies.
In summary, in this study, we developed and assessed a fusion protein system containing a known rapamycin-mediated protein-protein interaction both in cancer cells and in living mice implanted with cancer cells. This fusion protein–based split reporter complementation assay can be further extended for studying other drug-mediated protein-protein interactions efficiently in living animals. The developed systems based on split-EGFP or split-RLuc reporters should make it possible for high-throughput screening for new protein-protein interaction–targeted drugs in cells along with further evaluation in living animals.
Acknowledgments
Grant support: NIH grants RO1 CA082214 (S.S. Gambhir) and ICMIC P50 CA114747 (S.S. Gambhir) and Department of Energy contract DE-FC03-87ER60615.
We thank Dr. P. Padmanabhan and Manishkumar Patel for their assistance in animal experiments and flow cytometry, respectively, and Andy Loening for his critical reading to improve the article.
References
- 1.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–6. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 2.Johnsson N, Varshavsky A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci U S A. 1994;91:10340–4. doi: 10.1073/pnas.91.22.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi F, Charlton CA, Blau HM. Monitoring protein-protein interactions in intact eukaryotic cells by β-galactosidase complementation. Proc Natl Acad Sci U S A. 1997;94:8405–10. doi: 10.1073/pnas.94.16.8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato M, Ozawa T, Inukai K, Asano T, Umezawa Y. Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat Biotechnol. 2002;20:287–94. doi: 10.1038/nbt0302-287. [DOI] [PubMed] [Google Scholar]
- 5.Pollok BA, Heim R. Using GFP in FRET-based applications. Trends Cell Biol. 1999;9:57–60. doi: 10.1016/s0962-8924(98)01434-2. [DOI] [PubMed] [Google Scholar]
- 6.Paulmurugan R, Ray P, De A, Chan CT, Gambhir SS. Imaging protein-protein interactions in living subjects. In: Golemis Erica., editor. Protein-protein interactions: a molecular cloning manual. 2. New York: CSHL Press; 2005. In press. [Google Scholar]
- 7.Paulmurugan R, Ray P, De A, Chan CT, Gambhir SS. Imaging protein-protein interactions in living subjects. TrAC Trends in Analytical Chemistry. 2005;24:446–58. [Google Scholar]
- 8.Pelletier JN, Arndt KM, Pluckthun A, Michnick SW. An in vivo library-versus-library selection of optimized protein-protein interactions. Nat Biotechnol. 1999;17:683–90. doi: 10.1038/10897. [DOI] [PubMed] [Google Scholar]
- 9.Pelletier JN, Campbell-Valois FX, Michnick SW. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc Natl Acad Sci U S A. 1998;95:12141–6. doi: 10.1073/pnas.95.21.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wehrman T, Kleaveland B, Her JH, Balint RF, Blau HM. Protein-protein interactions monitored in mammalian cells via complementation of β-lactamase enzyme fragments. Proc Natl Acad Sci U S A. 2002;99:3469–74. doi: 10.1073/pnas.062043699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paulmurugan R, Gambhir SS. Monitoring protein-protein interactions using split synthetic Renilla luciferase protein-fragment-assisted complementation. Anal Chem. 2003;75:1584–9. doi: 10.1021/ac020731c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh I, Hamilton AD, Regan L. Antiparallel leucine-zipper directed protein reassembly: application to the green fluorescent protein. J Am Chem Soc. 2000;122:5658–9. [Google Scholar]
- 13.Paulmurugan R, Massoud TF, Huang J, Gambhir SS. Molecular imaging of drug-modulated protein-protein interactions in living subjects. Cancer Res. 2004;64:2113–9. doi: 10.1158/0008-5472.can-03-2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulmurugan R, Umezawa Y, Gambhir SS. Noninvasive imaging of protein-protein interactions in living subjects using reporter protein complementation and reconstitution strategies. Proc Natl Acad Sci U S A. 2002;99:15608–13. doi: 10.1073/pnas.242594299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luker KE, Piwnica-Worms D. Optimizing luciferase protein fragment complementation for bioluminescent imaging of protein-protein interactions in live cells and animals. Methods Enzymol. 2004;385:349–60. doi: 10.1016/S0076-6879(04)85019-5. [DOI] [PubMed] [Google Scholar]
- 16.Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci U S A. 2004;101:12288–93. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhaumik S, Gambhir SS. Optical imaging of Renilla luciferase reporter gene expression in living mice. Proc Natl Acad Sci U S A. 2002;99:377–82. doi: 10.1073/pnas.012611099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 2003;17:545–80. doi: 10.1101/gad.1047403. [DOI] [PubMed] [Google Scholar]
- 19.Rossi FM, Blakely BT, Blau HM. Interaction blues: protein interactions monitored in live mammalian cells by β-galactosidase complementation. Trends Cell Biol. 2000;10:119–22. doi: 10.1016/s0962-8924(99)01707-9. [DOI] [PubMed] [Google Scholar]
- 20.Ray P, Pimenta H, Paulmurugan R, et al. Noninvasive quantitative imaging of protein-protein interactions in living subjects. Proc Natl Acad Sci U S A. 2002;99:3105–10. doi: 10.1073/pnas.052710999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massoud TF, Paulmurugan R, Gambhir SS. Molecular imaging of homodimeric protein-protein interactions in living subjects. FASEB J. 2004;18:1105–7. doi: 10.1096/fj.03-1128fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozawa T, Kaihara A, Sato M, Tachihara K, Umezawa Y. Split luciferase as an optical probe for detecting protein-protein interactions in mammalian cells based on protein splicing. Anal Chem. 2001;73:2516–21. doi: 10.1021/ac0013296. [DOI] [PubMed] [Google Scholar]