

Abstract
Induction of fetal hemoglobin (HbF) has therapeutic importance for patients with sickle cell disease (SCD) and the beta-thalassemias. It was recently reported that increased expression of LIN28 proteins or decreased expression of its target let-7 miRNAs enhances HbF levels in cultured primary human erythroblasts from adult healthy donors. Here LIN28A effects were studied further using erythrocytes cultured from peripheral blood progenitor cells of pediatric subjects with SCD. Transgenic expression of LIN28A was accomplished by lentiviral transduction in CD34(+) sickle cells cultivated ex vivo in serum-free medium. LIN28A over-expression (LIN28A-OE) increased HbF, reduced beta (sickle)-globin, and strongly suppressed all members of the let-7 family of miRNAs. LIN28A-OE did not affect erythroblast differentiation or prevent enucleation, but it significantly reduced or ameliorated the sickling morphologies of the enucleated erythrocytes.
Introduction
Sickle Cell Disease (SCD) and the beta-thalassemias are among the most prevalent genetic disorders worldwide with high levels of morbidity and mortality [1], [2]. Our understanding of these beta-hemoglobinopathies at both the clinical and molecular levels has increased greatly in recent decades [3]. However, despite a homogeneous genetic mutation, clinical outcomes and response to therapy vary widely among patients with SCD [4].
Pharmaceutical induction of fetal hemoglobin (HbF) is an effective approach toward therapy [1]. HbF levels of 20% or greater are associated with improvements in the SCD patients' clinical status [5]. Therefore, manipulation of HbF levels in human erythrocytes remains an intense topic of benign hematology research. As a result, significant knowledge regarding the molecular mechanisms underlying HbF regulation has been uncovered. For instance, gene linkage studies associated BCL11A, HSB1L-MYB and HBB regions in the genome with increased HbF expression in adults [6]–[9], and suppression of BCL11A causes an increase in HbF levels and reverses the SCD phenotype in model systems [10], [11].
Recently, LIN28A and LIN28B proteins as well as let-7 microRNAs (miRNAs) were shown to cause a partial reversal of ontogeny-related changes in the human erythroid phenotype including increased expression of HbF in adult cells [12]. The highly conserved RNA-binding LIN28 proteins are known to control developmentally-timed events in multicellular organisms by negatively regulating the biogenesis of let-7 miRNAs [13]. Human let-7 miRNAs demonstrate increased expression in adult human reticulocytes when compared to cord blood reticulocytes [14].
Here we study erythrocytes cultured from purified CD34(+) cells from children with SCD to further explore the potential for transgenic erythroid LIN28A expression to decrease let-7 miRNAs levels and increase HbF expression. We also report a novel assay for exploring the effects of increased HbF expression upon the sickling phenomenon using purified erythrocytes generated in serum-free culture.
Methods
Ethics Statement
Institutional Review Board approval for the study and the informed consent process was obtained from Children's National Medical Center and the National Institute of Diabetes and Digestive and Kidney Diseases. Written informed consent was obtained from the parent or legal guardian for all research participants less than 18 years of age and from all research subjects 18 years of age or older prior to participation in this study. Written assent was obtained from all participants between the ages of 7 and 17 years of age.
Patients and samples
Discarded whole blood from partial manual exchange transfusions was collected from five pediatric research subjects with HbSS genotype (ages 9–16 years) who were at steady state and not being treated with hydroxyurea. The partial manual exchange transfusions were performed as primary (Donors 1–4) or secondary (Donor 5) stroke prophylaxis every 4 to 5 weeks. The chronic transfusion regimens had been in place for a range of 3 to 11 years at the time of this study. Patient's clinical features are described in Table 1. After the RBCs were lysed (ACK Lysing Buffer, Lonza, Walkersville, MD), CD34(+) cells were isolated using CD34 antibodies conjugated to magnetic microbeads (CliniMACS CD34 Reagent, Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) and a magnetic column (CliniMACS Instrument, Miltenyi Biotec).
Table 1. Clinical features from the five HbSS patients used in this study.
| Donor # | Gender | Age (years) | Hgb (g/dL) | %S | %F | %A | %A2 |
| 1 | M | 15 | 9.9 | 37.2 | 4.1 | 55.8 | 2.9 |
| 2 | M | 13 | 9.8 | 36.3 | 3.4 | 57.1 | 3.2 |
| 3 | M | 9 | 8.5 | 20.0 | 1.3 | 76.1 | 2.6 |
| 4 | M | 9 | 8.7 | 34.3 | 3.1 | 59.6 | 3.0 |
| 5 | M | 16 | 10.5 | 38.3 | 7.1 | 51.3 | 3.3 |
Cell culture
Ex vivo culture was performed in a 3-week serum-free system consisting of three phases (phase I from day 0 to 7, phase II from day 7 to 14 and phase III from day 14 to 21) as previously described [12]. Throughout phase III, cells were kept in a 2% oxygen environment.
Recombinant viral transduction
Human LIN28A over-expression and empty vector control lentiviral particles were purchased from Qiagen (Valencia, CA). On culture day 3 of phase I, CD34(+) cells were transduced with LIN28A viral particles (estimated MOI 12). After 24 hours, puromycin (Sigma Aldrich, St. Louis, MO) was added to the culture. On culture day 7, cells were transferred to phase II medium containing EPO and puromycin until culture day 9. After culture day 9, cells were cultivated at the conditions previously described without puromycin.
Flow cytometry analyses
On days 14 and 21, erythroid differentiation was assessed with antibodies directed against CD71 and glycophorin A (Invitrogen, Carlsbad, CA) using the BD FACSAria I flow cytometer (BD Biosciences, San Jose, CA) as previously described [15]. Enucleation was quantitated by thiazole orange (TO) staining (Sigma) on day 21. Enucleated sickle erythrocytes [TO(−) population] were sorted and imaged as described below. Flow cytometric analyses of HbF were performed in cells fixed with paraformaldehyde and stained with antibody against HbF as previously reported [16].
Quantitative PCR and Western analyses
Q-RT-PCR assays and Western analyses were performed as previously described [12], [14], [17].
HPLC for sickle and fetal hemoglobins
Samples for HPLC analysis were prepared and analyzed as previously described [15].
Low oxygen exposure and assessment of cellular morphologies
Cells were cultured in a 3-week serum-free system as previously described [12]. During the final week in culture, cells were incubated in a 2% oxygen environment consistent with the lower oxygen range estimated for human bone marrow [18]. Initial experiments using cells from healthy volunteers demonstrated that 2% oxygen conditions significantly increased enucleation compared to matched cells cultured in a 21% environment (21% oxygen: 29.5±10.5%; 2% oxygen: 44.0±13.9%; p = 0.019; Figure S1). Enucleated cells detected by thiazole orange staining were sorted directly into wells containing culture medium [duplicate plates for control and LIN28A over-expression (LIN28A-OE) transductions]. Sorted cells as well as control erythrocytes from individuals with HbSS were incubated for 16 hours at 2% oxygen prior to imaging studies. All images were taken within three minutes after removal from 2% oxygen without further manipulation of the cells. Cell imaging was accomplished using an inverted microscope (32X magnification) equipped with a Zeiss AxioCam MRc5 camera (Carl Zeiss, Oberkochen, Germany). Four random fields were imaged from each well, and cellular morphologies were scored by blinded observers.
Statistical analysis
Mean ± SD values were used for calculating statistical significance by Student's t-test.
Results
LIN28A suppresses the let-7 family of miRNAs in human CD34(+) sickle cells
Increased LIN28 in cultured erythroblasts from adult healthy donors causes increased gamma-globin gene and protein expression as they differentiate in culture [12]. As a result, the HbF content of the enucleated erythrocytes that are produced is also elevated. To explore these effects in primary sickle cells, we investigated LIN28A-OE using erythroblasts from five children with SCD. CD34(+) cells were transduced with a LIN28A encoding lentivirus, and LIN28A-OE was confirmed by Q-RT-PCR (control: 8.6E+00±8.1E+00 copies/ng, LIN28A-OE: 2.3E+05±2.1E+05 copies/ng, p = 0.033) and Western blot analyses (Figure 1). LIN28A-OE did not affect the expression of its homolog LIN28B as assessed by Q-RT-PCR on culture day 14 (data not shown). As expected, LIN28A-OE strongly suppressed the levels of all let-7 miRNA family members, with average reductions from 64–96% for let-7a, let-7b, let-7c, let-7d, let-7e, let-7f-2, let-7g, let-7i and miR-98 (Figure 2A). LIN28A-OE mediated changes in the expression of BCL11A, KLF1 and SOX6, erythroid transcription factors involved in the regulation of HbF [10], [19], were not statistically significant (Figure 2B–D). Examination of BCL11A expression in each donor's cells revealed variable changes associated with LIN28A-OE when compared with controls (Figure S2).
Figure 1. LIN28A over-expression in human sickle erythroblasts was confirmed by (A) Q-RT-PCR quantitation of copy number per nanogram of complementary DNA (cDNA) (copies/ng cDNA) and (B) Western blot analysis.

Analyses were performed at culture day 14. Open bars represent control and black bars represent LIN28A over-expression. C: empty vector control; OE: LIN28A over-expression.
Figure 2. LIN28A over-expression strongly suppresses all members of the let-7 family of miRNAs in cultured human sickle erythroblasts.

(A) LIN28A over-expression compared to control samples in the relative expression levels of the let-7 family of miRNAs. The miRNA relative expression levels (y-axis) in the control cells were defined as a level of one for comparison. LIN28A over-expression compared to control samples in the mRNA expression levels of (B) BCL11A, (C) KLF1 and (D) SOX6. Analyses were performed at culture day 14. Open bars represent control and black bars represent LIN28A over-expression. Mean value ± SD of five independent research subjects for each condition. P values were calculated using Student's t-test. C: empty vector control; OE: LIN28A over-expression. *p<0.05.
LIN28A regulates fetal hemoglobin expression but does not affect erythroid differentiation or enucleation of cultured sickle erythrocytes
Expression of the globin genes demonstrated no major differences in alpha-, mu-, theta-, zeta-, delta- and epsilon-globins mRNA levels among LIN28A-OE cells (Figure 3A). However, gamma-globin mRNA expression levels increased significantly in LIN28A-OE samples (control: 2.0E+06±7.0E+05 copies/ng, LIN28A-OE: 1.5E+07±6.0E+06 copies/ng, p = 0.006), and beta (sickle)-globin mRNA decreased in LIN28A-OE samples (control: 2.0E+07±5.2E+06 copies/ng, LIN28A-OE: 1.6E+07±6.3E+06 copies/ng, p = 0.024; Figure 3B).
Figure 3. LIN28A regulates gamma-globin mRNA levels in cultured human sickle erythroblasts.

LIN28A over-expression compared to control samples in the mRNA expression levels of (A) alpha-, mu-, theta- and zeta-globins and (B) beta-, delta-, gamma- and epsilon-globins. Analyses were performed at culture day 14. Open bars represent control and black bars represent LIN28A over-expression. Mean value ± SD of five independent research subjects for each condition. P values were calculated using Student's t-test. *p<0.05.
Consistent with the changes in mRNA levels, LIN28A-OE increased the percentage of HbF compared to controls. Increased HbF was detected by HPLC for each donor (Figure 4A–J). The mean increase in HbF was statistically significant (control: 10.8±7.1%, range 2.3 to 22%; LIN28A-OE: 40.1±14.0%, range 17.6 to 51.2%; p = 0.003). The absence of HbA or other major peaks confirmed the HbSS phenotype. Fetal hemoglobin staining demonstrated a pancellular shift in HbF staining when compared to the isotypic controls (Figure S3). A bimodal distribution of HbF-mediated fluorescence intensity was noted in the controls. By comparison, LIN28A-OE caused a significant increase of cells demonstrating higher level fluorescence (control: 45.0±17.6%, LIN28A-OE: 66.5±18.6%, p = 0.007).
Figure 4. LIN28A regulates fetal hemoglobin levels in cultured human sickle erythroblasts.
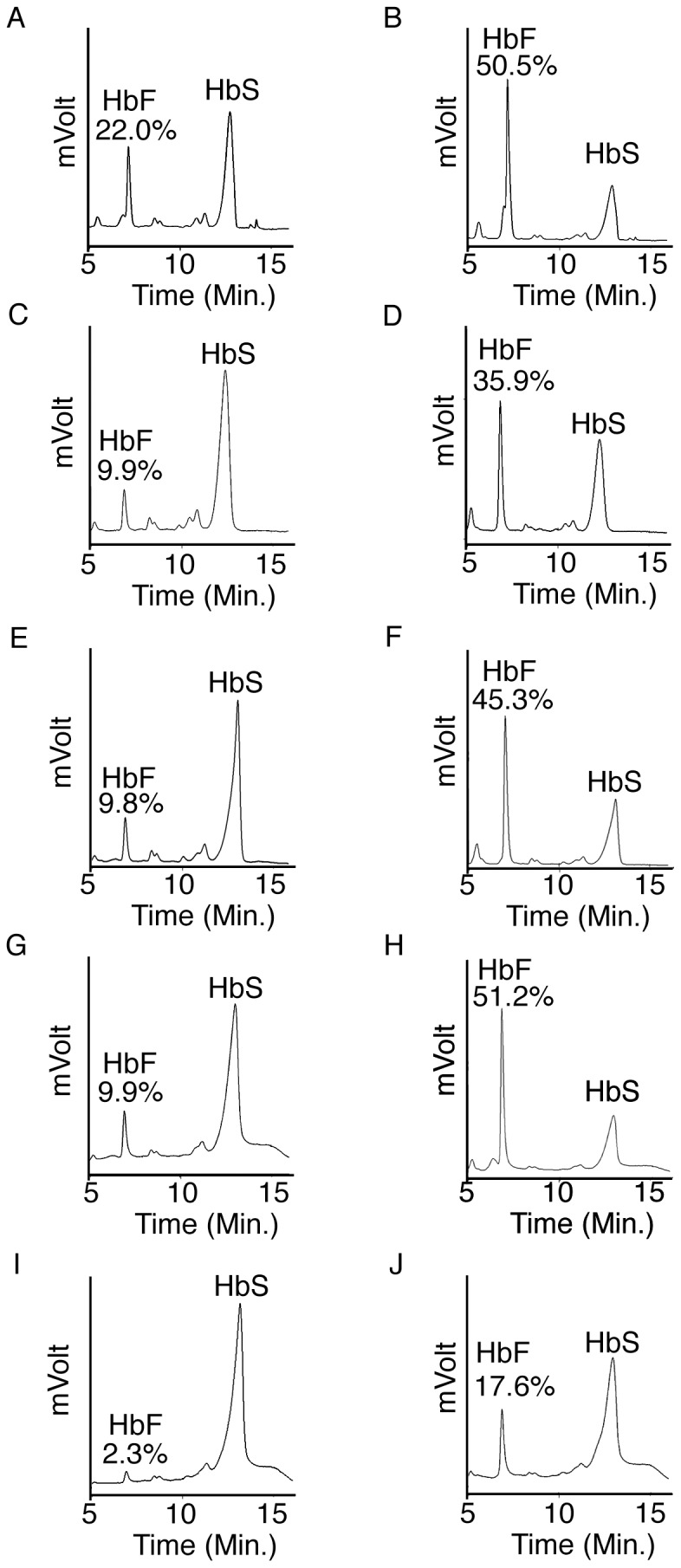
HPLC analysis of hemoglobin from control and LIN28A-OE samples were performed at culture day 21. Donor 1 (A) Control and (B) LIN28A-OE, Donor 2 (C) Control and (D) LIN28A-OE, Donor 3 (E) Control and (F) LIN28A-OE, Donor 4 (G) Control and (H) LIN28A-OE and Donor 5 (I) Control and (J) LIN28A-OE. HbF and HbS peaks are labeled with the ratio of HbF/HbF+HbS expressed as a percentage above each HbF peak (y-axis, mVolts; x-axis, elution time in minutes).
According to flow cytometry analyses of transferrin receptor (CD71) and glycophorin A (GPA), erythroblast differentiation was not affected by LIN28A-OE (Figure S4A). Furthermore, thiazole orange (TO) staining, showed equivalent enucleation achieved between the LIN28A-OE cells and the control transductions at culture day 21 (LIN28A-OE enucleation 40.8±17.0% compared to control 49.9±23.4%, p = 0.19; Figure S4B). Upon completion of the culture period (day 21), cell counts in LIN28A-OE and control transductions were equivalent (data not shown). Taken together, these data show that transgenic expression of LIN28A in erythroblasts from pediatric subjects with sickle cell anemia regulates both fetal and beta (sickle)-globin expression with comparable ex vivo differentiation of the cells.
LIN28A reduces hypoxia-related sickling
Based upon the demonstration of robust enucleation, we next determined whether sickle cell morphologies could be identified among the cultured erythroblasts. The assay was developed using cells from the first three donors, and the morphologies were scored for comparison using Donor 4 and Donor 5 cells. Microscopic examination showed morphologies that were characteristically similar to those reported from human blood including elongated, maple-leaf, and the classic “sickle” shaped cells [20] (See Figure 5 and Figure S5). By comparison, images from healthy HbAA donor cells produced round morphologies in culture (Figure S6). As a result of these differences, the cellular populations were scored according to two general categories: round cells (no evidence of sickle morphology) versus cells with non-round shapes defined in this study as possessing variable sickle morphologies. Further attempts to subcategorize were not made due to the broad range of abnormal morphologies. According to this scoring method, control erythrocytes obtained from HbSS donors' peripheral blood that were maintained under the same 2% oxygen environment in culture medium demonstrated sickled morphologies in 88.8±5.4% of the cells.
Figure 5. LIN28A over-expression ameliorates abnormal morphologies associated with sickle mature erythrocytes.

Cell images are representative from two independent research subjects as follows: Donor 4 (A) control and (C) LIN28A-OE, and Donor 5 (B) control and (D) LIN28A-OE. Cells were scored according to round versus variable sickle morphologies (non-round). Average cell counts of variable sickle morphologies from Donor 4 (E) and Donor 5 (F). Error bars denote ± SD of 8 fields for each condition per donor. P values were calculated using Student's t-test. Control: empty vector control; LIN28A-OE: LIN28A over-expression. *p<0.05.
The same morphology scoring method (round versus non-round) was utilized to quantitate LIN28A-OE effects upon the morphological sickling phenomenon. Images from the control and LIN28A-OE transductions were assigned random numbers, and then scored by blinded observers. Cellular debris or non-hemoglobinized cellular ghosts (less than 10% of particles) were not scored. The percentages of cells from each donor that demonstrated variable sickled morphologies (non-round) were compared between LIN28A-OE and control transductions. Representative fields are shown in Figure 5A–D (all microscopic fields are shown in Figure S5). LIN28A-OE caused significant reductions in the variable sickle morphologies in both donors' cells. In Donor 4, LIN28A-OE caused HbF expression to increase in the cells to 51.2% (versus control HbF 9.9%), with a marked reduction in the variable sickle morphologies of the cultured erythrocytes (control: 79±7.8%, LIN28A-OE: 41.6±11.1%, p = 0.00004; Figure 5E). In Donor 5 cells, LIN28A-OE caused increased HbF levels to 17.6% (versus control HbF 2.3%), and caused a less intense reduction in the variable sickle morphologies (control: 94.1±2.3%, LIN28A-OE: 81.8±4.4%, p = 0.0003; Figure 5F). While both donors demonstrated decreases in sickling, the more robust reduction in Donor 4 cells occurred in association with the greater increase in HbF.
Discussion
The lin28 gene was first described in the nematode Caenorhabditis elegans as a heterochronic gene that regulates timing and sequence of developmental events [21]. Two homologs of the lin28 gene – LIN28A and LIN28B – have been identified in humans and were correlated with the pluripotency of stem cells [22], differentiation of skeletal muscle cells [23], and developmental timing characteristics such as variation in height [24], timing of puberty [25], [26] and age at natural menopause [26]. LIN28 genes are known to regulate the let-7 family of miRNAs, and the expression of LIN28 transcripts is associated with the inhibition of let-7 [13]. The LIN28-let-7 axis is highly conserved across evolution, serving as a regulator of cell growth and development in a cellular/tissue specific manner. In erythroid biology, the let-7 family of miRNAs was associated with the developmental transition from fetal to adult erythropoiesis, since several members of the let-7 family are up-regulated in adult compared to cord blood human reticulocytes samples [14]. Recently, expression of either LIN28 protein in adult erythroblasts caused reductions in let-7 miRNAs as well as increases in HbF expression [12]. Here our studies with LIN28A over-expression demonstrate similar effects in cultured human sickle cells. LIN28A-OE was also shown to reduce the cell-sickling phenomenon in these cultured cells.
Potential effects of LIN28A-OE upon the expression of BCL11A, SOX6 and KLF1 were also studied [12], [27]. BCL11A was previously associated with the human genetic variation in HbF levels [6], [7], [9], [24], the regulation of HbF in adult erythroid cells [10], and the amelioration of the SCD phenotype in adult mice models of the disease [11]. In SCD patients, BCL11A has been associated with the baseline levels of fetal hemoglobin [28] and was down-regulated by hydroxyurea treatment in early reticulocytes [29]. In cooperation with BCL11A, the transcription factor SOX6 was also reported to occupy the human beta-globin cluster and play a role in regulating HbF in adult human erythroid cells [30]. KLF1 was identified as a regulator of BCL11A expression in human erythroid cells [19]. Significant reductions in BCL11A expression upon the transgenic over-expression of LIN28 were previously detected in cells from healthy donors [12]. In this study, the suppression of BCL11A expression was not consistent among the 5 donors (Figure S2), and LIN28A over-expression with let-7 miRNAs suppression did not significantly change the mean expression levels of BCL11A, KLF1 or SOX6 (Figure 2B–D). Further, all the samples demonstrated increased levels of HbF, but those increases were not well correlated with the levels of LIN28A over-expression (compare Figures 1 and 4). These results emphasize the current lack of mechanistic understanding for how LIN28/let-7 mediates increases in HbF expression. Future studies of LIN28A, including examination of alternate model systems and determination of the minimal LIN28A levels required for its regulation of globin gene expression, are needed.
The sickling morphology of erythrocytes is due to polymerization of hemoglobin S with formation of intracellular fibers [31]. For decades, a combination of basic and clinical research has been aimed toward augmentation of HbF because of its sparing effects on sickle hemoglobin polymerization [5], [32]. We developed a novel approach for studies of the sickling phenomenon using culture-generated erythrocytes. During the terminal stages of differentiation, the erythroblasts were cultured in a 2% oxygen environment consistent with levels in bone marrow [17], [33]. Highly purified populations of enucleated cells were then imaged to demonstrate sickle morphologies. LIN28A-OE significantly reduced the morphological sickling in both donors' cells. While several factors are involved in the cellular sickling phenomenon [34], [35], increased HbF expression in LIN28A-OE cells is thought to play a critical role in reducing the morphological abnormalities.
In summary, LIN28A-OE reduced let-7 miRNAs levels and increased HbF expression in erythroblasts cultured from CD34(+) cells from pediatric sickle cell anemia donors' blood at magnitudes similar to those previously reported using cells from healthy adults. The ex vivo culture model was explored in the context of tissue engineering to demonstrate reduction of the sickling phenomenon among the enucleated cells. As such, these data support further studies of the LIN28/let-7 regulatory axis in erythroid biology with the eventual goal of identifying new therapeutic interventions for beta-hemoglobinopathies.
Supporting Information
Low oxygen culture conditions (2% oxygen) compared to 21% oxygen culture conditions increases erythroid enucleation of cultured CD34+ cells from healthy volunteers. Enucleation was assessed by thiazole orange staining. Flow cytometry analyses of three independent healthy volunteers were performed at culture day 21. HV = healthy volunteer.
(TIF)
Effect of LIN28A over-expression upon the expression levels of BCL11A . BCL11A mRNA levels were investigated by Q-RT-PCR quantitation of copy number per nanogram of complementary DNA (cDNA) (copies/ng cDNA) at culture day 14. Open bars represent control and black bars represent LIN28A over-expression. C: empty vector control; OE: LIN28A over-expression.
(TIF)
LIN28A over-expression increases the percentage of cells with high-levels of fetal hemoglobin. Fetal hemoglobin staining was assessed by flow cytometry analysis of control and LIN28A-OE samples on culture day 21. Cell analyses from (A) Donor 1, (B) Donor 2, (C) Donor 3, (D) Donor 4 and (E) Donor 5 are shown. Fluorescence units (FU) for cells stained with the isotypic control antibody (IgG1) are shown in the shaded histograms. Fluorescence from the anti-HbF stained, control and LIN28A-OE transduced cells are demonstrated by dotted- and bold-lined histograms, respectively. The bar in the upper right of each panel denotes the fluorescence within the upper two decades (defined here and described in the text as high-level fluorescence).
(TIF)
LIN28A over-expression in human sickle erythroblasts does not affect erythroblast differentiation and enucleation. Flow cytometry analyses of (A) control and LIN28A-OE at culture day 14 and day 21 stained with anti-transferrin receptor (CD71) and anti-glycophorin A (GPA) antibodies. (B) Enucleation was assessed by thiazole orange staining of culture day 21 for control and LIN28A-OE cells. Data are representative of five independent research subjects. Control: empty vector control; LIN28A-OE: LIN28A over-expression.
(TIF)
Microscopic fields of HbSS cells. Enucleated cells from Donor 4 (A) control and (B) LIN28A-OE , and Donor 5 (C) control and (D) LIN28A-OE . The square in the first field from each image corresponds to the representative field shown in Figure 5. Control: empty vector control; LIN28A-OE: LIN28A over-expression.
(TIF)
Enucleated erythroid cells from healthy HbAA donor. Representative fields from (A) control (non-transduced cells) and (B) transduced cells. Culture conditions, cell imaging and counting procedures were the same as used for the sickle erythroid cells.
(TIF)
Acknowledgments
We wish to thank the patients and families for participation in this clinical research project.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
The Intramural Research Programs of the National Institute of Diabetes and Digestive and Kidney Diseases supported this work. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ley TJ, DeSimone J, Anagnou NP, Keller GH, Humphries RK, et al. (1982) 5-azacytidine selectively increases gamma-globin synthesis in a patient with beta+ thalassemia. N Engl J Med 307: 1469–1475. [DOI] [PubMed] [Google Scholar]
- 2. Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, et al. (1984) Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest 74: 652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weatherall DJ (2013) The role of the inherited disorders of hemoglobin, the first “molecular diseases,” in the future of human genetics. Annu Rev Genomics Hum Genet 14: 1–24. [DOI] [PubMed] [Google Scholar]
- 4. Schechter AN (2008) Hemoglobin research and the origins of molecular medicine. Blood 112: 3927–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Noguchi CT, Rodgers GP, Serjeant G, Schechter AN (1988) Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med 318: 96–99. [DOI] [PubMed] [Google Scholar]
- 6. Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, et al. (2007) A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 39: 1197–1199. [DOI] [PubMed] [Google Scholar]
- 7. Thein SL, Menzel S, Peng X, Best S, Jiang J, et al. (2007) Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci U S A. 104: 11346–11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sedgewick AE, Timofeev N, Sebastiani P, So JC, Ma ES, et al. (2008) BCL11A is a major HbF quantitative trait locus in three different populations with beta-hemoglobinopathies. Blood Cells Mol Dis 41: 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, et al. (2008) Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 105: 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, et al. (2008) Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322: 1839–1842. [DOI] [PubMed] [Google Scholar]
- 11. Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, et al. (2011) Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 334: 993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee YT, de Vasconcellos JF, Yuan J, Byrnes C, Noh SJ, et al. (2013) LIN28B-mediated expression of fetal hemoglobin and production of fetal-like erythrocytes from adult human erythroblasts ex vivo. Blood 122: 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Viswanathan SR, Daley GQ (2010) Lin28: A microRNA regulator with a macro role. Cell 140: 445–449. [DOI] [PubMed] [Google Scholar]
- 14. Noh SJ, Miller SH, Lee YT, Goh SH, Marincola FM, et al. (2009) Let-7 microRNAs are developmentally regulated in circulating human erythroid cells. J Transl Med 7: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tanno T, Porayette P, Sripichai O, Noh SJ, Byrnes C, et al. (2009) Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 114: 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meier ER, Byrnes C, Weissman M, Noel P, Luban NL, et al. (2011) Expression patterns of fetal hemoglobin in sickle cell erythrocytes are both patient- and treatment-specific during childhood. Pediatr Blood Cancer 56: 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sripichai O, Kiefer CM, Bhanu NV, Tanno T, Noh SJ, et al. (2009) Cytokine-mediated increases in fetal hemoglobin are associated with globin gene histone modification and transcription factor reprogramming. Blood 114: 2299–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holzwarth C, Vaegler M, Gieseke F, Pfister SM, Handgretinger R, et al. (2010) Low physiologic oxygen tensions reduce proliferation and differentiation of human multipotent mesenchymal stromal cells. BMC Cell Biol 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou D, Liu K, Sun CW, Pawlik KM, Townes TM (2010) KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat Genet 42: 742–744. [DOI] [PubMed] [Google Scholar]
- 20. Horiuchi K, Ohata J, Hirano Y, Asakura T (1990) Morphologic studies of sickle erythrocytes by image analysis. J Lab Clin Med 115: 613–620. [PubMed] [Google Scholar]
- 21. Moss EG, Lee RC, Ambros V (1997) The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell 88: 637–646. [DOI] [PubMed] [Google Scholar]
- 22. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, et al. (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318: 1917–1920. [DOI] [PubMed] [Google Scholar]
- 23. Polesskaya A, Cuvellier S, Naguibneva I, Duquet A, Moss EG, et al. (2007) Lin-28 binds IGF-2 mRNA and participates in skeletal myogenesis by increasing translation efficiency. Genes Dev 21: 1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, et al. (2008) Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet 40: 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ong KK, Elks CE, Li S, Zhao JH, Luan J, et al. (2009) Genetic variation in LIN28B is associated with the timing of puberty. Nat Genet 41: 729–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He C, Kraft P, Chen C, Buring JE, Pare G, et al. (2009) Genome-wide association studies identify loci associated with age at menarche and age at natural menopause. Nat Genet 41: 724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sankaran VG, Orkin SH (2013) The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med 3: a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Green NS, Ender KL, Pashankar F, Driscoll C, Giardina PJ, et al. (2013) Candidate sequence variants and fetal hemoglobin in children with sickle cell disease treated with hydroxyurea. PLoS One 8: e55709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flanagan JM, Steward S, Howard TA, Mortier NA, Kimble AC, et al. (2012) Hydroxycarbamide alters erythroid gene expression in children with sickle cell anaemia. Br J Haematol 157: 240–248. [DOI] [PubMed] [Google Scholar]
- 30. Xu J, Sankaran VG, Ni M, Menne TF, Puram RV, et al. (2010) Transcriptional silencing of {gamma}-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev 24: 783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Crepeau RH, Dykes G, Garrell R, Edelstein SJ (1978) Diameter of haemoglobin S fibres in sickled cells. Nature 274: 616–617. [DOI] [PubMed] [Google Scholar]
- 32. Noguchi CT, Schechter AN (1981) The intracellular polymerization of sickle hemoglobin and its relevance to sickle cell disease. Blood 58: 1057–1068. [PubMed] [Google Scholar]
- 33. Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, et al. (2014) Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 508: 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barabino GA, Platt MO, Kaul DK (2010) Sickle cell biomechanics. Annu Rev Biomed Eng 12: 345–367. [DOI] [PubMed] [Google Scholar]
- 35. Brittenham GM, Schechter AN, Noguchi CT (1985) Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 65: 183–189. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Low oxygen culture conditions (2% oxygen) compared to 21% oxygen culture conditions increases erythroid enucleation of cultured CD34+ cells from healthy volunteers. Enucleation was assessed by thiazole orange staining. Flow cytometry analyses of three independent healthy volunteers were performed at culture day 21. HV = healthy volunteer.
(TIF)
Effect of LIN28A over-expression upon the expression levels of BCL11A . BCL11A mRNA levels were investigated by Q-RT-PCR quantitation of copy number per nanogram of complementary DNA (cDNA) (copies/ng cDNA) at culture day 14. Open bars represent control and black bars represent LIN28A over-expression. C: empty vector control; OE: LIN28A over-expression.
(TIF)
LIN28A over-expression increases the percentage of cells with high-levels of fetal hemoglobin. Fetal hemoglobin staining was assessed by flow cytometry analysis of control and LIN28A-OE samples on culture day 21. Cell analyses from (A) Donor 1, (B) Donor 2, (C) Donor 3, (D) Donor 4 and (E) Donor 5 are shown. Fluorescence units (FU) for cells stained with the isotypic control antibody (IgG1) are shown in the shaded histograms. Fluorescence from the anti-HbF stained, control and LIN28A-OE transduced cells are demonstrated by dotted- and bold-lined histograms, respectively. The bar in the upper right of each panel denotes the fluorescence within the upper two decades (defined here and described in the text as high-level fluorescence).
(TIF)
LIN28A over-expression in human sickle erythroblasts does not affect erythroblast differentiation and enucleation. Flow cytometry analyses of (A) control and LIN28A-OE at culture day 14 and day 21 stained with anti-transferrin receptor (CD71) and anti-glycophorin A (GPA) antibodies. (B) Enucleation was assessed by thiazole orange staining of culture day 21 for control and LIN28A-OE cells. Data are representative of five independent research subjects. Control: empty vector control; LIN28A-OE: LIN28A over-expression.
(TIF)
Microscopic fields of HbSS cells. Enucleated cells from Donor 4 (A) control and (B) LIN28A-OE , and Donor 5 (C) control and (D) LIN28A-OE . The square in the first field from each image corresponds to the representative field shown in Figure 5. Control: empty vector control; LIN28A-OE: LIN28A over-expression.
(TIF)
Enucleated erythroid cells from healthy HbAA donor. Representative fields from (A) control (non-transduced cells) and (B) transduced cells. Culture conditions, cell imaging and counting procedures were the same as used for the sickle erythroid cells.
(TIF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.