

Abstract
Human epidermal growth factor receptor type 2 (HER2) is a well-established tumor biomarker that is overexpressed in a wide variety of cancers and that serves as a molecular target for therapeutic intervention. HER2 also serves as a prognostic indicator of patient survival and as a predictive marker of the response to antineoplastic therapy. The development of 18F-labeled biomolecules for PET imaging of HER2 (HER2 PET) is very important because it may provide a powerful tool for the early detection of HER2-positive tumor recurrence and for the monitoring of HER2-based tumor treatment.
Methods
In this study, anti-HER2 monomeric and dimeric protein scaffold molecules [ZHER2:477 and (ZHER2:477)2, respectively] were radiofluorinated at a reasonable radiochemical yield (13%–18%) by use of site-specific oxime chemistry. The resulting radiofluorinated protein scaffold molecules were then evaluated as potential molecular probes for small-animal HER2 PET by use of a SKOV3 tumor–bearing mouse model.
Results
The 4-18F-fluorobenzaldehyde conjugated aminooxy-protein scaffolds [18F-N-(4-fluorobenzylidene)oxime (FBO)-ZHER2:477 and 18F-FBO-(ZHER2:477)2] both displayed specific HER2-binding ability in vitro. Biodistribution and small-animal PET imaging studies further revealed that 18F-FBO-ZHER2:477 showed rapid and high SKOV3 tumor accumulation and quick clearance from normal tissues, whereas 18F-FBO-(ZHER2:477)2 showed poor in vivo performance (low tumor uptake and tumor-to-normal tissue ratios). The specificity of 18F-FBO-ZHER2:477 for SKOV3 tumors was confirmed by its lower uptake on pretreatment of tumor-bearing mice with the HER2-targeting agents ZHER2 and trastuzumab. Moreover, small-animal PET imaging studies revealed that 18F-FBO-ZHER2:477 produced higher-quality tumor imaging than 18F-FBO-(ZHER2:477)2. 18F-FBO-ZHER2:477 could clearly identify HER2-positive tumors with good contrast.
Conclusion
Overall, these data demonstrate that 18F-FBO-ZHER2:477 is a promising PET probe for imaging HER2 expression in living mice. It has a high potential for translation to clinical applications. The radiofluorination method developed can also be used as a general strategy for the site-specific labeling of other proteins with 18F. The protein scaffold molecules used here are attractive for the further development of PET probes for other molecular targets.
Keywords: Affibody, HER2, PET, imaging, 18F
Human epidermal growth factor receptor type 2 (HER2), a transmembrane protein and a member of the erbB family of receptor tyrosine kinase proteins, is a well-established tumor biomarker that is overexpressed in a wide variety of cancers, including breast, ovarian, lung, gastric, and oral cancers (1,2). HER2 amplification and activation promote tumor cell proliferation, growth, migration, adhesion, and invasiveness. HER2 upregulation also contributes to the resistance of cancer cells to apoptosis, thereby leading to an improvement in the survival of cancer cells. It has been found that HER2 overexpression is usually associated with high-grade aggressive tumors and a poor prognosis (2). Therefore, HER2 has great value as a molecular target for therapeutic intervention, as a prognostic indicator of patient survival, and as a predictive marker of the response to antineoplastic therapy.
Over the last decade, noninvasive molecular imaging of HER2 expression with various imaging modalities has been extensively studied; these modalities include radionuclide imaging with PET and SPECT (3–15), MRI (16), and optical imaging (17,18). PET imaging of HER2 (HER2 PET) is particularly useful because of its high sensitivity, high spatial resolution, strong quantification ability, and great potential for translation to clinical applications. Compared with the fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC) tests currently used in clinical practice for the determination of HER2 status, PET provides a real-time assay of overall tumor HER2 expression in patients; therefore, it is expected to avoid false-negative results, which are caused by sampling errors and heterogeneity of expression in biopsied tumor samples. Furthermore, HER2 PET may be very helpful for the identification of HER2 expression in tumors over time, the selection of patients for HER-targeted treatment (such as trastuzumab-based therapy), prediction of the response to therapy, the evaluation of drug efficacy, and many other applications.
Although various HER2 PET techniques have been evaluated with preclinical small-animal tumor models, developing a PET probe for routine clinical use has not yet been successfully achieved, mainly because of the limited availability of HER2-binding ligands to the molecular imaging field. Indeed, the majority of efforts for HER2 imaging agent development have been focused on using HER2-avid antibodies or antibody-derived fragments (3,4,17,18). Unfortunately, the slow tumor targeting and slow normal tissue clearance of antibody-based probes severely hamper their clinical applications. Moreover, the slow pharmacokinetic clearance of antibody-based probes generally leads to the selection of radioisotopes with relatively long half-lives, such as 64Cu (12.7 h), 86Y (14.7 h), 124I (4.18 d), and 76Br (16.0 h), as radiolabels, potentially resulting in elevated radiation doses to patients. The limited availability and high cost of those radioisotopes also become significant problems and hinder the further development of antibody-based HER2 imaging agents.
Recently, molecules that were derived from one of the IgG-binding domains of staphylococcal protein A (Affibody molecules) have received a lot of attention (5–15,19). Affibody molecules are composed of a relatively small engineered protein scaffold with 58 amino acid residues and a 3-helix-bundle scaffold structure. They possess some favorable properties, including ease of preparation, possible production by chemical synthesis, high affinity and specificity, rapid blood clearance, and rapid and good tumor tissue penetration and accumulation. Moreover, Affibody binders against desired targets can be quickly identified and selected by use of phage display technology (20). Thus, Affibody molecules have great potential to become a universal platform for developing imaging or therapeutic agents for various biomarkers.
High-affinity HER2-binding Affibody molecules were obtained by use of phage display technology and affinity maturation (8,20). Subsequently, these proteogenic derivatives were radiolabeled with various radioisotopes, including 99mTc, 111/114mIn, 90Y, 177Lu, 68Ga, 125I, 76/77/82Br, and 211At (5–15), and were evaluated as therapeutic and imaging agents for malignant tumors overexpressing HER2. These studies clearly demonstrated that Affibody molecules are a promising new class of HER2-targeting ligands worthy of further evaluation in the development of PET probes for clinical HER2 imaging.
We hypothesized that Affibody binders would be ideal candidates for developing 18F-labeled PET molecular probes because of their fast tumor-targeting ability, fast pharmacokinetic clearance, and ability to recognize a wide variety of molecular targets. 18F, the most commonly used PET radionuclide, is widely available and has almost ideal imaging properties (half-life: 110 min; emits β+-particles at an energy of 635 keV; 97% abundant) (21); these features make this radionuclide highly clinically relevant. Our goal is to ultimately apply Affibody-based PET probes for imaging patients. In this study, we developed a method for the site-specific radiofluorination of Affibody molecules, using anti-HER2 Affibody molecules [ZHER2:477 and (ZHER2:477)2] as model proteins. The in vitro and in vivo biologic profiles of these 18F-labeled anti-HER2 Affibody molecules were evaluated in cultured SKOV3 cells with high HER2 expression levels and nude mice bearing subcutaneous SKOV3 tumors. Finally, the ability of 18F-labeled ZHER2:477 and (ZHER2:477)2 to image HER2 expression in living mice was evaluated with small-animal PET.
MATERIALS AND METHODS
General
The anti-HER2 imaging agent, Affibody molecule ZHER2:477, and the anti-HER2, unconjugated bivalent Affibody molecule, (ZHER2:477)2, were purchased from Affibody AB. All of the other general materials and instruments and the synthesis of the bifunctional linker [2-(aminooxy)-N-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)acetamide hydrochloride] (Mal-AO), aminooxy-functionalized Affibody molecules [ZHER2:477-ONH2 and (ZHER2:477)2-ONH2], and 4-fluorobenzaldehyde (4-FBA)–conjugated aminooxy-functionalized Affibody molecules [N-(4-fluorobenzylidene)oxime (FBO)-ZHER2:477 and FBO-(ZHER2:477)2], are described in the supplemental materials (available online only at http://jnm.snmjournals.org). The procedures for in vitro analysis of Affibody bioconjugates are also described in the supplemental materials.
Radiosynthesis of 18F-Labeled Affibody Molecules: 18F-FBO-ZHER2:477 and 18F-FBO-(ZHER2:477)2
The Affibody molecules ZHER2:477-ONH2 and (ZHER2:477)2-ONH2 were radiofluorinated with an 18F-labeled prosthetic group (4-18F-FBA) by use of a slight modification of a procedure reported before (22). 4-18F-FBA was first prepared by use of a GE TRACERlab FX-FN synthetic module (GE Healthcare) (Fig. 1). In brief, non–carrier-added 18F– trapped on a quaternary ammonium anion exchange cartridge (11.1–37 GBq [300–1,000 mCi]) was washed with a solution of K2CO3 and Kryptofix 2.2.2 (Sigma) in water and acetonitrile. After evaporation of the solution and azeotropic drying of the residue, a solution of a 4-formyl-N,N,N-trimethylanilinium triflate precursor (4–6 mg) in dimethyl sulfoxide was added to the reactor, and the mixture was heated for 10 min at 85°C. Purification of the reaction mixture with a C18 cartridge system afforded the product 4-18F-FBA at a 50%–70% radiochemical yield (decay corrected) and an overall synthesis time of about 40 min (specific activity: 592–1,480 MBq/μg [16–40 mCi/μg]).
FIGURE 1.
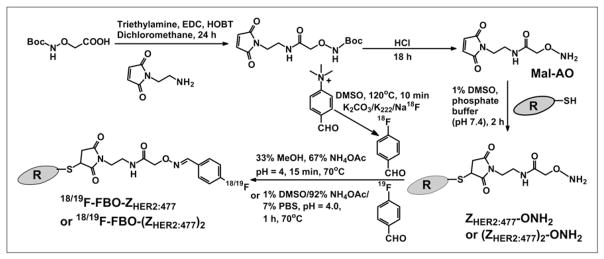
Synthetic schemes for bifunctional linker Mal-AO, ZHER2:477-ONH2, (ZHER2:477)2-ONH2, 18F-FBO-ZHER2:477, and 18F-FBO-(ZHER2:477)2. Boc = tert-butoxycarbonyl; DMSO = dimethyl sulfoxide; EDC = 1-ethyl-3-(3-dimethyl-aminopropyl)carbodiimide; HOBT = N-hy-droxybenzotriazole; MeOH = methanol; NH4OAc = ammonium acetate; PBS = phosphate-buffered saline.
Subsequently, ZHER2:477-ONH2 or (ZHER2:477)2-ONH2 (0.25 or 0.5 mg, respectively, in 100 μL of ammonium acetate [pH 4]) (Fig. 1) was incubated with 4-18F-FBA (0.185–0.259 GBq [5–7 mCi] in 50 μL of methanol) at 70°C for 15 min. The reaction solution was cooled and purified by use of an analytic high-performance liquid chromatography (HPLC) column (flow rate, 1 mL/min). For the purification of 18F-FBO-ZHER2:477, the mobile phases were 95% solvent A and 5% solvent B (0–3 min), 80% solvent A and 20% solvent B (at 25 min), and then 85% solvent B and 15% solvent A (25–36 min); the last solvent composition was maintained for another 3 min (36–39 min), and then the solvent composition was returned to the initial composition by 42 min. For the purification of 18F-FBO-(ZHER2:477)2, the mobile phases were 95% solvent A and 5% solvent B (0–3 min), 35% solvent A and 65% solvent B (at 30 min), and then 85% solvent B and 15% solvent A (33–36 min); the last solvent composition was maintained for another 3 min (36–39 min), and then the solvent composition was returned to the initial composition by 42 min. The HPLC fractions containing the 18F-labeled Affibody molecules were collected, combined, and treated with a rotary evaporator to dry the product. The radiolabeled Affibody molecules were reconstituted in phosphate-buffered saline and passed through a 0.22-μm Millipore filter into a sterile vial for in vitro and animal experiments.
Cell Assays
SKOV3 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (Invitrogen Life Technologies). The cells were maintained in a humidified atmosphere of 5% CO2 at 37°C, and the medium was changed every other day. A confluent monolayer was detached with trypsin and dissociated into a single-cell suspension for further cell culturing.
In vitro cell uptake assays for 18F-FBO-ZHER2:477 and 18F-FBO-(ZHER2:477)2 were performed with SKOV3 cells as previously described (23). In brief, the cells were used to seed 24-well tissue culture plates at a density of 0.2 × 106 cells per well and were allowed to attach overnight. After a wash with RPMI 1640 medium, the cells were incubated at 37°C for 0.5–2 h with the radioactive probes (1.11–6.29 kBq [0.03–0.17 μCi]) in 0.5 mL of medium. Nonspecific binding of the probes to the SKOV3 cells was determined by coincubation with nonradiolabeled Affibody molecules [10 μg of (ZHER2:477)2 was added to each sample for 18F-FBO-(ZHER2:477)2, for a final concentration of 1.43 μM; 1 μg of ZHER2 was added to each sample for 18F-FBO-ZHER2:477, for a final concentration of 0.28 μM]. The cells were rinsed 3 times with 0.01 M phosphate-buffered saline (pH 7.4) and 0.2% bovine serum albumin and lysed in 0.5 mL of 1.0 M NaOH for 5 min, and the radioactivity of the cells was measured. The protein concentrations of the samples were measured by use of the Bradford assay (Bio-Rad). All of the data were analyzed with the ORI-GIN6.0 computer program (Microcal Software Inc.).
Biodistribution Studies
All animal studies were performed in compliance with federal and local institutional rules for animal experimentation. Approximately 106 SKOV3 cells suspended in Matrigel (BD Biosciences) were implanted subcutaneously in the flanks of nude mice. Tumors were allowed to grow to a size of 500–750 mg (2–3 wk), and then the tumor-bearing mice were subjected to in vivo biodistribution and imaging studies.
For biodistribution studies, SKOV3 tumor–bearing mice (n =3 for each group) were injected with 18F-FBO-ZHER2:477 (0.37–1.11 MBq [10–30 μCi]; 0.125–0.375 μg) or 18F-FBO-(ZHER2:477)2 (0.74–1.665 MBq [20–45 μCi]; 0.5–1.125 μg) via the tail vein and sacrificed at various time points from 30 min to 3 h after injection. Tumor and normal tissues of interest were removed and weighed, and their radioactivity was measured with a γ-counter. The radioactivity uptake in the tumor and normal tissues was expressed as a percentage of the injected radioactive dose per gram of tissue (%ID/g). For testing of the in vivo HER2-targeting specificity of 18F-FBO-ZHER2, nude mice bearing SKOV3 tumors (n =3 for each group) were treated with either 500 μg of trastuzumab (tail vein injection, 72 h before radiolabeled probe injection) or 300 μg of ZHER2:477 (tail vein injection, 1 h before radiolabeled probe injection). The mice were injected with 18F-FBO-ZHER2:477, and biodistribution at 1 h after injection was examined.
Small-Animal PET Imaging
PET of tumor-bearing mice was performed by use of a micro-PET R4 rodent-model scanner (Siemens Medical Solutions). Mice bearing SKOV3 tumors were injected with 18F-FBO-ZHER2:477 (0.444–0.814 MBq [12–22 μCi]; 0.15–0.275 μg) or 18F-FBO-(ZHER2:477)2 (1.85–2.035 MBq [50–55 μCi]; 1.25–1.375 μg) via the tail vein. At various times after injection (0.5 and 1 h), the mice were anesthetized with 2% isoflurane and placed in the prone position and near the center of the field of view of the scanner. Static scans (5 min) were obtained, and the images were reconstructed by use of a 2-dimensional ordered-subsets expectation maximization algorithm. No background correction was performed. Regions of interest (ROIs; 5 pixels for coronal and transaxial slices) were drawn over the tumors on decay-corrected whole-body coronal images. The maximum counts per pixel per minute were obtained from the ROIs and converted to counts per milliliter per minute by use of a calibration constant. On the basis of the assumption of a tissue density of 1 g/mL, the ROIs were converted to counts per gram per minute. Image ROI–derived %ID/g values were determined by dividing counts per gram per minute by injected dose. No attenuation correction was performed.
Statistical Methods
Statistical analysis was performed by use of the Student t test for unpaired data. A 95% confidence level was chosen to determine the significance between groups, with P values of less than 0.05 indicating significant differences.
RESULTS
Chemistry and Radiochemistry
A distal C-terminal cysteine residue in the engineered anti-HER2 Affibody molecules [ZHER2:477 and (ZHER2:477)2] provides a thiol moiety that can be used for site-specific modification of the proteins. A bifunctional linker (Mal-AO) consisting of 2 orthogonal groups, a thiol-reactive maleimide group for conjugation to the engineered cysteine and an 18/19F-aldehyde–reactive aminooxy group, was synthesized. This linker was prepared by reacting N-(2-aminoethyl) malemide with 2-(tert-butoxycarbonylaminooxy)acetic acid under carbodiimide-mediated coupling conditions, yielding the protected form of the linker. The aminooxy group was deprotected by acid cleavage of the tert-butoxycarbonyl group to yield the final product at a quantitative yield (Fig. 1).
The monomeric and dimeric Affibody molecules [ZHER2:477 and (ZHER2:477)2, respectively] were selectively conjugated to the maleimide–aminooxy linker, with quantitative coupling yields. For both constructs, the reactions were completed in 2 h. The monomeric and dimeric aminooxy conjugates [ZHER2:477-ONH2 and (ZHER2:477)2-ONH2, respectively] were easily purified by dialysis, with recovery yields of 82% and 79%, respectively. Electrospray ionization–mass spectrometry (ESI-MS) analysis of the final products (purity of >95%, as determined by HPLC analysis) confirmed the absence of starting material, and only the products expected, with molecular masses of 6,962.58 Da and 14,431.45 Da, were obtained for the monomeric and dimeric constructs, respectively [calculated molecular weights (MWs) for constructs ZHER2:477-ONH2 and (ZHER2:477)2-ONH2 were 6,963.55 and 14,431.26, respectively]. The aminooxy-functionalized Affibody molecules were reacted with 4-FBA to prepare nonradioactive FBO-ZHER2:477 and FBO-(ZHER2:477)2 as standard compounds. For both preparations, recovery yields were typically 70%–90%, and coupling yields were quantitatively determined by ESI-MS characterization (i.e., no detected starting material). Purified FBO-ZHER2:477 and FBO-(ZHER2:477)2 were characterized by ESI-MS. The measured MWs for both constructs were consistent with the expected MWs [for FBO-ZHER, calculated MW = 7,069.64 and found MW = 7,068.94; for FBO-(ZHER2)2, calculated MW = 14,537.67 and found MW = 14,538.59].
Similarly, 4-18F-FBA was prepared and conjugated to the Affibody molecules [ZHER2:477-ONH2 and (ZHER2:477)2-ONH2] (coupling yields: 30%–40%). Under the HPLC conditions used, the retention times of 18F-FBO-ZHER2 and 18F-FBO-(ZHER2:477)2 were found to be 33.6 and 29.0 min, respectively. The HPLC radiochromatogram of purified 18F-FBO-ZHER2:477 is shown in Figure 2. Both products were found to be greater than 95% radiochemically and chemically pure, as determined by analytic HPLC. The overall radiochemical yield ranged from 13% to 18% (end of synthesis; corrected for decay), and the specific activities were 10.4–20.8 MBq/nmol (20–40 μCi/μg) and 13.0–20.7 MBq/nmol (50–80 μCi/μg) for the 14-kDa and 7-kDa Affibody bioconjugates, respectively. The total time needed for the synthesis of the 18F-labeled Affibody molecules was about 100 min.
FIGURE 2.

HPLC radiochromatogram of purified 18F-FBO-ZHER2:477.
HER2-Binding Affinities of Affibody Bioconjugates
The binding affinities of the various anti-HER2 Affibody constructs were evaluated in vitro by surface plasmon resonance. The sensorgrams for ZHER2:477 and FBO-(ZHER2:477)2 are shown in Figure 3. The binding affinities of ZHER2:477-ONH2, FBO-ZHER2:477, (ZHER2:477)2-ONH2, and FBO-(ZHER2:477)2 were 400, 430, 180, and 200 pM, respectively. In general, the binding affinity of ZHER2:477 was slightly lower than the binding affinity of the dimeric construct, (ZHER2:477)2, but was still in the picomolar range. In addition, conjugation of a small molecule like 4-FBA to the Affibody had a minor effect on the affinity of the precursor, arguably considered nonsignificant. An advantageous combination of the properties of these molecules is their very fast “on rate,” with association constants in the range of 105–106 for the FBO monomer and the FBO dimer (association time is over 2 min), and their very slow “off rate,” with dissociation constants in the range of 10–4–10–5 (disassociation time is over 15 min), close to the sensitivity limits of the surface plasmon resonance assay.
FIGURE 3.

Biosensor binding studies of ZHER2:477 (A) and FBO-(ZHER2:477)2 (B). Sensorgrams were obtained after injection of various concentrations of purified Affibody molecules onto sensor chip flow cell surface containing amine-coupled Fc–HER2 chimeric protein. Resp. Diff. = response difference.
In Vitro Cell Assays of Affibody Bioconjugates
Tumor cell line SKOV3, with a high level of HER2 expression (Abhijit De, unpublished data, January 2007), was used for evaluation of the HER2-targeting ability of 18F-labeled Affibody molecules. Levels of cell uptake of 18F-FBO-ZHER2:477 and 18F-FBO-(ZHER2:477)2 at 37°C over a 2-h incubation period are shown in Figures 4A and 4B, respectively. 18F-FBO-ZHER2:477 quickly accumulated in SKOV3 cells and reached a value of 0.00476 (cell counts per minute [cpm]/medium cpm/μg of protein/μL) at 0.5 h. The uptake was maintained at almost the same level until 2 h. A similar cell uptake pattern was observed for 18F-FBO-(ZHER2:477)2, but it showed a level of uptake 3-fold higher than that of the monomeric Affibody at 0.5 h: 0.01447 (cell cpm/medium cpm/μg of protein/μL). When both probes were incubated with large excesses of nonradioactive Affibody molecules, their levels of uptake in SKOV3 cells were inhibited significantly (P < 0.05) at all incubation time points. For example, uptake values of only 0.00329 ± 0.00027 (mean ± SD) and 0.00623 ± 0.00046 at 0.5 h of incubation were observed for 18F-FBO-ZHER2:477 and 18F-FBO-(ZHER2:477)2, respectively. Overall, these results strongly suggested the HER2-binding specificity of 18F-FBO-ZHER2:477 and 18F-FBO-(ZHER2:477)2.
FIGURE 4.
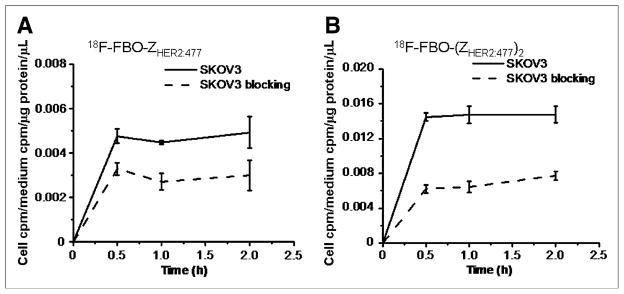
Uptake of 18F-FBO-ZHER2:477 (A) and 18F-FBO-(ZHER2:477)2 (B) in SKOV3 tumors over time at 37°C in presence or absence of nonradioactive Affibody molecules. All results, expressed as percentage of activity, are mean of triplicate measurements ± SD.
In Vivo Biodistribution Studies
The in vivo biodistribution of 18F-FBO-ZHER2:477 and 18F-FBO-(ZHER2:477)2 was examined by use of a SKOV3 human ovarian tumor–bearing mouse model. The biodistributions of 18F-FBO-ZHER2:477 at 0.5, 1, and 3 h and of 18F-FBO-(ZHER2:477)2 at 0.5, 1, and 2.7 h are shown in Figure 5. For 18F-FBO-ZHER2:477, rapid and high levels of radioactivity accumulation in the HER2-overexpressing SKOV3 tumors were observed at early time points (4.77 ± 0.78 %ID/g at 0.5 h after injection). Tumor uptake was slightly reduced, to 3.61 ± 0.29 %ID/g, at 3 h after injection (Fig. 5A). However, the uptake of 18F-FBO-(ZHER2:477)2 in SKOV3 tumors measured only 2.03 ± 0.31 and 1.08 ± 0.15 %ID/g at 0.5 and 2.7 h after injection, respectively; these values were significantly lower than the values for the 18F-labeled monomeric construct (P < 0.05) (Fig. 5D).
FIGURE 5.

Biodistribution results (A, B, D, and E) and tumor-to-normal tissue ratios (C and F) for 18F-FBO-ZHER2:477 (A, B, and C) and 18F-FBO-(ZHER2:477)2 (D, E, and F) in nude mice bearing subcutaneously xenotransplanted SKOV3 tumors (human ovarian cancer) (T). Data are expressed as %ID/g at various times after intravenous injection of 18F-FBO-ZHER2:477 (0.37–1.11 MBq [10–30 μCi]) or 18F-FBO-(ZHER2:477)2 (0.74–1.67 MBq [20–45 μCi]) (n = 3 for each group). 18F-FBO-ZHER2:477 showed significantly higher uptake in SKOV3 tumors than 18F-FBO-(ZHER2:477)2 (P < 0.05).
Both 18F-labeled Affibody bioconjugates displayed relatively rapid blood clearance; blood uptake values of 1.50 ± 0.35 and 1.56 ± 0.29 %ID/g were observed at 1 h after injection for 18F-FBO-ZHER2:477 and 18F-FBO-(ZHER2:477)2, respectively. However, at 0.5 h after injection, the 18F-labeled dimeric construct exhibited a higher level of blood uptake than the monomeric construct (3.81 ± 0.50 and 1.97 ± 0.24 %ID/g, respectively; P < 0.05). Moreover, 18F-FBO-(ZHER2:477)2 also displayed a significantly higher level of muscle uptake than 18F-FBO-ZHER2:477 at 0.5 h after injection but displayed similar levels of uptake at later time points (Figs. 5A and 5D). Both radiolabeled proteins displayed high levels of renal uptake at early time points (e.g., 0.5 h after injection; over 20 %ID/g) but moderate levels of uptake at later time points (Figs. 5B and 5E). The monomeric construct also demonstrated a higher level of radioactivity accumulation in the kidneys than the dimeric construct (7.37 ± 0.85 and 2.08 ± 0.45 %ID/g at 3 and 2.7 h after injection, respectively; P < 0.05). 18F-FBO-(ZHER2:477)2 showed a level of liver uptake about 2-fold higher than that of 18F-FBO-ZHER2:477 at 0.5 h after injection and showed a similar level at 1 h after injection. These data clearly suggested that 18F-labeled dimeric and monomeric Affibody constructs were cleared through the hepatobiliary, renal, and urinary systems. Importantly, it was found that 18F-FBO-ZHER2:477 showed significantly higher SKOV3 tumor-to-normal tissue (blood, muscle, liver, and lung) ratios than 18F-FBO-(ZHER2:477)2 at all time points investigated (Figs. 5C and 5F). At 3 h after injection, the tumor-to-blood and tumor-to-muscle ratios of 18F-FBO-ZHER2:477 were 12.72 ± 2.94 and 28.03 ± 8.28, respectively.
Because 18F-FBO-ZHER2:477 demonstrated high levels of tumor accumulation and favorable pharmacokinetic properties, its in vivo tumor-targeting specificity was further evaluated. It was found that the pretreatment of SKOV3 tumor–bearing mice with ZHER2:477 specifically reduced the SKOV3 tumor uptake of 18F-FBO-ZHER2:477 to about 36% of the corresponding control tumor uptake (4.77 ± 0.88 and 1.73 ± 0.37 %ID/g, respectively; P < 0.05). However, no significant difference was observed for uptake in all of the other normal tissues (Fig. 6A). Similarly, the pretreatment of SKOV3 tumor–bearing mice with a HER2-binding protein, trastuzumab (500 μg each), for 72 h also specifically inhibited the uptake of 18F-FBO-ZHER2:477, to about 47% of the control tumor uptake (4.77 ± 0.88 and 2.25 ± 0.49 %ID/g, respectively; P < 0.05), but did not alter its accumulation in most of the other normal organs, except for the kidneys (Fig. 6A). Consequently, the SKOV3 tumor-to-normal tissue ratios were all significantly reduced by ZHER2 and trastuzumab treatments (P < 0.05) (Fig. 6B).
FIGURE 6.

In vivo HER2 tumor–targeting specificity of 18F-FBO-ZHER2:477. One-hour biodistribution results (A) and tumor-to-normal tissue ratios (B) are shown for 18F-FBO-ZHER2:477 with or without pretreatment with either 300 μg of ZHER2:477 or 500 μg of trastuzumab. 18F-FBO-ZHER2:477 SKOV3 tumor (T) uptake was significantly inhibited by ZHER2:477 or trastuzumab (P < 0.05) (n = 3).
Small-Animal PET Imaging
Decay-corrected coronal (top row) and transaxial (bottom row) small-animal PET images of a SKOV3 tumor–bearing mouse at 0.5, 1, 2, and 4 h after tail vein injection of 18F-FBO-ZHER2:477 or 18F-FBO-(ZHER2:477)2 are shown in Figures 7A and 7B, respectively. For 18F-FBO-(ZHER2:477)2, the SKOV3 tumor was visible, especially at later time points, but with low tumor-to-background contrast. For 18F-FBO-ZHER2:477, the SKOV3 tumor was clearly visualized, with much better tumor-to-background contrast, at 0.5–4 h after injection. Also observed were high levels of activity accumulation in the kidneys and part of the gastrointestinal tract for both tracers. Quantification of activity accumulation in the SKOV3 tumor and in the contralateral muscle tissue showed that 18F-FBO-ZHER2:477 had about 2-fold-higher tumor uptake but lower muscle uptake than 18F-FBO-(ZHER2:477)2 at 0.5–4 h. The absolute tumor and muscle uptake values (in %ID/g) obtained from PET image data were consistent with the findings obtained in the biodistribution studies.
FIGURE 7.

Decay-corrected coronal (top) and transaxial (bottom) small-animal PET images of nude mice bearing SKOV3 tumors on right shoulder at 0.5, 1, 2, and 4 h after tail vein injection of 18F-FBO-ZHER2:477 (A) and 18F-FBO-(ZHER2:477)2 (B). Arrows indicate locations of tumors. (C) Tumor and muscle time–activity curves derived from multiple-time-point small-animal PET images of mice bearing SKOV3 tumors. Data are shown as mean ± SD %ID/g (n = 3).
DISCUSSION
The development of PET molecular probes is an important area of molecular imaging that also represents a great challenge, as evidenced by the fact that only a small number of PET probes are clinically available. It is expected that the combination of various general strategies should dramatically facilitate the process of imaging probe development. These translatable approaches include, but are not limited to, high-throughput technologies for the identification of ligands for specific disease biomarkers, reliable and site-specific conjugation chemistries for coupling reporting moieties (such as radionuclides and fluorescent molecules) to binding ligands, and techniques for rapid evaluation of the in vitro and in vivo properties of the resulting bioconjugates. Antibodies and their fragments traditionally have been used as valuable platform molecules for imaging and therapeutic agent development. A vast number of antibodies against a variety of antigens can be easily generated by use of conventional hybridoma technology. Antibodies labeled with long-lived PET radioisotopes have been investigated and have been shown to have broad applications in tumor detection, tumor target validation, radiation dose estimation, and optimization and individualization of antibody-based therapies (24). However, the slow pharmacokinetic properties of antibodies makes them less than ideal as a targeting platform for PET probes, especially for 18F-based imaging agents. More recently, many in vitro display technologies (phages, bacteria, yeasts, ribosomes, and messenger RNA) in conjunction with rational protein engineering have been used to construct nonimmunoglobulin protein libraries with a defined scaffold. The rapid progress of protein display technologies has led to the discovery of many novel small proteins and peptides with high affinity and specificity for a variety of molecular targets (25,26). These proteins are superior sources for the development of biodrugs and molecular probes (27). Importantly, their relatively small size generally leads to fast clearance, rapid tumor accumulation, and relatively short in vivo biologic half-life (6–13), which potentially matches well the physical half-life of 18F, one of the most widely available PET radioisotopes. We hypothesized that this small protein scaffold would have great potential for the development of novel PET probes for a variety of targets. Thus, Affibody molecules with a 3-helix-bundle scaffold structure were selected for radiolabeling with 18F and evaluated in this work as a proof-of-concept study.
A strategy based on oxime chemistry has been developed for the radiofluorination of Affibody molecules. Site-specific labeling of a protein is critical to preserving the biologic activity of the biomolecule. A cysteine residue was engineered into the Affibody molecule C terminus, providing a thiol moiety to which site-selective chemistry could be applied. A bifunctional linker (Mal-AO) consisting of a thiol-reactive maleimide group and an aldehyde-reactive aminooxy group was prepared. It was then specifically conjugated to the Affibody molecules via the well-documented thiol-maleimide reaction, resulting in a protein with amino-oxy functionality (Fig. 1). This aminooxy-functionalized protein could react efficiently and specifically with an aldehyde-functionalized 18F-synthon, such as 4-18F-FBA, under slightly acidic conditions, thereby forming a stable, nonreversible oxime linkage (22). Simultaneously, the acidic conditions avoided the potential reaction of the aldehyde with amine groups present on the Affibody molecules to form Schiff bases (Mohammad Namavari, unpublished data, March 2007). The aminooxy functionalization of the Affibody molecules allowed the opportunity for radiochemistry with fewer steps and thereby higher yield through a scheme that left the radiosynthesis step until the end of the procedure but still enabled selective conjugation. Only 2 radiosynthesis steps were involved in this procedure, and a nonoptimized radiochemical yield (decay corrected) of 13%–18% was achieved in a total preparation time of 100 min. The radiochemical yield reported here was lower than that of the 18F-labeled peptides (60%–80%) reported by Poethko et al. (22), likely for the following reasons: the concentration of the aminooxy precursor [ZHER2:477-ONH2 or (ZHER2:477)2-ONH2; 0.238 mmol/L] was lower than that of the aminooxy–peptide precursor (0.5 mmol/L), and the pH used for Affibody molecule radio-fluorination (pH 4) was higher than those found to be optimal for 18F-peptides (pH 2–pH 3). Thus, further optimization of our current labeling procedure is under investigation and may result in a higher radiolabeling yield. Compared with an amine-reactive 18F-synthon, such as N-succinimidyl-4-18F-fluorobenzoate (18F-SFB), 4-18F-FBA–aminooxy chemistry has some distinctive advantages. First, 4-18F-FBA–aminooxy chemistry provides a method for the site-specific radiofluorination of a protein, whereas 18F-SFB can react with any lysine presenting in the protein, a property that may compromise the biologic activity of the protein. Second, 18F-SFB labeling takes 173 min for the 4-step radiosynthesis of an 18F-SFB-labeled diabody. The overall decay-corrected radiochemical yield is only 1.4% (28). The strategy for the site-specific radiofluorination of an aminooxy-functionalized protein with an aldehyde-functionalized 18F-synthon developed in the present study has a relatively short radiosynthesis time and a moderate yield (100 min; 13%–18% yield). It can be used for the 18F labeling of many other proteins and has potential for generating PET probes for clinical applications.
The biologic properties of 18F-FBO-ZHER2:477 and 18F-FBO-(ZHER2:477)2 were evaluated by in vitro cell assays, biodistribution studies, and small-animal PET imaging studies. Both 18F-FBO-ZHER2:477 and 18F-FBO-(ZHER2:477)2 showed significantly higher uptake in SKOV3 cells than ZHER2:477, demonstrating their HER2-binding specificity in vitro. Furthermore, 18F-FBO-ZHER2:477 showed lower cell uptake than 18F-FBO-(ZHER2:477)2. This result was likely caused by the higher HER2-binding affinity of 18F-FBO-(ZHER2:477)2 (200 vs. 430 pM), indicating that multimerization of proteins does help to improve the binding affinity and cell uptake of ligands. This result was also consistent with other find-ings reported in the literature (29). However, nonspecific SKOV3 cell uptake was about 2-fold higher with 18F-FBO-(ZHER2:477)2 than with 18F-FBO-ZHER2:477 (Fig. 4).
Evaluation of both probes in mice demonstrated that 18F-FBO-ZHER2:477 is a promising agent for HER2 imaging. It rapidly localized in SKOV3 tumors and showed good tumor uptake, retention, and tumor-to-muscle ratios (Figs. 5 and 7). Even at 0.5 h after injection, SKOV3 tumors could be clearly visualized by PET, with good contrast. Higher levels of tumor-to-normal tissue contrast were achieved at later times, such as 1 and 3 h. The in vivo HER2-binding specificity of 18F-FBO-ZHER2:477 was also verified by the reduced SKOV3 tumor uptake obtained after pretreatment with a known HER2-targeting ligand (ZHER2 or trastuzumab). Overall, the relatively easy preparation, favorable pharmacokinetic properties (such as rapid and high tumor uptake and good tumor contrast), and high specificity of 18F-FBO-ZHER2:477 render it a useful agent for HER2 PET and related applications.
In contrast, although 18F-FBO-(ZHER2:477)2 showed higher affinity and cell uptake, its in vivo performance was quite poor: low tumor uptake and relatively high uptake in and slow clearance from normal tissues (such as blood, muscle, liver, lung, and spleen). These data suggest that the high binding affinity of a probe does not necessarily correspond to high tumor uptake in vivo. Multimerization of proteins may not always be an effective way to improve the tumor-targeting ability of a probe. Compared with a monomer, the larger size of a dimer may deter its tumor penetration ability and prolong its clearance. Moreover, the charge and polarity of a probe may also be altered by dimerization and thus affect its biodistribution substantially.
Many other radiohalogens (125I, 76/77/82Br, and 211At) and radiometals (99mTc, 111/114mIn, 90Y, 177Lu, and 68Ga) have been used to label Affibody molecules (5–15). Our data are generally consistent with the findings reported in these published studies. For example, it was reported that 99mTc-CGG-ZHER2:342 displayed uptake levels of 3.36 ± 0.78 and 4.23 ± 1.03 %ID/g in SKOV3 tumors at 1 and 4 h after injection, respectively, whereas the corresponding levels in the kidneys were 67.62 ± 12.85 and 58.84 ± 8.76 %ID/g (9). In the present study, the levels of uptake of 18F-FBO-ZHER2:477 in SKOV3 tumors at 1 and 3 h after injection were 4.77 ± 0.88 and 3.61 ± 0.29 %ID/g, respectively, and the corresponding levels in the kidneys were 18.81 ± 1.09 and 7.37 ± 0.85 %ID/g. Overall, radiolabeled Affibody molecules rapidly accumulated in tumors, with high uptake, but they typically showed high uptake in the kidneys as well. Good tumor-to-normal tissue ratios were achieved at early times after injection of the probes. It was also found that radiolabeled dimeric Affibody molecules showed poorer performance (tumor targeting and clearance) than monomeric Affibody molecules (8). In a previously published study, trastuzumab failed to block SKOV3 tumor uptake of 177Lu-DOTA-ZHER2:342-pep2 (6), whereas it blocked 18F-FBO-ZHER2:477 tumor uptake in the present study. Although the cause of these observations is not completely clear, different routes of administration of the antibodies in these 2 studies (subcutaneous in the previously published study vs. tail vein injection in the present study) may be a factor. Moreover, different clones of anti-HER2 Affibody molecules used in these 2 studies may be another explanation.
CONCLUSION
A strategy for radiolabeling Affibody molecules has been successfully developed with oxime chemistry and anti-HER2 Affibody molecules as a model platform. This strategy can potentially be translated to a universal approach for other applications. Moderate radiochemical yield and specific activity of the probes can be reliably obtained in a relatively short time. Biodistribution and small-animal PET imaging studies further demonstrated that 18F-FBO-ZHER2:477 is a promising PET probe for imaging HER2 expression in living mice. The difference in performance between the monomer and the dimer demonstrated the importance of the fine balance between good affinity and size effects, which could influence penetration and profile properties. In addition, the combination of the high resolution, sensitivity, and quantitative ability of PET with the high specificity, selectivity, and fast pharma-cokinetic profile of the Affibody protein scaffold make engineered protein–based PET an attractive strategy for further exploration.
Acknowledgments
This work was supported, in part, by Medical Diagnostics, GE Healthcare, the National Cancer Institute (NCI), Small-Animal Imaging Resource Program (SAIRP) grant R24 CA93862, and NCI In Vivo Cellular Molecular Imaging Center (ICMIC) grant P50 CA114747. We thank Dr. David Dick for 18F production, Dr. Frederick T. Chin for modification of a GE TRACERlab FX-FN synthetic module for the radiosynthesis of 4-18F-FBA, Dr. Joshua Hoerner and Hans Grade of GE Global Research for mass spectrometry analysis, and Dr. Alan Cuthbertson and Dr. Alex Gibson of GE Healthcare for their helpful suggestions and reviewing of the article.
References
- 1.Meric-Bernstam F, Hung MC. Advances in targeting human epidermal growth factor receptor-2 signaling for cancer therapy. Clin Cancer Res. 2006;12:6326–6330. doi: 10.1158/1078-0432.CCR-06-1732. [DOI] [PubMed] [Google Scholar]
- 2.Ferretti G, Felici A, Papaldo P, Fabi A, Cognetti F. HER2/neu role in breast cancer: from a prognostic foe to a predictive friend. Curr Opin Obstet Gynecol. 2007;19:56–62. doi: 10.1097/GCO.0b013e328012980a. [DOI] [PubMed] [Google Scholar]
- 3.Smith-Jones PM, Solit DB, Akhurst T, Afroze F, Rosen N, Larson SM. Imaging the pharmacodynamics of HER2 degradation in response to Hsp90 inhibitors. Nat Biotechnol. 2004;22:701–706. doi: 10.1038/nbt968. [DOI] [PubMed] [Google Scholar]
- 4.Tang Y, Wang J, Scollard DA, et al. Imaging of HER2/neu-positive BT-474 human breast cancer xenografts in athymic mice using 111In-trastuzumab (Herceptin) Fab fragments. Nucl Med Biol. 2005;32:51–58. doi: 10.1016/j.nucmedbio.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Baum R, Orlova A, Tolmachev V, Feldwisch J. Receptor PET/CT and SPECT using an Affibody molecule for targeting and molecular imaging of HER2-positive cancer in animal xenografts and human breast cancer patients [abstract] J Nucl Med. 2006;47(suppl):108P. [Google Scholar]
- 6.Orlova A, Tolmachev V, Pehrson R, et al. Synthetic Affibody molecules: a novel class of affinity ligands for molecular imaging of HER2-expressing malignant tumors. Cancer Res. 2007;67:2178–2186. doi: 10.1158/0008-5472.CAN-06-2887. [DOI] [PubMed] [Google Scholar]
- 7.Tolmachev V, Orlova A, Pehrson R, et al. Radionuclide therapy of HER2-positive microxenografts using a 177Lu-labeled HER2-specific Affibody molecule. Cancer Res. 2007;67:2773–2782. doi: 10.1158/0008-5472.CAN-06-1630. [DOI] [PubMed] [Google Scholar]
- 8.Orlova A, Magnusson M, Eriksson TL, et al. Tumor imaging using a picomolar affinity HER2 binding Affibody molecule. Cancer Res. 2006;66:4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 9.Tran T, Engfeldt T, Orlova A, et al. In vivo evaluation of cysteine-based chelators for attachment of 99mTc to tumor-targeting Affibody molecules. Bioconjug Chem. 2007;18:549–558. doi: 10.1021/bc060291m. [DOI] [PubMed] [Google Scholar]
- 10.Tolmachev V, Nilsson FY, Widström C, et al. 111In-Benzyl-DTPA-ZHER2:342, an Affibody-based conjugate for in vivo imaging of HER2 expression in malignant tumors. J Nucl Med. 2006;47:846–853. [PubMed] [Google Scholar]
- 11.Orlova A, Nilsson FY, Wikman M, et al. Comparative in vivo evaluation of technetium and iodine labels on an anti-HER2 Affibody for single-photon imaging of HER2 expression in tumors. J Nucl Med. 2006;47:512–519. [PubMed] [Google Scholar]
- 12.Orlova A, Rosik D, Sandstrom M, Lundqvist H, Einarsson L, Tolmachev V. Evaluation of [(111/114m)In]CHX-A″-DTPA-Z(HER2:342), an Affibody ligand conjugate for targeting of HER2-expressing malignant tumors. Q J Nucl Med Mol Imaging. 2007;51:1–10. [PubMed] [Google Scholar]
- 13.Fortin MA, Orlova A, Malmstrom PU, Tolmachev V. Labelling chemistry and characterization of [90Y/177Lu]-DOTA-ZHER2:342-3 Affibody molecule, a candidate agent for locoregional treatment of urinary bladder carcinoma. Int J Mol Med. 2007;19:285–291. [PubMed] [Google Scholar]
- 14.Mume E, Orlova A, Larsson B, et al. Evaluation of ((4-hydroxyphenyl)ethyl) maleimide for site-specific radiobromination of anti-HER2 Affibody. Bioconjug Chem. 2005;16:1547–1555. doi: 10.1021/bc050056o. [DOI] [PubMed] [Google Scholar]
- 15.Steffen AC, Almqvist Y, Chyan MK, et al. Biodistribution of 211At labeled HER-2 binding Affibody molecules in mice. Oncol Rep. 2007;17:1141–1147. [PubMed] [Google Scholar]
- 16.Artemov D, Mori N, Ravi R, Bhujwalla ZM. Magnetic resonance molecular imaging of the HER-2/neu receptor. Cancer Res. 2003;63:2723–2727. [PubMed] [Google Scholar]
- 17.Hilger I, Leistner Y, Berndt A, et al. Near-infrared fluorescence imaging of HER-2 protein over-expression in tumour cells. Eur Radiol. 2004;14:1124–1129. doi: 10.1007/s00330-004-2257-9. [DOI] [PubMed] [Google Scholar]
- 18.Tada H, Higuchi H, Wanatabe TM, Ohuchi N. In vivo real-time tracking of single quantum dots conjugated with monoclonal anti-HER2 antibody in tumors of mice. Cancer Res. 2007;67:1138–1144. doi: 10.1158/0008-5472.CAN-06-1185. [DOI] [PubMed] [Google Scholar]
- 19.Nilsson FY, Tolmachev V. Affibody molecules: new protein domains for molecular imaging and targeted tumor therapy. Curr Opin Drug Discov Devel. 2007;10:167–175. [PubMed] [Google Scholar]
- 20.Wikman M, Steffen AC, Gunneriusson E, et al. Selection and characterization of HER2/neu-binding Affibody ligands. Protein Eng Des Sel. 2004;17:455–462. doi: 10.1093/protein/gzh053. [DOI] [PubMed] [Google Scholar]
- 21.Okarvi SM. Recent progress in fluorine-18 labelled peptide radiopharmaceuticals. Eur J Nucl Med. 2001;28:929–938. doi: 10.1007/s002590100508. [DOI] [PubMed] [Google Scholar]
- 22.Poethko T, Schottelius M, Thumshirn G, et al. Two-step methodology for high-yield routine radiohalogenation of peptides: 18F-labeled RGD and octreotide analogs. J Nucl Med. 2004;45:892–902. [PubMed] [Google Scholar]
- 23.Cheng Z, Xiong Z, Subbarayan M, Chen X, Gambhir SS. 64Cu-Labeled alpha-melanocyte-stimulating hormone analog for microPET imaging of melanocortin 1 receptor expression. Bioconjug Chem. 2007;18:765–772. doi: 10.1021/bc060306g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verel I, Visser GW, van Dongen GA. The promise of immuno-PET in radioimmunotherapy. J Nucl Med. 2005;46(suppl):164S–171S. [PubMed] [Google Scholar]
- 25.Rothe A, Hosse RJ, Power BE. In vitro display technologies reveal novel biopharmaceutics. FASEB J. 2006;20:1599–1610. doi: 10.1096/fj.05-5650rev. [DOI] [PubMed] [Google Scholar]
- 26.Uchiyama F, Tanaka Y, Minari Y, Tokui N. Designing scaffolds of peptides for phage display libraries. J Biosci Bioeng. 2005;99:448–456. doi: 10.1263/jbb.99.448. [DOI] [PubMed] [Google Scholar]
- 27.Ladner RC. Polypeptides from phage display: a superior source of in vivo imaging agents. Q J Nucl Med. 1999;43:119–124. [PubMed] [Google Scholar]
- 28.Cai W, Olafsen T, Zhang X, et al. PET imaging of colorectal cancer in xenograft-bearing mice by use of an 18F-labeled T84.66 anti–carcinoembryonic antigen diabody. J Nucl Med. 2007;48:304–310. [PubMed] [Google Scholar]
- 29.Cheng Z, Wu Y, Xiong Z, Gambhir SS, Chen X. Near-infrared fluorescent RGD peptides for optical imaging of integrin αvβ3 expression in living mice. Bioconjug Chem. 2005;16:1433–1441. doi: 10.1021/bc0501698. [DOI] [PMC free article] [PubMed] [Google Scholar]