

Abstract
Introduction:
The information regarding therapeutically relevant genomic alterations in small cell lung cancer (SCLC) is not well developed. We analyzed the SCLC genome using an integrative approach to stratify the targetable alterations.
Methods:
We performed whole exon sequencing (n = 51) and copy number analysis (n =47) on surgically resected tumors and matched normal tissue samples from treatment-naive Japanese SCLC patients.
Results:
The demographics of the 51 patients included in this study were as follows: median age, 67 years (range, 42–86 years); female, 9 (18%); history of smoking, 50 (98%); and pathological stage I/II/III/IV, 28/13/9/1, respectively. The average number of nonsynonymous mutations was 209 (range, 41–639; standard deviation, 130). We repeatedly confirmed the high prevalence of inactivating mutations in TP53 and RB1, and the amplification of MYC family members. In addition, genetic alterations in the PI3K/AKT/mTOR pathway were detected in 36% of the tumors: PIK3CA, 6%; PTEN, 4%; AKT2, 9%; AKT3, 4%; RICTOR, 9%; and mTOR, 4%. Furthermore, the individual changes in this pathway were mutually exclusive. Importantly, the SCLC cells harboring active PIK3CA mutations were potentially targetable with currently available PI3K inhibitors.
Conclusions:
The PI3K/AKT/mTOR pathway is distinguishable in SCLC genomic alterations. Therefore, a sequencing-based comprehensive analysis could stratify SCLC patients by potential therapeutic targets.
Keywords: Lung cancer, Small cell, Genome, Comprehensive, PI3K/AKT/mTOR
Small cell lung cancer (SCLC) comprises approximately 15% of all lung cancers,1 and it is an exceptionally aggressive malignancy with a high proliferative index and an unusually strong predilection for early metastasis.2 Despite extensive basic and clinical research over the past 30 years, little progress has been made2 in treating this disease.
A better understanding of the genomic changes in SCLC is essential to identify new therapeutic targets. Genomic analyses have revealed genetically altered therapeutic targets in lung adenocarcinoma3–5 and squamous cell lung carcinoma.6 However, a systematic genomic analysis of SCLC is difficult because this cancer subtype is rarely treated surgically, resulting in the lack of suitable tumor specimens for comprehensive analysis.
Two reports regarding the comprehensive genomic analysis of SCLC with a relatively small number of samples have been published recently. These reports suggested that transcriptional deregulation (i.e., via RB1, SOX2, MYC family members and chromatin modifiers) might play a role in SCLC biology.7,8 However, to date, attempts to develop targeted therapies toward these transcriptional deregulations have had limited success.
Activating alterations to oncogenes, such as receptor tyrosine kinases (RTKs) and PI3K/AKT/mTOR pathway proteins,9–13 are regarded as successful therapeutic targets. We conducted a comprehensive genomic study in over 50 SCLC cases, and we found a higher penetrance of activating alterations of the PI3K/AKT/mTOR pathway that act in a mutually exclusive manner.
PATIENTS AND METHODS
Samples
This study was approved by the Institutional Review Board (IRB) of the National Cancer Center, Japan (IRB number: 2011-201). All data used in this study were obtained from a database at the Division of Thoracic Oncology, National Cancer Center Hospital East, Kashiwa, Japan.
From July 1992 to March 2012, we consecutively collected 1042 SCLC cases at our hospital. Fifty-five of these cases were included in the current study based on the following criteria: a surgical resection or mediastinoscopy was performed; a re-review confirmed a pathological diagnosis of SCLC; the tumor specimens contained a minimum of 70% tumor cells; enough tissue was obtained for a comprehensive analysis; the patient did not receive any neoadjuvant treatment; and the corresponding normal tissue, which was obtained from paraffin-embedded blocks of resected lung tissue that was microscopically free of cancer cells, was also available for analysis. We analyzed the exomes of these 55 samples to assess their mutational burden.
Depending on the tissue size, three to six sections (10 μm thickness) were cut. For the tumors showing a combined SCLC and other histology, only the SCLC compartment was dissected and used for analysis. Total DNA was obtained from formalin- (n = 43) or methanol-fixed (n = 12) paraffin-embedded tumors and matched normal tissue samples. All patients (100%) were Japanese.
Among these 55 cases, four exome data sets did not meet the sequence quality requirement and were excluded from further analyses. In addition, 48 samples received copy number analysis using single nucleotide polymorphism array data.
Procedures
The detailed experimental procedures are described in the Supplemental Information section (Supplemental Digital Content 1, http://links.lww.com/JTO/A625)
Whole Exon Sequencing and Copy Number Analysis
The Absolutely RNA FFPE kit (modified protocol for DNA extraction, Agilent Technologies, Santa Clara, CA) was used to prepare the DNA. Using 1 μg of dsDNA, quantified by Quant-iT PicoGreen dsDNA Reagent and Kits (Life Technologies, Carlsbad, CA), the exome-sequencing libraries were prepared. All exomes were captured using the SureSelect Human All Exon V4+UTRs Kit (Agilent Technologies) (71 mb). The exome capture libraries were sequenced by HiSeq 2000 (Illumina, San Diego, CA) to generate 100-bp paired-end data.
The Illumina HumanOmniExpress-FFPE BeadChip assay was used to analyze the genotype, DNA copy number, and loss of heterozygosity (LOH) in 48 primary-normal paired samples. All samples, except for 1 (n = 47), passed our quality control metrics for sample identity and data quality. A subset of 693,000 high-quality single nucleotide polymorphisms was selected for all analyses (Supplemental Figure 1, Supplemental Digital Content 2, http://links.lww.com/JTO/A626). A gene was considered copy number amplified if the calculated copy number in a sample was more than or equal to 4, and a gene was considered copy loss if the copy number in a sample was 0. Recurrent genomic regions with DNA copy gain and loss were identified using GISTIC, version 2.0.14,15
Identification of Significantly Mutated Genes
Significantly mutated genes were identified according to a previously reported protocol.16 The length of the total coding sequence regions was represented as N (approximately 39.8 mb). When a patient (patient i) harbored a total of mi single nucleotide variants (SNVs), the probability that the patient harbored SNVs in gene t (length: n) was calculated as follows:
The sum of Pt,i in 51 samples was represented as the expected number of cases with SNVs in gene t:
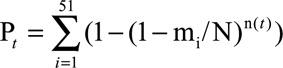 |
The p values of the observed number were calculated using the binomial probability function with R pbinom.
Cancer Census Genes and Analysis of Hot Spot Mutations
We defined the cancer census genes as follows: 487 genes listed in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (release version 64; http://cancer.sanger.ac.uk/cancergenome/progects/cosmic/) and 13 genes reported by Peifer et al.7 were considered candidate driver genes. To analyze hot spot mutations, mutation data from the SCLC cases were downloaded from the COSMIC database (release version 64 or 68).
Cell Lines and Assays
The cell lines were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) or hydrocortisone, insulin, transferrin, estradiol, and selenium (HITES) medium with 5% FBS. Then, 10,000 cells were plated in three replicates into 96-well plates. After 72 hours incubation with inhibitors, cell viability was analyzed with a WST-8 assay and a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Western blotting was performed as described in the Supplemental Information section (Supplemental Digital Content 1, http://links.lww.com/JTO/A625).
RESULTS
Patient Characteristics
The characteristics of the available patient exome data (n = 51) are summarized in Table 1 and Supplemental Table 3 (Supplemental Digital Content 3, http://links.lww.com/JTO/A627). Fifty patients received surgical resections and one patient received a mediastinoscopy. Forty-two patients were male and nine were female. The median age at the time of surgical resection was 67 years (range, 42–86 years). Of the 51 patients, 50 (98%) had a history of smoking, and the pathological stages were distributed as follows: stage I, 28 patients; stage II, 13 patients; stage III, nine patients; and stage IV, one patient. All patients were positive for at least one of the following neuroendocrine markers: CD56, chromogranin A, or synaptophysin.
TABLE 1.
Patient Characteristics

Somatic Point Mutations
The exome capture, sequencing, and analysis of the 51 SCLC tumor–normal tissue pairs identified 10,640 protein-altering somatic mutations, including 9376 missense, 707 nonsense, and 557 protein-altering insertions and/or deletions (INDEL) (Supplemental Table 4, Supplemental Digital Content 3, http://links.lww.com/JTO/A627). The SCLC tumors had an average of 209 protein-altering SNVs (range, 41–639) per case, with a mean nonsynonymous mutation rate of 6.15 mutations per mega-base (Supplemental Figure 2, Supplemental Digital Content 2, http://links.lww.com/JTO/A626). Significantly mutated genes are determined as Supplemental Table 5 (Supplemental Digital Content 3, http://links.lww.com/JTO/A627). Overall, 414 genes had a p value of less than 0.01 and 1321 genes had a p value of less than 0.05.
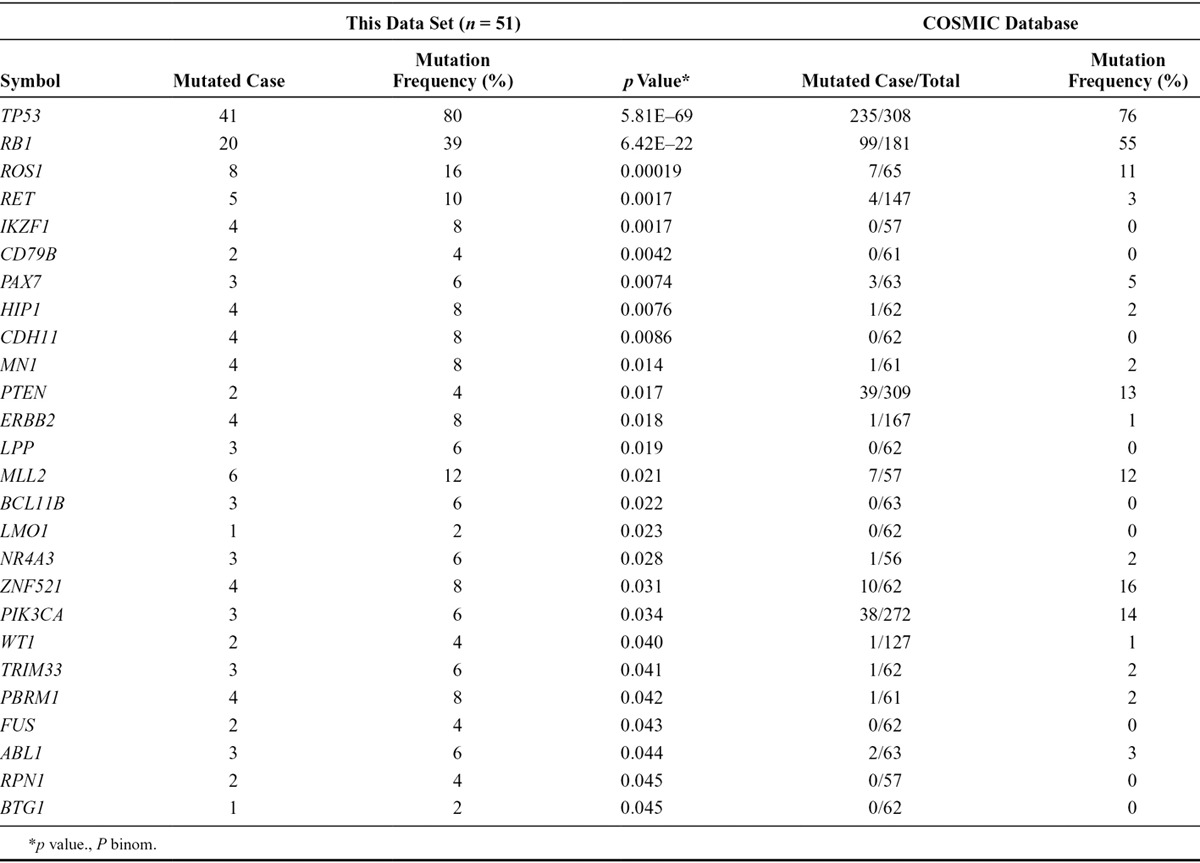
A description of the significantly mutated cancer census genes (p value <0.05 in our data set) is provided in Table 2. Notably, TP53 was the most frequently mutated gene (mutation frequency of 80%, p value of 5.81E–69). The mutation frequencies and p values of cancer census genes were 39% and 6.42E–22, 16% and 0.00019, 10% and 0.0017 for RB1, ROS1, and RET, respectively. Mutations of histone modifiers were also recurrently identified in this study; CREBBP was mutated in 6% of the patients, and EP300 was mutated in 4% of the patients (Fig. 1). Recently reported candidate driver genes were also recurrently identified in the PI3K/AKT/mTOR signaling pathway. Three patients (6%) had mutations in PIK3CA (one E545K, two others), and two patients (4%) had mutations in the PTEN C2 domain (Supplemental Figure 3, Supplemental Digital Content 2, http://links.lww.com/JTO/A626).
TABLE 2.
Significantly Mutated Cancer Census Genes (*p < 0.05 in This Study)
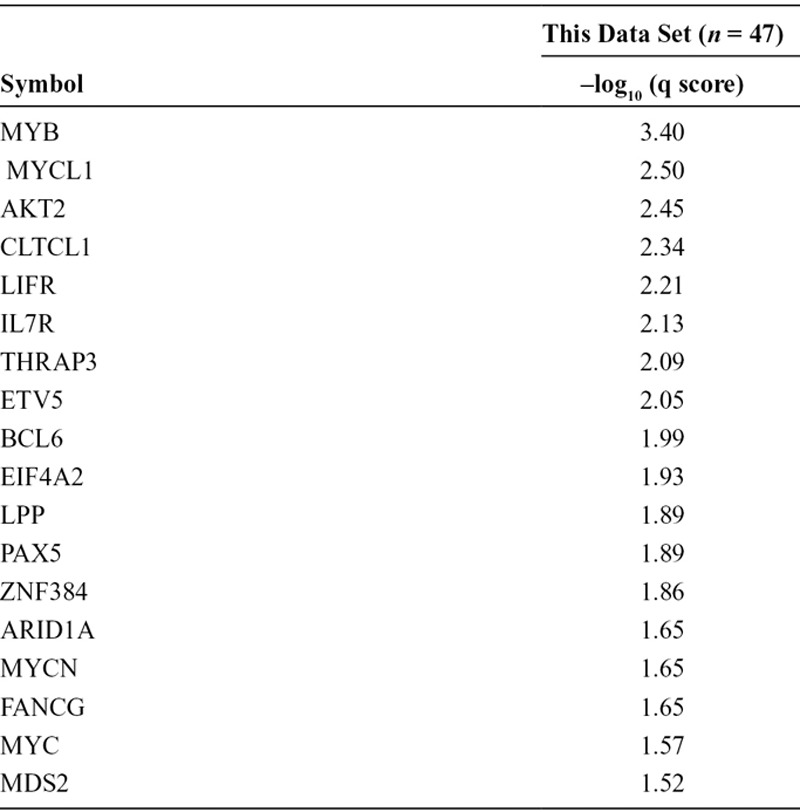
FIGURE 1.
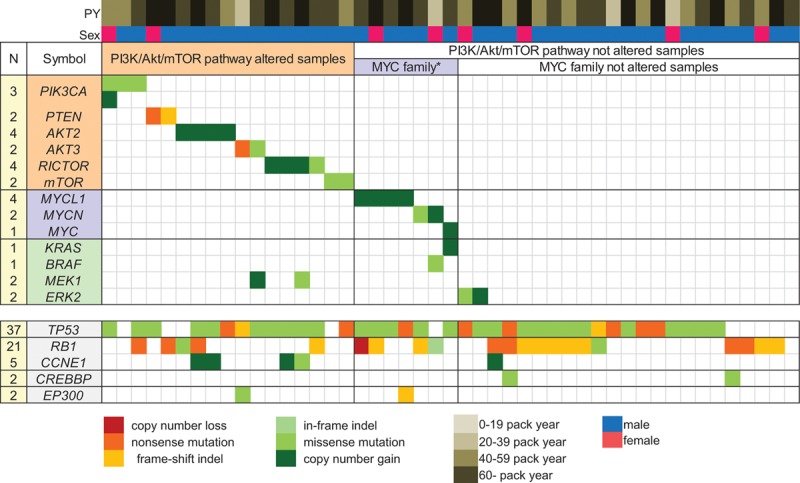
An overview of the key driver mutations and major associated clinical features of 47 SCLC samples. The number of events per gene is noted on the left. The genes are displayed as rows, and the samples are displayed as columns, with major associated clinical features.
PY, pack years; MYC family*, MYC family altered samples.
To validate the whole exon sequencing data, we performed Sanger sequencing for the variants including four SNVs of PIK3CA and ROS1 and a deletion of KIT in five individual tumor samples. All the variants detected using whole exon sequencing were reproduced using conventional Sanger sequencing (Supplemental Figure 4, Supplemental Digital Content 2, http://links.lww.com/JTO/A626). Furthermore, we designed a custom target-capturing panel containing all the coding exons of 244 genes. Four tumor samples were applied to the target resequencing, and all 41 SNVs or indels in these tumor genomes were reproducibly identified (Supplemental Table 6, Supplemental Digital Content 3, http://links.lww.com/JTO/A627).
Copy Number Analysis
Next, we applied a novel algorithm to identify the significant somatic copy number alterations (Supplemental Figure 5, Supplemental Digital Content 2, http://links.lww.com/JTO/A626). A description of frequently amplified cancer census genes (GISTIC –log10 q score ≥1.50) is provided in Table 3. MYC family members were frequently amplified (GISTIC q scores were 2.50, 1.65, and 1.57 for MYCL1, MYCN, and MYC, respectively). The amplifications affected MYCL1 (4/47 cases), MYC (1/47 cases), and MYCN (1/47 cases). All MYC family member amplifications (13% of cases) were mutually exclusive (Fig. 1). In addition, gene amplifications were frequently found in the PI3K/AKT/mTOR signaling pathway (GISTIC q scores were 2.45 and 1.22 for AKT2 and RICTOR, respectively). The gene amplifications in PI3K/AKT/mTOR signaling were observed in AKT2 (4/47 cases) and RICTOR (3/47 cases), and they were also mutually exclusive (Fig. 1). Previously reported amplifications involving SOX2 (1/47 cases) and KIT (1/47 cases) were also identified.
TABLE 3.
Frequently Mutated Cancer Census Genes (–log10 q score ≥1.5)
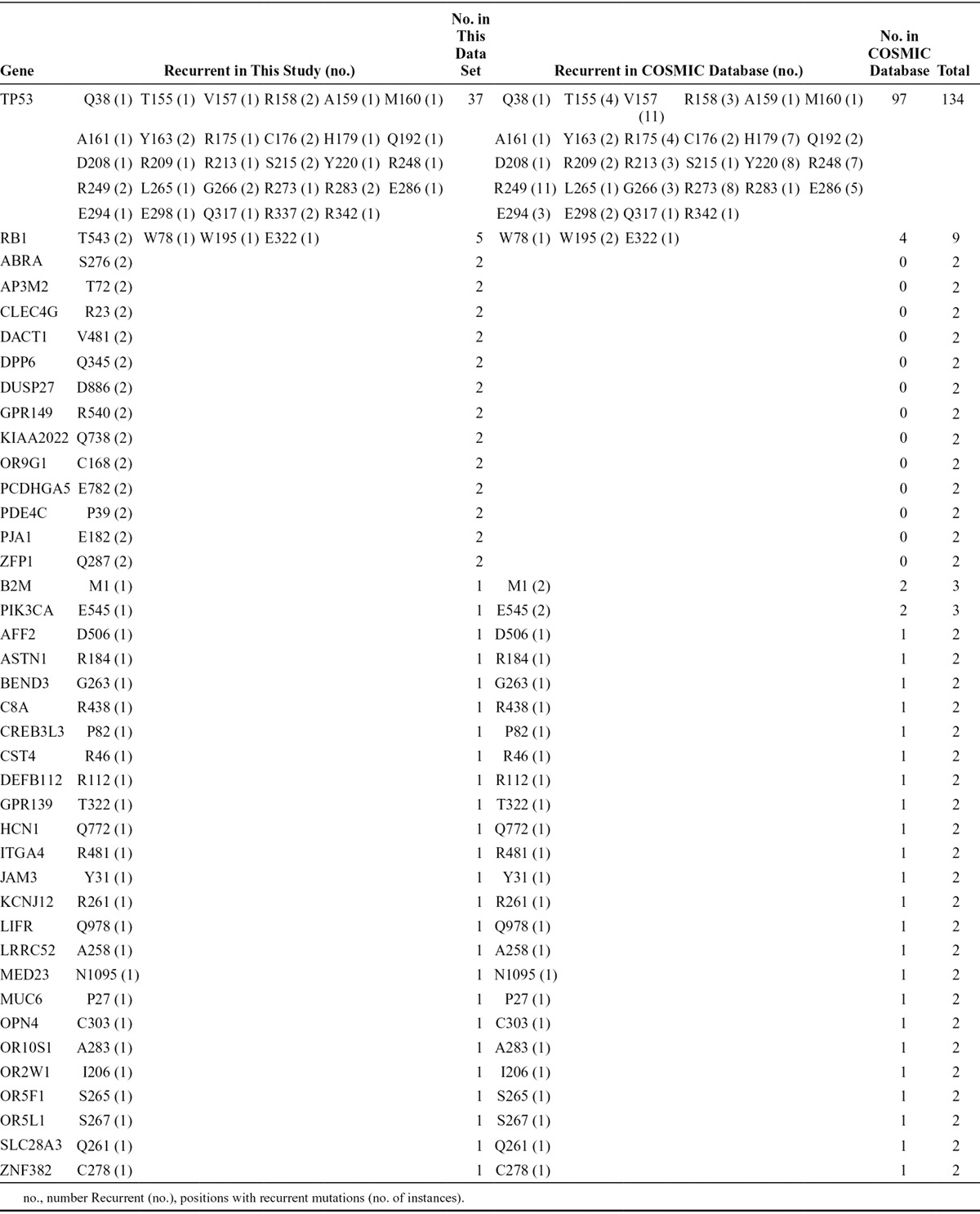
Recurrent Mutations at the Same Position
Forty genes with recurrent somatic mutations at the same position were identified in this study and the COSMIC database (Table 4). TP53, the well-characterized tumor suppressor gene, had 29 different positions that mutated more than or equal to two times (total recurrent samples, 134). RB1 had four different positions that mutated two or three times. The remaining 38 genes had one position that mutated two or three times. Well-established activating mutations in PIK3CA, the catalytic subunit of phosphoinositide-3 kinase (E545), were also detected in another SCLC cohort.
TABLE 4.
The Recurrent Mutations Detected at the Same Position in This Study and the COSMIC Database
PI3K/AKT/mTOR Pathway Alteration
Because of the large number of somatic point mutations and focal amplifications found in the PI3K/AKT/mTOR signaling pathway (e.g., PIK3CA, PTEN, AKT2, and RICTOR), we focused our investigation on the changes in the PI3K/AKT/mTOR pathway. We observed that the PI3K/AKT/mTOR pathway was altered in 17/47 (36%) of the SCLC tumors (Fig. 1), and all altered genes in the PI3K/AKT/mTOR pathway were mutually exclusive. There was no difference in the clinical characteristics, such as smoking status, gender, and age, between the PI3K/AKT/mTOR pathway-affected group (Group A) and the PI3K/AKT/mTOR pathway-unaffected group (Group B). The frequencies of TP53 and RB1 mutations were identical between Group A and Group B. However, more MYC family genes tended to be amplified in Group B; Group A did not harbor KRAS or BRAF mutations, and most patients in Group A did not have MAPK/ERK pathway changes.
The correlation between the PI3K/AKT/mTOR pathway changes and RTKs is shown in Supplemental Figure 6 (Supplemental Digital Content 2, http://links.lww.com/JTO/A626). The changes in various targetable RTK genes were detected, such as ERBB2 (n = 4), KIT (n = 2), PDGFRA (n = 3), PDGFRB (n = 2), KDR (n = 3), MET (n = 1), ROS1 (n = 8), and RET (n = 5). However, none of these genes showed a recurrent mutation at the same point in this data set or the COSMIC database. The PI3K/AKT/mTOR pathway status did not correlate with the RTK changes.
Drug Sensitivity
To further investigate whether the PI3K/AKT/mTOR pathway could be a feasible therapeutic target in SCLC, we tested the in vitro drug sensitivity of the SCLC cell lines using the clinically developed compounds targeting this pathway (Fig. 2 and Supplemental Figure 7, Supplemental Digital Content 2, http://links.lww.com/JTO/A626). We selected three SCLC cell lines with genetic alterations in the PI3K/AKT/mTOR pathway: H446 (PTEN-loss, MYC-amplified), H1048 (PIK3CA mutation), and H1694 (AKT3-amplified). We also examined H82 (MYC-amplified) and H209, which do not display activation of the PI3K/AKT/mTOR pathway. Using these cell lines, we assessed the efficacy of four compounds that inhibit the PI3K/AKT/mTOR pathway and are in on-going phase I/II trials: BEZ235 (PI3K and mTOR inhibitor), BKM120 (PI3K inhibitor), INK128 (mTOR inhibitor), and MK2206 (AKT inhibitor), as well as one cytotoxic agent, cisplatin. None of the cell lines showed apparent cytotoxicity in response to doses up to 1 μM cisplatin. Conversely, all PI3K/AKT/mTOR inhibitors significantly impaired the proliferation of the SCLC cell lines. H1048, which harbors a PIK3CA mutation (H1047R), was the most sensitive to all of the PI3K/AKT/mTOR inhibitors, with IC50 values of 3.8, 5.4, 99.9, and 195.4 nM for INK128, BEZ235, MK2206, and BKM120, respectively. BEZ235 was the most effective compound to specifically inhibit H1048 cell growth (IC50 = 5.4), with an IC50 value greater than 10-fold lower than that of H82 (IC50 = 58.3 nM) and fivefold lower than that of H209 (IC50 = 29.7 nM). In contrast, H446 (IC50 = 33.3 nM) and H1694 cells (IC50 = 52.5 nM) were relatively resistant to BEZ235 treatment.
FIGURE 2.
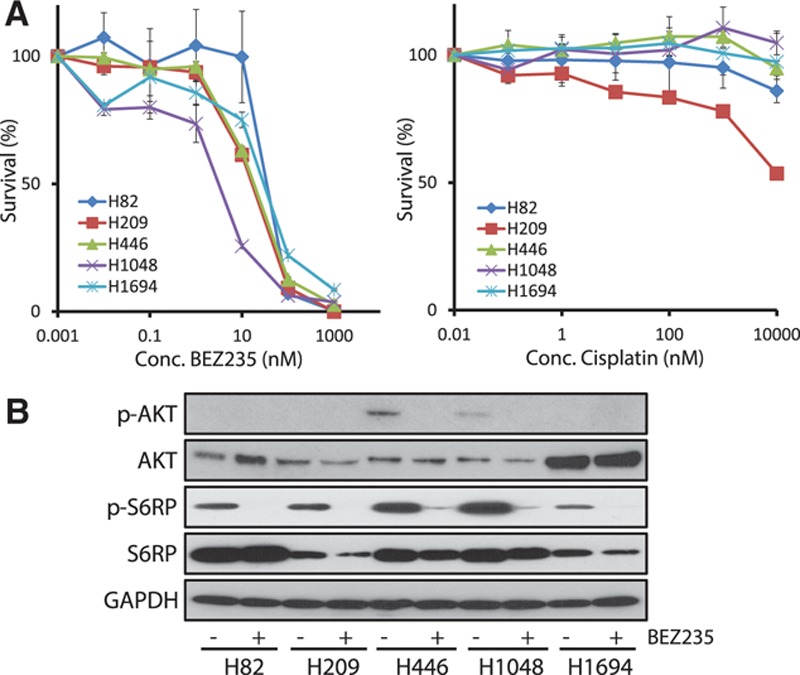
(A) The concentration–response cell survival curves of SCLC cell lines with or without genetic alteration in the PI3K/AKT/mTOR pathway in response to BEZ235 (nM) and Cisplatin (nM). The PIK3CA mutation positive cell line, H1048, is relatively sensitive to BEZ235. The H82 and H209 cell lines are negative controls. (B) Western blotting was used to investigate the impact of BEZ235 on AKT phosphorylation and S6RP phosphorylation in the SCLC cells. AKT was activated in H446 and H1048 cells, and it was inhibited after being treated with 10 nM BEZ235. AKT was amplified but not constitutively phosphorylated in the H1694 cells. AKT phosphorylation was not detected in the negative control cell lines, H82 and H209. With regard to factors located downstream of mTOR, S6RP was phosphorylated in all five SCLC cell lines. Especially, the phosphorylation level was high in AKT-activated H446 and H1048 cells. BEZ235 significantly reduced the phosphorylation of S6RP in all the cells.
The impact of BEZ235 on AKT phosphorylation in SCLC cells was investigated using Western blot analysis. AKT was activated in the H446 and H1048 cells under these culture conditions, and it was effectively inhibited after being treated with 10 nM BEZ235. Conversely, constitutive phosphorylation of AKT was not observed in H1694 cells, even when pan-AKT was over-expressed. In addition, AKT phosphorylation was not detected in the H82 and H209 cells. Regarding factors located downstream of mTOR, S6RP was phosphorylated in all five SCLC cell lines. Especially, the phosphorylation level was high in AKT-activated H446 and H1048 cells. BEZ235 significantly reduced the phosphorylation of S6RP in all the cells.
To evaluate the contribution of PI3K/AKT/mTOR signaling to SCLC cell proliferation, we used RNA interference (RNAi) to down-regulate the expression of PIK3CA in H1048 cells. The transient silencing of PIK3CA impaired the phosphorylation of AKT and S6RP (Supplemental Figure 8, Supplemental Digital Content 2, http://links.lww.com/JTO/A626). In addition, PIK3CA silencing induced a decrease in the proliferation of H1048 cells.
DISCUSSION
We performed an integrative genomic analysis of SCLC in Japanese patients. The SCLC tumors had a significantly high mutation rate. An analysis of the base-level transitions and transversions showed that G-to-T transversions were predominant (Supplemental Figure 2, Supplemental Digital Content 2, http://links.lww.com/JTO/A626), which was consistent with the demonstrated effects of tobacco smoke carcinogens on DNA.8,17 A high prevalence of inactivating mutations in TP53 and RB1 and recently reported candidate driver genes, including the mutations of histone modifiers (CREBBP7 and EP3007), were recurrently observed along with the amplification of MYC family members.7,14,18,19 These data indicate that the genomic landscape of SCLC is equivalent between Asian and Caucasian populations.7,8,17,18
SCLC is characterized by aggressive growth and a poor prognosis, and no single molecular targeted drug has shown any clinical efficacy over an extended period. A number of inhibitors targeting changes in RTKs are currently used in clinical use. Alterations in well-known, targetable RTK genes, such as ERBB2, KIT, PDGFRA, PDGFRB, KDR, MET, ROS1, and RET, were detected in this study. However, these alterations did not overlap with previously reported activating mutations.
The PI3K/AKT/mTOR signaling pathway is involved in the survival, proliferation, and migration of SCLC cell lines.13 We confirmed the activation of the PI3K pathway in the SCLC-derived cell lines. AKT protein overexpression was observed in the AKT3-amplified H1694 cells, and phosphorylated-AKT and S6RP were increased in the PTEN-lacking H446 cells and PIK3CA-mutated H1048 cells. In addition, the significant decrease in the proliferation of H1048 cells induced by PIK3CA silencing suggested that the proliferation of these cells was strongly dependent on the PI3K/AKT/mTOR pathway (Supplemental Figure 8, Supplemental Digital Content 2, http://links.lww.com/JTO/A626). Consistently, genetic changes in the PI3K/AKT/mTOR pathway were detected in approximately 40% of our clinical samples. In addition to high penetrance, these alterations occurred in a mutually exclusive manner. A similar trend was observed in another Japanese cohort of primary SCLC (Supplemental Table 7, Supplemental Digital Content 3, http://links.lww.com/JTO/A627). In addition to SCLC, a significant exclusion pattern among PI3K pathway molecules was observed in the systematic analysis of breast cancer genomes.20 Together, these data suggest indispensable roles for this pathway in tumorigenesis.
Two specific inhibitors of mTORC1, everolimus10 and temsirolimus,11 were tested against SCLC in a Phase II study. However, single-agent antitumor activity was limited in unselected patients; the response rate in these studies was less than 10%. To improve the response to these inhibitors, the addition of PI3K inhibition has been suggested. The dual inhibition of PI3K and mTOR might be advantageous over single inhibition by suppressing a S6K feedback loop that leads to the pathway reactivation.21 Based on this idea, an on-going phase I study of the PI3K and mTORC1/2 dual inhibitor, BEZ235, was designed for the patients with advanced solid tumors harboring PIK3CA or PTEN alteration (NCT01195376). In this study, we showed that the survival of the cisplatin-resistant SCLC cell lines was well suppressed by BEZ235, accompanied by the suppression of S6RP phosphorylation. Notably, the effect was most significant against H1048 cells, which harbor a PIK3CA-activating mutation.
However, we found that not all SCLC cell lines harboring PI3K/AKT/mTOR pathway alterations exhibited a similar sensitivity to BEZ235. Although AKT phosphorylation was significantly inhibited by BEZ235 in both the H446 cells and H1048 cells, the sensitivity of the H446 cells was less than that of the H1048 cells. MYC gene amplification reportedly evades PI3K-targeted therapy.22 MYC amplification was demonstrated in H446 cells,23,24 and this co-alteration could be one cause of the observed low sensitivity. Thus, to determine the most beneficial concentrations for patients, both direct target molecules and other interfering signaling pathways should be simultaneously assessed. In this study, no surgically resected tumors harbored co-alterations of the PI3K/AKT/mTOR pathway and MYC gene amplification. However, the sample size of this study and other published systemic analyses remained small, and many of the samples were obtained from relatively early-stage tumors. We should expand the sample size and further analyze samples of advanced tumors using biopsy, necropsy, and autopsy specimens to clarify the coexistence of oncogenic alterations in SCLC.
Although further large-scale validation studies are needed, our data suggest that evaluating the genetic status of molecules that modify the PI3K/AKT/mTOR signaling pathway, such as MYC family and MAPK pathway molecules, is essential to select patients with potential sensitivity to PI3K/AKT/mTOR inhibitors. In other words, enriching the study population by performing the integrative genomic analysis is essential when performing phase studies of PI3K/AKT/mTOR inhibitors in SCLC.
In conclusion, the SCLC genome possesses distinguishable genetic features in the PI3K/AKT/mTOR pathway. Genetic alterations in the PI3K/AKT/mTOR pathway were noted as a top therapeutic priority in SCLC. In addition to surgically resected samples, advanced tumors should be examined for comprehensive genomic analysis.
ACKNOWLEDGMENTS
This study was performed as a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct), Ministry of Education, Culture, Sports, Science and Technology of Japan, and it was supported by JSPS KAKENHI Grant Number 24300346 and National Cancer Center Research and Development Fund (23-A-8, 15). The authors thank Ms. Fumiko Koh, Drs. Masao Yamaguchi, Hideki Okada, Keiju Aokage, Tomoyuki Hishida, Junji Yoshida, Keisuke Kirita, Eri Sugiyama, Yoshitaka Zenke, Tatsuya Yoshida, Yuji Matsumoto, and Yuuki Matsumura for their support and comments.
Footnotes
This work was partly presented at the Annual Meeting of the American Society of Clinical Oncology, May 31–June 4, 2013, Chicago, IL. This study was performed as a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct), Ministry of Education, Culture, Sports, Science and Technology of Japan, and it was supported by JSPS KAKENHI Grant Number 24300346, 26870876 and National Cancer Center Research and Development Fund (23-A-8, 15).
Disclosures: The authors declare no conflicts of interest.
REFERENCES
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.William WN, Jr, Glisson BS. Novel strategies for the treatment of small-cell lung carcinoma. Nat Rev Clin Oncol. 2011;8:611–619. doi: 10.1038/nrclinonc.2011.90. [DOI] [PubMed] [Google Scholar]
- 3.Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network. Comprehensive genomic characterisation of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peifer M, Fernández-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudin CM, Durinck S, Stawiski EW, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pietanza MC, Ladanyi M. Bringing the genomic landscape of small-cell lung cancer into focus. Nat Genet. 2012;44:1074–1075. doi: 10.1038/ng.2415. [DOI] [PubMed] [Google Scholar]
- 10.Tarhini A, Kotsakis A, Gooding W, et al. Phase II study of everolimus (RAD001) in previously treated small cell lung cancer. Clin Cancer Res. 2010;16:5900–5907. doi: 10.1158/1078-0432.CCR-10-0802. [DOI] [PubMed] [Google Scholar]
- 11.Pandya KJ, Dahlberg S, Hidalgo M, et al. A randomised, phase II trial of two dose levels of temsirolimus (CCI-779) in patients with extensive-stage small-cell lung cancer who have responding or stable disease after induction chemotherapy: a trial of the Eastern Cooperative Oncology Group (E1500). J Thorac Oncol. 2007;2:1036–1041. doi: 10.1097/JTO.0b013e318155a439. [DOI] [PubMed] [Google Scholar]
- 12.Voortman J, Lee JH, Killian JK, et al. Array comparative genomic hybridization-based characterization of genetic alterations in pulmonary neuroendocrine tumors. Proc Natl Acad Sci U S A. 2010;107:13040–13045. doi: 10.1073/pnas.1008132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wojtalla A, Fischer B, Kotelevets N, et al. Targeting the phosphoinositide 3-kinase p110-α isoform impairs cell proliferation, survival, and tumor growth in small cell lung cancer. Clin Cancer Res. 2013;19:96–105. doi: 10.1158/1078-0432.CCR-12-1138. [DOI] [PubMed] [Google Scholar]
- 14.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki A, Mimaki S, Yamane Y, et al. Identification and characterization of cancer mutations in Japanese lung adenocarcinoma without sequencing of normal tissue counterparts. PLoS One. 2013;8:e73484. doi: 10.1371/journal.pone.0073484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arriola E, Cañadas I, Arumí M, Rojo F, Rovira A, Albanell J. Genetic changes in small cell lung carcinoma. Clin Transl Oncol. 2008;10:189–197. doi: 10.1007/s12094-008-0181-1. [DOI] [PubMed] [Google Scholar]
- 19.Iwakawa R, Takenaka M, Kohno T, et al. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes Cancer. 2013;52:802–816. doi: 10.1002/gcc.22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gadgeel SM, Wozniak A. Preclinical rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer. Clin Lung Cancer. 2013;14:322–332. doi: 10.1016/j.cllc.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci U S A. 2011;108:E699–E708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brennan J, O’Connor T, Makuch RW, et al. myc family DNA amplification in 107 tumors and tumor cell lines from patients with small cell lung cancer treated with different combination chemotherapy regimens. Cancer Res. 1991;51:1708–1712. [PubMed] [Google Scholar]
- 24.Sos ML, Dietlein F, Peifer M, et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A. 2012;109:17034–17039. doi: 10.1073/pnas.1207310109. [DOI] [PMC free article] [PubMed] [Google Scholar]